

Abstract
CASP2/caspase 2 plays a role in aging, neurodegeneration, and cancer. The contributions of CASP2 have been attributed to its regulatory role in apoptotic and nonapoptotic processes including the cell cycle, DNA repair, lipid biosynthesis, and regulation of oxidant levels in the cells. Previously, our lab demonstrated CASP2-mediated modulation of autophagy during oxidative stress. Here we report the novel finding that CASP2 is an endogenous repressor of autophagy. Knockout or knockdown of CASP2 resulted in upregulation of autophagy in a variety of cell types and tissues. Reinsertion of Caspase-2 gene (Casp2) in mouse embryonic fibroblast (MEFs) lacking Casp2 (casp2−/−) suppresses autophagy, suggesting its role as a negative regulator of autophagy. Loss of CASP2-mediated autophagy involved AMP-activated protein kinase, mechanistic target of rapamycin, mitogen-activated protein kinase, and autophagy-related proteins, indicating the involvement of the canonical pathway of autophagy. The present study also demonstrates an important role for loss of CASP2-induced enhanced reactive oxygen species production as an upstream event in autophagy induction. Additionally, in response to a variety of stressors that induce CASP2-mediated apoptosis, casp2−/− cells demonstrate a further upregulation of autophagy compared with wild-type MEFs, and upregulated autophagy provides a survival advantage. In conclusion, we document a novel role for CASP2 as a negative regulator of autophagy, which may provide important insight into the role of CASP2 in various processes including aging, neurodegeneration, and cancer.
Keywords: apoptosis, autophagy, autophagy regulators, caspase 2, caspases, reactive oxygen species
Introduction
Macroautophagy (hereafter referred to as autophagy) is a stress-induced adaptation process and is required for the long-term survival of eukaryotic cells. During autophagic process, double-membrane structures sequester portions of cytoplasm, and are known as autophagosomes; these autophagosomes fuses with lysosomes leading to formation of autolysosomes, and this results in bulk degradation. Autophagy leads to the elimination of damaged or harmful cellular components, out of which some are recycled to maintain nutrient and energy homeostasis. Thus, autophagy is important in the maintenance of cells in a healthy state.1-3 In addition, autophagy also plays a role in the regulation of a variety of cellular processes that include cell growth, development, and homeostasis.4 It is further being recognized as a participant in programmed cell death.5-7 Growing evidence now links deregulated autophagy with many human pathophysiologies including cancer, neurodegeneration, and age-associated disorders.8-10 Therefore, the identification of regulatory mechanisms of autophagy machinery continues to be an important and evolving field.
Autophagy is a highly conserved process and its regulation is complex.11 Over the past decade, various proteins have been characterized as positive or negative regulators of autophagy.12 Pathways modulated by molecules such as phosphatidylinositol 3-kinase, mechanistic target of rapamycin (MTOR), GTPases, and calcium, play important roles in regulating autophagy. Autophagy is suppressed by MTOR activation (mediated by AKT and MAPK [mitogen-activated protein kinase] signaling), whereas negative regulators of MTOR like AMPK (AMP-activated protein kinase) and the TP53 signaling pathway promote autophagy.13,14 Furthermore, the phosphatidylinositol 3-kinase complex that contains autophagy-related (ATG) proteins or UVRAG (UV radiation-resistance associated), is required for the induction of autophagy. Also, important roles for the proteins encoded by the autophagy-related (Atg) genes (essential proteins of the autophagic machinery) have been well characterized in the regulation of autophagy.12,15
Interestingly, a growing amount of evidence also suggests that autophagy may be controlled by apoptosis, and extensive crosstalk exists between autophagy and apoptosis.16,17 For instance, regulators of apoptosis, that include BCL2 family members (BCL2, BCLXL,18,19 and BAX20,21) can regulate autophagy; proteins involved in autophagy, such as ATG5, BECN1/Beclin 1 and ATG7, can also modulate apoptosis.7,22,23 Furthermore, CASP (caspase) proteins, cysteine-rich proteases that play a major role in the apoptotic process, also regulate autophagy.24 For example, loss of CASP8 results in autophagic cell death,24 whereas CASP9 acts as a positive regulator of autophagy.25 Recently, studies from our lab identified CASP2 in the modulation of the autophagic response of neurons during mitochondrial oxidative stress.26 Interestingly, our data also indicated a role for CASP2 in the regulation of autophagy in the absence of an external stressor. These observations led us to ask whether CASP2 can modulate autophagy under normal as well as stress conditions and, if so, dissect out molecular mechanisms involved in this modulation.
CASP2, the most conserved member of the CASP family, is best known for its role as a regulator of apoptosis.27,28 CASP2 initiates apoptosis during various stress conditions that include oxidative stress,26 cytoskeleton disruption,29 and heat shock (HS).30 In addition to its role in apoptosis, evidence suggests a potential role for CASP2 in nonapoptotic processes, including cell cycle regulation,31 DNA repair,32 lipid sensing,33 tumor suppressor,34 metabolic regulation,35 and regulation of oxidant levels in cells.36 Previously, our lab demonstrated that the loss of CASP2 results in accumulation of oxidatively damaged proteins.37 These observations have recently been confirmed by Kumar and colleagues, where they also define the mechanism of reactive oxygen species (ROS) generation resulting from loss of CASP2. Their study identifies that the loss of CASP2 results in modulation of FOXO3 (forkhead box O3) signaling thereby leading to downregulation of the antioxidant enzyme SOD2 (superoxide dismutase 2, mitochondrial) and an increase in ROS (superoxide) levels.36 The present study identifies a novel nonapoptotic role for CASP2, as a negative regulator of autophagy. CASP2-mediated regulation of ROS played an important role in autophagy regulation. This finding contributes to the further understanding of a complex homeostatic regulation whereby CASP2 and autophagy are interconnected and may play a role in determining the outcome of various normal and pathological processes.
Results
Loss of CASP2 upregulates endogenous levels of autophagy under normal conditions
Our previous study demonstrated that CASP2 modulates the autophagic response against mitochondrial oxidative stress in primary neurons.26 Of note, we also observed that even the basal level of autophagy was higher in the primary neurons cultured from Casp2 knockout (casp2−/−) mice compared with wild type (WT). These data raised an interesting possibility that CASP2 may play a role as a negative regulator of autophagy even under normal (i.e., in the absence of an external stressor) conditions. Therefore in the present study we investigated a role for CASP2 as a modulator of autophagy under normal conditions (in the absence of external stressors) and the underlying mechanism involved. Since mouse embryonic fibroblasts (MEFs) are a good model system to perform mechanistic studies, we used cultures of MEFs derived in our lab from WT and casp2−/− mice and examined endogenous (basal) levels of autophagy. Autophagy was monitored following different approaches (as recommended by Klionsky et al).38 First, the conversion of microtubule-associated protein 1 light chain 3 (LC3)-I to LC3-II, a marker for autophagosome formation and the expression level of the SQSTM1/p62 protein, which is degraded by autolysosomes and serves as a marker for autophagic flux,38 were determined by western blotting. Cells lacking Casp2 maintained significantly higher levels of LC3-II and lower levels of SQSTM1 than the WT cells (Fig. 1A, ratio between LC3-II and SQSTM1 is demonstrated in Fig. 1B), suggesting that the loss of CASP2 led to upregulation of autophagy. Furthermore, no significant change was observed in the Sqstm1 mRNA level between WT and casp2−/−, suggesting that the downregulation of SQSTM1 observed at the protein level was not due to downregulation of Sqstm1 mRNA (Fig. 1C and D).

Figure 1. Loss of CASP2 upregulates endogenous levels of autophagy in unstressed cells. (A) Western blot analysis of LC3 (an increase in LC3-II indicates an increase in autophagosome formation) and SQSTM1 to determine autophagic flux in cell lysates prepared from 4 different batches of WT and casp2−/− MEFs (loaded in separate lanes 1 to 4) cultured in complete medium in the absence of stressors. The same blots were reprobed with tubulin, α (TUBA) as a loading control. Shown are the representative blots. The experiment was repeated at least 3 times. (B) Densitometry was performed to determine the level of expression using ImageJ analysis. A ratio of expression levels of LC3-II/SQSTM1 as a % of tubulin, α as determined by densitometric analysis demonstrates autophagic flux. Error bar represents ± SEM. Statistical significance was determined by the Student t test. n = 4, **P ≤ 0.01, *P ≤ 0.05. (C and D) The Sqstm1 mRNA levels were determined in the cell lysates prepared from 3 different batches of WT and casp2−/− MEFs (loaded in separate lanes 1 to 3); Gapdh served as the experimental control. (D) Densitometry was performed to determine the level of expression as % of the control using ImageJ. Error bar represents ± SEM. Statistical significance was determined by the Student t test. n = 3, **P ≤ 0.01, *P ≤ 0.05. (E–H) Cells (WT and casp2−/− MEFs) were treated with PepA (1 µM) and EST (10 µM) (inhibits lysosomal proteases) for the indicated times to determine autophagic flux. (E and F) Cells were stained with LC3 antibody (green), LysoTracker Red (red, to detect lysosomes) and Hoechst 33258 (blue, to detect nuclei) and analyzed by confocal microscopy. (E) Confocal microscopy analysis of LC3 puncta (determines autophagosomes) and colocalization with LysoTracker Red (stains lysosomes, yellowing due to colocalization of red and green) (determines autolysosomes) in WT and casp2−/− MEFs. Shown are the representative confocal images (60x with 2 times optical zoom). (F) Quantification of the confocal images for the cells carrying more than 10 LC3 puncta per cell (both autophagosomes and autolysosomes) as a % of the total number of cells counted. At least 150 to 200 cells were counted for each set; n = 3. Error bar represents ± SEM. Statistical significance was determined by performing 2-way ANOVA. **P ≤ 0.01, *P ≤ 0.05. The experiment was repeated at least 3 times. (G) Western blotting for LC3 to detect autophagic flux as described earlier. LC3-II/LC3-I ratios were determined by probing the blot with an antibody specific to LC3, GAPDH was used as loading control. Shown are the representative blots. (H) Densitometric analysis to determine expression levels indicates LC3-II/LC3-I ratio as % of loading control (GAPDH). Error bar represents ± SEM. Statistical significance was determined by the Student t test. **P ≤ 0.01, *P ≤ 0.05. (I) Long-lived protein degradation assay to determine autophagic flux. WT and casp2−/− MEFs were untreated, treated with EST+PepA or starved (medium was replaced by HBSS containing calcium and magnesium). Long-lived protein degradation % of control was calculated as described in the Materials and Methods section. Error bar represents ± SEM. Statistical significance was determined by performing 2-way ANOVA. n = 4, **P ≤ 0.01, *P ≤ 0.05.
Next, we investigated whether elevation of LC3-II in the absence of CASP2 was a result of increased initiation and progression of autophagy or defective autophagosome-lysosome fusion or degradation.39 The cells were treated with lysosomal protease inhibitors pepstatin A (PepA) and EST (to prevent degradation within autolysosomes) at different time points and autophagy was assessed by monitoring colocalization of LC3 with the lysosomal marker (LysoTracker Red) by confocal microscopy as well as western blotting for LC3 (Fig. 1E–H). Lysosomal protease inhibitors further enhanced the loss of CASP2-triggered accumulation of autophagosomes (an increase in number and size of LC3 puncta [green]), as well as an accumulation of autolysosomes (colocalization of LC3 with lysosomes) as compared with the WT cells (Fig. 1E). Interestingly, the size of accumulated autophagosomes and autolysosomes was also exacerbated in casp2−/− MEFs compared with the WT cells (Fig. 1E). Similarly, western blotting for LC3 demonstrated that the lysosomal protease inhibitors further increased the loss of Casp2-triggered induction of LC3-II in a time-dependent manner (Fig. 1G and H). These findings were further confirmed by expressing a tandem mCherry-GFP-tagged LC3 chimera in WT and casp2−/− MEFs, which enabled simultaneous quantification of autophagosome induction (GFP + mCherry) LC3 puncta and autolysosome maturation (mCherry single-positive puncta due to the sensitivity of GFP to acidic pH). Significantly higher numbers of both autophagosomes and autolysosomes were observed in casp2−/− MEFs compared with the WT in the presence of LY294002, an early-stage autophagy inhibitor (data not shown).
