

Abstract
Chagas disease is a chronic infection caused by the protozoan parasite Trypanosoma cruzi, manifested in progressive cardiomyopathy and/or gastrointestinal dysfunction. Therapeutic options to prevent or treat Chagas disease are limited. CYP51, the enzyme key to the biosynthesis of eukaryotic membrane sterols, is a validated drug target in both fungi and T. cruzi. Sulfonamide derivatives of 4-aminopyridyl-based inhibitors of T. cruzi CYP51 (TcCYP51), including the sub-nanomolar compound 3, have molecular structures distinct from other validated CYP51 inhibitors. They augment the biologically relevant chemical space of molecules targeting TcCYP51. In a 2.08 Å x-ray structure, TcCYP51 is in a compound 3-induced conformation distinct from the previously characterized ground-state conformation of CYP51 drug-target complexes. That the binding site was modulated in response to an incoming inhibitor for the first time characterizes TcCYP51 as a flexible target rather than a rigid template.
Keywords: Drug design, protein structure, inhibitors, Chagas disease, CYP51
Introduction
Chagas disease, a parasitic disease prevalent in Latin America, is caused by chronic infection by the protozoan parasite Trypanosoma cruzi, which is transmitted by an insect vector of the family Triatominae. Following an initially symptomatic acute stage, T. cruzi invades the heart, gastrointestinal tract or nervous system, where it may persist asymptomatically for years before manifesting itself in cardiomyopathy, megacolon or megaoesophagus syndromes.[1] Antiparasitic treatment is now recommended for all acute and chronic patients, including those with the indeterminate chronic form[1-2]. The only two drugs available for treatment of Chagas disease, nifurtimox and benznidazole, date from the late 1960s. Although they show considerable efficacy in the acute stage, their use is controversial in the chronic stage, in which they are also associated with adverse side effects such as dermatitis, gastrointestinal, and neurologic toxicities[3]. There is a need for new therapeutics with better safety profiles and improved efficacy to treat T. cruzi infections and prevent or cure cardiovascular Chagas disease.
Sterol 14-demethylase (CYP51) is a validated therapeutic target for both fungal and parasitic infections due to its key role in the biosynthesis of ergosterol, an essential cell membrane component of these pathogenic organisms[4]. Because the anti-fungal CYP51 inhibitors posaconazole and ravuconazole failed to achieve sustainable parasitological cure in recently completed clinical trials[5], the quest for better chemotherapy for Chagas disease must continue. We applied de novo structure-aided chemical tailoring to the original 4-aminopyridinyl-based hit, LP10, which was identified by a high-throughput screen and then modified specifically to target T. cruzi CYP51[6]. Subsequent medicinal chemistry efforts have improved, by up to four orders of magnitude, the EC50 of the scaffold series members compared to that of the parental hit.[7] Both the pharmacokinetics and pharmacodynamics of new compounds were monitored during the course of hit-to-lead optimization, with the goal of obtaining at the outlet of the development pipeline a class of molecules that are orally deliverable, potent, safe and affordable.
Our hypothesis concerning the binding mode of the 4-aminopyridyl scaffold[7a] first relied on co-crystal structures of early stage LP10 analogs with CYP51 from Mycobacterium tuberculosis[8]. A decision to focus on the S-absolute configuration at the chiral carbon center was made based on the binding affinity of first generation analogs which were also evaluated in the cell-based T. cruzi assay that has largely propelled our hit-to-lead optimization program[7a]. When the potency of the S-enantiomer series of LP10 analogs increased to EC50 values in the hundred nanomolar range, we were able to resolve the first co-crystal structure of the S-1 analog carrying the biaryl substituent at the chiral center at a resolution of 2.67 Å (PDB ID 4BJK)[7a], although that structure was for the Trypanosoma brucei CYP51 ortholog (TbCYP51) (Figure 1).
Figure 1. Co-crystal structures of CYP51 with 4-aminopyridyl analogs.

The asterisk marks the chiral carbon center. The biaryl scaffold variants in R- and S-configuration have been characterized previously.[7]
The S-configuration analogs synthesized and tested attained maximum efficacy around 10 nM,[7a] which encouraged us to continue our efforts to achieve more potent alternatives. The structure of the S-1•TbCYP51 complex (PDB ID 4BJK) demonstrated that the biaryl substituent at the chiral carbon extends into a different space than that occupied by posaconazole[9], inspiring us to also test the enantiomer of S-1, namely compound R-2 (Figure 1). This step led to an increase in inhibitor potency of four orders of magnitude, and to the first co-crystal structure of the R-2 analog with the ultimate therapeutic target, T. cruzi CYP51, at a resolution of 3.1 Å (4BY0)[7b].
Based on the consistently superior potency, the R-configuration of this class of the inhibitors was used as a template for follow-up optimizations of inhibitor R-2, including those of the sulfonamide derivatives 3-11 reported in this article (Table 1). The SAR optimization of the R-sulfonamide series yielded two potent inhibitors, compounds 3 and 4, whose in vitro efficacy significantly exceeded that of the S-sulfonamide analog, 27r, (Fig. S1), reported previously.[7a] A sub-nanomolar inhibitor of T. cruzi in the cell-based assay, compound 3 has been structurally characterized in complex with the drug target to a resolution of 2.08 Å, the highest yet reported for the TcCYP51 drug-target complexes. To accommodate the unusual shape of compound 3, TcCYP51 undergoes conformational changes not previously observed for this target, thus defining compound 3 as the first CYP51 inhibitor perturbing the “ground-state” conformation of TcCYP51 in the crystal. This high resolution x-ray structure revealed features key to further refinement of the pharmacodynamic properties of the 4-aminopyridyl-based inhibitors.
Table 1. Ranking sulphonamide analogs by in vitro potency.
| Cpd | R= | EC50, nM Cell-based assay[a] |
KD, nM UV-vis assay[b] |
|---|---|---|---|
|
| |||
| 3 |
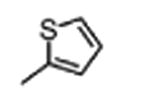
|
0.82±0.01 | ≤10.0 |
| 4 |
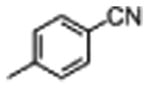
|
2.6±0.5 | 47±19 |
| 5 |
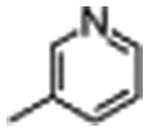
|
2400±800 | 15±6 |
| 6 |
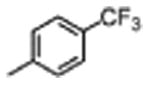
|
2800±3300 | ≤10.0 |
| 7 |
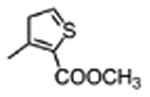
|
3100±1200 | 20±7 |
| 8 |
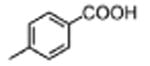
|
NE[c] | 20±7 |
| 9 |
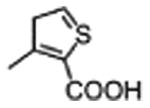
|
NE | ≤10.0 |
| 10 |
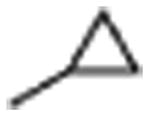
|
NE | 25±6 |
| 11 |

|
NE | 19±4 |
each measurement performed in triplicate;
apparent values,
not effective at ≤10 μM
Results and Discussion
Synthesis of inhibitors
A series of 4-aminopyridyl-based analogs containing sulfonamide units (Table 1) were synthesized according to Scheme 1. The N-Boc-protected aryl ester 12 was synthesized by following the previously reported method[7a]. The ester group was hydrolyzed under basic conditions to yield intermediate 13. Amide coupling of 13 and 14[7b] followed by N-Boc deprotection led to the key intermediate 16. Inhibitors 3-11 were generated by the acylation reaction of 16 and various aryl sulfonyl chloride reagents.
Scheme 1.
