

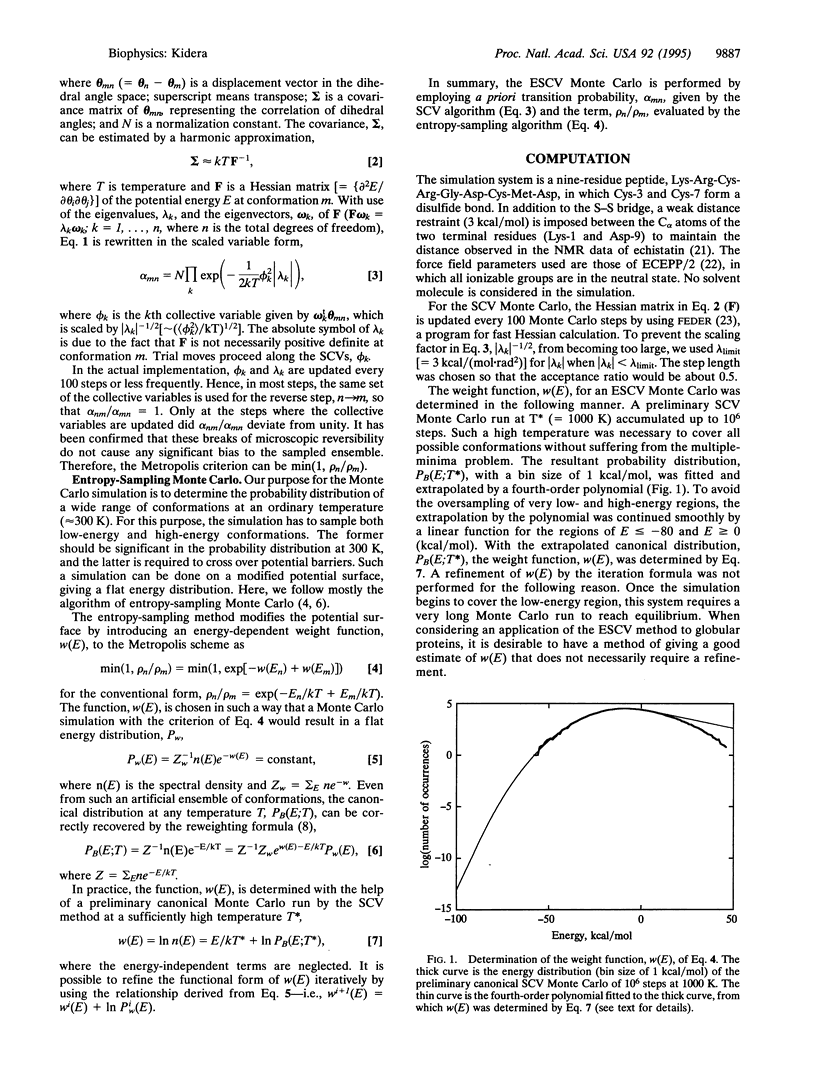
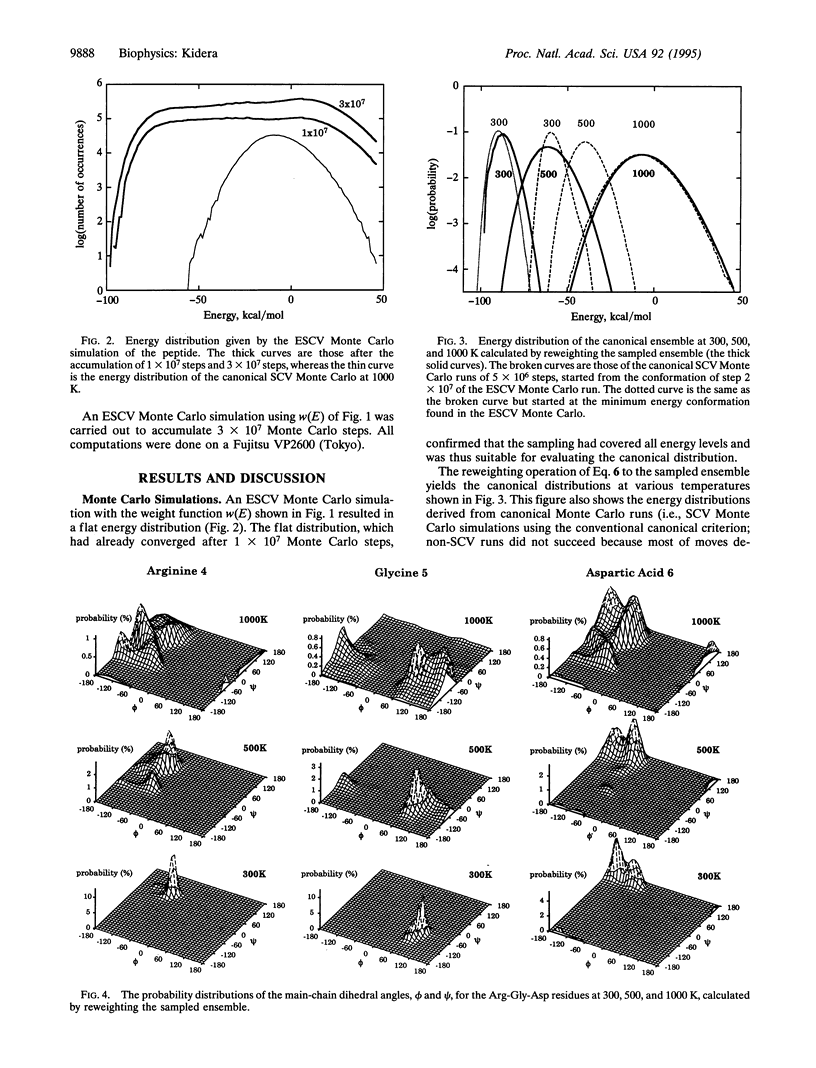
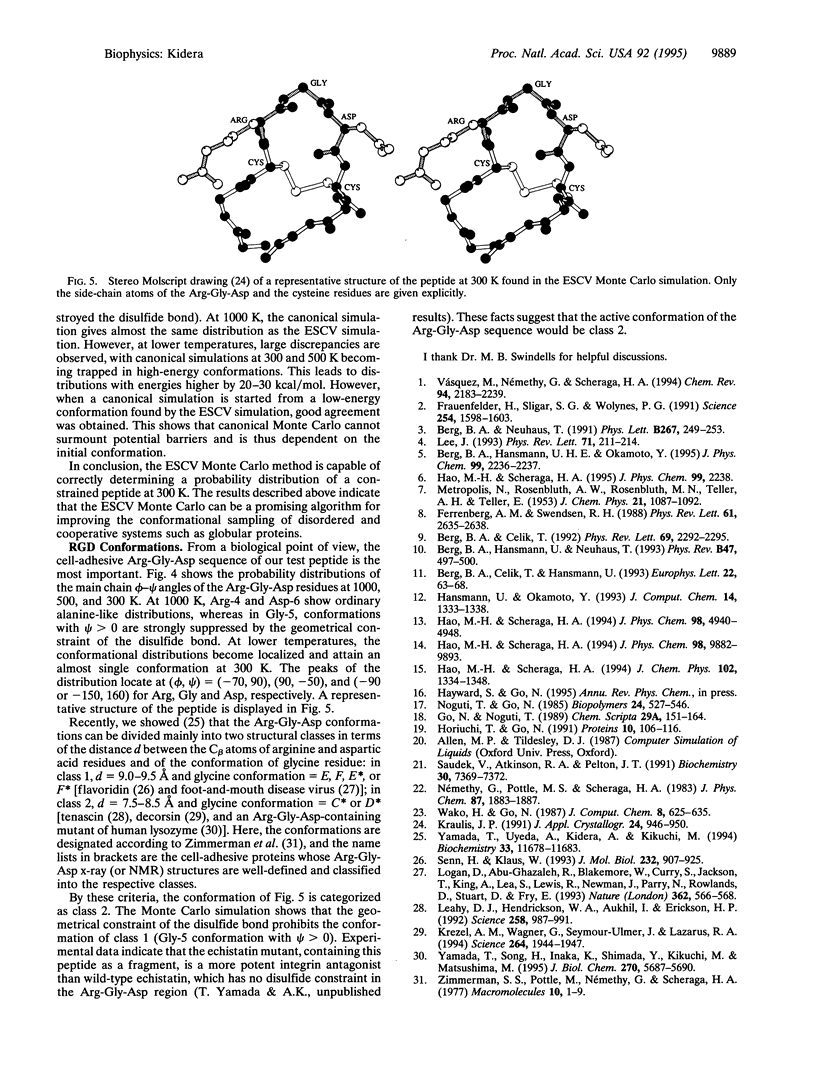
Abstract
A Monte Carlo simulation method for globular proteins, called extended-scaled-collective-variable (ESCV) Monte Carlo, is proposed. This method combines two Monte Carlo algorithms known as entropy-sampling and scaled-collective-variable algorithms. Entropy-sampling Monte Carlo is able to sample a large configurational space even in a disordered system that has a large number of potential barriers. In contrast, scaled-collective-variable Monte Carlo provides an efficient sampling for a system whose dynamics is highly cooperative. Because a globular protein is a disordered system whose dynamics is characterized by collective motions, a combination of these two algorithms could provide an optimal Monte Carlo simulation for a globular protein. As a test case, we have carried out an ESCV Monte Carlo simulation for a cell adhesive Arg-Gly-Asp-containing peptide, Lys-Arg-Cys-Arg-Gly-Asp-Cys-Met-Asp, and determined the conformational distribution at 300 K. The peptide contains a disulfide bridge between the two cysteine residues. This bond mimics the strong geometrical constraints that result from a protein's globular nature and give rise to highly cooperative dynamics. Computation results show that the ESCV Monte Carlo was not trapped at any local minimum and that the canonical distribution was correctly determined.