We also assessed autophagic flux by measuring the rate of turnover of long-lived proteins that are normally metabolized via autophagy by measuring the release of TCA-soluble [14C]valine from cells. Lysosomal protein degradation was estimated by measuring [14C]valine release from cells treated with and without EST+PepA as well as during starvation, a classical inducer of autophagy (Fig. 1I). In casp2−/− cells, protein degradation was significantly higher than that observed in WT MEFs. Also, after starvation, the casp2−/− cells still exhibited significantly higher activity compared with the WT, whereas in the presence of EST+PepA, protein degradation was significantly inhibited and no significant difference was observed between casp2−/− and WT cells (Fig. 1I).
To further confirm a role for CASP2 in regulation of autophagy, we employed 2 different approaches: i) Casp2 was knocked down in the WT cells using a prevalidated specific short interfering (si) RNA against Casp2, and ii) Casp2 (expressing normal and catalytically inactive mutant at C303), was reinserted in Casp2-deficient cells using a plasmid vector pcDNA3. Western blotting for LC3 was performed to detect autophagy (Fig. 2A–D). Knockdown of Casp2 resulted in an upregulation of autophagy as indicated by an increase in LC3-II levels (Fig. 2A and B). Conversely, reinsertion of CASP2 (both normal and active site mutant: cysteine [C303] is mutated to alanine) inhibited autophagy, confirming a role for CASP2 as a negative regulator of autophagy, which was independent of the presence of the catalytic active site (C303) (Fig. 2C and D).

Figure 2.Casp2 knockdown or reinsertion can modulate autophagy. (A and B) WT MEFs were transiently transfected with prevalidated siRNA against Casp2. (C and D) casp2−/− MEFs were transiently transfected with Casp2 expression vector. (A and C) Western blot demonstrating the expression of CASP2 and autophagy was detected by determining the levels of LC3-II. The same blots were reprobed for determining tubulin, α (TUBA) levels that served as loading control. Shown are the representative blots. (B and D) Densitometric analysis for the level of expression was performed using ImageJ analysis. Expression levels of LC3-II were normalized by tubulin, α and expressed as LC3-II levels, as % of loading control. Each experiment was repeated at least 3 times. Error bar represents ± SEM. Statistical significance was determined by the Student t test. **P ≤ 0.01, *P ≤ 0.05. (E) Fluorescence confocal microscopy analysis mice of cerebellar sections from GFP-LC3 transgenic mice crossed with WT and casp2−/− mice. Images were acquired at 60X magnification. At least 12 to 13 images were analyzed to detect LC3 puncta formation that indicates autophagy. Shown is the percent of cells with more than 8 detectable LC3 puncta per image. Level of significance was determined by performing the Student t test, n = 12 images from 2 different mice. *P < 0.01. (F) Primary culture from astrocytes, neurons, and osteoclasts were established from WT and casp2−/− mice. The protein samples were prepared and western blotting for LC3 was performed to detect autophagy. Western blots for LC3 levels (an increase in LC3-II indicates an increase in autophagy) in casp2−/− cells compared with the WT. The same blots were reprobed for GAPDH that served as the protein loading control.
We next examined the extent of autophagy in tissue sections obtained from crossing green fluorescent protein (GFP)-LC3 mice with WT and casp2−/− mice. As shown in Figure 2E, casp2−/− brain sections (cortex) showed a significantly higher number of cells with GFP-LC3 puncta (green), indicating higher autophagy compared with WT brain sections. Higher autophagy was also observed in other tissues including liver and kidney (data not shown). Furthermore, we also assessed autophagy in primary cultures of different cell types from WT and casp2−/−, and autophagy was monitored by determining the levels of LC3. Higher levels of autophagy were observed in casp2−/− cell types compared with the WT (Fig. 2F). Thus, data from our lab suggest that loss of CASP2 upregulates autophagy in a variety of cell or tissue types. Further detailed analysis on other tissue and cell types is required to determine the generality of our findings.
CASP2 levels are not regulated by autophagy
Since we observed that CASP2 functions as a negative regulator of autophagy, we also examined whether CASP2 levels were modulated by autophagy upregulation. WT MEFs were treated with an autophagy inducer (rapamycin) as well as an early and a late stage inhibitor of autophagy. Western blotting was performed to determine the levels of CASP2 and the same blots were probed with LC3 antibody (to detect autophagy) to validate the effectiveness of the inhibitors or autophagy inducer (Fig. S1). Enhancement of autophagy or inhibition of autophagy at an early or late stage did not modulate the levels of CASP2 (Fig. S1A). In addition, there was no change in the levels of cleaved forms of CASP2 (indicating CASP2 activation) that corroborated our data demonstrating no change in CASP2 activity (determined by a CASP2 activity assay) upon enhancement or inhibition of autophagy (data not shown). Furthermore, immunocytochemistry data also indicated a lack of colocalization between CASP2 with LC3 puncta (determined by Pearson Coefficient) under normal conditions as well as during autophagy induction (Fig. S1B). These results indicated that although CASP2 plays a role in the modulation of autophagy under normal conditions, autophagy does not regulate the levels of CASP2 in the absence of external stressors.
AMPK and MTOR are involved in CASP2-modulated autophagy
Autophagy is controlled by several kinases including MTOR, which suppresses autophagy40 and AMPK, which induces autophagy.41 Western blotting results indicated that in casp2−/− cells, PRKAA/AMPKα was hyperphosphorylated (which indicates AMPK activation), whereas MTOR as well as its substrates RPS6KB/p70S6K and EIF4EBP1/4-EBP1 were hypophosphorylated (which reflects MTOR inhibition) compared with WT MEFs (Fig. 3A and B). Also, siRNA-mediated depletion of PRKAA1/AMPKα1 and PRKAA2/AMPKα2 inhibited autophagy in casp2−/− cells suggesting an involvement of PRKAA in loss of CASP2-mediated autophagy (Fig. 3C). Inhibition of MTOR with rapamycin eradicated the difference in autophagy between the WT and casp2−/− cells as determined by western blotting for LC3 (Fig. 3D). These results suggest that the loss of CASP2-mediated upregulation of autophagy involves an MTOR-AMPK-dependent pathway.

Figure 3. Role of AMPK and MTOR in the CASP2-mediated modulation of autophagy. (A) Western blotting was performed to detect the phosphorylation status of PRKAA, MTOR, RPS6KB, and EIF4EBP1 vs. total (unphosphorylated) protein in WT and casp2−/− MEFs. The same blots were reprobed for GAPDH or tubulin, α (TUBA) that served as loading controls. (B) Densitometric analysis was performed using ImageJ to determine the expression levels of active (phosphorylated) vs. total (unphosphorylated) PRKAA, MTOR, RPS6KB and EIF4EBP1 and the values are expressed as active vs. total and the values were further normalized as percent of loading control. Error bar represents ± SEM. Statistical significance was determined by the Student t test. **, P ≤ 0.01, *, P ≤ 0.05, NS, P ≥ 0.05, the experiment was repeated 3 times. (C) casp2−/− MEFs were transfected with siRNAs specific for Prkaa1 and Prkaa2. The representative western blot demonstrates the efficiency of siRNA-mediated downregulation of PRKAA1 and PRKAA2, as assessed by western blot analysis. Western blotting for LC3 demonstrates the effect of Prkaa1 and Prkaa2 siRNA on loss of CASP2-mediated autophagy. (D) Effect of 500 nM rapamycin (inhibitor of MTOR) on autophagy in WT and casp2−/− MEFs. Autophagy was detected by LC3 (an increase in LC3-II levels). Shown are the representative blots; the experiment was repeated at least 3 times.
To further assess the involvement of the canonical pathway of autophagy,42 we utilized atg5−/− and atg7−/− MEFs with their respective WT controls. Casp2 was selectively silenced using prevalidated siRNAs, followed by western blot analysis for LC3 (I and II) levels to assess autophagy (Fig. 4). Interestingly, loss of CASP2 induced autophagy in WT MEFs but did not induce autophagy in cells lacking ATG5 and ATG7, indicating an involvement of ATG5 and ATG7 in loss of CASP2-induced autophagy (Fig. 4A and B). It is important to mention that siRNA-mediated depletion of Atg5 or Atg7 was not sufficient to inhibit loss of CASP2-mediated autophagy (data not shown), suggesting that even a lower level of ATG5 and ATG7 was sufficient to mediate autophagy induced by loss of CASP2.
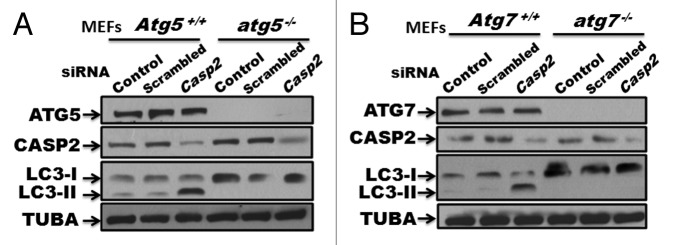
Figure 4. Role of ATG5 and ATG7 in loss of Casp2-induced autophagy. (A) atg5−/− and Atg5+/+ MEFs, and (B) atg7−/− and Atg7+/+ MEFs were treated with prevalidated siRNA against Casp2. Western blotting was performed to validate the efficiency of the siRNA by probing the blots for protein levels of CASP2. Modulation of autophagy was detected by determining LC3 levels (LC3-I and LC3-II). The same blots were probed for ATG5 or ATG7 to confirm the genotype and tubulin, α (TUBA) that served as a loading control. Shown is a representative blot. Each experiment was repeated at least 3 times.
Role of MAPK in CASP2-mediated regulation of autophagy
To identify other mediators involved in autophagy induced by loss of CASP2 further studies were conducted. Regulation of autophagy by the members of the MAPK family, including MAPK1/ERK2 (mitogen-activated protein kinase 1) and MAPK3/ERK1, MAPK11/12/13/14 (p38 β, γ, δ and α, respectively) and MAPK8/9/10 (JNK1/2/3, respectively) has been documented.43-47 Furthermore, previous reports have identified a regulatory association between CASP2 and MAPKs.48-52 Thus, we studied the involvement of MAPK pathways in CASP2-mediated autophagy regulation. First, constitutive levels of activation for MAPK1/3, MAPK14, and MAPK8/9 were determined in WT and casp2−/− cells by performing western blotting for their active (i.e., by detecting their phosphorylation status) vs. total forms (Fig. 5A–F). Western blotting results demonstrated that casp2−/− cells maintained significantly lower levels of active MAPK14 (Fig. 5A) and a significantly higher level of active MAPK1/3 than the WT MEFs (Fig. 5B). No significant differences were observed in the levels of active MAPK8/9 between WT and casp2−/− MEFs (Fig. 5C). To identify the role of these MAPKs in regulation of autophagy due to loss of CASP2, WT and casp2−/− MEFs were treated with SP600125 (MAPK8/9/10 inhibitor), SB203580 (MAPK14 inhibitor) and U0126 (an inhibitor of MAP2K1/2 [MEK1/2], and also a MAPK1/3 inhibitor) and autophagy was assessed. Western blotting for LC3 was performed to detect changes in autophagy (Fig. 5D). There was no significant effect of MAPK8/9 inhibition on loss of CASP2-induced autophagy. Whereas, exposure of MEFs to U0126 reduced the total levels of LC3 protein and LC3-I to LC3-II conversion in casp2−/− MEFs; the levels of LC3-I and LC3-II were brought back to the levels observed in the WT controls. Interestingly, the MAPK14 inhibitor did not induce any further change in autophagy in casp2−/− cells, whereas it upregulated autophagy in the WT, similar to casp2−/− control cells. An involvement of MAPK1/3 and MAPK14 in loss of CASP2-mediated autophagy was further confirmed by performing experiments using siRNA against Mapk1/3 and Mapk14 (Fig. S2). These results indicate a possible involvement of MAPK1/3 activation and downregulation of MAPK14 in loss of CASP2-mediated autophagy.

