

Abstract
Members of Gram-positive Actinobacteria cause economically important diseases to plants. Within the Rhodococcus genus, some members can cause growth deformities and persist as pathogens on a wide range of host plants. The current model predicts that phytopathogenic isolates require a cluster of three loci present on a linear plasmid, with the fas operon central to virulence. The Fas proteins synthesize, modify, and activate a mixture of growth regulating cytokinins, which cause a hormonal imbalance in plants, resulting in abnormal growth. We sequenced and compared the genomes of 20 isolates of Rhodococcus to gain insights into the mechanisms and evolution of virulence in these bacteria. Horizontal gene transfer was identified as critical but limited in the scale of virulence evolution, as few loci are conserved and exclusive to phytopathogenic isolates. Although the fas operon is present in most phytopathogenic isolates, it is absent from phytopathogenic isolate A21d2. Instead, this isolate has a horizontally acquired gene chimera that encodes a novel fusion protein with isopentyltransferase and phosphoribohydrolase domains, predicted to be capable of catalyzing and activating cytokinins, respectively. Cytokinin profiling of the archetypal D188 isolate revealed only one activate cytokinin type that was specifically synthesized in a fas-dependent manner. These results suggest that only the isopentenyladenine cytokinin type is synthesized and necessary for Rhodococcus phytopathogenicity, which is not consistent with the extant model stating that a mixture of cytokinins is necessary for Rhodococcus to cause leafy gall symptoms. In all, data indicate that only four horizontally acquired functions are sufficient to confer the trait of phytopathogenicity to members of the genetically diverse clade of Rhodococcus.
Introduction
Plant pathogenic bacteria employ an array of molecules to dampen immunity, alter plant physiological responses, and mimic plant hormones to modulate host-signaling pathways [1], [2]. Collectively and often coordinately, the virulence molecules manipulate host cells to give pathogens access to host tissues and resources as well as facilitate egress and spread. Because of the intimate interactions of these pathogen-synthesized molecules with host cells, virulence genes are subject to strong selective pressures and are often dynamic, exhibiting patterns of high genetic diversity [3]. Horizontal gene transfer (HGT) is one mechanism that contributes to evolutionary dynamism, as virulence genes are often found on plasmids, associated with mobile genetic elements, and/or located within large stretches of genomic islands that have signatures indicative of HGT [4]. Pathoadaptation, a process whereby mutations modify traits and improve virulence, has been implicated as another mechanism in the evolution of pathogenicity [5], [6].
Actinobacteria is one of the largest taxonomical units within the domain Bacteria and its members inhabit a diversity of ecosystems. A very small number of genera within Actinobacteria have members that are pathogenic to plants. Rhodococcus are non-spore forming, non-motile, mycolic acid-containing bacteria within Actinobacteria [7]. Its members are best known as environmental bacteria with a wide range of catabolic functions and large genomes ranging from ∼4 megabases (Mb) to ∼9 Mb [8]. Rhodococcus fascians is the first species in the genus characterized as a plant pathogen and it infects plants in an unusual manner [9], [10]. R. fascians grows epiphytically on the surface of leaves. During the transition to an endophyte, the pathogen breaches the host cuticle, collapses the epidermal layer, and forms ingression sites beneath epiphytic colonies [11]. The bacterium then grows inside the host tissue and provokes cell differentiation and de novo organogenesis, resulting in proliferations and abnormal growths called witches’ brooms or leafy galls [12]. Rhodococcus is a persistent pathogen and can remain associated with the plant throughout its life [13]. Its host range is exceedingly large and includes more than 120 species representing both monocots and dicots, herbaceous and woody plants [12].
