

Abstract
Congenital human cytomegalovirus (HCMV) infection can cause severe brain abnormalities. Apoptotic HCMV-infected brain cells have been detected in a congenitally infected infant. In biologically relevant human neural precursor cells (hNPCs), cultured in physiological oxygen tensions, HCMV infection (m.o.i. of 1 or 3) induced cell death within 3 days post-infection (p.i.) and increased thereafter. Surprisingly, its known anti-apoptotic genes, including the potent UL37 exon 1 protein (pUL37x1) or viral mitochondria-localized inhibitor of apoptosis (vMIA), which protects infected human fibroblasts (HFFs) from apoptosis and from caspase-independent, mitochondrial serine protease-mediated cell death, were expressed by 2 days p.i. Consistent with this finding, an HCMV UL37x1 mutant, BADsubstitutionUL37x1 (BADsubUL37x1) induced cell death in hNPCs (m.o.i. = 1) to level which were indistinguishable from parental virus (BADwild-type)-infected hNPCs. Surprisingly, although BADsubUL37x1 is growth defective in permissive HFFs, it produced infectious progeny in hNPCs with similar kinetics and to levels comparable to BADwild-type-infected hNPCs (m.o.i. = 1). While delayed at a lower multiplicity (m.o.i. = 0.3), the BADsubUL37x1 mutant reached similar levels to revertant within 12 days, in contrast to its phenotype in HFFs. The inability of pUL37x1/vMIA to protect hNPCs from HCMV-induced cell death did not result from impaired trafficking as pUL37x1/vMIA trafficked efficiently to mitochondria in transfected hNPCs and in HCMV-infected hNPCs. These results establish that pUL37x1/vMIA, although protective in permissive HFFs, does not protect HCMV-infected hNPCs from cell death under physiologically relevant oxygen tensions. They further suggest that pUL37x1/vMIA is not essential for HCMV growth in hNPCs and has different cell type-specific roles.
Introduction
In developed countries, congenital human cytomegalovirus (HCMV) infection is the leading viral cause of neurological birth defects, including non-genetic sensorineural hearing loss, post-natal seizures and epilepsy (Bristow et al., 2011; Revello & Gerna, 2004; Rosenthal et al., 2009; Suzuki et al., 2008). The neuropathology associated with congenital HCMV infections of the central nervous system (CNS) is not well understood; however, prominent abnormal morphologies include polymicrogyria and microcephaly (Becroft, 1981; Malm & Engman, 2007; Perlman & Argyle, 1992). Abnormal neural development and loss of nervous tissue associated with HCMV infection suggests that its pathology involves death of neural stem and progenitor cells.
HCMV preferentially infects cells in the ventricular zone and subventricular zone (SVZ) of the developing human brain (Becroft, 1981; Perlman & Argyle, 1992). The SVZ is a neurogenesis site in mammals through the differentiation of neural stem cells into neuronal and glial progenitors (Doetsch et al., 1999; Lim & Alvarez-Buylla, 1999; Menn et al., 2006; Quiñones-Hinojosa et al., 2006; Schwartz et al., 2003). The recent isolation and culture of human neural precursor cells (hNPCs) from aborted fetuses (Cheeran et al., 2005; Lokensgard et al., 1999; Odeberg et al., 2006) and from neonatal autopsy tissue (Schwartz et al., 2003) have provided biologically relevant in vitro models for studying HCMV infection of hNPCs (Luo et al., 2008, 2010).
HCMV infection of hNPCs can alter cell fate determination. Differentiation along a neuronal lineage is inhibited by HCMV infection of differentiating hNPCs at early stages of fate determination (Odeberg et al., 2006). HCMV infection of hNPCs can cause premature and abnormal neuronal differentiation (Luo et al., 2010). Furthermore, HCMV infection of hNPCs inhibits astrocytic differentiation through its late proteins (Odeberg et al., 2007).
Programmed cell death of hNPCs and neurons occurs during normal CNS development (Lossi et al., 2005; Narayanan, 1997). Disruptions in the balance of survival and death of neural cell lineages can negatively impact the developing CNS (Blomgren et al., 2007). Thus, HCMV-induced cell death of hNPCs could alter the balance of progenitor and committed cells, which would predictably alter the cellular composition of the developing brain.
Apoptotic cells closely associated with acute infection were observed in a CNS specimen from an infant with congenital HCMV encephalitis (DeBiasi et al., 2002). Consistently, HCMV infection induced apoptosis in primary hNPCs, following their differentiation along neuronal lineage (Odeberg et al., 2006). Apoptosis was also observed in HCMV-infected primary astrocytes (Lokensgard et al., 1999). In a conflicting study, HCMV infection was shown to cause neuronal injury within differentiating human neuroepithelial cultures, but evidence of apoptosis was not found (McCarthy et al., 2000). Thus, the ability of HCMV to induce apoptosis in neural stem cell populations and their differentiated derivatives remains controversial.
HCMV encodes multiple anti-apoptotic proteins, including the UL37 exon 1 protein (pUL37x1), also known as viral mitochondria-localized inhibitor of apoptosis (vMIA), UL36, UL38, major immediate-early (IE) proteins as well as an anti-apoptotic beta 2.7 (β2.7) RNA (Goldmacher et al., 1999; Hayajneh et al., 2001; Reeves et al., 2007; Skaletskaya et al., 2001; Terhune et al., 2007; Zhu et al., 1995). Of these, pUL37x1/vMIA and β2.7 RNA inhibit mitochondrial-mediated apoptosis. pUL37x1/vMIA traffics from the endoplasmic reticulum to the outer mitochondrial membrane (OMM), where it binds and blocks Bax pro-apoptotic activity in human and mouse fibroblasts and HeLa cells (Arnoult et al., 2004; Bozidis et al., 2010; Mavinakere et al., 2006; Norris & Youle, 2008; Poncet et al., 2004; Williamson & Colberg-Poley, 2010a). pUL37x1/vMIA also protects infected cells from late caspase-independent cell death induced by mitochondrial HtrA2/Omi (McCormick et al., 2008). In contrast, β2.7 RNA interacts with mitochondrial complex I, which prevents its relocalization from the inner mitochondrial membrane to discrete perinuclear sites (Reeves et al., 2007). HCMV pUL37x1/vMIA ensures efficient replication in permissive human foreskin fibroblasts (HFFs) during its protracted growth (Dunn et al., 2003; Reboredo et al., 2004; Sharon-Friling et al., 2006; Yu et al., 2003).
Because of the findings of HCMV-infected apoptotic cells in the CNS of a congenitally infected infant, we hypothesized that HCMV infection causes cell death of hNPCs despite the production of its potent anti-apoptotic product, pUL37x1/vMIA. We, therefore, examined cell death during HCMV infection of hNPCs, a biologically relevant model of HCMV infection of the CNS, further strengthened by their culture in physiological oxygen tensions (Panchision, 2009). Indeed, we found that HCMV infection detectably induced cell death of hNPCs even though its known anti-apoptotic products were produced prior to HCMV-induced apoptosis. Further, while pUL37x1/vMIA was produced and trafficked efficiently to mitochondria throughout HCMV infection of hNPCs, it was not protective against cell death during HCMV growth in hNPCs, in contrast to HFFs.
Results
HCMV infection induces cell death in biologically relevant hNPCs grown in physiological oxygen tensions
Cell death was examined in hNPCs infected with bacterial artificial chromosome HCMV strain AD169 wild-type (BADwt) virus (Sharon-Friling et al., 2006) grown in physiological oxygen tensions. Flow cytometry analysis using annexin V (AV) and propidium iodide (PI) staining allows for the detection of viable cells (AV−/PI−), early (AV+/PI−) or late apoptosis (AV+/PI+), as well as non-apoptotic cell death (AV−/PI+). hNPCs were mock- or HCMV-infected (m.o.i. of 3) and examined at 5 days post-infection (p.i.) (Fig. 1a). At 5 days p.i., we observed late apoptosis in BADwt- (26.1 %) and mock-infected hNPCs (3.6 %).
Fig. 1.