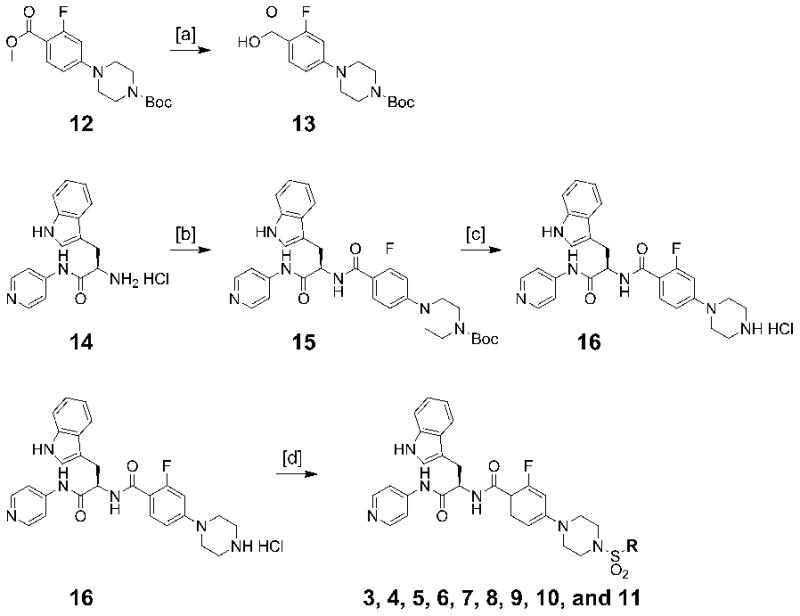
Synthesis of Inhibitors, 3, 4, 5, 6, 7, 8, 9, 10, and 11 (Table 1). Reagents and conditions: [a] 10% NaOH (aq), MeOH, THF, 60 °C, 1 h, 94% [b]13, PyBOP, HOBt, Et3N, CH2Cl2, 23 °C, 1h, 79% [c] 4N HCl in dioxane, 23 °C, 12 h, 94% [d] appropriate sulfonyl chloride, Et3N, CH2Cl2, 0 °C to 23 °C, 1h, 23-75%.
In vitro potency against T. cruzi
The EC50 of compounds was assessed in T. cruzi-infected mouse C2C12 myoblasts as previously described.[7] In this new scaffold series, substituents at the sulfonamide unit strongly influenced the level of anti-T.cruzi activity. The potency of the analogs varied from sub-nanomolar (3) to inactive at ≤10 μM (8-11), depending on the nature of the substituent at the sulfonamide group (Table 1). The dose-response curves for tested compounds are shown in Fig. 2 and Fig. S2 (see Supporting information). The analogs carrying small aliphatic substituents (10, 11) and those featuring ionizable groups (8, 9) failed to elicit T. cruzi inhibition in cell culture at ≤10 μM. The analogs featuring aromatic rings carrying non-ionizable groups (6, 7) or a 3-pyridinyl moiety (5) inhibited T. cruzi at the low micromolar concentrations. Finally, analogs containing thiophenyl (3) or 4-cyanophenyl (4) moieties appended to the sulfonyl group were the most potent in this scaffold series (high pM and low nM, respectively).
Figure 2. Dose-response curves (A) and KD titration curves (B) for compounds 3 and 4.
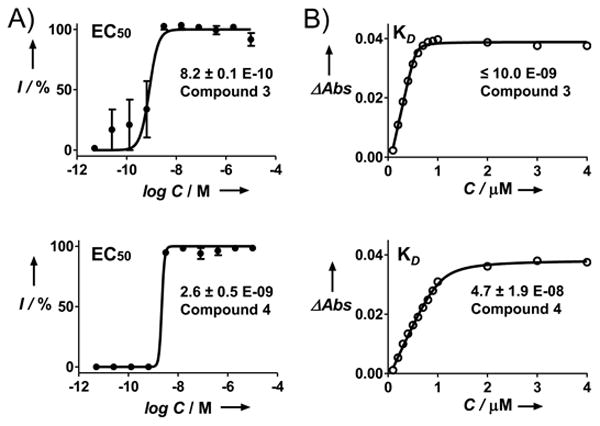
EC50 of compounds were determined in triplicate in the T.cruzi-infected mouse myoblasts. Apparent KD values were determined spectrophotometrically, using 1 μM TcCYP51. Efficacy of 3 is consistent with compound's tight binding affinity. EC50 of 4 is more than an order of magnitude lower than measured KD value. Axes: I – inhibition; ΔAbs – absorbance shift between 425 nm and 390 nm; C – inhibitorconcentration.
Binding affinity to the target
The binding affinity of compounds was assessed at 1 μM TcCYP51 concentration by the UV-vis P450 assay described previously[10]. The low sensitivity of the assay does not allow discrimination among tightly binding inhibitors below 10 nM, and thus provides estimates rather than absolute values of binding affinity. Also, compound binding properties are affected by their solubility in aqueous solution. Subject to multiple and often difficult to dissect factors, the KD values obtained from the titration curves are thus range-bound rather than absolute. With that said, all the compounds demonstrated virtually stoichiometric linear saturation of the binding site at sub-micromolar concentrations reaching a plateau at a 1:1 enzyme:inhibitor ratio (Fig. 2B and Fig. S3), thus providing a KD estimate of ca. ≤10 nM (Table 1). High efficacy of compound 3 in the cell-based assay was consistent with the tight binding predicted by the titration curve (Fig. 2A). In contrast, the KD for compounds 5-11 were estimated to be much lower than the observed EC50, if any, suggesting that factors other than interaction with the target, such as permeability through the cell and parasite membranes, may limit efficacy of these compounds in the cell-based assay. Finally, the titration curve for compound 4 failed to plateau at 1:1 ratio, resulting in a KD of 47 nM, more than an order of magnitude higher than the observed EC50 of 2.6 nM, allowing us to speculate that low solubility in aqueous solution may account for poor saturation of the TcCYP51 active site, at the same time favoring membrane permeability in the cell-based assays.
Overall structure of CYP51 drug-target complexes
A drug target in several human pathogens, CYP51 has been intensively studied over the last decade. Significant progress has been made in characterization of the CYP51 orthologs from Mycobacterium tuberculosis,[6b, 8, 11] kinetoplastids, including Trypanosoma cruzi [7b, 9, 12], Trypanosoma brucei,[7a, 9, 13] and Leishmania infantum 13b, 14, as well as of the human host [15]. This accumulated knowledge supports structure-aided drug development against protozoan parasites, including T. cruzi. A limitation to progress, however, is the low resolution of TcCYP51 co-crystal structures; the highest being around 2.3 Å for the TcCYP51-fluconazole complex, 2WX2[9]. The accuracy of low-resolution structures largely relies on the topology/parameter restraints used during refinement, particularly as they pertain to new small-molecule ligands, which are not part of the default computer databases used by the structure determination software. Yet if not misinterpreted, low-resolution structures can deliver information of high scientific and practical value. A complicating factor, however, can be scientific errors associated with inaccurate restraint refinement against low-resolution data. The erroneous assignment of sp3 rather than sp2 electronic configurations to aromatic nitrogen atoms of the triazole ring result in deformed fluconazole ligands in 3KHM[12a] and 3L4D[14] CYP51 structures; also erroneously assigned is the absolute configuration of one of the chiral centers in posaconazole in 3K10[12a].
TcCYP51 belongs to a category of proteins whose crystallization propensity entirely relies on interactions with ligands bound in the active site. We made inroads on that problem in the course of our efforts to obtain potent TcCYP51 inhibitors. For the first time resolution of the TcCYP51 complex approached the 2 Å barrier, deepening the level of atomic information available for structure-aided drug discovery. Data collection and refinement statistics are shown in Table 2. The binding mode of compound 3, 2-fluoranyl-N-[(2R)-3-(1H-indol-3-yl)-1-oxidanylidene-1-(pyridin-4-ylamino)propan-2-yl]-4-(4-thiophen-2-ylsulfonylpiperazin-1-yl)benzamide, has been characterized to a resolution of 2.08 Å, the highest currently available for this therapeutic target. Consistent with the previously analyzed 4-aminopyridyl-based analogs[7], the pyridinyl moiety of compound 3 coordinates to the heme iron, while the indole ring points at the heme macrocycle. The longest substituent at the chiral carbon center extends along the cleft marking the α- and β-domain interface. The sulfonyl group-enabled 90° bend allows the thiophene ring to be positioned virtually orthogonal to the piperazine ring and facing the interior of the β-domain, while the sulfonyl group points to the exterior through the cleft between the domains (Fig. 3A).
Table 2. Data collection and refinement statistics.
| Protein | TcCYP51 |
| PDB ID | 4COH |
| Small molecule ID | T9H |
|
| |
| Data Collection | |
| Space group | P212121 |
| Cell dimensions: | |
| a, b, c (Å) | 79.2, 96.4, 137.1 |
| α, β, γ (°) | 90, 90, 90 |
| Molecules in AU | 2 |
| Wavelength | 1.11587 |
| Resolution (Å) | 2.08 |
| Rsym or Rmerge (%) | 14.7 (88.7)[a] |
| I/σI | 9.2 (1.8) |
| Completeness (%) | 95.6 (76.0) |
| Redundancy | 7.2 (4.9) |
| Refinement | |
| No. reflections | 57134 |
| Rwork/Rfree (%) | 17.4/24.3 |
| No. atoms | |
| Protein | 7084 |
| Heme | 86 |
| Inhibitor | 88 |
| Solvent | 676 |
| Mean B value | 25.1 |
| B-factors | |
| Protein | 24.7 |
| Heme | 20.9 |
| Inhibitor | 17.4 |
| Solvent | 32.5 |
| R.m.s deviations | |
| Bond lengths (Å) | 0.022 |
| Bond angles (°) | 2.012 |
Values in parentheses are for highest-resolution shell
Figure 3. Overall structure of TcCYP51.