Phytopathogenicity of R. fascians D188 requires three virulence loci clustered on the conjugative linear plasmid, pFiD188 [14]–[19]. The fasA-F operon is the primary virulence operon and is implicated in the synthesis and modification of cytokinins, a class of plant growth regulating hormones (Fig. 1; [15], [18], [20]). The collective functions of the Fas proteins are hypothesized to be necessary for the pathogen to synthesize a mixture of cytokinins to upset homeostatic levels and cause and maintain leafy gall disease symptoms [10]. FasD is an isopentenyltransferase (IPT) and the key enzyme that transfers an isoprenoid moiety to adenine, the limiting step in cytokinin biosynthesis [14], [21]. A loss-of-function fasD mutant is non-pathogenic [14]. FasF is a homolog of LONELY GUY (LOG; a phosphoribohydrolase) of plants and functions to release activated cytokinins from their riboside forms [18], [22]. FasA is predicted to produce trans-zeatin (tZ)-types of cytokinins that are hypothesized to be important constituents of the bacterial-synthesized mixture of cytokinins [18], [20], [23]. The second locus is fasR, which encodes a predicted member of the AraC-like transcriptional regulatory protein family that is hypothesized to indirectly influence the transcription of fasA-F [10], [16], [18]. Finally, the att locus is also necessary for full virulence of R. fascians D188 [17]. The translated sequences for some of the att genes are homologous to antibiotic biosynthesis enzymes and thus predicted to be involved in secondary metabolism, though the specific metabolite(s) has yet to be identified.
Figure 1. Predicted pathway of cytokinin metabolism in phytopathogenic Rhodococcus.
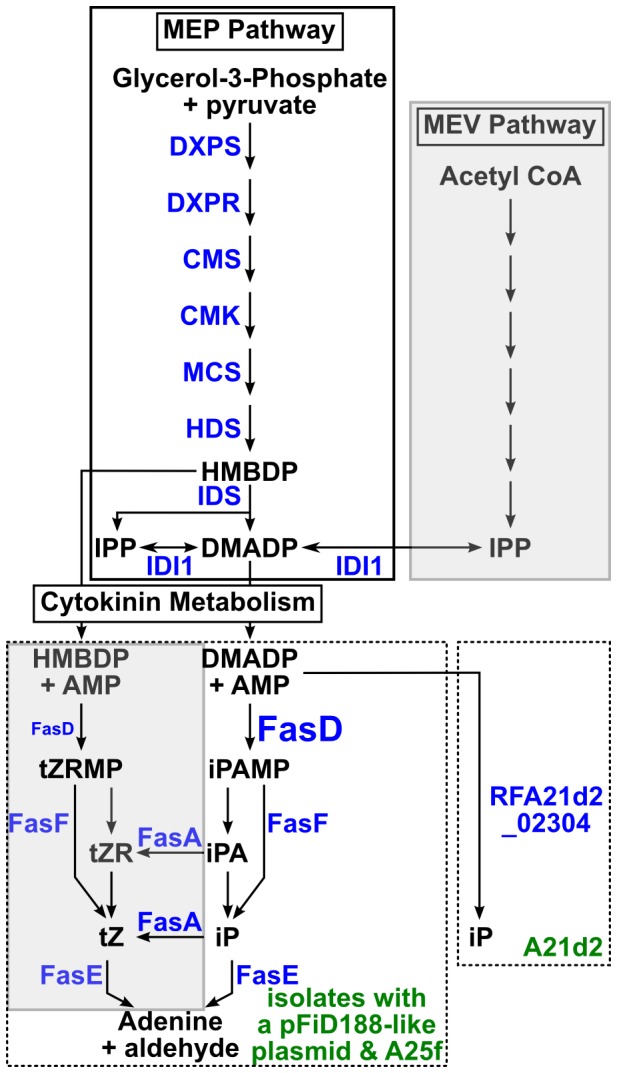
Pathways for the biosynthesis of cytokinin precursors are presented within boxes with solid lines. Abbreviations: MEP = methylerythritol phosphate; HMBDP = (E)-4-hydroxy-3-methyl-but-2-enyl diphosphate; DMADP = dimethylallyl diphosphate; IPP = isopentenyl diphosphate; MEV = mevalonate. Abbreviations in blue indicate enzymes with homologs encoded by the 20 isolates of Rhodococcus: DXPS = Deoxyxylulose 5-phosphate synthase; DXPR = DXP reductiosomerase; CMS = 4-diphosphocytidyl-2C-methyl-D-erythirol synthase; CMS = 4-(cytidine 5′-diphospho)-2-C-methyl-D-erythritol kinase; MCS = 2-C-methyl-D-erythritol 2,4-cyclodiphosphate synthase; (HDS = hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate synthase; IDS = hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate reductase; IDI1 = isopentenyl diphosphate isomerase. The predicted cytokinin metabolism steps are presented within boxes with dotted lines. Left = products of the Fas operon (the difference in sizes of FasD symbolizes in vitro substrate preference). FasD is an isopentenyltransferase that transfers an isoprenoid moiety to adenine; FasA is a homolog of P450-type cytochrome monooxygenases that hydroxylate the isopentenyl side chain to produce trans-zeatin; FasF removes the ribose 5′-monophosphate and releases the corresponding activate free base cytokinin; FasE is a cytokinin oxidase/dehydrogenase that degrades and inactivates cytokinins. The functions