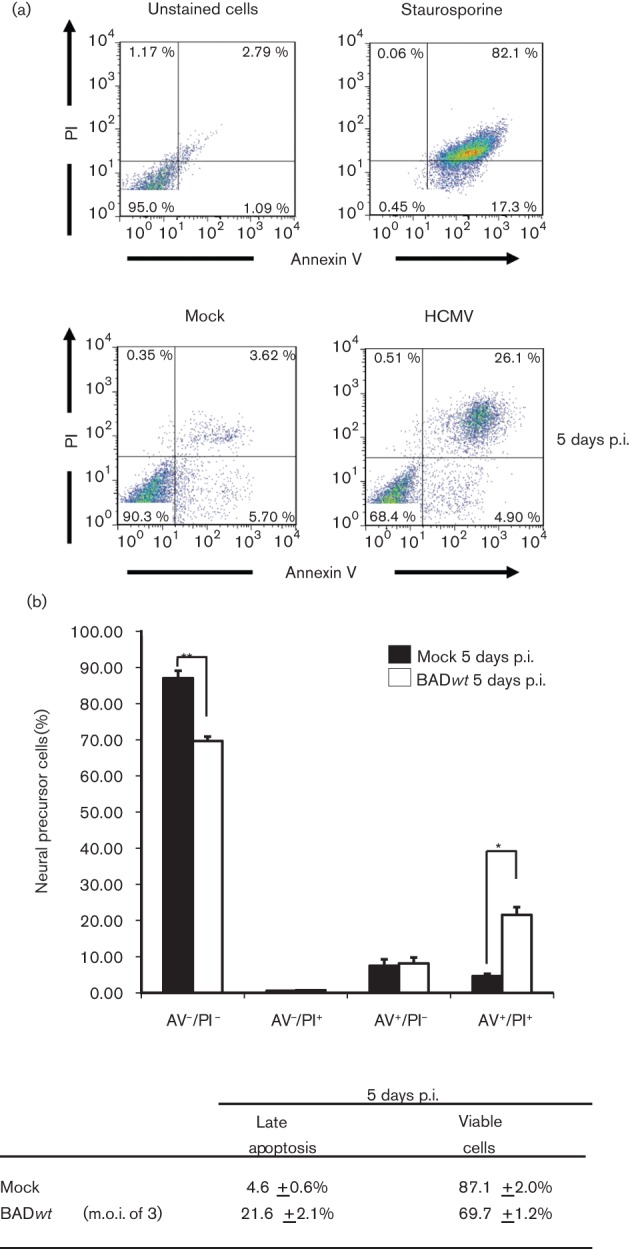
HCMV-induced apoptosis of hNPCs under physiological oxygen tensions. (a) Flow cytometry analyses. (Top) Control hNPCs were unstained or staurosporine-treated and analysed for apoptosis by flow cytometry as described in Methods. (Bottom) hNPCs, cultured in 5 % oxygen, were mock-infected (left) or BADwt-infected (right, m.o.i. of 3), harvested at 5 days p.i. and analysed by flow cytometry. (b) Quantification of HCMV-induced apoptosis of hNPCs under physiological oxygen tensions. Apoptosis induction and cell viability of HCMV-infected hNPCs were analysed in three independent experiments as in (a). Results of each experiment were averaged based on the population of cells in each quadrant. Results are displayed for mock-infected hNPCs (black) and BADwt-infected hNPCs (white) at 5 days p.i. Analysis is shown as ±sem (n = 3, *P<0.05, **P<0.01, Student’s unpaired t-test). The average values for late (AV+/PI+) apoptosis and viable cell numbers are listed in the table below.
These experiments were repeated and cell death was quantified by flow cytometry (Fig. 1a, b). By 5 days p.i., there was a significant increase in late apoptosis in BADwt- versus mock-infected hNPCs. Total AV+ cells at 5 days p.i. in BADwt- (29.8 %) and mock-infected (12.1 %) hNPCs verified the induction of cell death. There was a commensurate decrease in viable cells at 5 days p.i. of BADwt-infected versus mock-infected hNPCs.
Detection of fragmented nuclei in HCMV-infected hNPCs
In a complementary approach, we examined chromatin condensation and changes in morphology associated with apoptosis (Fig. 2). Nuclear fragmentation (indicated by arrowheads) and membrane blebbing (highlighted by arrows) were clearly detected in HCMV-infected hNPCs but not in mock-infected hNPCs at 3 days p.i. These morphological changes in the HCMV-infected hNPCs are consistent with HCMV induction of apoptosis within 3 days p.i.
Fig. 2.
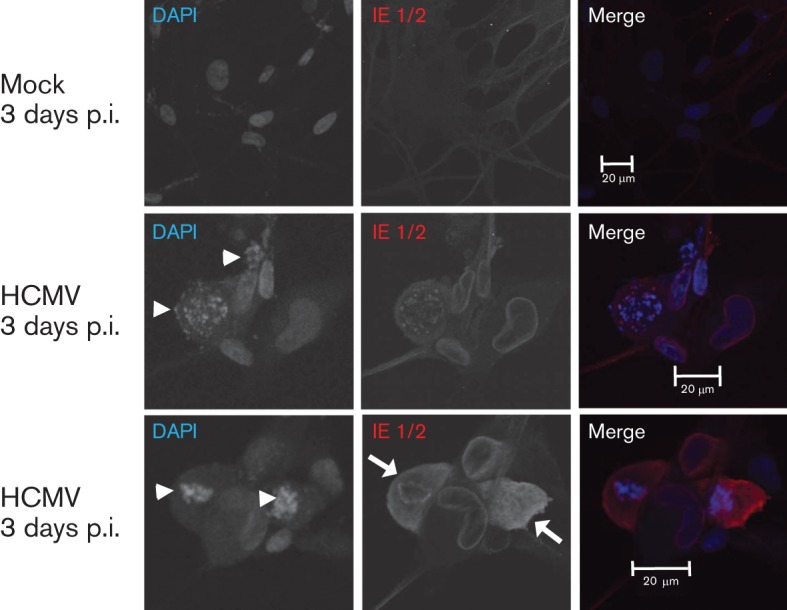
Nuclear fragmentation and membrane blebbing of HCMV-infected hNPCs at 3 days p.i. hNPCs were mock- or HCMV-infected (strain AD169) at an m.o.i. of 3. Cells were fixed at 3 days p.i., stained with DAPI (blue) and with anti-IE1/2 antibody (red). The arrowheads and arrows indicate nuclear fragmentation and membrane blebbing, respectively.
Transcription of HCMV anti-apoptotic genes in infected hNPCs
The HCMV genome encodes multiple anti-apoptotic products (Goldmacher et al., 1999; Hayajneh et al., 2001; Skaletskaya et al., 2001; Terhune et al., 2007; Zhu et al., 1995). Using RT-PCR, we examined whether these anti-apoptotic genes are expressed in infected hNPCs (Fig. 3a). UL37x1 RNA was detected by 2 h p.i. and increased thereafter (left) in BADwt- but not in mock-infected hNPCs. UL36, UL37x3, UL38, IE1/2 and β2.7 RNAs were also detected in BADwt-infected hNPCs (right). Thus, all known HCMV anti-apoptotic genes are transcribed in infected hNPCs by 2 days p.i., prior to detected apoptosis in the HCMV-infected hNPCs within 3 days p.i. (Fig. 2). Thus, the anti-apoptotic products do not protect infected hNPCs from HCMV-induced cell death.
Fig. 3.
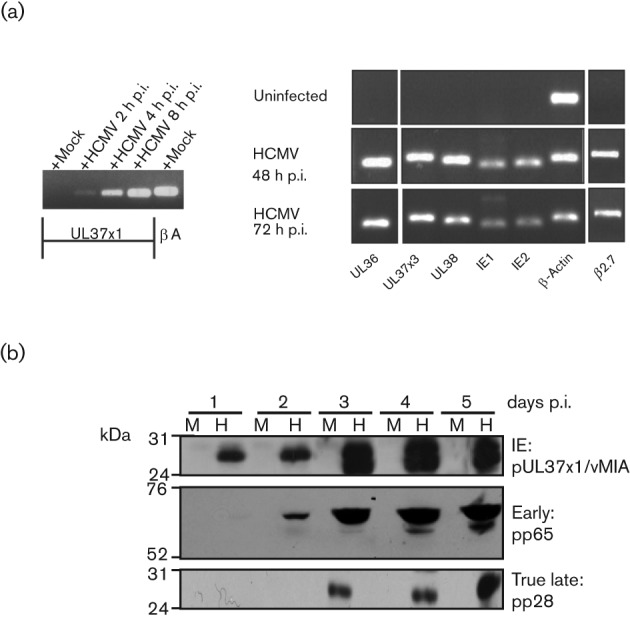
HCMV anti-apoptotic genes are expressed in hNPCs. (a) Transcription of HCMV anti-apoptotic genes in infected hNPCs. hNPCs were mock- or BADwt-infected (m.o.i. of 3) and cultured under low oxygen conditions. Total RNA was isolated from mock- and HCMV-infected hNPCs at the indicated times of infection and analysed by RT-PCR using UL37x1 or β-actin (left) and UL36, UL37x3, UL38, IE1, IE2 and β2.7 primers (right). (b) Kinetics of HCMV IE, early and true late protein production within HCMV-infected hNPCs. hNPCs were mock- (M) or BADwt-infected (H) (m.o.i. of 3) and cultured under low oxygen conditions. Total protein lysates were collected at the indicated times following infection. Total proteins (20 µg) were resolved by SDS-PAGE and analysed by Western blots for IE (pUL37x1/vMIA), early (pp65) and true late (pp28) proteins.