Full text
PDF

Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Berg BA, Celik T. New approach to spin-glass simulations. Phys Rev Lett. 1992 Oct 12;69(15):2292–2295. doi: 10.1103/PhysRevLett.69.2292. [DOI] [PubMed] [Google Scholar]
- Berg BA, Hansmann U, Neuhaus T. Simulation of an ensemble with varying magnetic field: A numerical determination of the order-order interface tension in the D=2 Ising model. Phys Rev B Condens Matter. 1993 Jan 1;47(1):497–500. doi: 10.1103/physrevb.47.497. [DOI] [PubMed] [Google Scholar]
- Ferrenberg AM, Swendsen RH. New Monte Carlo technique for studying phase transitions. Phys Rev Lett. 1988 Dec 5;61(23):2635–2638. doi: 10.1103/PhysRevLett.61.2635. [DOI] [PubMed] [Google Scholar]
- Frauenfelder H., Sligar S. G., Wolynes P. G. The energy landscapes and motions of proteins. Science. 1991 Dec 13;254(5038):1598–1603. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- Horiuchi T., Go N. Projection of Monte Carlo and molecular dynamics trajectories onto the normal mode axes: human lysozyme. Proteins. 1991;10(2):106–116. doi: 10.1002/prot.340100204. [DOI] [PubMed] [Google Scholar]
- Krezel A. M., Wagner G., Seymour-Ulmer J., Lazarus R. A. Structure of the RGD protein decorsin: conserved motif and distinct function in leech proteins that affect blood clotting. Science. 1994 Jun 24;264(5167):1944–1947. doi: 10.1126/science.8009227. [DOI] [PubMed] [Google Scholar]
- Leahy D. J., Hendrickson W. A., Aukhil I., Erickson H. P. Structure of a fibronectin type III domain from tenascin phased by MAD analysis of the selenomethionyl protein. Science. 1992 Nov 6;258(5084):987–991. doi: 10.1126/science.1279805. [DOI] [PubMed] [Google Scholar]
- Logan D., Abu-Ghazaleh R., Blakemore W., Curry S., Jackson T., King A., Lea S., Lewis R., Newman J., Parry N. Structure of a major immunogenic site on foot-and-mouth disease virus. Nature. 1993 Apr 8;362(6420):566–568. doi: 10.1038/362566a0. [DOI] [PubMed] [Google Scholar]
- Noguti T., Go N. Efficient Monte Carlo method for simulation of fluctuating conformations of native proteins. Biopolymers. 1985 Mar;24(3):527–546. doi: 10.1002/bip.360240308. [DOI] [PubMed] [Google Scholar]
- Saudek V., Atkinson R. A., Pelton J. T. Three-dimensional structure of echistatin, the smallest active RGD protein. Biochemistry. 1991 Jul 30;30(30):7369–7372. doi: 10.1021/bi00244a003. [DOI] [PubMed] [Google Scholar]
- Senn H., Klaus W. The nuclear magnetic resonance solution structure of flavoridin, an antagonist of the platelet GP IIb-IIIa receptor. J Mol Biol. 1993 Aug 5;232(3):907–925. doi: 10.1006/jmbi.1993.1439. [DOI] [PubMed] [Google Scholar]
- Yamada T., Song H., Inaka K., Shimada Y., Kikuchi M., Matsushima M. Structure of a conformationally constrained Arg-Gly-Asp sequence inserted into human lysozyme. J Biol Chem. 1995 Mar 17;270(11):5687–5690. doi: 10.1074/jbc.270.11.5687. [DOI] [PubMed] [Google Scholar]
- Yamada T., Uyeda A., Kidera A., Kikuchi M. Functional analysis and modeling of a conformationally constrained Arg-Gly-Asp sequence inserted into human lysozyme. Biochemistry. 1994 Oct 4;33(39):11678–11683. doi: 10.1021/bi00205a002. [DOI] [PubMed] [Google Scholar]
- Zimmerman S. S., Pottle M. S., Némethy G., Scheraga H. A. Conformational analysis of the 20 naturally occurring amino acid residues using ECEPP. Macromolecules. 1977 Jan-Feb;10(1):1–9. doi: 10.1021/ma60055a001. [DOI] [PubMed] [Google Scholar]