for FasB and FasC (not shown) are not known, but are suggested to be accessory proteins that provide energy for cytokinin metabolism. See associated text for references. Right = predicted pathway by a novel protein fusion encoded in isolate A21d2. RFA21d2_02304 is predicted to encode a fusion protein with FasD- and FasF-like functions. Abbreviations: iPAMP = isopentenyladenine ribotide; tZRMP = trans-zeatin ribotide; tZR = trans-zeatin riboside; tZ = trans-zeatin; iP = isopentenyladenine; iPA = isopentenyladenosine. Shaded boxes = not supported by results of this study.
Because of the absence of genomic resources and the focus on a single isolate, the evolution and contribution of other functions in the virulence of phytopathogenic Rhodococcus are poorly understood. However, insights into virulence evolution may be derived from characterizations of the Rhodococcus equi genome sequence [24]. R. equi infects mammals and is the only other species of this genus that is well documented as being pathogenic [25]. Comparisons of the R. equi 103S genome sequence to those of environmental species of Rhodococcus revealed little evidence for large-scale acquisition of niche-adaptation genes by HGT and instead suggested that pathogenicity of R. equi evolved through a limited number of key acquisition events coupled with co-option of genes core to Rhodococcus [24].
Here, we report the sequence and characterization of the genomes of 20 isolates of Rhodococcus. Our data indicate that HGT is important in virulence evolution but only four functions need to be acquired by members of Rhodococcus to gain the trait of phytopathogenicity. A striking discovery was made in isolate A21d2. This phytopathogenic isolate lacks the fas operon, which is replaced by a horizontally acquired and novel gene chimera. The protein fusion is predicted to be sufficient for the minimal functions of cytokinin catalysis and activation, typically provided by fasD and fasF of the fas operon. The absence of two-thirds of the fas operon from A21d2 is not consistent with the cytokinin mixture model. We profiled cytokinins in the wild type isolate D188 and its mutants, ΔfasD and ΔpFiD188, and could detect only one active cytokinin type that was synthesized in a fasD-dependent manner. Therefore, only one cytokinin type appears necessary for phytopathogenicity.
Results
Twenty Rhodococcus isolates were selected for genome sequencing
To quantify the virulence of the 20 selected isolates, we measured their effects on root growth of Nicotiana benthamiana seedlings [13]. First, we used wild type D188 and key mutants previously shown to be non-pathogenic or compromised in virulence to standardize the root inhibition assay (Figs. 2A and 2B). Of the 20 isolates, 15 isolates significantly inhibited root growth of N. benthamiana (Fig. 2C). The isolates we selected represent multiple clades of Rhodococcus (Creason and Chang, data not shown). Therefore, isolates, representing the genetic diversity of the sequenced samples, were tested and demonstrated to cause leafy galls to N. benthamiana (Fig. 2D).
Figure 2. Sequenced isolates of Rhodococcus vary in phytopathogenicity.