Transition of HCMV temporal gene expression in infected hNPCs
We next examined HCMV temporal gene expression in infected hNPCs (Fig. 3b). Consistent with the results above, pUL37x1/vMIA, an IE protein (Al-Barazi & Colberg-Poley, 1996; Kouzarides et al., 1988), was detected in BADwt-infected hNPCs by 1 day p.i. and increased thereafter. The early protein (pp65) and the true late protein (pp28) were detected by 2 and 3 days p.i., respectively, and increased through 5 days p.i. These results show temporally regulated HCMV gene expression in infected hNPCs and transition to early (2 days p.i.) and late (3 days p.i.) phases. Further, pUL37x1/vMIA was detected through all temporal phases in HCMV-infected hNPCs.
Cell death induction by BADsubUL37x1 and BADwt viruses
pUL37x1/vMIA is strongly anti-apoptotic in HeLa cells or HFFs and its activity is essential in HCMV-infected HFFs (Goldmacher et al., 1999; Hayajneh et al., 2001; Reboredo et al., 2004; Sharon-Friling et al., 2006). Furthermore, pUL37x1/vMIA protects HFFs from caspase-independent cell death caused by mitochondrial HtrA2/Omi at very late times of infection (McCormick et al., 2008). To determine if pUL37x1/vMIA has protective roles in infected hNPCs, hNPCs were infected with BADwt or BADsubUL37x1 at a lower m.o.i. of 1 and analysed by flow cytometry (Fig. 4a). At 5 days p.i., late apoptosis was slightly increased in BADwt-infected cells (8.5 %), while early and late apoptosis were increased in BADsubUL37x1-infected cells (7.9 and 5.98 %, respectively) above mock-infected cells (3.5 and 1.9 %, respectively). At 8 days p.i., early and late apoptosis levels were further increased in hNPCs in BADwt (6.6 and 40.4 %, respectively) or BADsubUL37x1 (11.0 and 26.5 %, respectively) above mock-infected hNPCs (3.0 and 3.3 %, respectively). Thus, cell death levels in BADsubUL37x1- and BADwt-infected hNPCs are not significantly different, suggesting that pUL37x1/vMIA does not have protective activities in hNPCs.
Fig. 4.
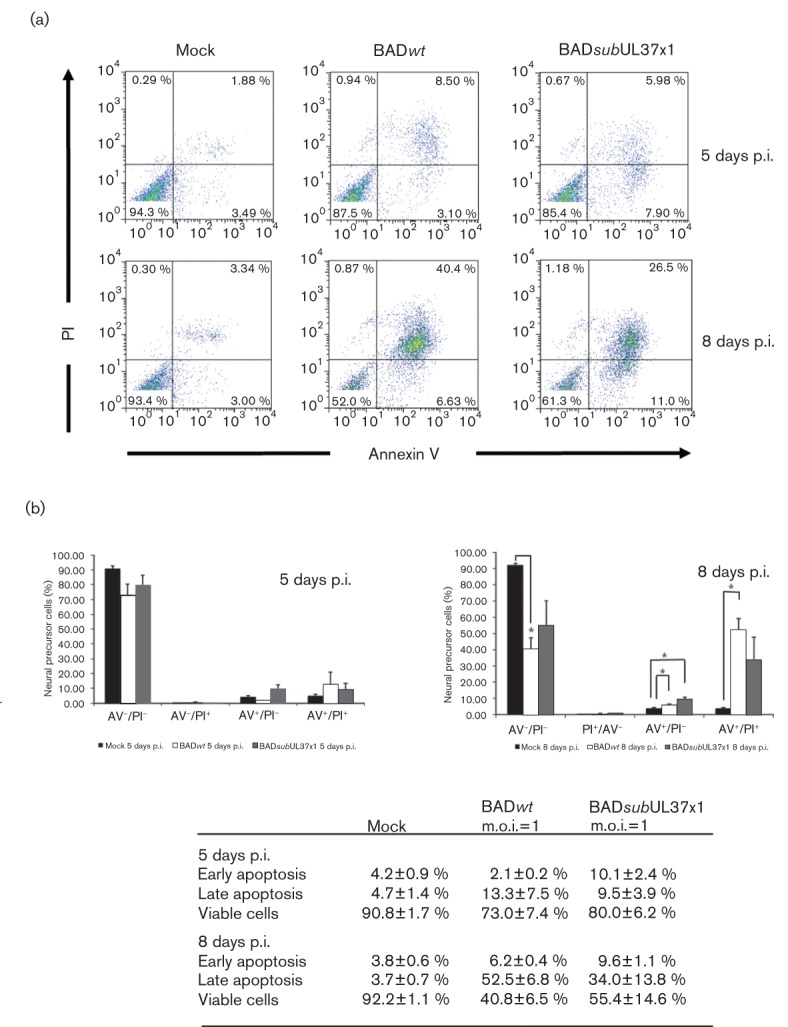
Induction of apoptosis in BADsubUL37x1-infected and in BADwt-infected hNPCs. (a) hNPCs were mock- (left), BADwt- (m.o.i. of 1) (middle) or BADsubUL37x1- (m.o.i. of 1) (right) infected. hNPCs were harvested at 5 and 8 days p.i. and analysed by flow cytometry. (b) Quantification of apoptosis induction in BADsubUL37x1-infected and in BADwt-infected hNPCs. Apoptosis induction was analysed in three independent experiments. hNPCs were mock- (black), BADwt- (white) or BADsubUL37x1- (grey) infected. The average values for early (AV+/PI−) and late apoptosis (AV+/PI+), viable cells (AV−/PI−), are listed on the table (bottom). Analysis is shown as ±sem (n = 3, *P<0.05, Student’s unpaired t-test).
Quantification of three independent experiments showed that early (BADsubUL37x1) and late (both) apoptosis of hNPCs was modestly induced by HCMV infection at 5 days p.i. (Fig. 4b). At 8 days p.i., there was a further increase in early and late apoptosis in HCMV-infected above mock-infected hNPCs and a commensurate decrease in viability of infected cells. This was again coupled with a significant decrease in cell viability in infected hNPCs versus mock-infected cells. Importantly, there was no significant difference in total AV+ cells in BADwt-infected hNPCs at 5 and 8 days p.i. (15.3 and 58.5 %, respectively) and BADsubUL37x1-infected hNPCs (19.6 and 43.6 %, respectively). These results suggest that the HCMV UL37x1 mutant induced cell death similarly to parental HCMV at the times studied. Together, these results suggest that pUL37x1/vMIA does not have protective activity in HCMV-infected hNPCs.
HCMV gene expression and infectious progeny production in BADsubUL37x1-infected hNPCs
To determine if temporal gene expression was affected by the absence of pUL37x1/vMIA during HCMV infection of hNPCs, the production of viral IE (IE1/2 and pUL37x1/vMIA), early (pp65) and true late (pp28 and major capsid protein, MCP) proteins was monitored in BADsubUL37x1-infected hNPCs (Fig. 5). BADwt-infected hNPCs were examined in parallel. HCMV IE1-72 was detected in BADwt- and BADsubUL37x1-infected hNPCs within 1 day p.i. and increased thereafter; whereas, IE2-86 was weakly detected at 1 (BADwt) and 2 days p.i. (BADsubUL37x1) and increased thereafter. pUL37x1/vMIA was detected within 1 day p.i. in BADwt-infected cells but not in the BADsubUL37x1-infected hNPCs, as expected. Early gene expression was observed within 2 days p.i. in BADsubUL37x1-infected hNPCs and by 3 days p.i. in BADwt-infected hNPCs. True late protein pp28 was detected by 4 days p.i. in BADwt-infected hNPCs, but severely reduced in BADsubUL37x1-infected hNPCs. Another true late gene product, MCP, was detected in BADwt- and in BADsubUL37x1-infected hNPCs, albeit at a somewhat reduced levels in UL37x1 mutant-infected hNPCs. This pattern of gene expression verifies the identity of BADsubUL37x1 and the temporal transition of HCMV gene expression with comparable kinetics to those observed in wt-infected hNPCs.