Figure 5. Role of MAPKs in CASP2-mediated modulation of autophagy. (A–F) Protein lysates were prepared from different batches of WT and casp2−/− MEFs (loaded in separate lanes 1 to 3 or 1 and 2 for MAPK8/9), cultured in complete medium in the absence of stressors. Western blotting was performed to detect the levels of active (phosphorylated) and total (non-phosphorylated) MAPKs using specific antibodies against (A) p-MAPK1/3, MAPK1/3 (C) p-MAPK14, MAPK14 and (E) p-MAPK8/9, and MAPK8/9 as described in the Materials and Methods. The same blots were reprobed for tubulin, α (TUBA) that served as a loading control. Shown are the representative blots. (B, D, and F) Densitometric analysis was performed using ImageJ to detect the expression levels. Y-axis represents expression levels of active vs. total MAPK as percent of the loading control. Error bar represents ± SEM. Statistical significance was determined by the Student t test, comparing WT vs. casp2−/−. **, P ≤ 0.01, *, P ≤ 0.05, NS, P ≥ 0.05, experiments repeated at least 3 times. (G) Effects of MAPK inhibitors: SP600125 (10 µM), SB203580 (10 µM), and U0126 (20 µM) on CASP2-mediated autophagy modulation. WT and casp2−/− MEFs were incubated with these inhibitors for 24 h followed by PepA (1 µM) and EST (10 µM) treatment for 6 h to determine autophagy flux. Autophagy was detected by western blotting to determine the levels of LC3 (LC3-I and LC3-II). GAPDH was used as a loading control. Shown is a representative blot, the experiment was repeated 3 times.
Loss of CASP2 promotes ROS generation and oxidative stress that leads to increased autophagy
We, along with others, have reported that the loss of CASP2 is associated with higher ROS generation and accumulation of oxidative stress;36,37 oxidative stress has been associated with an increase in autophagy.53-55 Thus, we investigated a possible link between the higher levels of oxidative stress in casp2−/− cells as a cause of higher autophagy. First, basal levels of ROS (mainly peroxides and superoxide), protein carbonylation (proteins are carbonylated during oxidative stress) and lipid peroxidation (western blotting for 4-HNE) were measured in WT and casp2−/− MEFs. In accordance with previous findings, our results demonstrate that casp2−/− cells maintained significantly higher basal levels of ROS (both superoxide and peroxide) (Fig. 6A and B) as well as protein carbonylation (Fig. 6D) than the WT cells. However, no significant difference was observed in the levels of mitochondrial superoxide levels and 4-HNE staining between WT and casp2−/− cells (data not shown). Next, the role of enhanced oxidative stress/ROS content in autophagy modulation was determined in the WT and casp2−/− cells. A previous study by Chen et al (2009) identifies that superoxide but not peroxide plays an important role in the modulation of autophagy.56 Thus, to distinguish between the 2 (superoxide and peroxide), we used inhibitors of peroxide as well as superoxide. WT and casp2−/− MEFs were treated with peroxide inhibitors (2.5 mM N-acetyl cysteine/NAC and 5 mM glutathione/GSH), or superoxide inhibitors (2.5 mM Tempol and10 µM Mn-CPX-3) and the extent of autophagy was determined (Fig. 6C). Scavenging of peroxide production resulted in only a marginal (not significant) inhibition of autophagy in the casp2−/− cells but not in the WT cells; whereas treatment with superoxide scavengers brought the level of autophagy in the casp2−/− cells to normal levels (similar to the WT levels) with no apparent change in the WT cells. These results indicated an important role for superoxide in CASP2-mediated modulation of autophagy.

Figure 6. Involvement of ROS in CASP2-mediated modulation of autophagy. WT and casp2−/− MEFs were loaded with (A) CM-H2DCFDA (25 µM for 30 min) to detect peroxide levels (by measuring DCF intensity) and (B) HE (10 µM for 30 min) to detect superoxide levels, as described in the Materials and Methods section. Mean fluorescence (of DCF or HE) was detected in a spectrofluorimeter and values were normalized by nuclear content (mean Hoechst 33258 fluorescence or protein concentration). Shown are the mean DCF intensities normalized by nuclear content (Hoechst 33258 mean intensity) ± SEM values, n = 6. The experiment was repeated 3 times with different set of MEFs obtaining similar results. Asterisk (*) represents P values ≤ 0.05 obtained by performing the Student t test. (C) Effect of antioxidants on the endogenous levels of autophagy in WT and casp2−/− MEFs. Cells were treated with different antioxidants for 48 h (GSH, NAC to inhibit peroxide and Tempol and Mn-CPX-3 to inhibit superoxide), followed by EST and PepA treatment for 6 h before the completion of the 48 h (to determine the flux). Western blotting was performed to detect LC3 levels (LC3-I and LC3-II). The blots were normalized by reprobing the same blot with tubulin, α (TUBA) antibody. Shown are the representative blots and the experiment was repeated 3 times. (D and E) Protein carbonylation in WT and casp2−/− MEFs in the presence or absence of PepA (1 µM) and EST (10 µM) (late stage autophagy inhibitors) for 6 h. (D) Western blot detection of carbonyl groups was performed after derivatization of the samples with 2,4-dinitrophenylhydrazine. The same blot was reprobed for GAPDH as a loading control. Shown is a representative blot. (E) Densitometric analysis to determine level of protein carbonylation was performed using ImageJ analysis. Protein carbonylation levels are shown as a percent of loading control. Error bar represents ± SEM. Statistical significance was determined by the Student t test. **P ≤ 0.01, *P ≤ 0.05. The experiment was repeated at least 3 times.
A role for autophagy has not only been suggested in removal of oxidative damage but also in the enhancement of ROS levels in certain cases.57,58 Thus, we investigated whether enhanced autophagy is a cause or a consequence of ROS in casp2−/− cells by studying protein carbonylation (as a measurement of oxidative stress) in cells treated with autophagy inhibitors. Inhibition of autophagy resulted in higher accumulation of oxidized proteins in casp2−/− compared with WT cells, suggesting a role for enhanced autophagy in removal of the damaged/oxidized protein instead of causing enhanced ROS/oxidative damage (Fig. 6D).
Increased ROS production following loss of CASP2 occurs upstream of AMPK, MTOR and MAPK1/3 activation
Loss of CASP2 results in an upregulation of ROS levels. A role for ROS is well established as an upstream mediator of autophagy by modulating activation of AMPK, MTOR, and MAPKs. Thus, we investigated a possible involvement of ROS as an upstream event in loss of CASP2-induced autophagy. Cells were treated in the presence or absence of Tempol and activation of PRKAA, MTOR, and MAPK1/3 was investigated by studying their phosphorylation status (Fig. 7A). In casp2−/− MEFs, treatment with Tempol resulted in a downregulation of PRKAA and MAPK1/3 phosphorylation levels and an upregulation of MTOR phosphorylation compared with the control. In contrast, in WT MEFs Tempol treatment had no significant effect on the phosphorylation status of PRKAA, MAPK1/3, or MTOR.

Figure 7. Role of ROS as an upstream event to AMPK, MTOR, and MAPK1/3 in the CASP2-mediated modulation of autophagy. (A) WT and casp2−/− MEFs were treated in the presence or absence of Tempol for 48 h and cell lysates were prepared. Western blotting was performed to detect the phosphorylation status of PRKAA, MTOR, RPS6KB, and MAPK1/3 vs. total in WT and casp2−/− MEFs. The same blots were reprobed for tubulin, α (TUBA) that served as a loading control. (B) WT and casp2−/− MEFs were transfected with siRNAs specific for the catalytic Prkaa1 and Prkaa2 subunits of Prkaa. The representative western blot demonstrates the efficiency of siRNA-mediated downregulation of PRKAA1 and PRKAA2, phosphorylation status of PRKAA, MTOR, RPS6KB, and MAPK1/3 vs. total in WT and casp2−/− MEFs. Shown are the representative blots; the experiment was repeated at least 3 times.
Various reports have suggested a role for AMPK activation as an upstream event to MTOR inactivation. Interestingly, a role for AMPK has also been identified in both MAPK1/3 activation as well as MAPK1/3 inactivation depending upon the cell type and stressor.59,60 Thus, a role for AMPK activation as an upstream event in CASP2-mediated modulation of autophagy was also investigated. Our results demonstrated that siRNA-mediated inhibition of Prkaa1 and Prkaa2 resulted in an upregulation of MTOR phosphorylation and downregulation MAPK1/3 phosphorylation in casp2−/− MEFs compared with the controls (Fig. 7B). Furthermore, given a role for MAPK1/3 in regulation of MTOR activation, we also studied MTOR activation in the presence of Mapk1/3 siRNA. Our results indicated no significant change in the levels of p-MTOR on siRNA-mediated inhibition of Mapk1/3 (data not shown), suggesting MTOR regulation as a MAPK1/3-independent event in the present context. Together, these results suggest i) an upstream role for CASP2-mediated ROS regulation in modulating autophagy and ii) AMPK activation as an upstream event regulating the activation of MTOR and MAPK1/3 during CASP2-regulated autophagy.
Loss of CASP2 leads to protection from oxidative stress and enhances autophagy compared with the WT
CASP2 is involved in induction of cell death via apoptosis under various conditions including oxidative stress,26,61-63 heat shock,30 and microtubule disruption.29 It has been established that the loss of CASP2 protects cells against theses stressors by the inhibition of apoptosis. Previously, we determined that CASP2 is involved in mitochondrial oxidative stress-induced apoptosis in neurons and the loss of CASP2 improved survival against mitochondrial oxidative stress, by upregulation of autophagy.26 Our data also indicate that casp2−/− cells maintained higher levels of autophagy than WT in the presence of the classical autophagy-inducer, starvation (Fig. 1E; Fig. S3). Thus, we next asked: 1) whether loss of CASP2 can further upregulate autophagy in the presence of variety of external stressors; and 2) whether CASP2-mediated regulation of autophagy affects survival of the cells in the presence of these external stressors. In order to address these questions, we included a variety of well-established inducers of CASP2-mediated cell death, namely oxidative stress (Rotenone [Rot] and hydrogen peroxide [H2O2]), HS, and microtubule disruption (Colchicine [COLC]). Appropriate dosages for all stressors were determined by performing cell death assays (data not shown). Treatment with these stressors at their determined effective (apoptosis inducing) concentration induced a higher extent of cell death in WT compared with casp2−/− MEFs (Fig. 8C). Since apoptosis induction and CASP2 activation are well established under these conditions, we focused on determining the role of CASP2 in autophagy regulation. Using similar concentrations, autophagy was determined by performing western blotting for LC3. Western blotting data demonstrated that the loss of CASP2 resulted in a higher extent of autophagy induction (as indicated by an increase in LC3-II) than the WT in the presence of oxidative stressors (Fig. 8A, i and ii) as well as HS (Fig. 8A, iii); whereas, no significant difference was observed between the WT and casp2−/− in autophagy induced by COLC (Fig. 8A, iv). Since upregulation of autophagy promotes survival by maintaining the levels of ATP during stress conditions, we further determined the levels of ATP in the presence and absence of the stressor in both the cell types. Our results demonstrated that casp2−/− cells maintained higher levels of ATP compared with WT in the presence of oxidative stressor and heat shock treatment; whereas, upon treatment within COLC, no significant difference in ATP levels was observed between the WT and casp2−/− MEFs (Fig. 8B).