(A) Inhibition of N. benthamiana root growth by phytopathogenic Rhodococcus. Seedlings were independently inoculated with D188, its genetic variants, or 10 mM MgCl2 buffer (mock). Photographs were taken at 7 days after inoculation; scale bar = 0.25 cm. (B and C) Average root growth of N. benthamiana seedlings infected with D188 and its genetic variants (B) or with Rhodococcus isolates (C). Root lengths were measured (cm) and averaged. Error bars indicate standard error of the mean (SEM); *significant (p-value threshold ≤0.01). All treatments were repeated at least three times with similar results. (D) Isolates of Rhodococcus cause leafy gall disease. N. benthamiana was infected with the ΔpFiD188 strain of D188, mock, or members that represent the diversity of the clade. The black scale bar = 1 cm.
A minimum of 17.5 million reads was generated for each genome sequence (Table 1). Isolates A44a and D188 were selected as references and were more deeply sequenced using hybrid approaches. The reads were processed and independently de novo assembled for each genome. The number of scaffolds ranged from 9–50. Isolate A44a had the fewest scaffolds, which we attribute to the use of mate pair sequencing. The A44a assembly had two scaffolds that corresponded to its linear and circular plasmids and seven for the chromosome. Based on the finished genomes of R. jostii RHA1 and R. equi, we infer that A44a has four rRNA-encoding loci that could account for three of the physical gaps in the sequence. The use of 454 reads helped reduce the number of scaffolds for D188 when assembled using Illumina reads alone, but relative to other assemblies, was not effective in dramatically improving the quality of the assemblies. All 20 sequenced isolates have a high GC% of around 64%. The average genome size and number of coding sequences (CDSs) for all 20 isolates was estimated at 5.8 Mb and 5,475, respectively.
Table 1. Summary statistics of Rhodococcus draft genome assemblies.
| Isolate* | Pathogen¥ | # usable reads | # scaffolds† | ∼Size (Mb) | GC% | # CDSs | # tRNAs | Linear plasmid¥ |
| GIC26 | NO | 18,924,378 | 49 | 5.3 | 64.45 | 5022 | 48 | NO |
| GIC36 | NO | 18,778,318 | 46 | 5.6 | 64.48 | 5255 | 52 | NO |
| 05-561-1 | NO | 19,807,810 | 30 | 5.6 | 64.49 | 5303 | 50 | NO |
| LMG3605 | YES | 19,206,754 | 27 | 5.3 | 64.53 | 4966 | 51 | YES |
| D188 | YES | 65,408,710§ | 49 | 5.4 | 64.56 | 5125 | 51 | YES |
| LMG3602 | NO | 18,978,318 | 25 | 5.4 | 64.50 | 5124 | 50 | NO |
| LMG3616 | YES | 19,617,964 | 42 | 5.8 | 64.33 | 5486 | 51 | YES |
| A3b | YES | 20,464,110 | 34 | 6.0 | 64.21 | 5793 | 51 | YES |
| LMG3623 | YES | 20,002,692 | 30 | 5.8 | 64.38 | 5426 | 48 | YES |
| A78 | YES | 22,374,616 | 41 | 6.0 | 64.33 | 5684 | 50 | YES |
| A21d2 | YES | 20,072,238 | 30 | 6.0 | 64.10 | 5626 | 54 | NO |
| 04-516 | NO | 19,136,324 | 23 | 5.8 | 64.20 | 5465 | 52 | NO |
| A25f | YES | 20,556,130 | 17 | 5.9 | 64.13 | 5544 | 50 | NO |
| LMG3625 | YES | 17,750,698 | 17 | 5.9 | 64.10 | 5754 | 56 | YES |
| 05-339-1 | YES | 20,611,418 | 22 | 5.7 | 64.71 | 5449 | 54 | YES |
| A76 | YES | 18,608,398 | 29 | 6.0 | 64.56 | 5720 | 53 | YES |
| A44a | YES | 87,282,290§ | 9 | 5.9 | 64.47 | 5584 | 54 | YES |
| 02-815 | YES | 22,244,434 | 30 | 6.2 | 64.28 | 5819 | 55 | YES |
| 02-816c | YES | 23,697,112 | 45 | 6.1 | 64.52 | 5843 | 58 | YES |
| A73a | YES | 23,823,844 | 23 | 5.9 | 64.37 | 5501 | 56 | YES |
*Isolates designated with LMG were obtained from Belgium co-ordinated collection of micro-organisms (BCCM); GIC isolates are from a Greenland glacial ice core; all remaining isolates except D188, were obtained from diseased plants submitted to the Oregon State University (OSU) Plant Clinic. Italicized isolates = first sequenced using a hybrid approach; bold = type strain.