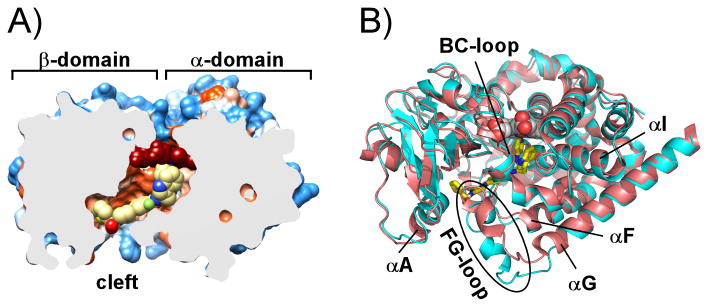
A) Slice through the binding site shows 3 (yellow spheres) in the hydrophobic tunnel of TcCYP51 (PDB ID 4COH) represented by solvent-accessible surface colored by hydrophobicity – hydrophilic areas in blue, hydrophobic areas in orange. Heme is in red spheres. B) TcCYP51 conformation induced by 3 in 4COH (cyan ribbon) compared to the biaryl analog structure 4BY0[7b] (red ribbon). The area of major conformational changes is enclosed in the oval shape; 3 is in yellow sticks, heme is in spheres. Images were generated using CHIMERA[16] or PYMOL.[17]
Topologically, cytochrome P450 enzymes, including CYP51, fold as a single domain with a core composed of a four-helix bundle carrying the trigonal prism-shaped structure of the P450 molecule[18]. Structurally, the scaffold is subdivided in two domains, the α-helical and β-sheet-rich, which are separated by a cleft on the surface of the protein distal with respect to the heme.[18] The cleft serves as a substrate binding site, with size and topology modulated by the concerted motion of the BC- and FG-loops, the F- and G-helices, and bending of the central and the longest P450 α-helix, the I-helix, running over the distal surface of the heme across the entire protein structure[18-19] (Fig. 3B). The secondary structure nomenclature is according to the generally accepted scheme introduced by Poulos et al.[20]
The bacterial CYP51 ortholog from Mycobacterium tuberculosis demonstrates the extreme solvent-exposure of the heme prosthetic group due to the bent I-helix and the unusual conformation of the BC-loop, which remains wide open even when inhibitors[6b, 8, 11a] or substrate analogs[11b, 11c] are bound in the active site. In eukaryotic CYP51, the substrate binding side is shaped as a tunnel buried in the protein interior and leading from the protein surface, to the heme group.[9, 15] Essentially the same ground-state close conformation is observed in all TcCYP51 drug-target complexes that have been characterized.[7b, 9, 12] Thus, compound 3 is the first inhibitor reported to perturb the ground-state conformation of TcCYP51, allowing the borders of inhibitor envelope to be expanded.
Compound 3 binding site in atomic details
The reported structure contains two molecules in the asymmetric unit, both with well-defined electron density. In chain A, the indole ring adopts a single conformation (Fig. 4A), while in chain B, the electron density map shows evidence of two alternative conformations of the indole ring (Fig. 4B). Superimposition of both chains in Fig. 4C demonstrates the ambiguity in indole ring binding, with good superimposition of the rest of the inhibitor molecule.
Figure 4. Compound 3 binding site.

A) A fragment of the electron density map (blue mesh) contoured at 1.2 σ corresponds to 3 (yellow sticks) in chain A of 4COH structure. B) Indole ring adopting alternative conformations in chain B. Protein in A) and B) is represented by a ribbon. C). 3-D alignment of chains A (cyan) and B (magenta) indicates subtle differences in position of amino acid residues surrounding the indole ring of 3 (black lines). D) A single water molecule (small red sphere) resolved in the binding site, H-bonds two amide nitrogen atoms of 3. Heme is in van der Waals spheres colored by atom type.
Altogether, 22 exclusively hydrophobic residues, I45, F48, I70, I72, Y103, I105, M106, F110, Y116, P210, A211, V213, F214, A287, F290, A291, G292, L356, M358, M360, M460 and V461, plus P450-conserved threonine, T295, constitute the binding site within 5 Å of 3 (Fig. 5). The indole ring binds between the bulky electron-rich residues Y103, M106, F110, Y116 and F290, most of which originate from the BC-loop. The 3-D alignment in Fig. 3B shows an efficient overlap between the side chain counterparts with the biggest shift of 0.8 Å observed for Y103. This modest perturbation is apparently sufficient to allow indole ring flipping in the active site.
Figure 5. Stereogram of 3 in the TcCYP51 active site.
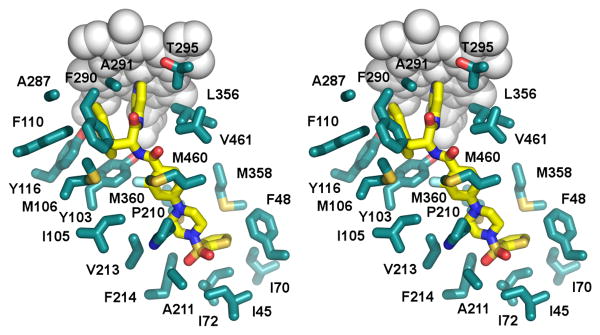
Amino acid side chains (cyan) are shown within 5 Å of 3 (yellow) in 4COH structure. Heme is in grey spheres.
The thiophene moiety of compound 3 is positioned invariantly in both protein chains pointing to the β-domain. To accommodate the orthogonal orientation of the thiophene ring, the FG-loop of TcCYP51 undergoes conformational changes, shifting about 7 Å away from the A-helix, thus widening the opening separating the α- and β-domains. Major points of contact are the aromatic side chains of F48 and F214, which are virtually in orthogonal orientation to the thiophene ring, at 3.7 Å and 3.5 Å to the ring edges, respectively (Fig. 5). As a result of induced conformational changes, the side chain of F214 is relocated 3.6 Å from its ground-state position. The aliphatic side chains I72 (3.7 Å away from the ring surface), I45, I70, V77, M358 and M360 (all within 4.2 to 5.5 Å distance), complete the hydrophobic cavity accommodating the thiophene moiety.
Finally, given the highly hydrophobic nature of the active site, no H-bonds mediating drug-target interactions, either directly or via water molecules, are suggested by the structure. A single water molecule observed in the binding site at 2.08 Å resolution H-bonds to two amide nitrogen atoms of compound 3 (Fig. 4D).
CYP51 inhibitor envelope
Large and hydrophobic, the TcCYP51 active site promiscuously permits small-molecule binding.[10] The inhibitor envelope, as shown by superimposed CYP51 drug-target complexes, branches into a Y-shape with an elongated “stem” and shorter “arms” (Fig. 6). The shortest arm orients toward the heme with the aromatic nitrogen atom on the azole or pyridyl moieties coordinating to the heme iron, while the longer arm extends into a hydrophobic channel presumably occupied by the aliphatic side chain of the sterol substrate. The elongated stem of the Y-shape extends into the hydrophobic tunnel at the interface of the α- and β-domains. Based on analysis of superimposed structures, each of the inhibitors only partially utilizes the binding envelope. Three inhibitors, VNF, VFV and NEE, extend into the channel occupied by the sterol aliphatic side chain; fluconazole (TPF) and tipifarnib (JKF) largely utilize the central space adjacent to the heme group, while posaconazole (X2N), VNI and VNF explore the stem of the envelope. Inhibitor nomenclature is according to small-molecule codes assigned by the PDB databank. The particularly long posaconazole moiety extends into the exterior space, adopting alternative conformations at the terminal unit, as revealed by the 2X2N structure[9] (Fig. 6B). Compared to other inhibitor classes, the 4-aminopyridyl-based inhibitors, 5PS, T9H and 18I, have a more elongated heme-coordinating arm, allowing the indole ring to bind at a more open angle to the heme macrocycle compared to the aromatic substituents in other inhibitor classes. This binding mode suggests that substituents at the C-5 of the indole ring might benefit from sharing the space presumably occupied by the sterol aliphatic chain. Such modifications would enhance interactions with the target and prevent indole ring from flipping in the active site.