Fig. 5.
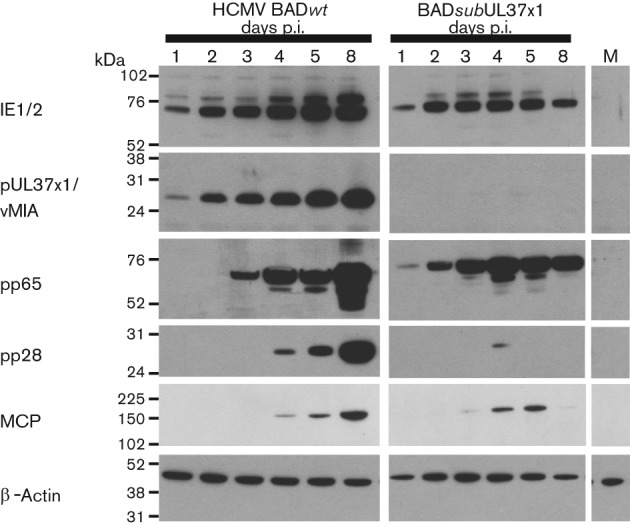
Regulated temporal HCMV gene expression in BADsubUL37x1-infected hNPCs. hNPCs were mock-, BADwt- or BADsubUL37x1- (m.o.i. of 1) -infected. Total protein was harvested at the indicated times. Total proteins (20 µg) were resolved by SDS-PAGE and analysed by Western blot analysis for IE (IE1/2, pUL37x1/vMIA), early (pp65) and true late (pp28, MCP) proteins. β-Actin was used as a loading control.
HCMV infectious progeny production in BADwt- and BADsubUL37x1-infected hNPCs
Because cell death and temporal gene expression were simultaneously observed, we examined production of infectious HCMV progeny in BADwt- and BADsubUL37x1-infected hNPCs at an m.o.i. of 1 (Fig. 6a). Production of BADwt progeny (left) was minimally detected in hNPCs by 3 days p.i. (6.4×104 p.f.u.), increased at 5 days p.i. (4.7×105 p.f.u.), but remained similar at 8 (3.9×105 p.f.u.) and 10 days p.i. (1.5×105 p.f.u.). These data demonstrate production of infectious HCMV progeny by BADwt-infected hNPCs, grown under physiological oxygen conditions.
Fig. 6.

Production of BADwt and BADsubUL37x1 infectious progeny in hNPCs. (a) hNPCs were infected with BADwt (left) or with BADsubUL37x1 (right) at an m.o.i. of 1. Supernatants were harvested at the indicated times and the levels of total infectious progeny per well were determined by plaque formation on indicator HFFs. Shown is a representative result of two independent experiments with duplicate samples for each. (b) hNPCs were infected with BADrev (left) or with BADsubUL37x1 (right) at an m.o.i. of 0.3 and infectious progeny was determined as in (a). Values from triplicate wells were averaged for each time point.
Similar to BADwt-infected hNPCs, BADsubUL37x1-infected hNPCs (right, m.o.i. of 1) produced infectious HCMV progeny (1×105 p.f.u. per well) by 5 days p.i. BADsubUL37x1 infectious progeny production increased to 3.6×105 p.f.u. per well by 8 days p.i. and then decreased to 1.2×105 p.f.u. per well by 10 days p.i. In contrast to HFFs (Sharon-Friling et al., 2006), these data demonstrate that BADsubUL37×1 is not growth defective in hNPCs. Thus, pUL37x1/vMIA appears to be non-essential for HCMV replication in hNPCs. Further, these results suggest that very low expression of pp28 is sufficient for HCMV growth in hNPCs. In contrast, when the m.o.i. was reduced to 0.3, BADsubUL37x1 grew more slowly than its BADrevertant (BADrev) (Fig. 6b). Infectious BADrev progeny was detected within 6 days p.i. (1.2×105 p.f.u.); whereas, BADsubUL37x1 levels were ~1000-fold lower at 6 (93 p.f.u.) and 7 days p.i. (557 p.f.u.). Nonetheless, in contrast to its replication in HFFs (Sharon-Friling et al., 2006), BADsubUL37x1 did eventually (by 12 days p.i.) produce infectious progeny to levels (1.5×105 p.f.u.) comparable to BADrev.
Trafficking of pUL37x1/vMIA to the mitochondria in transfected hNPCs
pUL37x1/vMIA functions as an anti-apoptotic protein at the OMM by blocking Bax-mediated permeabilization of the OMM and by inhibiting the mitochondrial serine protease HtrA2/Omi at very late times of infection (Arnoult et al., 2004; Goldmacher et al., 1999; Hayajneh et al., 2001; McCormick et al., 2008; Poncet et al., 2004; Reboredo et al., 2004). To determine whether pUL37x1/vMIA is defective for its protective role in hNPCs due to an inability to traffic properly to mitochondria, its co-localization with a mitochondrial marker was examined in transfected hNPCs expressing pUL37x1/vMIA wt-monomeric enhanced green fluorescent protein (mEGFP) (Fig. 7a). pUL37x1/vMIA wt-mEGFP showed extensive co-localization with a mitochondrial marker (dsRed1Mito) in transfected hNPCs. This result demonstrated that pUL37x1/vMIA traffics to mitochondria in transfected hNPCs comparably to HFFs (Bozidis et al., 2008; Williamson & Colberg-Poley, 2010b) where it has anti-apoptotic and protective activities (McCormick et al., 2008; Poncet et al., 2006; Reboredo et al., 2004).
Fig. 7.
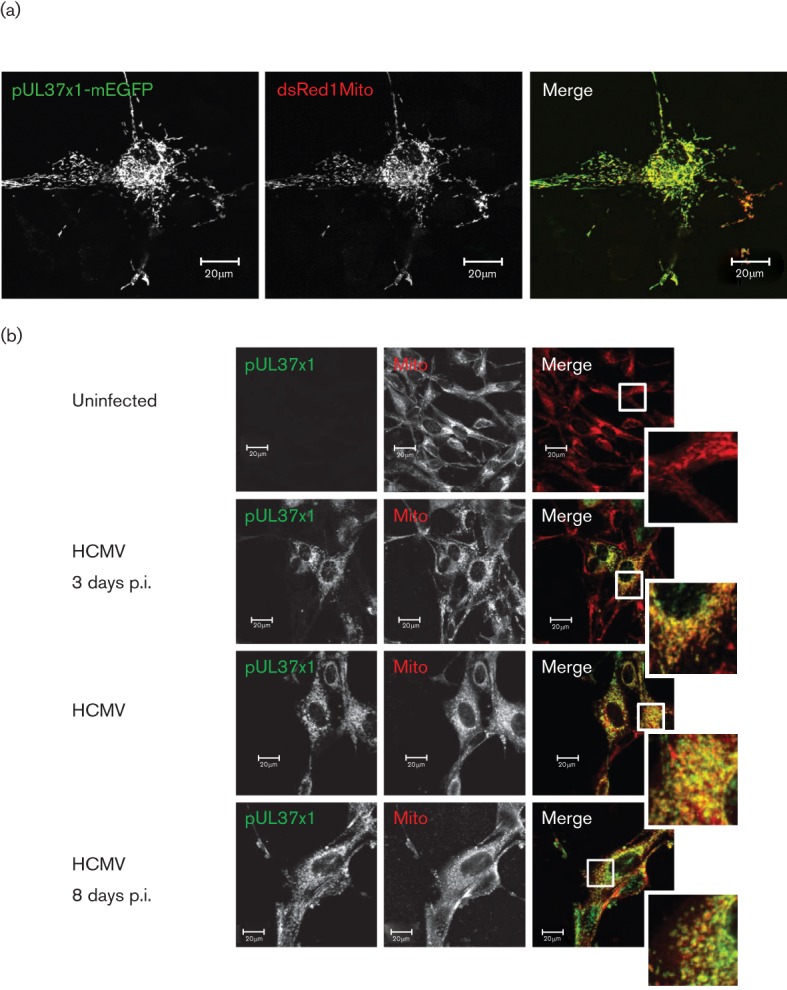
(a) Co-localization of pUL37x1/vMIA and a mitochondrial marker in transfected hNPCs. hNPCs were nucleofected with dsRed1Mito (red) and pUL37x1-mEGFP (green) expression vectors. Cells were fixed and imaged by confocal microscopy. (b) Co-localization of pUL37x1/vMIA with a mitochondrial marker in HCMV-infected hNPCs. hNPCs were uninfected (top) or HCMV-infected (strain AD169, m.o.i. = 1). Cells were fixed at the indicated times and stained with anti-pUL37x1/vMIA (green) and -mitochondrial (red) antibodies.