Determined based on leafy gall and root inhibition assays described in a separate study (see associated text for reference).
D188 had 65,301,274 PE Illumina reads and 107,436 454 Jr. reads and A44a had 25,093,452 PE Illumina reads and 62,188,838 Illumina mate pair reads from a 3.0 kb library.
Number greater than 1.0 kb in length.
A pFiD188-like plasmid is present in most, but not all, phytopathogenic isolates of Rhodococcus
The linear plasmid, pFiD188 is necessary for the virulence of R. fascians D188 [14]. We therefore used the sequence of pFiD188 as a query to search the genome assemblies to examine its necessity for Rhodococcus virulence [19]. CDSs homologous to those along the entire length of pFiD188 are present in 13 of 15 genome sequences of phytopathogenic isolates (Fig. 3A). The sequences of att, fasR, and fas are conserved (85% of the 204 CDSs in this cluster of virulence CDSs are identical and the remaining 15% are >99% identical to corresponding loci of the pFiD188 sequence generated in this study; Fig. 3B). However, within these loci, we identified 24 sequence discrepancies that differed in comparison to the previously reported pFiD188 sequence [19]. Because all linear plasmid sequences from this study were identical at these positions, we concluded the published sequence had errors. Of the 18 affecting the translated sequences of CDSs, 17 resulted in single amino acid changes. One was an insertion of a guanine residue in fasR, relative to the published sequence, that leads to substantially longer translated sequences because of an upstream, in-frame ATG start codon.
Figure 3. The att, fasR, and fas virulence loci are variable in organization among phytopathogenic isolates of Rhodococcus.

(A) GenomeRing of the 13 pFiD188-like sequences. Colored lines, corresponding to each isolate, indicate presence of a block. Transparent lines skip over absent blocks and connect co-linear blocks. A minimum block size of 1 kb was used. The inner portion defines conserved (R) and unique (U) regions of pFiD188 from isolate D188, as previously reported. The att-fas virulence loci and the NRPS-encoding region are also indicated by dotted regions. (B) Homology and co-linearity of the pFi_55-pFi_84 regions in the 15 phytopathogenic isolates. Top: structure of the region of pFiD188 from pFi_55 to pFi_84, with arrows depicting CDSs and the direction indicating the strand in which they are encoded. For isolates with a linear plasmid (Other 12), A25f, and A21d2, bars indicate the presence and range of sequence similarity. Relevant locus ID numbers are shown without RF25f_ and RFA21d2_ tags. The two cyan bars connected by a single line represent a gene fusion. (C) PCR-based detection of virulence loci (fasD) and pFiD188-like (pFi-013) sequences. The 16s rDNA gene was used as a positive control. Products were resolved on a 1%, 1X TAE agarose gel. Estimated product sizes (kb) are listed along the side.
A linear plasmid sequence is absent in two other pathogenic isolates, A25f and A21d2, and the five non-pathogenic isolates. PCR for a CDS located in the R1 regions of the pFiD188-like plasmids and implicated in their maintenance confirmed in silico predictions (Fig. 3C). Therefore, the linear plasmid is correlated with phytopathogenicity, but is not strictly necessary.
Between ∼40%–80% of the CDSs associated with the 13 linear plasmid sequences were identified as being acquired by HGT. Most are located in or at the border of the three “U” regions that were previously classified based on regions within pFiD188 of D188 that are unique in sequence relative to linear plasmids of other members of Rhodococcus [19]. Additionally, the gene gain/loss polymorphisms largely corresponded to the U regions and also correlated with evidence for HGT (Fig. 3A; Table S1). The pattern of insertions and deletions in the NRPS-encoding CDS of the U2 region is likely a reflection of limitations in assembling short reads exacerbated by the modularity of NRPS genes. The predicted functions for the polymorphic genes in the U regions did not provide strong evidence for a role in pathogen virulence (Table S1). Of the four conserved R1, R2, R3, and R6 regions, previously defined based on sequence similarities to other linear plasmids of Rhodococcus, there were fewer gene gain/loss polymorphisms but R3 and to some degree, R1 had evidence for HGT. In summary, results from the analysis of 13 linear plasmid sequences were consistent with previous reports in regards to the presence of conserved and unique regions, with higher incidences of recombination in the unique regions [19].