Figure 6. Inhibitor envelope.

Inhibitor envelope derived from the 3-D alignments of seven T. cruzi (A) and seven T. brucei (B, D) CYP51 drug-target complexes. All superimposed ligands are shown in black lines. Ligand highlighted in yellow is labeled by the small-molecule code, followed by resolution and the PDB ID of the corresponding structure (in parenthesis); heme is in red sticks. B) Two different posaconazole (X2N) conformers in 2X2N structure are shown. C) Inhibitor envelope represented by the van der Waals spheres colored by atom type: carbon in grey, nitrogen in blue, oxygen in red, sulfur in yellow, chlorine in green, fluorine in cyan. Compound 3 carbons are highlighted in a darker shade of grey. *The substrate analog LNP is in a bent conformation of the 5β-skeleton and is not included in the superimposition.
CYP51 substrate envelope
In contrast to the inhibitor envelope deduced from the 3-D alignments of multiple drug-target complexes, the substrate envelope is yet to be convincingly defined in CYP51. The co-crystal structure featuring the substrate lanosterol analog methylenecyclopropyl-Δ7-24,25-dihydrolanosterol (LNP) bound to TbCYP51 (PDB ID 3P99)[13b] depicts, at resolution of >3 Å, the sterol ligand with unnatural 5β-configuration of the H-atom at the C-5 bridgehead position, which is not consistent with the 5α-configuration of the lanosterol precursors used in the synthesis of the substrate analog[13b, 21]. This causes the sterol tetracycle in LNP to adopt a bent conformation of the 5β-skeleton, in contrast to the flat shape of the biogenic sterol precursor, lanosterol, due to its 5α-configuration[22]. Functionally significant in nature, the difference in shapes of both sterol skeletons was not commented on by the authors13b. We infer that these structural differences may be again an artifact of refinement. Furthermore, position of the lanosterol substrate is flipped in the recently reported x-ray structure of the Saccharomyces cerevisiae CYP51 (PDB ID 4LXJ)[23] with the aliphatic chain pointing in the oposite direction compared to that in T. brucei CYP51.
Nevertheless, with some degree of confidence, one can speculate that posaconazole (X2N) extends beyond the substrate envelope (Fig. 6B), making contact with residues at the tunnel entrance that are not significant for catalytic function[9]. As a result, the only mechanism of resistance reported for posaconazole is associated with target mutagenesis[24]. Based on this observation, a strategy to minimize opportunities for drug resistance is to optimize the binding of drugs within the substrate envelope.[25] Sulfonamide analogs of the type described here offer envelope extension within the interior space and may serve as fertile starting points for further inhibitor optimization.
Conclusion
The sulfonamide series of 4-aminopyridyl-based inhibitors delivered high picomolar and low nanomolar TcCYP51 inhibitors 3 and 4, which allowed the first high-resolution co-crystal structure for any TcCYP51 inhibitor complex to be generated. For the first time fit induced by the inhibitor has been observed in CYP51 and has been recognized as a factor affecting SAR for this therapeutic target. The 2.08 Å structure also revealed ambiguity in the binding of the indole ring, an essential and invariable portion of the 4-aminopyridyl-based scaffold series. Sulfonamide analog 3 marks an expansion of the inhibitor envelope within the interior of the CYP51 binding site which may be targeted in future analog series to reduce the propensity for drug resistance to develop due to drug-target interactions involving functionally non-essential residues.
Experimental Section
Chemistry, General Methods
All reaction solvents were purified before use. Dichloromethane, tetrahydrofuran, dimethylformamide and toluene were purified by passing through a column of activated A-1 alumina. All other reagents purchased from commercial suppliers were used as received. All reactions sensitive to moisture or oxygen were conducted under an argon atmosphere using flame-dried (under vacuum) or oven-dried (overnight) glassware. Removal of solvents was accomplished by using a rotary evaporator under reduced pressure and a water bath below 30 °C, followed by exposure to high vacuum using a vacuum pump.
Proton nuclear magnetic resonance (1H NMR) spectra and carbon (13C) NMR spectra were recorded on commercially available NMR spectrometers at 400 MHz and 100 or 176 MHz, respectively. The proton signal for non-deuterated solvent (δ 7.26 for CHCl3 or δ 2.50 for DMSO) was used as an internal reference for 1H NMR chemical shifts. Coupling constants (J) are reported in Hertz (Hz). 13C chemical shifts are reported relative to the δ 77.16 resonance of CDCl3 or the δ 39.52 resonance of DMSO-d6.
Analytical thin layer chromatography (TLC) was performed using glass plates precoated with a 0.25-mm thickness of silica gel. The TLC plates were visualized with UV light. Column chromatography was performed using a Biotage® Isolera flash purification system using Biotage® SNAP HP-SIL cartridge (30 μm silica, 10 g to 100 g size). Unless noted otherwise, all compounds isolated by flash chromatography were sufficiently pure by 1H NMR analysis for use in subsequent reactions. Polar compounds were purified using preparative high performance liquid chromatography (HPLC) using a SunFire column (30 mm × 250 mm) with a linear gradient elution ranging from 10% to 100% of CH3CN/CH3OH (1/1) in H2O (containing 0.1% TFA) at 60 mL/min flow rate.
The purity of all final compounds (typically ≥96%) was assayed at 254 nm wavelength by using analytical HPLC (Varian 1100 series) on a reverse phase ZORBAX Eclipse XDB-C18 column (4.6 × 150 mm, 5 μm). A linear gradient elution ranging from 2% to 98% CH3CN and H2O (containing 0.1% TFA and 1% CH3CN) at 1.5 mL/min was used. Compounds were lyophilized before dissolution in DMSO to give 10 mM stock solutions for use in biochemical and cell-based assays.
General procedures for the 3, 4, 5, 6, 7, 8, 9, 10, and 11 synthesis
To a solution of the appropriate sulfonyl chloride (ca. 1.1 eq.) in dry CH2Cl2 (5 mL) was slowly added 16 and triethylamine (ca. 3 eq.) at 0 °C. After being stirred for 15 min., the reaction mixture was warmed to ambient temperature and stirred for an additional 1 h. The solvent was removed under reduced pressure, and ethyl acetate (10 mL) was added to the crude product mixture. The solution was washed with saturated aqueous NaHCO3 (2 mL × 2) and brine (2 mL × 2). The organic layer was concentrated in vacuo and directly subjected to purification by flash chromatography or by high performance liquid chromatography to provide the titled products in 23-75% yield.
(R)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-2-fluoro-4-(4-(thiophen-2-ylsulfonyl)piperazin-1-yl)benzamide TFA salt (3)
The general procedure was followed using 2-thiophenesulfonyl chloride. The crude product was purified by HPLC to afford 3 as a white solid (47%): 1H NMR (400 MHz, DMSO-d6) δ 11.69 (s, 1H), 10.92 (d, J = 2.5 Hz, 1H), 8.77 – 8.60 (m, 2H), 8.16 – 8.02 (m, 3H), 7.99 (t, J = 6.7 Hz, 1H), 7.68 (dd, J = 3.8, 1.3 Hz, 1H), 7.65 – 7.49 (m, 2H), 7.40 – 7.27 (m, 2H), 7.25 (d, J = 2.3 Hz, 1H), 7.04 (ddd, J = 8.2, 6.9, 1.2 Hz, 1H), 6.99 – 6.87 (m, 1H), 6.84 – 6.67 (m, 2H), 4.86 (dt, J = 8.0, 6.0 Hz, 1H), 3.44 (t, J = 5.1 Hz, 4H), 3.38 – 3.22 (m, 2H), 3.02 (t, J = 5.0 Hz, 4H); 13C NMR (101 MHz, DMSO-d6) δ 173.04, 160.07, 152.17, 143.10, 136.08, 134.31, 134.24, 133.42, 128.44, 127.06, 121.02, 118.31, 114.44, 111.39, 110.61, 110.22, 108.86, 55.56, 46.17, 45.34; MS (ESI) m/z 633.2 [M+H]+.