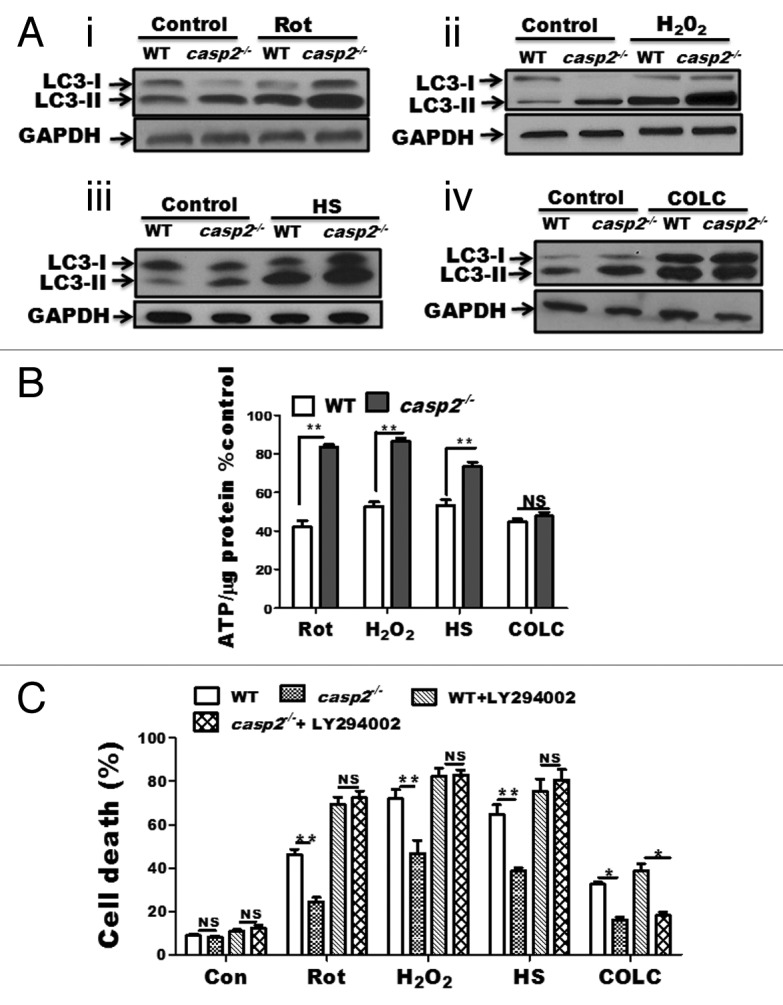
Figure 8. Role of CASP2 in regulation of autophagy in the presence of a variety of stressors. (A) WT and casp2−/− MEFs were untreated or exposed for 6 h to variety of stressors. Rot (10 µM), H2O2 (750 µM) HS (post-recovery), and COLC (2.5 µM) in the presence of PepA (1 µM) and EST (10 µM) to determine autophagy flux. Protein lysates were prepared and western blotting was performed to detect the levels of LC3, an increase in LC3-II indicated upregulation of autophagy. The same blots were reprobed for GAPDH that served as a loading control. Shown are the representative blots. (B and C) WT and casp2−/− MEFs where untreated or exposed to Rot (10 µM, 48 h), H2O2 (750 µM, 24 h), HS (24 h post recovery) and COLC (2.5 µM, 48 h). (B) Effect of these stressors on ATP levels was determined. ATP measurements were performed as described in the Materials and Methods section. The cellular ATP levels were normalized by cellular protein content and values were converted to percentage of untreated cells (control). (C) WT and casp2−/− MEFs cells were treated with stressors as described in the presence or absence of autophagy inhibitor. Cell viability was determined by trypan blue staining as described in the Materials and methods section and shown as percent cell death vs. total. (B and C) Level of significance was analyzed by 2-way ANOVA followed by the Bonferroni post-test, n = 3 and P ≥ 0.05 NS, **P ≤ 0.01, *P ≤ 0.05. All the experiments were repeated at least 3 times, obtaining similar results.
Last, we examined the role of autophagy in mediating cell survival due to loss of CASP2 in the presence of a variety of stressors. Cell viability data indicated that inhibition of autophagy resulted in the loss of survival advantage in casp2−/− cells against oxidative stress and HS, suggesting a role of upregulated autophagy in promoting survival (Fig. 8C). On the other hand, autophagy inhibition did not influence the extent of cell death in both WT and casp2−/− cells or eliminate the survival advantage offered by loss of CASP2 in response to COLC (Fig. 8C). Thus, our results indicate that CASP2-mediated modulation of autophagy plays a significant role in protecting against HS and oxidative stress; however, in the case of COLC, autophagy plays no significant role in the modulation of cell death.
Discussion
Autophagy, an adaptation response to stress, can be deleterious if dysregulated4 and has been associated with a variety of pathologies.8-10 Thus, identification of the regulators of the autophagic processes becomes important for understanding pathogenesis of a disease as well as developing therapeutic interventions. In this regard, the present study identifies CASP2 as a negative regulator of autophagy, which is a novel role for this member of the CASP family. Our results demonstrate that the loss of CASP2 leads to an increase in the steady-state basal level of autophagy in different cells types as well as tissues compared with WT. These findings support the emerging role for CASPs, best known for their role in apoptosis, in the regulation of autophagy and supports the idea that the same molecules may be involved in regulation of autophagic and apoptotic processes.7,64,65 A role for CASPs in the regulation of autophagy has been suggested that includes CASP8, CASP9, and CASP3.7,25,66 Lenardo and colleagues have identified a role for CASP8 as a negative regulator of autophagy where inhibition of CASP8 induces autophagic cell death.7 Also, activation of CASP3 downregulates autophagy under a variety of stress conditions and thus promotes apoptosis.67 Conversely, CASP9 promotes cytoprotective autophagy.25 Our results suggest that the loss of CASP2 promotes endogenous autophagy, defining its role as a negative regulator of autophagy similar to CASP8 and CASP3. Conversely, unlike CASP8, loss of CASP2 does not result in loss of cell survival; rather, in the presence of a stressor, upregulated autophagy due to loss of CASP2 promotes survival. Interestingly, CASP8 and CASP3 levels can be regulated by autophagic clearance.65,68 However, in the present study, our data indicated that autophagy upregulation did not affect the levels of CASP2. At this point, we do not know if autophagy regulates levels of active CASP2 in the presence of various stressors, and this needs further investigation. Furthermore, it is important to note that in our study a CASP2 mutant can reconstitute the autophagy regulatory function; whereas, previous reports suggest that this CASP2 mutant cannot restore the apoptotic functions. This suggests that CASP2-mediated modulation of autophagy is independent of its apoptotic function. However, because these processes (autophagy and apoptosis) regulate each other, a possibility that CASP2 can be a link between apoptosis and autophagy cannot be disregarded.
The present study also determined the mechanism by which CASP2 modulates autophagy. Induction of autophagy by the loss of CASP2 involved downregulation of the MTOR pathway and an upregulation of AMPK activation. Although in the present study, a role for CASP2-mediated ROS regulation was identified in AMPK and MTOR regulation, it is still not very clear how loss of CASP2 leads to AMPK activation or inactivation of MTOR. Given that AMPK, a positive regulator of autophagy, and MTOR, a negative regulator of autophagy, are involved in cellular energy homeostasis,13,14 it is interesting to note that the links exist indicating an involvement of CASP2 during energy crises in the cells.35 In this regard, identifying a role for the ATM-STK11/LKB1 pathway that plays an important role in cellular metabolism, in modulation of CASP2-mediated AMPK regulation becomes interesting and forms the basis of future investigation.69
A role for TP53/TRP53 in the modulation of CASP2 activation and apoptosis induction under various stress conditions is well established.70,71 Moreover, a role for TP53/TRP53 in the regulation of autophagy is emerging; of note inhibition of TP53 leads to upregulation of autophagy in unstressed cells.72,73 Thus we questioned if CASP2-mediated regulation of endogenous levels of autophagy involved a TP53-mediated pathway. However, as shown in the supplementary data (Fig. S4A), nonsignificant differences were observed in the levels of TRP53 or the active form of TRP53 (phosphorylation at serine 15, involved during autophagy) in the presence or absence of CASP2. Interestingly, our results indicate that in the absence of CASP2, the autophagy-inducing effect of a TRP53 inhibitor (Pifithrin α) was highly enhanced compared with WT MEFs, suggesting a synergistic effect on autophagy induction by inhibition of both proteins together (TRP53 and CASP2) (Fig. S4B). These findings suggest that CASP2 and TRP53 may work through different pathways to modulate autophagy; thus, inhibition of CASP2 and TRP53 together leads to a synergistic effect. At the same time, since a positive cross-regulation between CASP2 and TRP53/TP53 exists during a variety of stress conditions,70,71,74 a strong possibility still exists that in the presence of stressors; CASP2-mediated modulation of autophagy might involve a TP53-mediated pathway and needs further investigation.
It has been recognized that in the mammalian system, autophagy can be regulated by an ATG5- and ATG7-dependent pathway, a canonical pathway, or can also follow an alternative noncanonical pathway that is independent of ATG5 and ATG7. Our results demonstrated that CASP2-mediated regulation of autophagy required ATG5 and ATG7; even low levels of ATG5 and ATG7 were sufficient to mediate loss of CASP2-induced autophagy, suggesting an involvement of the canonical pathway of autophagy. In the present study, we further identified a role for MAPKs, specifically MAPK14 and MAPK1/3 in CASP2-mediated regulation of autophagy. Our results demonstrated that casp2−/− cells maintained lower levels of active MAPK14 and a constitutive upregulation of MAPK1/3 activation. In accordance with our findings, it has been shown that CASP2 is required for activation of MAPK14 under normal conditions.49 Currently, it is not clear how loss of CASP2 results in higher constitutive levels of MAPK1/3 activation. Furthermore, our results demonstrated that MAPK14 downregulation and MAPK1/3 upregulation were involved in enhancing autophagy in cells lacking CASP2, which is in accordance with previous reports suggesting a role for MAPK14 as a negative regulator of autophagy and MAPK1/3 as a positive regulator of autophagy.43,44,46,47,75 Interestingly, it has been suggested that the MAPK1/3 and MAPK14 pathways tightly control autophagy at the maturation stage. It has been proposed that MAPK14 limits constitutive autophagy activity, by reducing the maturation of autophagosomes. Conversely transiently activated MAPK1/3 reaches a critical threshold that might relieve this blockade and stimulate the maturation of autophagosomes.76 Thus in casp2−/− cells, lower levels of MAPK14 activation and higher levels of MAPK1/3 activation may work together to coordinate autophagy.