We further examined pUL37x1/vMIA trafficking to mitochondria in HCMV-infected hNPCs at multiple times of infection (Fig. 7b). pUL37x1/vMIA partially co-localized with a mitochondrial marker at 3, 5 and 8 days p.i. Quantification of co-localization using MetaMorph showed that ≥90 % of the pUL37x1-mEGFP signal co-localized with the mitochondrial marker at 3 (90.0 %), 5 (92.4 %) and 8 (91.5 %) days p.i. These results show that pUL37x1/vMIA traffics efficiently to mitochondria from early through late times of infection in HCMV-infected hNPCs, similarly to HCMV-infected HFFs (Bozidis et al., 2010; Mavinakere & Colberg-Poley, 2004).
Discussion
Our studies examine HCMV-induced cell death of uncommitted primary hNPCs, grown in physiological oxygen tensions, and the potential requirement for its protective product, pUL37x1/vMIA, for HCMV growth in this biologically relevant model of congenital HCMV infection. While HCMV infection can protect a neuronal cell line from rotenone-induced cell death (Reeves et al., 2007), HCMV infection induced apoptosis of primary differentiating hNPCs under ambient oxygen conditions (Lokensgard et al., 1999; Odeberg et al., 2006), as well as apoptosis-independent cell death of neural epithelial cells (McCarthy et al., 2000). We detected cell death in HCMV-infected hNPCs cultured in low oxygen conditions, which more accurately reflect physiological conditions during brain development. HCMV infection also induced cell death of hNPCs under ambient oxygen conditions (R. L. Hildreth and A. M. Colberg-Poley, unpublished results). Tellingly, apoptosis in uninfected hNPCs grown in low oxygen conditions remained lower than in uninfected hNPCs grown in ambient oxygen. Thus, the potential confounding effects of ambient oxygen on hNPC cell death were reduced by our low oxygen culture conditions.
Induction of apoptosis of undifferentiated hNPCs by HCMV infection was measured by AV/PI binding and nuclear fragmentation. Odeberg and co-workers found that HCMV infection induced apoptosis of differentiating neuroblasts (Odeberg et al., 2006) and of differentiating astrocytes (Odeberg et al., 2007). In contrast, Cheeran et al. (2005) did not detect apoptosis in HCMV-infected (20–30 % infected) immature cells from fetal brains as measured by fragmented DNA up to 7 days (168 h) p.i., although they detected apoptosis in astrocytes by AV binding by 3 days p.i. (Lokensgard et al., 1999). Our detection of cell death in HCMV-infected hNPCs may have resulted from the use of earlier markers of apoptosis (AV binding), the higher efficiency of HCMV infection (>90 %), and the expanded time frames (from 1 to 8 days p.i.) used in our experiments. We verified apoptosis by examining the appearance of nuclear fragmentation from 3 days p.i. in the HCMV-infected hNPCs. Because the cells were isolated at distinct gestational stages and their culture conditions were dissimilar (Cheeran et al., 2005; McCarthy et al., 2000), the hNPCs are probably at different developmental stages and may respond distinctly to HCMV infection.
The HCMV temporal cascade of gene expression was observed when cell death occurred. BADwt virus gave similar gene expression profiles and levels of progeny viruses as strain Towne infection of the same hNPCs (SC30) (Luo et al., 2008). BADsubUL37x1 is markedly growth defective in HFFs with a 5 day delay and a 103–104 reduction in infectious progeny over an extended time frame of 11 days (Sharon-Friling et al., 2006). Surprisingly, the HCMV UL37x1/vMIA mutant also showed regulation of temporal gene expression and production of infectious progeny in hNPCs, despite the absence of pUL37x1 and reduced levels of pp28. Although there was an initial delay in the production of BADsubUL37x1 mutant progeny at a lower (0.3) multiplicity, within 12 days the mutant progeny reached levels comparable to those of the revertant virus. Thus, in contrast to its requirement for HCMV growth in HFFs (Sharon-Friling et al., 2006), we conclude that pUL37x1/vMIA is not essential for HCMV replication in hNPCs. Because reduced pp28 levels were detected in UL37x1 mutant-infected hNPCs, it appears low levels of pp28 suffice for HCMV growth in hNPCs. Conversely, pp28 accumulation in HFFs is a prerequisite for its multimerization within the assembly compartment, an essential step in the envelopment and production of infectious virions (Britt et al., 2004; Seo & Britt, 2008). Thus, cell type-specific roles of HCMV pUL37x1/vMIA and pp28 are now emerging.
Expression of all the HCMV anti-apoptotic loci was detected in infected hNPCs. Of these, pUL37x1/vMIA protects infected HFFs from Bax-induced mitochondrial membrane permeabilization from early through late times of infection (Arnoult et al., 2004; Poncet et al., 2004). In addition, pUL37x1/vMIA also protects HFFs at very late times of infection from caspase-independent cell death induced by the mitochondrial serine protease, HtrA2/Omi (McCormick et al., 2008). At very late times (12–13 days p.i.) of HCMV infection, we detected increasing levels of AV+/PI+ cells (65–90 %) in the infected hNPCs (R. L. Hildreth and A. M. Colberg-Poley, unpublished results), suggesting that pUL37x1/vMIA does not block either apoptosis (during early times of infection) or caspase-independent cell death (at very late times). We also detected the anti-apoptotic IE1/IE2 proteins (Zhu et al., 1995) throughout HCMV infection of hNPCs, even when cell death was increasing. Thus, HCMV anti-apoptotic gene products did not effectively protect hNPCs from HCMV-induced apoptosis or from later caspase-independent cell death.
The inability of pUL37x1/vMIA to protect hNPCs was substantiated by examining cell death following infection with BADsubUL37x1, which lacks the UL37x1 ORF. A lower multiplicity (m.o.i. of 1) was used for these experiments, resulting in initially lower levels of apoptosis. Nonetheless, the UL37x1 mutant induced cell death of hNPCs as measured by AV binding to levels similar to the parental virus at the same multiplicity. Importantly, the UL37x1 mutant and wild-type under these conditions were statistically indistinguishable throughout the time-course studied. We therefore conclude that pUL37x1/vMIA does not have anti-apoptotic or pro-apoptotic activities in infected hNPCs.
This lack of its protective effects could result from the inability of pUL37x1/vMIA to traffic to the OMM. However, we found that pUL37x1/vMIA traffics efficiently to mitochondria from early through late times of infection when apoptosis was detected. Alternatively, pUL37x1/vMIA may not efficiently bind Bax or regulate mitochondrial HtrA2/Omi in hNPCs. An alternative explanation might be that pUL37x1/vMIA binding of Bax may result in a deleterious effect on hNPCs. Intriguingly, Bax can inhibit viral induced neuronal apoptosis (Lewis et al., 1999), and cleavage of the loop regions of Bcl-xL by calpains switches this protein from anti-apoptotic to a pro-apoptotic regulator (Nakagawa & Yuan, 2000). It is not known whether Bax is pro-apoptotic or anti-apoptotic in hNPCs. If Bax has anti-apoptotic activities in hNPCs, binding by pUL37x1/vMIA may affect this function and cause apoptosis. However, use of BADsubUL37x1, which did not alter the percentages of apoptotic hNPCs, suggests that pUL37x1/vMIA does not have pro-apoptotic or anti-apoptotic functions in hNPCs.