(R)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-4-(4-((4-cyanophenyl)sulfonyl)piperazin-1-yl)-2-fluorobenzamide TFA salt (4)
The general procedure was followed using 4-cyanobenzene-1-sulfonyl chloride. The crude product was purified by HPLC to afford 4 as a white solid (58%): 1H NMR (400 MHz, DMSO-d6) δ 11.58 (s, 1H), 10.91 (d, J = 2.5 Hz, 1H), 8.78 – 8.60 (m, 2H), 8.21 – 8.09 (m, 2H), 8.06 – 8.01 (m, 2H), 8.01 – 7.90 (m, 3H), 7.59 (d, J = 7.9 Hz, 1H), 7.54 (t, J = 9.1 Hz, 1H), 7.32 (d, J = 8.1 Hz, 1H), 7.24 (d, J = 2.4 Hz, 1H), 7.04 (ddd, J = 8.2, 6.9, 1.2 Hz, 1H), 6.97 – 6.89 (m, 1H), 6.80 – 6.69 (m, 2H), 4.95 – 4.79 (m, 1H), 3.40 (t, J = 5.0 Hz, 4H), 3.36 – 3.18 (m, 2H), 3.05 (t, J = 5.0 Hz, 4H); 13C NMR (101 MHz, DMSO-d6) δ 173.05, 152.14, 143.19, 139.10, 136.09, 133.66, 128.29, 127.04, 124.11, 121.04, 118.32, 117.59, 115.83, 114.45, 111.41, 110.21, 108.85, 55.54, 46.31, 45.23; MS (ESI) m/z 652.3 [M+H]+.
(R)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-2-fluoro-4-(4-(pyridin-3-ylsulfonyl)piperazin-1-yl)benzamide TFA salt (5)
The general procedure was followed using pyridine-3-sulfonyl chloride. The crude product was purified by HPLC to afford 5 as a white solid (75%): 1H NMR (400 MHz, DMSO-d6) δ 12.22 (s, 1H), 10.95 (d, J = 2.5 Hz, 1H), 8.95 (d, J = 2.3 Hz, 1H), 8.90 (dd, J = 4.9, 1.6 Hz, 1H), 8.70 (d, J = 7.0 Hz, 2H), 8.25 – 8.14 (m, 3H), 7.96 (t, J = 6.8 Hz, 1H), 7.71 (dd, J = 8.1, 4.8 Hz, 1H), 7.64 (d, J = 7.9 Hz, 1H), 7.55 (t, J = 9.1 Hz, 1H), 7.31 (d, J = 8.1 Hz, 1H), 7.26 (d, J = 2.3 Hz, 1H), 7.03 (t, J = 7.4 Hz, 1H), 6.90 (t, J = 7.5 Hz, 1H), 6.81 – 6.69 (m, 2H), 4.90 (q, J = 6.8 Hz, 1H), 3.40 (t, J = 4.9 Hz, 4H), 3.37 – 3.24 (m, 2H), 3.06 (t, J = 4.9 Hz, 4H); 13C NMR (101 MHz, DMSO-d6) δ 173.20, 163.09, 162.56, 160.10, 153.74, 153.51, 153.02, 147.61, 142.08, 136.08, 135.98, 131.67, 127.10, 124.71, 124.17, 120.99, 118.48, 118.30, 114.54, 111.38, 110.52, 110.23, 108.87, 101.19, 55.71, 46.29, 45.20, 26.97; MS (ESI) m/z 628.3 [M+H]+.
(R)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-2-fluoro-4-(4-((4-(trifluoromethyl)phenyl)sulfonyl)piperazin-1-yl)benzamide (6)
The general procedure was followed using 4-(trifluoromethyl)benzene-1-sulfonyl chloride to provide 6 as a light yellow solid (51%): 1H NMR (400 MHz, DMSO-d6) δ 10.86 (d, J = 2.5 Hz, 1H), 10.67 (s, 1H), 8.51 – 8.37 (m, 2H), 8.12 – 7.93 (m, 4H), 7.81 (t, J = 7.3 Hz, 1H), 7.69 – 7.51 (m, 4H), 7.31 (d, J = 8.1 Hz, 1H), 7.19 (d, J = 2.3 Hz, 1H), 7.04 (t, J = 7.5 Hz, 1H), 6.92 (t, J = 7.4 Hz, 1H), 6.83 – 6.64 (m, 2H), 4.95 – 4.77 (m, 1H), 3.40 (t, J = 5.0 Hz, 4H), 3.25 (qd, J = 14.6, 6.7 Hz, 2H), 3.05 (t, J = 4.9 Hz, 4H); 13C NMR (101 MHz, DMSO-d6) δ 171.70, 162.83, 162.49, 160.04, 153.56, 153.45, 149.91, 145.85, 138.85, 136.06, 133.13, 132.80, 131.62, 128.57, 127.17, 126.74, 126.70, 124.75, 123.89, 122.04, 120.97, 118.41, 118.24, 113.46, 111.33, 110.86, 110.73, 110.22, 109.17, 101.42, 54.93, 46.32, 45.30, 27.49; MS (ESI) m/z 695.4 [M+H]+.
(R)-methyl 3-((4-(4-((3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)carbamoyl)-3-fluorophenyl)piperazin-1-yl)sulfonyl)thiophene-2-carboxylate (7)
The general procedure was followed using methyl 3-(chlorosulfonyl)thiophene-2-carboxylate to provide 7 as a light yellow solid (67%): 1H NMR (400 MHz, DMSO-d6) δ 10.86 (d, J = 2.5 Hz, 1H), 10.65 (s, 1H), 8.51 – 8.39 (m, 2H), 8.00 (d, J = 5.3 Hz, 1H), 7.81 (t, J = 7.3 Hz, 1H), 7.68 – 7.52 (m, 4H), 7.47 (d, J = 5.3 Hz, 1H), 7.38 – 7.27 (m, 1H), 7.19 (d, J = 2.3 Hz, 1H), 7.04 (ddd, J = 8.2, 7.0, 1.1 Hz, 1H), 6.92 (ddd, J = 8.0, 7.0, 1.0 Hz, 1H), 6.87 – 6.70 (m, 2H), 4.93 – 4.79 (m, 1H), 3.85 (s, 3H), 3.39 (dd, J = 6.7, 3.5 Hz, 4H), 3.34 – 3.17 (m, 6H); 13C NMR (101 MHz, DMSO-d6) δ 171.70, 162.89, 162.55, 160.00, 153.59, 150.01, 145.76, 138.49, 136.06, 134.17, 131.40, 130.13, 127.17, 123.89, 120.98, 118.42, 118.25, 113.45, 111.34, 109.18, 63.47, 54.92, 53.15, 46.62, 45.21, 27.50; MS (ESI) m/z 691.4 [M+H]+.
(R)-4-((4-(4-((3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)carbamoyl)-3-fluorophenyl)piperazin-1-yl)sulfonyl)benzoic acid TFA salt (8)
The general procedure was followed using 4-(chlorosulfonyl)benzoic acid. The crude product was purified by HPLC to afford 8 as a white solid (23%): 1H NMR (400 MHz, DMSO-d6) δ 13.58 (s, 1H), 11.36 (s, 1H), 10.91 (d, J = 2.4 Hz, 1H), 8.64 (s, 2H), 8.21 – 8.13 (m, 2H), 8.00 – 7.91 (m, 3H), 7.91 – 7.85 (m, 2H), 7.59 (d, J = 7.9 Hz, 1H), 7.53 (t, J = 9.1 Hz, 1H), 7.31 (d, J = 8.1 Hz, 1H), 7.23 (d, J = 2.3 Hz, 1H), 7.04 (ddd, J = 8.0, 6.9, 1.1 Hz, 1H), 6.92 (t, J = 7.5 Hz, 1H), 6.81 – 6.69 (m, 2H), 4.83 (q, J = 6.7 Hz, 1H), 3.40 (t, J = 5.0 Hz, 4H), 3.35 – 3.18 (m, 2H), 3.02 (t, J = 4.9 Hz, 4H); 13C NMR (176 MHz, DMSO-d6) δ 172.75, 166.11, 163.15, 161.99, 160.58, 157.96, 157.78, 153.59, 153.53, 150.65, 144.76, 138.41, 136.08, 135.01, 131.61, 131.59, 130.32, 127.90, 127.07, 124.07, 121.03, 118.33, 118.31, 114.26, 111.40, 110.72, 110.65, 110.21, 108.92, 101.39, 101.23, 55.40, 46.32, 45.31, 27.08 ;MS (ESI) m/z 669.0 [M-H]-.