In an attempt to delineate a more direct link between CASP2 and autophagy, the present study identified an important role for CASP2-mediated ROS regulation in modulating the levels of autophagy. Previously, our lab along with Kumar and colleagues has demonstrated that CASP2 modulates ROS levels.36,37 A growing amount of evidence indicates an essential role for ROS in the activation of autophagy.53-55 Regardless of whether the outcome of autophagy results in cell survival or death, ROS are invariably involved. Here, we demonstrate an important role for loss of CASP2-mediated ROS upregulation as an upstream event (relative to AMPK and MAPK1/3 activation and upregulation of MTOR activation) in the induction of autophagy. Based on these findings it appears that CASP2-mediated autophagy regulation may be a secondary event to ROS modulation by CASP2. These findings distinguish CASP2-mediated regulation of autophagy from other CASPs that regulate autophagy in a more direct way. However, the possibility of a more direct regulation of autophagy by CASP2 cannot be completely ruled out. Our results further suggest that superoxide but not peroxide levels were mainly involved in loss of CASP2-mediated autophagy. Previously, studies by Gibson and colleagues have determined that superoxide is the major ROS species regulating autophagy.56 Our data is in accordance with these observations and suggest that the endogenous levels of autophagy were regulated by superoxide levels. It is interesting to note that besides a role for autophagy in the clearance of oxidatively damaged molecules, previous studies have also identified a role for autophagy in the upregulation of ROS levels and accumulation of oxidative damage.57,58 However, our data demonstrated that autophagy upregulation in casp2−/− cells was responsible for the clearance of oxidatively damaged proteins and might be a compensatory response (protective) against oxidative stress and not a cause for the accumulation of oxidative stress.
Besides the identification of a role for CASP2 in the regulation of autophagy under normal conditions, the present study also detected involvement of CASP2 in the regulation of autophagy in response to a variety of stressors. Our data indicated that although the autophagy modulation and apoptosis induction by CASP2 are 2 independent functions, loss of CASP2-mediated upregulation of autophagy does play a role in promoting survival during apoptosis-inducing stress. In the present study, we utilized different stressors that mediate CASP2-dependent apoptosis: oxidative stress (mitochondrial as well as general), HS,30 and microtubule disruption.29 Loss of CASP2-mediated protection against oxidative stress and HS involved autophagy upregulation, whereas loss of CASP2-mediated protection (higher survival) against cytoskeletal disruption did not involve autophagy upregulation. It is interesting to note that in the case of COLC, the higher survival of the cells lacking CASP2 was not mediated by autophagy and some other survival mechanisms seems to play a role in this case. Presently, we do not know the molecular mechanism by which CASP2 regulates the stress-mediated autophagic response. It will be interesting to delineate the molecular pathways regulated by CASP2 and determine how the presence of CASP2 determines the cell’s choice toward apoptosis and autophagy. It will be further useful to determine whether CASP2-mediated modulation of autophagy plays a role in modulation of cell death signals that are not dependent on CASP2. It is to be noted that in contrast to its role in downregulation of apoptosis, autophagy itself is required for cell death in certain cases and may be an upstream event to apoptosis.77,78 Since loss of CASP2 leads to upregulation of autophagy, the possibility exists that certain stressors require autophagy as an upstream event for cell death; the loss of CASP2 may make cells more sensitive for those stressors.
Overall, the present study identifies a novel role for CASP2 in the regulation of autophagy (Fig. 9). Since regulation of autophagy by CASP2 is an unexplored and yet important area, the present study raises several possibilities and critical questions that form the basis of future investigations. Currently, we do not know whether CASP2 can also regulate autophagy in tumor cells and whether this biological effect of CASP2 plays a role in tumor suppression. Also, since autophagy plays a role in senescence, it will be interesting to identify whether CASP2 may also play a role in this process. Understanding these complex processes and interrelationships that exist between autophagy and apoptosis becomes important since the interplay between these 2 processes determines whether a cell will live or die and could regulate therapeutic outcomes and disease progression.
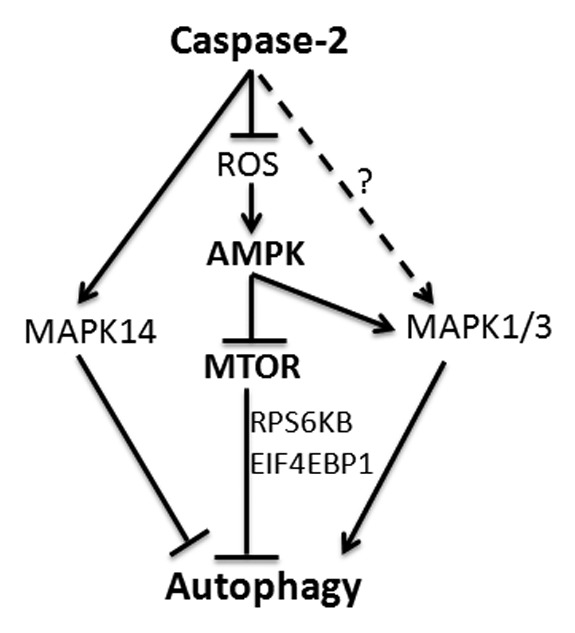
Figure 9. CASP2-mediated modulation of autophagy. Loss of CASP2 upregulates ROS that leads to upregulation of AMPK and downregulation of MTOR. CASP2 also regulates MAPK14 and MAPK1/3 that may play a role in autophagy modulation.
Material and Methods
Reagents
Dulbecco’s modified Eagle’s medium (11995), OPTIMEM (3198-070), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (15630), PBS (10010-049), 0.25% trypsin EDTA (25200-056) and penicillin/streptomycin (15140-122) were from Gibco (Invitrogen Corporation). Fetal bovine serum was procured from Thermo Scientific, HyClone (SH3091993). Rapamycin (R0395), COLC (C9754), GSH (G4251), H2O2 (H1009), NAC (A7250), Rot (R8857), trypan blue (T8154), SP600125 (S5567) and U0126 (U120) were purchased from Sigma. LY294002 (440204), PepA (516481), EST (330005), SB20580 (559398) and Pifithrin α (63208-82-2) were from Calbiochem. Alexa Fluor 488 IgG anti-rat (A-11006), Alexa Fluor 488 IgG anti-goat (A-11055), Alexa Fluor 568 IgG anti goat (A-11078), Hoechst 33258 (H-3569), hydroethidine (HE) (D-1168), 5,6-carboxy-2',7'-dichlorofluorescein diacetate (CM-H2DCFDA) (C6827) and LysoTracker Red DND-99 (L7529) were purchased from Molecular Probes/Invitrogen. Vectashield mounting medium (H-1000) was from Vector Laboratories. RIPA buffer (sc-24948), goat polyclonal LC3 (sc-16756) (for immunocytochemistry), SQSTM1/p62 (sc-25575) and HRP-labeled anti-mouse (sc-2096) or HRP-labeled anti-rabbit IgG (sc-2030) secondary antibodies, Tempol (sc-200825) and Mn-CPX-3 (sc-221951) were from Santa Cruz Biotechnology. CASP2 monoclonal antibodies (clone 11B4 for western blotting and 10C6 for immunocytochemistry) were from Alexis Biochemicals. Primary antibodies for GAPDH (2118), MTOR (2972), p-MTOR (2971), LC3 (2775) (for western blotting), ATG7 (8558), ATG5 (8540), MAPK8/9 (9258P), p-MAP8/9 (9255S), MAPK14 (9212P), p-MAPK14 (4551P), MAPK1/3 (4695P), p-MAPK1/3 (4370P), RPS6KB/p70S6K (2708), p-RPS6KB/p70S6K (9204), EIF4EBP1 (9644), p-EIF4EBP1 (9451), TP53 (2524) and p-TP53 (9284) were procured from Cell Signaling Technology. Primary antibodies for tubulin, α (ab6160), PRKAA/AMPKα (ab80039) and p-PRKAA/p-AMPKα (3930-1) were from Abcam. LipofectamineTM 2000 (11668) was from InvitrogenTM. Flag tag pcDNA3-Casp2 (11811), Flag tag cDNA3-Casp2 C303A (11812), the catalytically mutant form of CASP2 (submitted by Dr Guy Salvesen) and pBABE-puro mCherry-EGFP-LC3B79 (22418) (submitted by Dr Eric J Brown) were from Addgene, Inc. OxyBlot™ Protein Oxidation Detection Kit (S7150) was obtained from Chemicon International. Polyacrylamide gels (4–20% gradient gel) (EC60285BOX) were from Invitrogen and PVDF membranes (162-0177) were from BioRad Laboratories. The BCA protein assay kit (23225) and WEST PICO Chemiluminescence substrate (34080) were from Thermo Scientific, Pierce. Hyperfilm-ECL films (28-9068-40) and l-valine-14C (921-10-8) were procured from Amersham (Biosciences). In case the source is not mentioned, the chemicals were purchased from Sigma. All the cell culture plastic and glassware were from Nunc (Nalgene Nunc International).
Mice
The casp2−/− mice were originally generated by Dr Junying Yuan of Harvard University and kindly provided by Dr Carol Troy of Columbia University with Dr Yuan’s consent.80 The mice were backcrossed with C57Bl/6 for ten generations. GFP-LC3 Mice generated by Dr Noboru Mizushima81 were kindly provided by RIKEN BioResource center and were crossed with WT and casp2−/− mice. All the mice were housed in micro-isolator-topped cages and maintained in a pathogen-free environment at the AAALAC-accredited UTHSCSA animal facility following the NIH Guidelines for the Care and Use of Laboratory Animals. The university’s Institutional Animal Care and Use Committee approved the protocols used in this study. Mice of all the genotypes were maintained in an animal room with controlled temperature and were exposed to a 12:12 light-dark regimen with the lights on daily from 06:00 to 18:00. Food was available ad libitum.
Culture of MEFs
MEFs were isolated from 13.5-d-old embryos of WT and casp2−/− littermates after trypsin digestion and were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 100 mM penicillin/streptomycin, nonessential amino acids, and 10% fetal bovine serum. atg5−/− and respective WT MEFs were kindly provided by Dr Shengkan Jin and atg7−/− and its respective WT MEFs were kindly provided by Dr Toren Finkel. Cells were cultured in a humidified incubator at 37 °C with 5% CO2. Following standard procedures, the MEFs (WT and casp2−/−) were cryopreserved after 1st passage. The cells were revived whenever required. To avoid variation in results due to increase in passage number, all the experiments were performed using (WT and casp2−/−) MEFs between 3rd to 7th passages.
Primary culture of astrocytes, neuron, and osteoclasts
Primary culture of cerebral cortical astrocytes was prepared from WT and casp2−/− young adult mice (1- to 2-mo-old). The whole brain was removed and suspended in DMEM. The cerebral cortex was cut into small cubes (0.5 to 1 mm3) and then incubated in media containing papain (12 mg/6 mL in DMEM) at 30 °C in a shaker for 30 min. The digested tissue was washed free of papain and triturated 12 to 15 times using a siliconized pasteur pipette. The cells were then cultured in DMEM containing 10% fetal bovine serum. Cells were allowed to grow for a week and then samples were prepared for western blotting as described below.