Our observations that the cell death preventing mechanisms of HCMV appear inoperative in hNPCs may be related to the apparently disparate observations of Luo et al. (2010) that HCMV causes a premature and abnormal differentiation of hNPCs. That is, it may be advantageous, during neural development, not only to prune away excess numbers of neurons, for example as happens during normal development (Kim & Sun, 2011; Miura, 2011) but to ensure elimination of differentiating populations whose differentiation programmes have gone awry. It may be, then, that there is an overt, but not yet described, mechanism in hNPCs that prevents the anti-apoptotic HCMV machinery for the express purpose of eliminating those very cells whose abnormal differentiation, due to the infection itself, may well significantly alter neural development. Thus, this inhibition of the anti-apoptotic effects of HCMV in hNPCs may be a protective mechanism.
The ability of HCMV to infect hNPCs, whose subsequent death could result in associated neurological pathologies, encourages their use under low oxygen conditions as a model system for HCMV congenital infection of the brain. The induction of cell death in hNPCs, combined with its ability to affect proliferation and differentiation, predictably will negatively impact the generation of precursor and committed lineages required for normal development of the CNS.
Methods
Cell culture and viruses.
The hNPC line, SC30, was derived from cortical tissue harvested from the forebrain of a premature neonate, who died of natural causes unrelated to HCMV infection (Schwartz et al., 2003). hNPCs were grown in Ham’s F-12 Dulbecco’s modified Eagle’s high glucose medium (Irvine Scientific), supplemented with 10 % BIT9500 (StemCell Technologies) and 1 % GlutaMAX (Sigma) and antibiotic-antimycotic (Invitrogen). hNPCs were maintained in complete medium supplemented daily with basic fibroblast growth factor (R&D Systems) and epidermal growth factor (R&D Systems) (10 ng ml−1). hNPCs were seeded onto tissue culture dishes coated at 37 °C with polyornithine (100 µg ml−1; Sigma) and fibronectin (5 µg ml−1; Roche) and fed daily, replacing half of the medium with fresh, mitogen-supplemented medium. Low oxygen conditions (5 %) were maintained by the use of an atmospheric-regulated remote chamber and glove box (Biospherix) as described previously (Pistollato et al., 2007). The atmosphere within the remote chambers was regulated at a constant level of 5 % for both O2 and CO2, with brief exposure (<30 min) during subculture to ambient conditions.
Primary HFFs (Viromed) were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10 % FBS (Gemini BioProducts) and 1 % penicillin/streptomycin (Invitrogen) as described previously (Colberg-Poley & Santomenna, 1988; Santomenna & Colberg-Poley, 1990).
HCMV recombinant viruses BADwt, BADrev and BADsubUL37x1 were kindly provided by Dr Tom Shenk for these studies (Sharon-Friling et al., 2006; Yu et al., 2002). Stocks of HCMV (strain AD169; ATCC), BADwt, BADrev or BADsubUL37x1 were generated and titrated as described previously (Colberg-Poley & Santomenna, 1988). Mock stocks, uninfected HFFs grown in DMEM with 2 % FBS, were harvested in parallel. Infectious progeny from HCMV-infected hNPCs was quantified by plaque assays using indicator HFFs.
Western blot analysis.
hNPCs were lysed using RIPA buffer (Santa Cruz Biotechnology), containing 1× lysis buffer, PMSF, protease inhibitor cocktail and sodium orthovanadate. Proteins (20 µg) were resolved by electrophoresis in NuPAGE 4–12 % Bistris gels (Invitrogen) at 120 V for 2 h. Proteins were transferred onto nitrocellulose membranes (GE Healthcare) at 50 V for 1 h (Su et al., 2003). Membranes were probed with antibodies against IE1/2 (1 : 500, mAb810; Millipore), pUL37x1/vMIA aa 27–40 (DC35, 1 : 1000 or 1 : 2500) (Mavinakere et al., 2006), pp65 (1 : 500, mAb28-19, mAb65-8; Bill Britt; University of Alabama at Birmingham, School of Medicine or 1 : 10 000; Virusys), pp28 (1 : 1000 or 1 : 10 000; Abcam), MCP (1 : 10, mAb28-4 supernatant; Bill Britt) and cellular β-actin (1 : 1000; USBiological) for 2 h at room temperature (RT). Secondary antibodies coupled to HRP (1 : 2500; Santa Cruz) were incubated for 1 h at RT. Bioluminescence was used to detect proteins by incubation with SuperSignal West Pico ECL Substrate (Thermo Scientific) for 2 min. Membranes were then exposed to Kodak Biomax MS.
RNA analysis and RT-PCR.
Total RNA was isolated from mock- or HCMV-infected hNPCs using TRIZOL (Invitrogen) according to manufacturer’s protocol. cDNA libraries were produced by RT-PCR with Superscript III (Invitrogen) according to manufacturer’s protocol. Gene-specific primers were used for the detection of UL36, UL37x1 and UL37x3, UL38, IE1/2, β2.7 and β-actin RNAs as described previously (Adair et al., 2004; Su et al., 2003). PCR was performed in a GeneAmp PCR System 3700 thermocycler (Applied Biosystems) with initial denaturation at 95 °C (5 min), 35 cycles of denaturation at 95 °C (30 s), primer annealing at 60 °C (30 s) and primer extension at 72 °C (45 s), and a final extension at 72 °C (10 min). PCR products were resolved by agarose gel electrophoresis in 1× Tris/borate-EDTA buffer (Invitrogen).
Flow cytometry analysis.
Mock- or HCMV-infected hNPCs were harvested and resuspended in 1× AV binding buffer at 1×106 cells ml−1. hNPCs (3–5×105) were stained with 5 µl of allophycocyanin-conjugated AV (BD Biosciences) and 2 µl of PI (50 µg ml−1; BD Biosciences) for 15 min at RT. As control, mitochondrial-mediated apoptosis of hNPCs was induced with staurosporine (0.5 µM; Sigma) treatment for 24 h. hNPCs were analysed within 1 h of staining on a FACS Caliber (BD Biosciences) and data acquisition files were analysed using FlowJo version 7.6 (TreeStar). Flow cytometry was performed in three independent experiments. Statistical analyses were performed using Student’s unpaired t-test.
Immunofluorescence.
hNPCs were co-transfected with pUL37x1/vMIA-mEGFP and dsRed1Mito expression vectors (Bozidis et al., 2008; Williamson & Colberg-Poley, 2010b) using the AMAXA Nucleofector. hNPCs (5×105) were nucleofected with 5 µg of DNA in Ingenio Electroporation Solution (Mirus) using Program A-033. hNPCs were fixed at 24 h with 4 % paraformaldehyde (Sigma) for 30 min and mounted with Prolong Gold anti-fade reagent (Invitrogen) and imaged by confocal microscopy.
hNPCs were uninfected or infected with HCMV strain AD169 or BADsubUL37x1 (m.o.i. of 1). Cells were fixed with ice-cold methanol or 4 % paraformaldehyde. Primary antibodies included mouse anti-IE1/2 (1 : 100, mAb810; Millipore), UL37x1 mAb4B-6B (undiluted cell supernatant) (Sharon-Friling et al., 2006) and human anti-mitochondrial (1 : 50; ImmunoVision) antibodies for 1 h at RT. Secondary staining was performed using the corresponding antibodies (647Alexa or 488Alexa Fluor antibodies, 1 : 1000; Invitrogen) for 1 h at RT. Coverslips were mounted using Prolong Gold anti-fade reagent (Invitrogen).
Confocal microscopy.
Cells were imaged by confocal microscopy, using a LSM 510 laser scanning confocal microscope (Zeiss) and image analysis was conducted as described previously (Williamson & Colberg-Poley, 2010b). Co-localization of pUL37x1-mEGFP and mitochondrial markers in >100 cells was quantified using MetaMorph version 7.7.1.0.
Acknowledgements
The authors thank Dr Tom Shenk for the gift of the recombinant HCMV viruses BADwt, BADrev, BADsubUL37x1 and antibody mAb4B-6B, Dr Bill Britt for the gift of mAb28-4, mAb28-19 and mAb65-8. We are grateful to Drs Lina Chakrabarti and Chad Williamson for help in analysing the flow cytometry and quantifying the MetaMorph co-localization signals, respectively. The studies were supported by NIH R01 AI057906, National Center for Research Resources UL1RR031988 and Children’s Research Institute funds to A. C. P. Confocal microscopy imaging was performed in the Cellular Imaging & Analysis Core at Children’s Research Institute and was supported by a core grant to the Children’s Intellectual and Developmental Disabilities Research Center (1P30HD40677). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health. These studies were performed by R. L. H. in partial fulfilment of his doctoral studies in the Molecular Medicine Program at George Washington University Institute of Biomedical Sciences.