(R)-3-((4-(4-((3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)carbamoyl)-3-fluorophenyl)piperazin-1-yl)sulfonyl)thiophene-2-carboxylic acid TFA salt (9)
To a solution of 7 (25.2 mg, 36 μmol) in MeOH (5 mL) was slowly added 10% NaOH (5 mL) at 0 °C. After stirring for 1 h at 0 °C, the reaction mixture was acidified with 2N HCl, and it was directly subjected to purification by high performance liquid chromatography to provide the titled product 9 as a white solid (10.7 mg, 16 μmol, 44%):1H NMR (400 MHz, DMSO-d6) δ 11.32 (s, 1H), 10.92 (d, J = 2.5 Hz, 1H), 8.63 (d, J = 6.2 Hz, 2H), 8.02 – 7.85 (m, 4H), 7.67 – 7.50 (m, 2H), 7.43 (d, J = 5.3 Hz, 1H), 7.32 (d, J = 8.1 Hz, 1H), 7.24 (d, J = 2.4 Hz, 1H), 7.04 (ddd, J = 8.0, 6.9, 1.1 Hz, 1H), 6.98 – 6.88 (m, 1H), 6.85 – 6.72 (m, 2H), 4.84 (q, J = 6.8 Hz, 1H), 3.45 – 3.18 (m, 10H), 2.55 (t, J = 5.5 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 172.75, 163.20, 160.94, 160.14, 157.84, 153.72, 145.18, 137.56, 136.68, 136.11, 130.37, 130.12, 127.09, 124.11, 121.06, 118.36, 114.20, 111.44, 110.64, 110.21, 108.97, 101.15, 55.40, 46.63, 45.27, 27.13; MS (ESI) m/z 677.4 [M+H]+.
(R)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-4-(4-(cyclopropylsulfonyl)piperazin-1-yl)-2-fluorobenzamide TFA salt (10)
The general procedure was followed using cyclopropanesulfonyl chloride. The crude product was further purified by HPLC to afford 10 as a white solid (61%): 1H NMR (400 MHz, DMSO-d6) δ 11.62 (s, 1H), 10.94 (d, J = 2.5 Hz, 1H), 8.77 – 8.60 (m, 2H), 8.08 – 7.94 (m, 3H), 7.68 – 7.49 (m, 2H), 7.33 (d, J = 8.1 Hz, 1H), 7.27 (d, J = 2.4 Hz, 1H), 7.05 (ddd, J = 8.1, 6.9, 1.1 Hz, 1H), 6.93 (ddd, J = 7.9, 6.9, 1.0 Hz, 1H), 6.89 – 6.72 (m, 2H), 4.87 (dt, J = 8.2, 5.8 Hz, 1H), 3.42 (dd, J = 6.8, 3.5 Hz, 4H), 3.39 – 3.13 (m, 6H), 2.64 (tt, J = 7.9, 4.9 Hz, 1H), 1.08 – 0.83 (m, 4H); 13C NMR (101 MHz, DMSO-d6) δ 173.13, 163.26, 162.66, 160.20, 153.93, 153.82, 152.16, 143.25, 136.12, 131.71, 127.07, 124.18, 121.08, 118.37, 114.47, 111.45, 110.56, 110.44, 110.22, 108.89, 101.42, 55.58, 46.67, 45.29, 27.01, 24.68, 4.29, 3.92; MS (ESI) m/z 591.3 [M+H]+.
(R)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-2-fluoro-4-(4-(methylsulfonyl)piperazin-1-yl)benzamide TFA salt (11)
The general procedure was followed using methanesulfonyl chloride. The crude product was purified by HPLC to afford 11 as a white solid (43%, ca. 35% reactant was recovered): 1H NMR (400 MHz, DMSO-d6) δ 11.71 (s, 1H), 10.93 (d, J = 2.5 Hz, 1H), 8.83 – 8.54 (m, 2H), 8.17 – 8.03 (m, 2H), 7.98 (t, J = 6.8 Hz, 1H), 7.74 – 7.52 (m, 2H), 7.32 (dd, J = 8.1, 0.9 Hz, 1H), 7.26 (d, J = 2.4 Hz, 1H), 7.05 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 6.93 (ddd, J = 8.0, 6.9, 1.0 Hz, 1H), 6.88 – 6.72 (m, 2H), 4.88 (q, J = 6.7 Hz, 1H), 3.43 (dd, J = 6.4, 3.8 Hz, 4H), 3.39 – 3.24 (m, 2H), 3.21 (dd, J = 6.3, 3.9 Hz, 4H), 2.91 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 173.08, 163.22, 153.87, 153.75, 152.24, 143.07, 136.10, 131.69, 131.65, 127.07, 124.13, 121.04, 118.36, 114.46, 111.41, 110.49, 110.36, 110.18, 108.87, 101.09, 55.56, 46.51, 44.82, 34.01, 26.99; MS (ESI) m/z 565.4 [M+H]+.
4-(4-(tert-Butoxycarbonyl)piperazin-1-yl)-2-fluorobenzoic acid (13)
To a solution of 12 (3.63 g, 11 mmol) in methanol/THF (10/10 mL) was added 10% NaOH (10 mL), and the reaction mixture was stirred for 1 h at 60 °C. After completion of the reaction as indicated by TLC analysis, the mixture was cooled to ambient temperature, and 2N HCl was added until a solid precipitated. The white solid was diluted with ethyl acetate (60 mL) and washed with brine (10 mL × 2). The organic layer was dried over magnesium sulfate, filtered, and concentrated in vacuo to give a crude 13 (3.28 g, 10 mmol, 94%) as a white solid: 1H NMR (400 MHz, DMSO-d6) δ 12.48 (s, 1H), 7.70 (t, J = 9.0 Hz, 1H), 6.94 – 6.51 (m, 2H), 3.43 (dd, J = 6.8, 3.7 Hz, 4H), 3.37 – 3.28 (m, 4H), 1.42 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 164.85, 164.81, 164.46, 161.93, 154.95, 154.84, 153.82, 133.15, 133.11, 109.31, 106.97, 106.87, 101.11, 100.85, 79.12, 46.20, 28.04; MS (ESI) m/z 323.2 [M-H]-.
(R)-tert-Butyl 4-(4-((3-(1H-indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)carbamoyl)-3-fluorophenyl)piperazine-1-carboxylate (15)
To a solution of 13 (0.788 g, 2.4 mmol), PyBOP (1.25 g, 2.4 mmol), and HOBt (ca. 10 mol%) in dry CH2Cl2 (20 mL) was slowly added triethylamine (1.3 mL, 4 eq.) at ambient temperature, and the reaction mixture was stirred for 15 min. After the reaction mixture became homogenous, D-tryptophan derivative 14 (0.662 g, 2.1 mmol)[7b] was added, and the reaction mixture was stirred at room temperature for 1 h. After confirming that the reaction was complete by using TLC analysis, the solvent was removed under reduced pressure. Ethyl acetate (60 mL) was added to the crude product mixture, and this solution was sequentially washed then with saturated aqueous NaHCO3 (10 mL × 2), saturated aqueous NH4Cl (10 mL × 2), and brine (10 mL × 2). The organic layer was dried over magnesium sulfate, filtered, and concentrated in vacuo. The product mixture was purified by flash chromatography to provide the titled product as a light yellow solid (0.972 g, 1.7 mmol, 79%): 1H NMR (400 MHz, CDCl3) δ 9.72 (s, 1H), 8.58 (s, 1H), 8.27 (d, J = 5.7 Hz, 2H), 7.85 (t, J = 9.1 Hz, 1H), 7.61 (d, J = 8.0 Hz, 1H), 7.45 (d, J = 5.7 Hz, 2H), 7.34 (dd, J = 25.7, 7.2 Hz, 2H), 7.18 – 7.06 (m, 2H), 6.96 (t, J = 7.5 Hz, 1H), 6.68 – 6.57 (m, 1H), 6.47 – 6.34 (m, 1H), 5.13 (q, J = 6.9 Hz, 1H), 3.54 (t, J = 5.3 Hz, 4H), 3.41 (d, J = 6.9 Hz, 2H), 3.25 (t, J = 5.3 Hz, 4H), 1.48 (s, 9H).; 13C NMR (176 MHz, CDCl3) δ 171.49, 164.43, 163.25, 161.85, 155.05, 154.99, 154.71, 148.15, 136.37, 132.92, 132.90, 127.25, 123.55, 122.44, 119.86, 118.79, 114.13, 111.51, 110.45, 110.13, 109.14, 109.08, 100.97, 100.81, 80.46, 55.59, 47.20, 28.54, 27.75; MS (ESI) m/z 587.4 [M+H]+.