Primary cultures of cortical neurons were prepared as described previously.26 Primary cultures of osteoclasts were prepared from WT and casp2−/− mice 4- to 6-wk of age. The mice were anesthetized with isofluorane and bones were excised. Marrow was flushed out with a 27G needle into Minimum Essential Medium Alpha Modification (Life Technologies, 12571-063). Nonadherent cells were removed after 24 h and plated with 30 ng/mL CSF1 (R&D Systems, 416-ML-050). After 3 d, osteoclast precursors were removed and replated at a density of 2 × 104 cells/cm2 onto 35-mm culture dishes with 30 ng/mL CSF1 and 10 ng/mL TNFSF11/RANKL (R&D Systems, 462-TEC-010). Media was changed after 3 d, and on d 4 cells were lysed into cold RIPA buffer.
Cell viability measurements
Depending upon the hours of incubation with the stressor (by the end of the experiment the confluence reaches ~80%), the MEFs were plated in 12-well plates in 1 ml of the complete growth medium and were cultured at 37 °C for 24 h. Next, the medium was changed and stressors were added for a defined time. At the end of the incubation time, both attached and nonattached cells were collected and pooled together. Cell counting and viability was performed using a cell viability analyzer (Beckman Coulter, Inc., California, USA) based on the trypan blue exclusion method. Cell death was calculated as a percent of dead cells in the total cell population. Experiments were performed in triplicates and repeated at least 3 times obtaining similar results.
Transient transfection
For siRNA transfection, MEFs were seeded at 0.5 × 106 cells/well in a 6-well plate and allowed to attach for 24 h. Cells were then transfected in 1.5 ml OPTIMEM containing 5 µL lipofectamineTM 2000, with 33 nM siRNA duplexes, following the manufacturer’s protocols. After 24 h of transfection, the medium was removed and replaced with complete growth medium and samples were collected after 72 h. For each transfection Qiagen siRNA duplex with following sequences were used: Ampkα1: Mm_Prkaa1_1 SI01388219 CACGAGTTGA CCGGACATAA A; Ampkα2: Mm_Prkaa2_1 SI01388247 CAGGGAAGCC TTAAATATTT A; Atg5: Mm_Apg5I_1 SI00230664 ATGGTTCTAG ATTCAATAAT A; Atg7: Mm_Apg7I_1 SI00900515 TAGCATCATC TTTGAAGTGA A; Casp2: Mm_Casp2_3 SI00941717 CAGGGTCACT TGGAAGACTT A; Mapk3: Mm_Mapk3_1 SI01300579 CAGAACATTC CTAAGTCTCA A; Mapk1: Mm_Mapk1_1 SI02672117 CTGAATTGTA TAATAAATTT A; Mapk14: Mm_Mapk14_2 SI01300530 TCAGATAATA CCACTGGTTA A.
After completion of the experiment, the cells were scraped, washed, and pelleted and protein lysates were prepared in RIPA buffer. Protein lysates were used for western blotting as described in the later section. Each experiment was performed in triplicate and repeated at least 3 times.
DNA transfection was performed using X-treme GENE HP DNA Transfection Reagent (Roche). casp2−/− MEFs (0.5 × 106 cells/well in a 6-well plate) were transfected with 2 µg of the plasmid DNA and 8 µL of the X-treme GENE reagent in OPTIMEM, following a protocol provided by the manufacturer. After 24 h of transfection, the medium was removed and replaced with complete growth medium and samples were collected after 72 h.
After completion of the experiment, the cells were scraped, washed, and pelleted and protein lysates were prepared in RIPA buffer. Protein lysates were used for western blotting as described in a later section. Each experiment was performed in triplicate and repeated at least 3 times.
Reverse transcription-PCR
RNA was extracted with the RNeasy kit (Qiagen, 74004). One microgram of total RNA from each sample was used as a template for cDNA synthesis with a QuantiTect reverse transcriptase kit (Qiagen, 205311). An equal volume of cDNA product was used in the PCR performed using the TopTaq Master Mix Kit (Qiagen, 200403). PCR amplification was performed using the following primers (purchased from Invitrogen): mouse Sqstm1 gene 5′-AGCTGCCCTC AGCCCTCTA’-3 (forward) and 5′-GGCTTCTCTT CCCTCCATGTT’-3 (reverse) and Gapdh 5′-ATGGTGAAGG TCGGTGTGAA’-3 (forward) and 5′ TTCACACCCA TCACAAACAT'-3 (reverse). The PCR conditions were set according to the standard protocol. The PCR products were resolved on an agarose gel containing GelRedTM nucleic acid gel stain (Biotium, 41002) and exposed using a Kodak Image Station 440CF (Eastman Kodak Company, New Haven, CT USA).
Measurement of long-lived protein degradation
Protein degradation was determined according to a method previously reported, with a few modifications.82 Briefly, MEFs (5 × 104) were plated into 6-well plates. After overnight recovery, cells were labeled with 0.5 μCi/ml L-[14C]valine in complete medium for 18 h. Unincorporated radioisotopes and degraded amino acids released from short-lived proteins were removed by rinsing with phosphate-buffered saline (PBS pH 7.4; Life Technologies, 10010-023) 3 times. Cells were then chased with the culture medium containing 10% FCS and 10 mM cold valine for 4 h to get rid of short-lived proteins. After a 4 h incubation, at which time short-lived proteins were being degraded, the chase medium was replaced with fresh medium or as indicated for another 8 h. At the end of the incubation period, 1 mL of the media was collected from each well and combined with trichloroacetic acid (TCA) to reach 10% TCA concentration. The samples were then centrifuged at 12,000 × g for 5 min; and 400 μL of the supernatant fraction was removed to measure 14C readout with a scintillation counter. For the cells, these were first washed with DPBS (Life Technologies, 14040-133), then 1 mL of 10% TCA was added to each well and incubated for 5 min at RT; after fixation, the cells were washed with 10% TCA and dissolved in 0.2 M NaOH; and 400 μL of the lysate were analyzed to measure 14C readout (the total 14C readout in the medium and total 14C readout within the cells). Then, the ratio of released 14C in the medium to the total 14C readout was calculated; this represents the degradation percentage in a given time duration. The experiment was performed in triplicate and repeated 3 times obtaining similar results.
Fluorescence staining of lysosomes, LC3, and CASP2
The cells (~2.5 × 104) were plated on glass coverslips in 24-well plates. To visualize lysosomes, the cells were stained with LysoTracker Red (500 nM) for 30 min in complete medium and were washed free of dye prior to fixation. The cells were fixed by the addition of 3.7% paraformaldehyde in PHEM buffer (60 mM PIPES, 25 mM HEPES, 10 mM EGTA, 2 mM MgCl2, pH 7.00) for 20 min at room temperature (RT). Cells were washed and permeabilized (0.3% Triton X-100). For goat polyclonal LC3 and rat monoclonal CASP2 (10C6, Alexis Biochemicals) blocking was performed in 10% goat serum for 1 h at RT and immunolabeled with the primary antibody (1:150 dilution and overnight incubation at 4 °C). Cells were then washed with PBS and incubated for 2 h with rat or goat Alexa Fluor 488 or goat Alexa Fluor 568 IgG secondary antibody (1:1000 dilutions) at RT (depending upon the combination). For nuclear staining, Hoechst 33258 (10 µg/mL) was added along with the secondary antibody. Cells were washed with PBS and then rinsed with water. The coverslips were mounted with Vectashield mounting medium and sealed. Approximately, 150 to 200 cells per coverslip were visualized on an LSM microscope (Olympus FV1000, Olympus Imaging America Inc. NY, USA), and images were digitally captured and scored blindly. Each experiment was repeated at least 3 times with separate batches of MEF cultures. ImageJ colocalization analysis was performed to calculate the Pearson Coefficient.
Fluorescence microscopy for tissue sections
To detect autophagy at the tissue level, tissue samples from GFP-LC3 WT and GFP-LC3 casp2−/− mice (2 to 3 mo of age) were prepared as follows: To prevent induction of autophagy during tissue preparation, mice were anesthetized with diethyl ether and immediately fixed by perfusion through the left ventricle with 4% paraformaldehyde in 0.1 M sodium phosphate buffer (NaH2PO4 * H2O, 3.1 g and Na2HPO4 (anhydrous) 10.9 g in 1 L water, pH 7.4). Tissues were harvested and further fixed with the same fixative for at least 4 h, followed by treatment with 15% sucrose in PBS for 4 h and then with 30% sucrose solution overnight. Tissue samples were embedded in Tissue-Tek OCT compound (Sakura Finetechnical Co., 25608-930) and stored at −70 °C. The samples were sectioned at 5- to 7-μm thickness with a cryostat (CM3050 S, Leica, Deerfield, IL), air-dried for 30 min, and stored at −20 °C until use. Then the tissue samples were stained with Hoechst 33258 (10 µM) for 20 min, washed in PBS and were mounted using FluorSave reagent from Calbiochem (345789). The slides were analyzed on LSM microscope (Olympus FV1000, Olympus Imaging America Inc. NY, USA). The number of cells showing more than 8 GFP-LC3 dots in each field (for different organs) was counted in 12 to 13 visual fields from individual mice. The results were expressed as the percent of cells showing LC3 puncta per field, ± SEM. The probability of statistical differences between experimental groups was determined by the Student t test.
Measurement of cellular ROS production
To determine ROS production, ~0.5 × 106 cells in 6-well plates were stained with 10 µM HE for superoxide production or 25 µM CM-H2DCFDA for peroxide production for 30 min at 37 °C in the dark. Upon oxidation by ROS, the nonfluorescent CM-H2DCFDA is converted to the highly fluorescent DCF (2',7'-dichlorofluorescein). Cells were trypsinized, washed, and resuspendend in PBS and immediately analyzed using a spectrofluorimeter (Becton-Dickinson, Mountain View, CA). Mean DCF intensity was normalized by DNA content (10 µM Hoechst 33258 counter staining) and mean HE intensity was normalized by protein concentration.
ATP determination
For ATP measurement, a commercially available Molecular Probes® ATP Determination Kit (A22066) was used that involves bioluminescence assay for quantitative determination of ATP with recombinant firefly luciferase and its substrate D-luciferin. Briefly, MEFs (WT and casp2−/−) were treated with various concentrations of stressors and then collected in 1 ml microcentrifuge tubes. After a single wash with ice-cold PBS, cells were lysed with the somatic cell ATP-releasing reagent provided by the kit. Luciferin substrate and luciferase enzyme were added and bioluminescence was assessed on a Perkin Elmer 3B spectroflurometer (Perkin-Elmer Inc., Waltham, MA, USA). Whole-cell ATP content was determined fluorescence units per milligram protein content. The cellular ATP level was converted to percentage of untreated cells (control).
Western blot analysis
Whole cell protein extracts (protein lysates) were prepared using RIPA lysis buffer (Santa Cruz Biotechnology, sc-24948). Protein estimation was performed using the BCA kit. Equal amounts of protein from the total cell lysates (22 to 50 µg/lane) were separated by SDS-PAGE (4–20% gradient gel), and transferred to PVDF membrane using wet transfer. Depending upon the primary antibody to be used, the membranes were blocked in 5% skim milk (Cell Signaling Technology, 9999) or 5% bovine serum albumin (Cell Signaling Technology, 9998) for 1 h. The membranes were then probed overnight at 4 °C with specific primary antibodies diluted in 1% bovine serum albumin or 1% skim milk. Then, the membranes were washed and probed with the respective secondary isotype-specific antibodies tagged with horseradish peroxidase. Bound immuno-complexes were detected using WEST PICO chemiluminescence substrate and exposed to Hyperfilm-ECL. The same blots were reprobed with GAPDH or tubulin, α antibody as a loading control. Densitometry analysis for determining the band intensity was performed using ImageJ software.