References
- Adair R., Liebisch G. W., Su Y., Colberg-Poley A. M. (2004). Alteration of cellular RNA splicing and polyadenylation machineries during productive human cytomegalovirus infection. J Gen Virol 85, 3541–3553 10.1099/vir.0.80450-0 [DOI] [PubMed] [Google Scholar]
- Al-Barazi H. O., Colberg-Poley A. M. (1996). The human cytomegalovirus UL37 immediate-early regulatory protein is an integral membrane N-glycoprotein which traffics through the endoplasmic reticulum and Golgi apparatus. J Virol 70, 7198–7208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnoult D., Bartle L. M., Skaletskaya A., Poncet D., Zamzami N., Park P. U., Sharpe J., Youle R. J., Goldmacher V. S. (2004). Cytomegalovirus cell death suppressor vMIA blocks Bax- but not Bak-mediated apoptosis by binding and sequestering Bax at mitochondria. Proc Natl Acad Sci U S A 101, 7988–7993 10.1073/pnas.0401897101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becroft D. M. (1981). Prenatal cytomegalovirus infection: epidemiology, pathology and pathogenesis. Perspect Pediatr Pathol 6, 203–241 [PubMed] [Google Scholar]
- Blomgren K., Leist M., Groc L. (2007). Pathological apoptosis in the developing brain. Apoptosis 12, 993–1010 10.1007/s10495-007-0754-4 [DOI] [PubMed] [Google Scholar]
- Bozidis P., Williamson C. D., Colberg-Poley A. M. (2008). Mitochondrial and secretory human cytomegalovirus UL37 proteins traffic into mitochondrion-associated membranes of human cells. J Virol 82, 2715–2726 10.1128/JVI.02456-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozidis P., Williamson C. D., Wong D. S., Colberg-Poley A. M. (2010). Trafficking of UL37 proteins into mitochondrion-associated membranes during permissive human cytomegalovirus infection. J Virol 84, 7898–7903 10.1128/JVI.00885-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bristow B. N., O’Keefe K. A., Shafir S. C., Sorvillo F. J. (2011). Congenital cytomegalovirus mortality in the United States, 1990-2006. PLoS Negl Trop Dis 5, e1140 10.1371/journal.pntd.0001140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britt W. J., Jarvis M., Seo J. Y., Drummond D., Nelson J. (2004). Rapid genetic engineering of human cytomegalovirus by using a lambda phage linear recombination system: demonstration that pp28 (UL99) is essential for production of infectious virus. J Virol 78, 539–543 10.1128/JVI.78.1.539-543.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeran M. C., Hu S., Ni H. T., Sheng W., Palmquist J. M., Peterson P. K., Lokensgard J. R. (2005). Neural precursor cell susceptibility to human cytomegalovirus diverges along glial or neuronal differentiation pathways. J Neurosci Res 82, 839–850 10.1002/jnr.20682 [DOI] [PubMed] [Google Scholar]
- Colberg-Poley A. M., Santomenna L. D. (1988). Selective induction of chromosomal gene expression by human cytomegalovirus. Virology 166, 217–228 10.1016/0042-6822(88)90163-8 [DOI] [PubMed] [Google Scholar]
- DeBiasi R. L., Kleinschmidt-DeMasters B. K., Richardson-Burns S., Tyler K. L. (2002). Central nervous system apoptosis in human herpes simplex virus and cytomegalovirus encephalitis. J Infect Dis 186, 1547–1557 10.1086/345375 [DOI] [PubMed] [Google Scholar]
- Doetsch F., Caillé I., Lim D. A., García-Verdugo J. M., Alvarez-Buylla A. (1999). Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 97, 703–716 10.1016/S0092-8674(00)80783-7 [DOI] [PubMed] [Google Scholar]
- Dunn W., Chou C., Li H., Hai R., Patterson D., Stolc V., Zhu H., Liu F. (2003). Functional profiling of a human cytomegalovirus genome. Proc Natl Acad Sci U S A 100, 14223–14228 10.1073/pnas.2334032100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmacher V. S., Bartle L. M., Skaletskaya A., Dionne C. A., Kedersha N. L., Vater C. A., Han J. W., Lutz R. J., Watanabe S. & other authors (1999). A cytomegalovirus-encoded mitochondria-localized inhibitor of apoptosis structurally unrelated to Bcl-2. Proc Natl Acad Sci U S A 96, 12536–12541 10.1073/pnas.96.22.12536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayajneh W. A., Colberg-Poley A. M., Skaletskaya A., Bartle L. M., Lesperance M. M., Contopoulos-Ioannidis D. G., Kedersha N. L., Goldmacher V. S. (2001). The sequence and antiapoptotic functional domains of the human cytomegalovirus UL37 exon 1 immediate early protein are conserved in multiple primary strains. Virology 279, 233–240 10.1006/viro.2000.0726 [DOI] [PubMed] [Google Scholar]
- Kim W. R., Sun W. (2011). Programmed cell death during postnatal development of the rodent nervous system. Dev Growth Differ 53, 225–235 10.1111/j.1440-169X.2010.01226.x [DOI] [PubMed] [Google Scholar]
- Kouzarides T., Bankier A. T., Satchwell S. C., Preddy E., Barrell B. G. (1988). An immediate early gene of human cytomegalovirus encodes a potential membrane glycoprotein. Virology 165, 151–164 10.1016/0042-6822(88)90668-X [DOI] [PubMed] [Google Scholar]
- Lewis J., Oyler G. A., Ueno K., Fannjiang Y. R., Chau B. N., Vornov J., Korsmeyer S. J., Zou S., Hardwick J. M. (1999). Inhibition of virus-induced neuronal apoptosis by Bax. Nat Med 5, 832–835 10.1038/10556 [DOI] [PubMed] [Google Scholar]
- Lim D. A., Alvarez-Buylla A. (1999). Interaction between astrocytes and adult subventricular zone precursors stimulates neurogenesis. Proc Natl Acad Sci U S A 96, 7526–7531 10.1073/pnas.96.13.7526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokensgard J. R., Cheeran M. C., Gekker G., Hu S., Chao C. C., Peterson P. K. (1999). Human cytomegalovirus replication and modulation of apoptosis in astrocytes. J Hum Virol 2, 91–101 [PubMed] [Google Scholar]
- Lossi L., Cantile C., Tamagno I., Merighi A. (2005). Apoptosis in the mammalian CNS: lessons from animal models. Vet J 170, 52–66 10.1016/j.tvjl.2004.05.010 [DOI] [PubMed] [Google Scholar]
- Luo M. H., Schwartz P. H., Fortunato E. A. (2008). Neonatal neural progenitor cells and their neuronal and glial cell derivatives are fully permissive for human cytomegalovirus infection. J Virol 82, 9994–10007 10.1128/JVI.00943-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M. H., Hannemann H., Kulkarni A. S., Schwartz P. H., O’Dowd J. M., Fortunato E. A. (2010). Human cytomegalovirus infection causes premature and abnormal differentiation of human neural progenitor cells. J Virol 84, 3528–3541 10.1128/JVI.02161-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malm G., Engman M. L. (2007). Congenital cytomegalovirus infections. Semin Fetal Neonatal Med 12, 154–159 10.1016/j.siny.2007.01.012 [DOI] [PubMed] [Google Scholar]
- Mavinakere M. S., Colberg-Poley A. M. (2004). Dual targeting of the human cytomegalovirus UL37 exon 1 protein during permissive infection. J Gen Virol 85, 323–329 10.1099/vir.0.19589-0 [DOI] [PubMed] [Google Scholar]
- Mavinakere M. S., Williamson C. D., Goldmacher V. S., Colberg-Poley A. M. (2006). Processing of human cytomegalovirus UL37 mutant glycoproteins in the endoplasmic reticulum lumen prior to mitochondrial importation. J Virol 80, 6771–6783 10.1128/JVI.00492-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy M., Auger D., Whittemore S. R. (2000). Human cytomegalovirus causes productive infection and neuronal injury in differentiating fetal human central nervous system neuroepithelial precursor cells. J Hum Virol 3, 215–228 [PubMed] [Google Scholar]