(R)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-2-fluoro-4-(piperazin-1-yl)benzamide hydrochloride (16)
To a solution of 15 (0.972 g, 1.7 mmol) in dioxane (10 mL) was added 4N HCl in dixoane (10 mL), and the reaction mixture was stirred at room temperature for 12 hours. The solvent was removed with a rotary evaporator, and water (50 mL) was added. Impurities or by-products were extracted with ether (20 mL × 2), and the aqueous solution was dried using a lyophilizer to obtain the crude product 16 (0.863 g, 1.6 mmol, 94%). 1H NMR (400 MHz, DMSO-d6) δ 12.24 (s, 1H), 10.98 (d, J = 2.5 Hz, 1H), 9.48 (s, 2H), 8.92 – 8.54 (m, 2H), 8.25 – 8.14 (m, 2H), 8.02 (t, J = 6.8 Hz, 1H), 7.73 – 7.55 (m, 2H), 7.37 – 7.24 (m, 2H), 7.04 (ddd, J = 8.1, 7.0, 1.1 Hz, 1H), 6.91 (ddd, J = 8.0, 6.9, 1.0 Hz, 1H), 6.88 – 6.75 (m, 2H), 4.93 (dt, J = 8.3, 6.0 Hz, 1H), 3.55 (t, J = 5.3 Hz, 4H), 3.36 (qd, J = 14.6, 6.9 Hz, 2H), 3.16 (dq, J = 9.2, 4.4 Hz, 4H); 13C NMR (101 MHz, DMSO-d6) δ 173.19, 163.09, 163.06, 162.60, 160.14, 153.45, 153.34, 152.98, 142.14, 136.09, 131.70, 127.11, 124.17, 121.00, 118.49, 118.31, 114.53, 111.38, 111.04, 110.91, 110.25, 108.88, 101.34, 55.74, 43.91, 42.03, 39.52, 26.97; MS (ESI) m/z 487.2 [M+H]+.
EC50 in cell-based assay
Efficacy of compounds was evaluated in cell-based assay using T. cruzi-infected mouse myoblasts as previously described.[7b] To determine the EC50 values, assay was performed in triplicate. Briefly, sterile, black 384-well plates with clear-bottom wells (Greiner Bio-One) were seeded with mouse C2C12 myoblasts (500 cells/well) and then were infected with CA-I/72 trypomastigotes (2500 parasites/well) in 50 μl of culture medium (DMEM H-21)/well. Culture plates were incubated at 37°C with 5% CO2. 24-hours post infection, culture medium was removed and test compounds were added in fresh medium. For this, an intermediate plate (384-well plate) was prepared by serial dilution for all the compounds in 100 % DMSO. Then, 50 nl of each sample were diluted in 50 μl media (DMEM H-21) to final concentrations of 10 μM, 2 μM, 400 nM, 80 nM, 16 nM, 3 nM, 128 pM, 25 pM and 5 pM, and added to the experimental plate followed by incubation at 37°C with 5% CO2 for 72h. Wells containing non-infected cells were used as a 100% cell survival reference, while T. cruzi-infected but untreated cells were used as a 0% cell survival reference.
Cells were then fixed for 2 h with 4% paraformaldehyde, and rinsed with a solution of 150 mM NaCl, 100 mM NH4Cl, 0.1% Triton X-100 and 0.1% NaN3. After that, they were treated for 4 h with 0.2 μg/ml of the DNA fluorescent dye, DAPI (4,6-diamidino-2-phenylindole), diluted in the same solution. Plates were kept at ambient temperature until image acquisition was performed. Images were acquired by an IN Cell Analyzer 2000 (GE Healthcare) and the procedure and analyses were performed according to previously described.[10, 26]
Binding affinity by UV-vis spectroscopy
Binding affinity was approximated from the spectrophotometry titration curves generated as previously described.[10] UV-vis absorption spectra were recorded on a Cary scanning spectrophotometer (Varian) in the 1-cm path length quartz cuvette at 23°C in 100 mM potassium phosphate buffer, pH 7.5, containing 10% glycerol, with TcCYP51 concentration of 1μM. Compound solutions used for titration were prepared in DMSO at 100 μM and added in 1-μl aliquots to 1 ml protein sample. To correct for organic solvent effect, the same volume of solvent was added into the reference cuvettes. To determine the KD values, titration data points were fitted to the quadratic tight-binding equation[27]:
| (1) |
where Aobs is the absorption shift between 425 nm and 390 nm determined at any ligand concentration; Amax is the maximal absorption shift obtained at saturation; KD is dissociation constant for the inhibitor-enzyme complex; S is the ligand concentration; Et is the total enzyme concentration.
X-ray structure analysis
To analyze the inhibitor binding mode, recombinant T. cruzi CYP51 modified by replacing the first 31 residues upstream of Pro32 with the fragment MAKKTSSKGKL and by inserting a His6-tag at the C-terminus was expressed and purified as described elsewhere[8]. Concentrated purified protein samples were stored at -80°C and diluted prior to crystallization to 0.1 mM by mixing with 10 mM K-PO4, pH 7.5, buffer supplemented with equimolar inhibitor. Crystallization conditions were determined using commercial high-throughput screening kits available in deep-well format (Hampton Research), a nanoliter drop-setting Mosquito robot (TTP LabTech) operating with 96-well plates, and a hanging drop crystallization protocol. Crystals were further optimized in 96-well plates for diffraction data collection and harvested directly from the 200-nl drop containing 0.05 M ammonium citrate, pH 7.0 and 14% PEG 3350. Prior to data collection, crystals were cryo-protected by plunging them into a drop of reservoir solution supplemented with 20% ethylene glycol, then flash frozen in liquid nitrogen.
Diffraction data were collected at 100-110 K at beamline 8.3.1, Advanced Light Source, Lawrence Berkeley National Laboratory, USA. Data indexing, integration, and scaling were conducted using MOSFLM[28] and the programs implemented in the ELVES software suite.[29]. The crystal structures were determined by molecular replacement using diffraction data processed in the corresponding space groups and atomic coordinates of T. cruzi CYP51 (PDB ID code: 2WX2) as a search model. The final model was built using COOT[30] and refinement was performed by using REFMAC5 software[31]. Data collection and refinement statistics are shown in Table 2.
Supplementary Material
Acknowledgments
We thank the staff members of beamline 8.3.1, James Holton, George Meigs and Jane Tanamachi, at the Advanced Light Source at Lawrence Berkeley National Laboratory, for assistance with data collection, Jair Lage Siqueira-Neto, Danielle Kellar and Jiri Gut (Center for Discovery and Innovation in Parasitic Diseases, UCSF) for assistance with the T. cruzi cell-based assay, and Potter Wickware for proof reading of the manuscript. This research was generously supported by NIH R01 grant AI095437. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Footnotes
Supporting information for this article is available
Contributor Information
Prof. Dr. William R. Roush, Email: roush@scripps.edu.
Prof. Dr. Larissa M. Podust, Email: larissa.podust@ucsf.edu.