The contribution of horizontal gene transfer to virulence evolution is limited in its scale
Virulence genes of plant pathogens can be located in islands within the chromosome. We therefore searched the genomes for regions with signatures of HGT [26]. The average size for the identified regions was 11.5 kb (Fig. 4A). The scale of HGT was variable between isolates, with no clear correlation to phytopathogenicity (Fig. 4B). In phytopathogenic isolate LMG3605 for example, only 240 CDSs were identified within regions acquired by HGT and their total size represented just 5.5% of the genome. In contrast, non-pathogenic isolate GIC36 has 78 regions with ∼800 CDSs representing 16.2% of the total size of the genome as potentially acquired by HGT. Of all identified regions, some of the outliers were substantial in size, highlighted by the ∼107.5 kb region, encoding 103 CDSs, in 05-339-1 (Fig. 4A).
Figure 4. The scale of horizontal gene transfer varies among isolates.

(A) Box-and-Whisker plots of regions acquired by HGT as a factor of size. Regions putatively acquired via HGT were identified using Alien Hunter, with minimum criteria of size ≥5 kb and minimum score ranging from 10.297 to 16.047. Bottom and top of the boxes indicate the first and third quartiles, respectively, with the yellow bar indicating the median. Whiskers delimit the lowest and highest data within 1.5 interquartile ranges of the lowest and highest quartiles, respectively. Red circles represent outliers. Pathogenic isolates are underlined. (B) Summary statistics of HGT for the sequenced isolates. Pathogenic isolates are underlined. (C) Histogram presenting the number of groups of CDSs as a factor of group size. All CDSs identified in regions putatively acquired by HGT were compared using BLAST analysis to determine groups of homologs. The numbers of groups of CDSs (y-axis) were enumerated and plotted according to the size of the groups (x-axis). Analysis was done for only the 15 phytopathogenic isolates.
We compared the non-redundant set of 6,867 CDSs present in the horizontally acquired regions from all 20 isolates to a custom database consisting of CDSs gathered from non-phytopathogenic Actinomycetes. Approximately 33% of the translated Rhodococcus CDSs did not have a BLASTP hit and were thus considered specific to this group of Rhodococcus isolates. The only two significantly enriched clusters of orthologous genes (COGs) categories were “General functional prediction only” and “Function unknown”, as would be expected for a group of relatively poorly studied bacteria. Of the CDSs horizontally acquired by only the phytopathogenic isolates, the significantly enriched functional categories related to energy, metabolism, and general functions (Fig. S1A). A parallel analysis using Gene Ontology (GO) terms yielded similar results, with general functions in metabolism, transport, as well as transcription and translation being the more prominently identified terms (Fig. S1B). The few clusters that are likely associated with virulence are cytokinin and isoprenoid biosynthesis and metabolic processes.
Next, we reasoned that horizontally acquired candidate virulence genes should be conserved across the majority of the phytopathogenic isolates. An overwhelming majority of the CDSs associated with regions putatively acquired by HGT were present in only one or two of the phytopathogenic isolates (Fig. 4C). Even when we lowered our criteria and considered CDS that had homologs in approximately one-half of the phytopathogenic isolates, fewer than 100 homologous families were identified. Each of the families had homologs present on pFiD188. Overall, data suggest that other than the linear plasmid, HGT does not play a large-scale role in virulence evolution of phytopathogenic Rhodococcus and likely contributes more to the outside-host lifestyle of the bacteria.