Immunoblot analysis of protein carbonylation
The assay was performed following the manufacturer’s protocol using OxyBlot™ Protein Oxidation Detection Kit (Chemicon International, S7150). Briefly, equal amounts (25 µg) of the protein lysates prepared in RIPA buffer were derivatized by adding 2,4-dinitrophenyl hydrazine (DNP) reagent to the sample in lysis buffer containing 6% SDS. The reaction was allowed to continue at RT for 15 min. The derivatization reaction was stopped by adding neutralizing solution followed by vortexing. Derivatized protein samples were then loaded onto a 12% SDS-PAGE gel, and electrophoresis was performed at 80 V for 2 h. Subsequently, the proteins were transferred onto a PVDF membrane. The membrane was blocked at RT and DNP bound protein was detected by using rabbit anti-DNP (supplied with the kit) as a primary antibody (1:150 dilution, incubated for 2 h at RT). The membrane was then incubated with goat anti-rabbit HRP polyclonal antibody for 1 h at RT. Bound immuno-complexes were detected as described above. Equal loading was confirmed by reprobing the same blot with GAPDH antibody. The protein carbonyl detection and quantification was done by using ImageJ.
Data analysis
Data in graphs are reported as ± standard error of the mean (SEM) and depict the average of at least 3 independent experiments. Morphological images are representative of at least 3 independent experiments with similar results. Statistical analysis was performed by the Student paired t test or a 2-way analysis of variance followed by a Bonferroni post-test when appropriate (P ≤ 0.05 was considered significant [*] and P ≤ 0.01 was considered highly significant [**]).
Supplementary Material
Disclosure of Potential Conflicts of Interest
Each author has participated and approved the contents of the submitted manuscript. There is no conflict of interest, direct or indirect, for any of the authors.
Acknowledgments
The authors wish to thank Dr Junying Yuan of Harvard University and Dr Carol Troy of Columbia University for providing the casp2−/− mice. The authors thank Dr Noboru Mizushima (Mizushima N et al., 2003)81 and RIKEN BioResource center for kindly providing GFP-LC3 Mice. The authors thank Dr Shengkan Jin from UMDNJ-Robert Wood Johnson Medical School (NJ USA) for kindly providing atg5−/− and Atg5+/+ MEFs and Dr Toren Finkel from the National Heart, Blood, and Lung institute (NIH, Bethesda, USA) for kindly providing atg7−/− and Atg7+/+ MEFs. The authors also thank Dr Victoria Centonze-Frohlich for her contributions and guidance in microscopy experiments. Images were generated in the Core Optical Imaging Facility, which is supported by UTHSCSA, NIH-NCI P30 CA54174 (San Antonio Cancer Institute), NIH-NIA P30 AG013319 (Nathan Shock Center) and NIH-NIA P01AG19316. The authors also thank Michelle E Bendele for assistance in mouse breeding and Sonya Karbch for her help in the experiments. This work has been supported by NIH grant 5 R37 AG007218-21.
Glossary
Abbreviations:
- CM-H2DCFDA
5,6-carboxy-2′,7′-dichlorofluorescein diacetate
- AMPK
AMP-activated protein kinase
- Atg
autophagy-related (gene)
- ATG
autophagy-related (protein)
- CASP
caspase protein
- Casp
caspase gene
- casp2−/−
Casp2 homozygous knockout
- COLC
colchicine
- GFP
green fluorescent protein
- GSH
glutathione
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HS
heat shock
- HE
hydroethidine
- MAPK
mitogen-activated protein kinase
- H202
hydrogen peroxide
- MEFs
mouse embryonic fibroblasts
- MTOR
mechanistic target of rapamycin
- MAP1LC3 (LC3)
microtubule-associated protein 1 light chain 3
- NAC
N-acetyl cysteine
- PepA
pepstatin A
- ROS
reactive oxygen species
- RT
room temperature
- Rot
rotenone
- SEM
standard error of the mean
- TCA
trichloroacetic acid
- WT
wild-type
References
- 1.Chang YY, Juhász G, Goraksha-Hicks P, Arsham AM, Mallin DR, Muller LK, Neufeld TP. Nutrient-dependent regulation of autophagy through the target of rapamycin pathway. Biochem Soc Trans. 2009;37:232–6. doi: 10.1042/BST0370232. [DOI] [PubMed] [Google Scholar]
- 2.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–6. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 3.Pattingre S, Espert L, Biard-Piechaczyk M, Codogno P. Regulation of macroautophagy by mTOR and Beclin 1 complexes. Biochimie. 2008;90:313–23. doi: 10.1016/j.biochi.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 4.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–73. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 5.Scott RC, Juhász G, Neufeld TP. Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr Biol. 2007;17:1–11. doi: 10.1016/j.cub.2006.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang M, Hossain MS, Tan W, Coolman B, Zhou J, Liu S, Casey PJ. Inhibition of isoprenylcysteine carboxylmethyltransferase induces autophagic-dependent apoptosis and impairs tumor growth. Oncogene. 2010;29:4959–70. doi: 10.1038/onc.2010.247. [DOI] [PubMed] [Google Scholar]
- 7.Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–2. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- 8.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007;6:304–12. doi: 10.1038/nrd2272. [DOI] [PubMed] [Google Scholar]
- 10.Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–5. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mizushima N, Ohsumi Y, Yoshimori T. Autophagosome formation in mammalian cells. Cell Struct Funct. 2002;27:421–9. doi: 10.1247/csf.27.421. [DOI] [PubMed] [Google Scholar]
- 12.Yang YP, Liang ZQ, Gu ZL, Qin ZH. Molecular mechanism and regulation of autophagy. Acta Pharmacol Sin. 2005;26:1421–34. doi: 10.1111/j.1745-7254.2005.00235.x. [DOI] [PubMed] [Google Scholar]
- 13.Alers S, Löffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol. 2012;32:2–11. doi: 10.1128/MCB.06159-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010;584:1287–95. doi: 10.1016/j.febslet.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9:1102–9. doi: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
- 16.Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A. Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009;16:966–75. doi: 10.1038/cdd.2009.33. [DOI] [PubMed] [Google Scholar]
- 17.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–52. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 18.Cárdenas-Aguayo MdelC, Santa-Olalla J, Baizabal JM, Salgado LM, Covarrubias L. Growth factor deprivation induces an alternative non-apoptotic death mechanism that is inhibited by Bcl2 in cells derived from neural precursor cells. J Hematother Stem Cell Res. 2003;12:735–48. doi: 10.1089/15258160360732759. [DOI] [PubMed] [Google Scholar]
- 19.Ciechomska IA, Goemans GC, Skepper JN, Tolkovsky AM. Bcl-2 complexed with Beclin-1 maintains full anti-apoptotic function. Oncogene. 2009;28:2128–41. doi: 10.1038/onc.2009.60. [DOI] [PubMed] [Google Scholar]
- 20.Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–48. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 21.Moretti L, Attia A, Kim KW, Lu B. Crosstalk between Bak/Bax and mTOR signaling regulates radiation-induced autophagy. Autophagy. 2007;3:142–4. doi: 10.4161/auto.3607. [DOI] [PubMed] [Google Scholar]
- 22.Lépine S, Allegood JC, Edmonds Y, Milstien S, Spiegel S. Autophagy induced by deficiency of sphingosine-1-phosphate phosphohydrolase 1 is switched to apoptosis by calpain-mediated autophagy-related gene 5 (Atg5) cleavage. J Biol Chem. 2011;286:44380–90. doi: 10.1074/jbc.M111.257519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Priault M, Hue E, Marhuenda F, Pilet P, Oliver L, Vallette FM. Differential dependence on Beclin 1 for the regulation of pro-survival autophagy by Bcl-2 and Bcl-xL in HCT116 colorectal cancer cells. PLoS One. 2010;5:e8755. doi: 10.1371/journal.pone.0008755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu L, Lenardo MJ, Baehrecke EH. Autophagy and caspases: a new cell death program. Cell Cycle. 2004;3:1124–6. doi: 10.4161/cc.3.9.1097. [DOI] [PubMed] [Google Scholar]
- 25.Jeong HS, Choi HY, Lee ER, Kim JH, Jeon K, Lee HJ, Cho SG. Involvement of caspase-9 in autophagy-mediated cell survival pathway. Biochim Biophys Acta. 2011;1813:80–90. doi: 10.1016/j.bbamcr.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 26.Tiwari M, Lopez-Cruzan M, Morgan WW, Herman B. Loss of caspase-2-dependent apoptosis induces autophagy after mitochondrial oxidative stress in primary cultures of young adult cortical neurons. J Biol Chem. 2011;286:8493–506. doi: 10.1074/jbc.M110.163824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bouchier-Hayes L, Green DR. Caspase-2: the orphan caspase. Cell Death Differ. 2012;19:51–7. doi: 10.1038/cdd.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumar S, Kinoshita M, Noda M, Copeland NG, Jenkins NA. Induction of apoptosis by the mouse Nedd2 gene, which encodes a protein similar to the product of the Caenorhabditis elegans cell death gene ced-3 and the mammalian IL-1 beta-converting enzyme. Genes Dev. 1994;8:1613–26. doi: 10.1101/gad.8.14.1613. [DOI] [PubMed] [Google Scholar]
- 29.Ho LH, Read SH, Dorstyn L, Lambrusco L, Kumar S. Caspase-2 is required for cell death induced by cytoskeletal disruption. Oncogene. 2008;27:3393–404. doi: 10.1038/sj.onc.1211005. [DOI] [PubMed] [Google Scholar]
- 30.Tu S, McStay GP, Boucher LM, Mak T, Beere HM, Green DR. In situ trapping of activated initiator caspases reveals a role for caspase-2 in heat shock-induced apoptosis. Nat Cell Biol. 2006;8:72–7. doi: 10.1038/ncb1340. [DOI] [PubMed] [Google Scholar]
- 31.Shi M, Vivian CJ, Lee KJ, Ge C, Morotomi-Yano K, Manzl C, Bock F, Sato S, Tomomori-Sato C, Zhu R, et al. DNA-PKcs-PIDDosome: a nuclear caspase-2-activating complex with role in G2/M checkpoint maintenance. Cell. 2009;136:508–20. doi: 10.1016/j.cell.2008.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Dorstyn L, Puccini J, Wilson CH, Shalini S, Nicola M, Moore S, Kumar S. Caspase-2 deficiency promotes aberrant DNA-damage response and genetic instability. Cell Death Differ. 2012;19:1288–98. doi: 10.1038/cdd.2012.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Logette E, Le Jossic-Corcos C, Masson D, Solier S, Sequeira-Legrand A, Dugail I, Lemaire-Ewing S, Desoche L, Solary E, Corcos L. Caspase-2, a novel lipid sensor under the control of sterol regulatory element binding protein 2. Mol Cell Biol. 2005;25:9621–31. doi: 10.1128/MCB.25.21.9621-9631.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ho LH, Taylor R, Dorstyn L, Cakouros D, Bouillet P, Kumar S. A tumor suppressor function for caspase-2. Proc Natl Acad Sci U S A. 2009;106:5336–41. doi: 10.1073/pnas.0811928106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nutt LK, Margolis SS, Jensen M, Herman CE, Dunphy WG, Rathmell JC, Kornbluth S. Metabolic regulation of oocyte cell death through the CaMKII-mediated phosphorylation of caspase-2. Cell. 2005;123:89–103. doi: 10.1016/j.cell.2005.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shalini S, Dorstyn L, Wilson C, Puccini J, Ho L, Kumar S. Impaired antioxidant defence and accumulation of oxidative stress in caspase-2-deficient mice. Cell Death Differ. 2012;19:1370–80. doi: 10.1038/cdd.2012.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y, Padalecki SS, Chaudhuri AR, De Waal E, Goins BA, Grubbs B, Ikeno Y, Richardson A, Mundy GR, Herman B. Caspase-2 deficiency enhances aging-related traits in mice. Mech Ageing Dev. 2007;128:213–21. doi: 10.1016/j.mad.2006.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–5. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 40.Bjedov I, Partridge L. A longer and healthier life with TOR down-regulation: genetics and drugs. Biochem Soc Trans. 2011;39:460–5. doi: 10.1042/BST0390460. [DOI] [PubMed] [Google Scholar]