References
- 1.Coura JR, Borges-Pereira J. Mem Inst Oswaldo Cruz. 2011;106:641–645. doi: 10.1590/s0074-02762011000600001. [DOI] [PubMed] [Google Scholar]
- 2.Viotti R, de Noya BA, Araujo-Jorge T, Grijalva MJ, Guhl F, Lopez MC, Ramsey JM, Ribeiro I, Schijman AG, Sosa-Estani S, Torrico F, Gascon J. Antimicrob Agents Chemother. 2013 doi: 10.1128/AAC.01662-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Castro JA, de Mecca MM, Bartel LC. Hum Exp Toxicol. 2006;25:471–479. doi: 10.1191/0960327106het653oa. [DOI] [PubMed] [Google Scholar]
- 4.Buckner FS, Urbina JA. Int J Parasitol Drugs Drug Resist. 2012;2:236–242. doi: 10.1016/j.ijpddr.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a Drugs for Neglected Diseases initiative (DNDi) press release. [accessed December 5, 2013];2013 http://www.dndi.org/media-centre/press-releases/1700-e1224.html.; b XVIII International Congress for Tropical Medicine and Malaria; Rio de Janejro, Brazil. September 23-27, 2012. [Google Scholar]
- 6.a Doyle PS, Chen CK, Johnston JB, Hopkins SD, Leung SSF, Jacobson MP, Engel JC, McKerrow JH, Podust LM. Antimicrob Agents Chemother. 2010;54:2480–2488. doi: 10.1128/AAC.00281-10. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Podust LM, von Kries JP, Nasser Eddine A, Kim Y, Yermalitskaya LV, Kuehne R, Ouellet H, Warrier T, Altekoster M, Lee JS, Rademann J, Oschkinat H, Kaufmann SHE, Waterman MR. Antimicrob Agents Chemother. 2007;51:3915–3923. doi: 10.1128/AAC.00311-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a Choi JY, Calvet CM, Gunatilleke SS, Ruiz C, Cameron MD, McKerrow JH, Podust LM, Roush WR. J Med Chem. 2013;56:7651–7668. doi: 10.1021/jm401067s. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Choi JY, Calvet CM, Vieira DF, Gunatelleke SS, Cameron MD, McKerrow JH, Podust LM, Roush WR. ACS Med Chem Lett. 2014 doi: 10.1021/ml500010m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen CK, Doyle PS, Yermalitskaya LV, Mackey ZB, Ang KKH, McKerrow JH, Podust LM. PLoS Negl Trop Dis. 2009;3:e372. doi: 10.1371/journal.pntd.0000372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen CK, Leung SSF, Guilbert C, Jacobson MP, McKerrow JH, Podust LM. PLoS Negl Trop Dis. 2010;4:e651. doi: 10.1371/journal.pntd.0000651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gunatilleke SS, Calvet CM, Johnston JB, Chen CK, Erenburg G, Gut J, Engel JC, Ang KK, Mulvaney J, Chen S, Arkin MR, McKerrow JH, Podust LM. PLoS Negl Trop Dis. 2012;6:e1736. doi: 10.1371/journal.pntd.0001736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a Podust LM, Poulos TL, Waterman MR. Proc Natl Acad Sci USA. 2001;98:3068–3073. doi: 10.1073/pnas.061562898. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Podust LM, Yermalitskaya LV, Lepesheva GI, Podust VN, Dalmasso EA, Waterman MR. Structure. 2004;12:1937–1945. doi: 10.1016/j.str.2004.08.009. [DOI] [PubMed] [Google Scholar]; c Nasser Eddine A, vonKries JP, Podust MV, Warrier T, Kaufmann SH, Podust LM. J Biol Chem. 2008;283:15152–15159. doi: 10.1074/jbc.M801145200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a Lepesheva GI, Hargrove TY, Anderson S, Kleshchenko Y, Furtak V, Wawrzak Z, Villalta F, Waterman MR. J Biol Chem. 2010;285:25582–25590. doi: 10.1074/jbc.M110.133215. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Andriani G, Amata E, Beatty J, Clements Z, Coffey BJ, Courtemanche G, Devine W, Erath J, Juda CE, Wawrzak Z, Wood JT, Lepesheva GI, Rodriguez A, Pollastri MP. J Med Chem. 2013;56:2556–2567. doi: 10.1021/jm400012e. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hargrove TY, Wawrzak Z, Alexander PW, Chaplin JH, Keenan M, Charman SA, Perez CJ, Waterman MR, Chatelain E, Lepesheva GI. J Biol Chem. 2013;288:31602–31615. doi: 10.1074/jbc.M113.497990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a Lepesheva GI, Park HW, Hargrove TY, Vanhollebeke B, Wawrzak Z, Harp JM, Sundaramoorthy M, Nes WD, Pays E, Chaudhuri M, Villalta F, Waterman MR. J Biol Chem. 2010;285:1773–1780. doi: 10.1074/jbc.M109.067470. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hargrove TY, Wawrzak Z, Liu J, Waterman MR, Nes WD, Lepesheva GI. J Lipid Res. 2012;53:311–320. doi: 10.1194/jlr.M021865. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Buckner FS, Bahia MT, Suryadevara PK, White KL, Shackleford DM, Chennamaneni NK, Hulverson MA, Laydbak JU, Chatelain E, Scandale I, Verlinde CL, Charman SA, Lepesheva GI, Gelb MH. Antimicrob Agents Chemother. 2012;56:4914–4921. doi: 10.1128/AAC.06244-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hargrove TY, Wawrzak Z, Liu J, Nes WD, Waterman MR, Lepesheva GI. J Biol Chem. 2011;286:26838–26848. doi: 10.1074/jbc.M111.237099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Strushkevich N, Usanov SA, Park HW. J Mol Biol. 2010;397:1067–1078. doi: 10.1016/j.jmb.2010.01.075. [DOI] [PubMed] [Google Scholar]
- 16.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 17.DeLano WL. The PyMOL molecular graphics system. DeLano Scientific, San Carlos, CA, USA; 2002. [Google Scholar]
- 18.a Li H, Poulos TL. Curr Top Med Chem. 2004;4:1789–1802. doi: 10.2174/1568026043387205. [DOI] [PubMed] [Google Scholar]; b Pochapsky TC, Kazanis S, Dang M. Antioxid Redox Signal. 2010;13:1273–1296. doi: 10.1089/ars.2010.3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Podust LM, Sherman DH. Nat Prod Rep. 2012;29:1251–1266. doi: 10.1039/c2np20020a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poulos TL, Finzel BC, Howard AJ. J Mol Biol. 1987;195:687–700. doi: 10.1016/0022-2836(87)90190-2. [DOI] [PubMed] [Google Scholar]
- 21.a Parish EJ, Schroepfer GJ., Jr J Lipid Res. 1981;22:859–868. [PubMed] [Google Scholar]; b Frye LL, Robinson CH. J Org Chem. 1990;55:1579–1584. [Google Scholar]
- 22.Moss GP. Queen Mary College, Pure & Appl Chem. 1989;61:1783–1822. [Google Scholar]
- 23.Monk BC, Tomasiak TM, Keniya MV, Huschmann FU, Tyndall JDA, O'Connell JD, III, Cannon RD, McDonald JG, Rodriguez A, Finer-Moore JS, Stroud RM. Proc Natl Acad Sci U S A. 2014;111:3865–3870. doi: 10.1073/pnas.1324245111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.a Howard SJ, Cerar D, Anderson MJ, Albarrag A, Fisher MC, Pasqualotto AC, Laverdiere M, Arendrup MC, Perlin DS, Denning DW. Emerg Infect Dis. 2009;15:1068–1076. doi: 10.3201/eid1507.090043. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Li X, Brown N, Chau AS, Lopez-Ribot JL, Ruesga MT, Quindos G, Mendrick CA, Hare RS, Loebenberg D, DiDomenico B, McNicholas PM. J Antimicrob Chemother. 2004;53:74–80. doi: 10.1093/jac/dkh027. [DOI] [PubMed] [Google Scholar]; c Pinto e Silva AT, Costa-de-Oliveira S, Silva-Dias A, Pina-Vaz C, Rodrigues AG. FEMS Yeast Res. 2009;9:626–633. doi: 10.1111/j.1567-1364.2009.00508.x. [DOI] [PubMed] [Google Scholar]; d Pfaller MA, Diekema DJ, Ghannoum MA, Rex JH, Alexander BD, Andes D, Brown SD, Chaturvedi V, Espinel-Ingroff A, Fowler CL, Johnson EM, Knapp CC, Motyl MR, Ostrosky-Zeichner L, Sheehan DJ, Walsh TJ. J Clin Microbiol. 2009;47:3142–3146. doi: 10.1128/JCM.00940-09. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Chau AS, Mendrick CA, Sabatelli FJ, Loebenberg D, McNicholas PM. Antimicrob Agents Chemother. 2004;48:2124–2131. doi: 10.1128/AAC.48.6.2124-2131.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ali A, Bandaranayake RM, Cai Y, King NM, Kolli M, Mittal S, Murzycki JF, Nalam MN, Nalivaika EA, Ozen A, Prabu-Jeyabalan MM, Thayer K, Schiffer CA. Viruses. 2010;2:2509–2535. doi: 10.3390/v2112509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Engel JC, Ang KKH, Chen S, Arkin MR, McKerrow JH, Doyle PS. Antimicrob Agents Chemother. 2010;54:3326–3334. doi: 10.1128/AAC.01777-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morrison JF. Biochim Biophys Acta. 1969;185:269–286. doi: 10.1016/0005-2744(69)90420-3. [DOI] [PubMed] [Google Scholar]
- 28.Leslie AGW. Joint CCP4 ESF-EAMCB Newslett Protein Crystallogr. 1992;26 [Google Scholar]
- 29.Holton J, Alber T. Proc Natl Acad Sci U S A. 2004;101:1537–1542. doi: 10.1073/pnas.0306241101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Emsley P, Cowtan K. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 31.a Murshudov GN, Vagin AA, Dodson EJ. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]; b Acta Crysallogr D. 1994;50:760–763. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.