- McCormick A. L., Roback L., Mocarski E. S. (2008). HtrA2/Omi terminates cytomegalovirus infection and is controlled by the viral mitochondrial inhibitor of apoptosis (vMIA). PLoS Pathog 4, e1000063 10.1371/journal.ppat.1000063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menn B., Garcia-Verdugo J. M., Yaschine C., Gonzalez-Perez O., Rowitch D., Alvarez-Buylla A. (2006). Origin of oligodendrocytes in the subventricular zone of the adult brain. J Neurosci 26, 7907–7918 10.1523/JNEUROSCI.1299-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura M. (2011). Apoptotic and non-apoptotic caspase functions in neural development. Neurochem Res 36, 1253–1260 10.1007/s11064-010-0341-x [DOI] [PubMed] [Google Scholar]
- Nakagawa T., Yuan J. (2000). Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J Cell Biol 150, 887–894 10.1083/jcb.150.4.887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan V. (1997). Apoptosis in development and disease of the nervous system: 1. Naturally occurring cell death in the developing nervous system. Pediatr Neurol 16, 9–13 10.1016/S0887-8994(96)00257-3 [DOI] [PubMed] [Google Scholar]
- Norris K. L., Youle R. J. (2008). Cytomegalovirus proteins vMIA and m38.5 link mitochondrial morphogenesis to Bcl-2 family proteins. J Virol 82, 6232–6243 10.1128/JVI.02710-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odeberg J., Wolmer N., Falci S., Westgren M., Seiger A., Söderberg-Nauclér C. (2006). Human cytomegalovirus inhibits neuronal differentiation and induces apoptosis in human neural precursor cells. J Virol 80, 8929–8939 10.1128/JVI.00676-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odeberg J., Wolmer N., Falci S., Westgren M., Sundtröm E., Seiger A., Söderberg-Nauclér C. (2007). Late human cytomegalovirus (HCMV) proteins inhibit differentiation of human neural precursor cells into astrocytes. J Neurosci Res 85, 583–593 10.1002/jnr.21144 [DOI] [PubMed] [Google Scholar]
- Panchision D. M. (2009). The role of oxygen in regulating neural stem cells in development and disease. J Cell Physiol 220, 562–568 10.1002/jcp.21812 [DOI] [PubMed] [Google Scholar]
- Perlman J. M., Argyle C. (1992). Lethal cytomegalovirus infection in preterm infants: clinical, radiological, and neuropathological findings. Ann Neurol 31, 64–68 10.1002/ana.410310112 [DOI] [PubMed] [Google Scholar]
- Pistollato F., Chen H. L., Schwartz P. H., Basso G., Panchision D. M. (2007). Oxygen tension controls the expansion of human CNS precursors and the generation of astrocytes and oligodendrocytes. Mol Cell Neurosci 35, 424–435 10.1016/j.mcn.2007.04.003 [DOI] [PubMed] [Google Scholar]
- Poncet D., Larochette N., Pauleau A. L., Boya P., Jalil A. A., Cartron P. F., Vallette F., Schnebelen C., Bartle L. M. & other authors (2004). An anti-apoptotic viral protein that recruits Bax to mitochondria. J Biol Chem 279, 22605–22614 10.1074/jbc.M308408200 [DOI] [PubMed] [Google Scholar]
- Poncet D., Pauleau A. L., Szabadkai G., Vozza A., Scholz S. R., Le Bras M., Brière J. J., Jalil A., Le Moigne R. & other authors (2006). Cytopathic effects of the cytomegalovirus-encoded apoptosis inhibitory protein vMIA. J Cell Biol 174, 985–996 10.1083/jcb.200604069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiñones-Hinojosa A., Sanai N., Soriano-Navarro M., Gonzalez-Perez O., Mirzadeh Z., Gil-Perotin S., Romero-Rodriguez R., Berger M. S., Garcia-Verdugo J. M., Alvarez-Buylla A. (2006). Cellular composition and cytoarchitecture of the adult human subventricular zone: a niche of neural stem cells. J Comp Neurol 494, 415–434 10.1002/cne.20798 [DOI] [PubMed] [Google Scholar]
- Reboredo M., Greaves R. F., Hahn G. (2004). Human cytomegalovirus proteins encoded by UL37 exon 1 protect infected fibroblasts against virus-induced apoptosis and are required for efficient virus replication. J Gen Virol 85, 3555–3567 10.1099/vir.0.80379-0 [DOI] [PubMed] [Google Scholar]
- Reeves M. B., Davies A. A., McSharry B. P., Wilkinson G. W., Sinclair J. H. (2007). Complex I binding by a virally encoded RNA regulates mitochondria-induced cell death. Science 316, 1345–1348 10.1126/science.1142984 [DOI] [PubMed] [Google Scholar]
- Revello M. G., Gerna G. (2004). Pathogenesis and prenatal diagnosis of human cytomegalovirus infection. J Clin Virol 29, 71–83 10.1016/j.jcv.2003.09.012 [DOI] [PubMed] [Google Scholar]
- Rosenthal L. S., Fowler K. B., Boppana S. B., Britt W. J., Pass R. F., Schmid S. D., Stagno S., Cannon M. J. (2009). Cytomegalovirus shedding and delayed sensorineural hearing loss: results from longitudinal follow-up of children with congenital infection. Pediatr Infect Dis J 28, 515–520 10.1097/INF.0b013e318198c724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santomenna L. D., Colberg-Poley A. M. (1990). Induction of cellular hsp70 expression by human cytomegalovirus. J Virol 64, 2033–2040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz P. H., Bryant P. J., Fuja T. J., Su H., O’Dowd D. K., Klassen H. (2003). Isolation and characterization of neural progenitor cells from post-mortem human cortex. J Neurosci Res 74, 838–851 10.1002/jnr.10854 [DOI] [PubMed] [Google Scholar]
- Seo J. Y., Britt W. J. (2008). Multimerization of tegument protein pp28 within the assembly compartment is required for cytoplasmic envelopment of human cytomegalovirus. J Virol 82, 6272–6287 10.1128/JVI.02345-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon-Friling R., Goodhouse J., Colberg-Poley A. M., Shenk T. (2006). Human cytomegalovirus pUL37x1 induces the release of endoplasmic reticulum calcium stores. Proc Natl Acad Sci U S A 103, 19117–19122 10.1073/pnas.0609353103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaletskaya A., Bartle L. M., Chittenden T., McCormick A. L., Mocarski E. S., Goldmacher V. S. (2001). A cytomegalovirus-encoded inhibitor of apoptosis that suppresses caspase-8 activation. Proc Natl Acad Sci U S A 98, 7829–7834 10.1073/pnas.141108798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y., Adair R., Davis C. N., DiFronzo N. L., Colberg-Poley A. M. (2003). Convergence of RNA cis elements and cellular polyadenylation factors in the regulation of human cytomegalovirus UL37 exon 1 unspliced RNA production. J Virol 77, 12729–12741 10.1128/JVI.77.23.12729-12741.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y., Toribe Y., Mogami Y., Yanagihara K., Nishikawa M. (2008). Epilepsy in patients with congenital cytomegalovirus infection. Brain Dev 30, 420–424 10.1016/j.braindev.2007.12.004 [DOI] [PubMed] [Google Scholar]
- Terhune S., Torigoi E., Moorman N., Silva M., Qian Z., Shenk T., Yu D. (2007). Human cytomegalovirus UL38 protein blocks apoptosis. J Virol 81, 3109–3123 10.1128/JVI.02124-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson C. D., Colberg-Poley A. M. (2010a). Intracellular sorting signals for sequential trafficking of human cytomegalovirus UL37 proteins to the endoplasmic reticulum and mitochondria. J Virol 84, 6400–6409 10.1128/JVI.00556-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson C. D., Colberg-Poley A. M. (2010b). Intracellular sorting signals for sequential trafficking of human cytomegalovirus UL37 proteins to the endoplasmic reticulum and mitochondria. J Virol 84, 6400–6409 10.1128/JVI.00556-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D., Smith G. A., Enquist L. W., Shenk T. (2002). Construction of a self-excisable bacterial artificial chromosome containing the human cytomegalovirus genome and mutagenesis of the diploid TRL/IRL13 gene. J Virol 76, 2316–2328 10.1128/jvi.76.5.2316-2328.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D., Silva M. C., Shenk T. (2003). Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc Natl Acad Sci U S A 100, 12396–12401 10.1073/pnas.1635160100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H., Shen Y., Shenk T. (1995). Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J Virol 69, 7960–7970 [DOI] [PMC free article] [PubMed] [Google Scholar]