- 41.Meley D, Bauvy C, Houben-Weerts JH, Dubbelhuis PF, Helmond MT, Codogno P, Meijer AJ. AMP-activated protein kinase and the regulation of autophagic proteolysis. J Biol Chem. 2006;281:34870–9. doi: 10.1074/jbc.M605488200. [DOI] [PubMed] [Google Scholar]
- 42.Codogno P, Mehrpour M, Proikas-Cezanne T. Canonical and non-canonical autophagy: variations on a common theme of self-eating? Nat Rev Mol Cell Biol. 2012;13:7–12. doi: 10.1038/nrm3249. [DOI] [PubMed] [Google Scholar]
- 43.Hu P, Lai D, Lu P, Gao J, He H. ERK and Akt signaling pathways are involved in advanced glycation end product-induced autophagy in rat vascular smooth muscle cells. Int J Mol Med. 2012;29:613–8. doi: 10.3892/ijmm.2012.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Keil E, Höcker R, Schuster M, Essmann F, Ueffing N, Hoffman B, Liebermann DA, Pfeffer K, Schulze-Osthoff K, Schmitz I. Phosphorylation of Atg5 by the Gadd45β-MEKK4-p38 pathway inhibits autophagy. Cell Death Differ. 2013;20:321–32. doi: 10.1038/cdd.2012.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lorin S, Pierron G, Ryan KM, Codogno P, Djavaheri-Mergny M. Evidence for the interplay between JNK and p53-DRAM signalling pathways in the regulation of autophagy. Autophagy. 2010;6:153–4. doi: 10.4161/auto.6.1.10537. [DOI] [PubMed] [Google Scholar]
- 46.Mao K, Wang K, Zhao M, Xu T, Klionsky DJ. Two MAPK-signaling pathways are required for mitophagy in Saccharomyces cerevisiae. J Cell Biol. 2011;193:755–67. doi: 10.1083/jcb.201102092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mei S, Gu H, Ward A, Yang X, Guo H, He K, Liu Z, Cao W. p38 mitogen-activated protein kinase (MAPK) promotes cholesterol ester accumulation in macrophages through inhibition of macroautophagy. J Biol Chem. 2012;287:11761–8. doi: 10.1074/jbc.M111.333575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dirsch VM, Kirschke SO, Estermeier M, Steffan B, Vollmar AM. Apoptosis signaling triggered by the marine alkaloid ascididemin is routed via caspase-2 and JNK to mitochondria. Oncogene. 2004;23:1586–93. doi: 10.1038/sj.onc.1207281. [DOI] [PubMed] [Google Scholar]
- 49.Lamkanfi M, D’hondt K, Vande Walle L, van Gurp M, Denecker G, Demeulemeester J, Kalai M, Declercq W, Saelens X, Vandenabeele P. A novel caspase-2 complex containing TRAF2 and RIP1. J Biol Chem. 2005;280:6923–32. doi: 10.1074/jbc.M411180200. [DOI] [PubMed] [Google Scholar]
- 50.Panaretakis T, Laane E, Pokrovskaja K, Björklund AC, Moustakas A, Zhivotovsky B, Heyman M, Shoshan MC, Grandér D. Doxorubicin requires the sequential activation of caspase-2, protein kinase Cdelta, and c-Jun NH2-terminal kinase to induce apoptosis. Mol Biol Cell. 2005;16:3821–31. doi: 10.1091/mbc.E04-10-0862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Troy CM, Rabacchi SA, Xu Z, Maroney AC, Connors TJ, Shelanski ML, Greene LA. beta-Amyloid-induced neuronal apoptosis requires c-Jun N-terminal kinase activation. J Neurochem. 2001;77:157–64. doi: 10.1046/j.1471-4159.2001.t01-1-00218.x. [DOI] [PubMed] [Google Scholar]
- 52.Yoo BH, Wang Y, Erdogan M, Sasazuki T, Shirasawa S, Corcos L, Sabapathy K, Rosen KV. Oncogenic ras-induced down-regulation of pro-apoptotic protease caspase-2 is required for malignant transformation of intestinal epithelial cells. J Biol Chem. 2011;286:38894–903. doi: 10.1074/jbc.M111.290692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dewaele M, Maes H, Agostinis P. ROS-mediated mechanisms of autophagy stimulation and their relevance in cancer therapy. Autophagy. 2010;6:838–54. doi: 10.4161/auto.6.7.12113. [DOI] [PubMed] [Google Scholar]
- 54.Lee J, Giordano S, Zhang J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J. 2012;441:523–40. doi: 10.1042/BJ20111451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–60. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen Y, Azad MB, Gibson SB. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009;16:1040–52. doi: 10.1038/cdd.2009.49. [DOI] [PubMed] [Google Scholar]
- 57.Chen SY, Chiu LY, Maa MC, Wang JS, Chien CL, Lin WW. zVAD-induced autophagic cell death requires c-Src-dependent ERK and JNK activation and reactive oxygen species generation. Autophagy. 2011;7:217–28. doi: 10.4161/auto.7.2.14212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E, Baehrecke EH, Lenardo M. Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci U S A. 2006;103:4952–7. doi: 10.1073/pnas.0511288103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Du J, Guan T, Zhang H, Xia Y, Liu F, Zhang Y. Inhibitory crosstalk between ERK and AMPK in the growth and proliferation of cardiac fibroblasts. Biochem Biophys Res Commun. 2008;368:402–7. doi: 10.1016/j.bbrc.2008.01.099. [DOI] [PubMed] [Google Scholar]
- 60.Wang J, Whiteman MW, Lian H, Wang G, Singh A, Huang D, Denmark T. A non-canonical MEK/ERK signaling pathway regulates autophagy via regulating Beclin 1. J Biol Chem. 2009;284:21412–24. doi: 10.1074/jbc.M109.026013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tamm C, Zhivotovsky B, Ceccatelli S. Caspase-2 activation in neural stem cells undergoing oxidative stress-induced apoptosis. Apoptosis. 2008;13:354–63. doi: 10.1007/s10495-007-0172-7. [DOI] [PubMed] [Google Scholar]
- 62.Tiwari M, Herman B, Morgan WW. A knockout of the caspase 2 gene produces increased resistance of the nigrostriatal dopaminergic pathway to MPTP-induced toxicity. Exp Neurol. 2011;229:421–8. doi: 10.1016/j.expneurol.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Uchibayashi R, Tsuruma K, Inokuchi Y, Shimazawa M, Hara H. Involvement of Bid and caspase-2 in endoplasmic reticulum stress- and oxidative stress-induced retinal ganglion cell death. J Neurosci Res. 2011;89:1783–94. doi: 10.1002/jnr.22691. [DOI] [PubMed] [Google Scholar]
- 64.Gordy C, He YW. The crosstalk between autophagy and apoptosis: where does this lead? Protein Cell. 2012;3:17–27. doi: 10.1007/s13238-011-1127-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hou W, Han J, Lu C, Goldstein LA, Rabinowich H. Autophagic degradation of active caspase-8: a crosstalk mechanism between autophagy and apoptosis. Autophagy. 2010;6:891–900. doi: 10.4161/auto.6.7.13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Djavaheri-Mergny M, Maiuri MC, Kroemer G. Cross talk between apoptosis and autophagy by caspase-mediated cleavage of Beclin 1. Oncogene. 2010;29:1717–9. doi: 10.1038/onc.2009.519. [DOI] [PubMed] [Google Scholar]
- 67.Zhu Y, Zhao L, Liu L, Gao P, Tian W, Wang X, Jin H, Xu H, Chen Q. Beclin 1 cleavage by caspase-3 inactivates autophagy and promotes apoptosis. Protein Cell. 2010;1:468–77. doi: 10.1007/s13238-010-0048-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Choi SI, Kim BY, Dadakhujaev S, Oh JY, Kim TI, Kim JY, Kim EK. Impaired autophagy and delayed autophagic clearance of transforming growth factor β-induced protein (TGFBI) in granular corneal dystrophy type 2. Autophagy. 2012;8:1782–97. doi: 10.4161/auto.22067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–75. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Janssens S, Tinel A. The PIDDosome, DNA-damage-induced apoptosis and beyond. Cell Death Differ. 2012;19:13–20. doi: 10.1038/cdd.2011.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhivotovsky B, Orrenius S. Caspase-2 function in response to DNA damage. Biochem Biophys Res Commun. 2005;331:859–67. doi: 10.1016/j.bbrc.2005.03.191. [DOI] [PubMed] [Google Scholar]
- 72.Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, Kroemer G. Autophagy regulation by p53. Curr Opin Cell Biol. 2010;22:181–5. doi: 10.1016/j.ceb.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 73.Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D’Amelio M, Criollo A, Morselli E, Zhu C, Harper F, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10:676–87. doi: 10.1038/ncb1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Oliver TG, Meylan E, Chang GP, Xue W, Burke JR, Humpton TJ, Hubbard D, Bhutkar A, Jacks T. Caspase-2-mediated cleavage of Mdm2 creates a p53-induced positive feedback loop. Mol Cell. 2011;43:57–71. doi: 10.1016/j.molcel.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pattingre S, Bauvy C, Codogno P. Amino acids interfere with the ERK1/2-dependent control of macroautophagy by controlling the activation of Raf-1 in human colon cancer HT-29 cells. J Biol Chem. 2003;278:16667–74. doi: 10.1074/jbc.M210998200. [DOI] [PubMed] [Google Scholar]
- 76.Corcelle E, Djerbi N, Mari M, Nebout M, Fiorini C, Fénichel P, Hofman P, Poujeol P, Mograbi B. Control of the autophagy maturation step by the MAPK ERK and p38: lessons from environmental carcinogens. Autophagy. 2007;3:57–9. doi: 10.4161/auto.3424. [DOI] [PubMed] [Google Scholar]
- 77.Laussmann MA, Passante E, Düssmann H, Rauen JA, Würstle ML, Delgado ME, Devocelle M, Prehn JH, Rehm M. Proteasome inhibition can induce an autophagy-dependent apical activation of caspase-8. Cell Death Differ. 2011;18:1584–97. doi: 10.1038/cdd.2011.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang Q, Chen Z, Diao X, Huang S. Induction of autophagy-dependent apoptosis by the survivin suppressant YM155 in prostate cancer cells. Cancer Lett. 2011;302:29–36. doi: 10.1016/j.canlet.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 79.N’Diaye EN, Kajihara KK, Hsieh I, Morisaki H, Debnath J, Brown EJ. PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep. 2009;10:173–9. doi: 10.1038/embor.2008.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bergeron L, Perez GI, Macdonald G, Shi L, Sun Y, Jurisicova A, Varmuza S, Latham KE, Flaws JA, Salter JC, et al. Defects in regulation of apoptosis in caspase-2-deficient mice. Genes Dev. 1998;12:1304–14. doi: 10.1101/gad.12.9.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–11. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bauvy C, Meijer AJ, Codogno P. Assaying of autophagic protein degradation. Methods Enzymol. 2009;452:47–61. doi: 10.1016/S0076-6879(08)03604-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.