Few genes are conserved and unique to phytopathogenic isolates
We hypothesized that, regardless of phylogenetic structure and HGT, the phytopathogenic isolates can be expected to have a core and exclusive set of CDS that distinguish them as pathogenic. We compared CDSs from 24 genome sequences, including the 20 from this study as well as Rhodococcus spp. AW25M09, JG-3, 29MFTsu3.1, and 114MTsu3.1 ([27]; GenBank BioProjects PRJNA200424, PRJNA195882, and PRJNA201196). The four additional isolates were identified from various environments, clustered with the phytopathogenic Rhodococcus, and lack loci known to be necessary for Rhodococcus phytopathogenicity (Creason and Chang, data not shown). Overall, we identified a “core” of 2,870 and a “pan” of 16,733 CDSs (Fig. 5). The COG categories in the “flexible” genome (13,863 CDSs) were enriched in categories related to energy and responding to the environment and its co-inhabitants (Fig. S2).
Figure 5. The pathogenic isolates have a small set of core coding sequences.
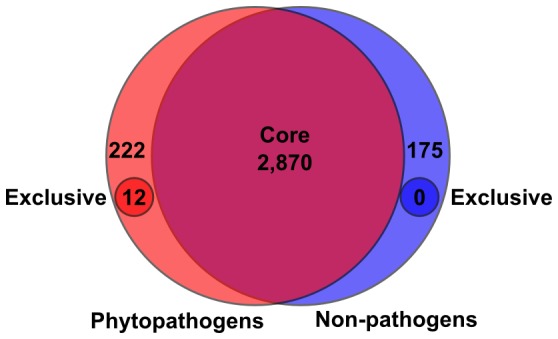
A total of 16,733-redundant CDSs are present in the “pan” genome of the 24 isolates of Rhodococcus that were examined. Based on reciprocal best-hit BLASTP analysis of the translated sequences, 2,870 are core. Coding sequences core to pathogenic (red) and non-pathogenic (blue) genomes were similarly identified for the 15 and 9 isolates, respectively.
Because we were interested in identifying CDSs that are exclusive to pathogens, we compared pan and core genomes of the 15 phytopathogenic isolates to the pan and core genomes of the 9 non-pathogenic isolates (Fig. 5). The 15 pathogenic isolates have a core of 234 CDSs that were not also identified as core to the nine non-pathogenic isolates. However, only 12 of the 234 were classified as exclusive based on their complete absence from the nine non-pathogenic isolates; the other 222 CDSs are part of the flexible genome of the non-pathogenic isolates. The 12 exclusive CDSs correspond to the att locus (10 CDSs), fasR, and a putative FAD-binding monooxygenase-encoding gene (pFi_057 of pFiD188), all of which have members on pFiD188. The reciprocal best hit for the FAD-binding monooxygenase-encoding gene of A21d2 barely exceeded threshold, with an identity of only 35.2% suggesting it may not be a bona fide member of the gene family, as each isolate has large numbers of monooxygenase-encoding genes (Davis and Chang, data not shown). Most notably, the six fas CDSs were not identified as core to the pathogenic isolates because of the absence of fas from isolate A21d2. PCRs using oligonucleotides specific to each CDSs of the fas operon were negative (Figs. 3B and 3C; Table S1).
The virulence loci of A25f and A21d2 are novel in structure and sequence
We used the pFiD188 sequence and reciprocal best-hit analysis to identify scaffolds of ∼190 kb and 18 kb in the A25f and A21d2 assemblies, respectively (Table S2). In A25f, 30 of the 33 best hit CDSs (RFA25f_04806-RFA25f_04843) are co-linear to their homologs on pFiD188, starting from nine CDSs upstream of the att locus and extending to two CDSs past the fas locus (Fig. 3B). The highest levels of sequence similarities were observed for att through fas (>95% identity; AttR = 94.5%). Despite that, the degree of sequence similarity was lower relative to the degree of similarity found among the 13 isolates that carry a linear plasmid like pFiD188. Analysis of flanking sequences did not provide any clues to the location of this scaffold in the genome because there were no regions of co-linearity with the D188 assembly. However, the scaffold lacked features typically associated with plasmids and the most parsimonious explanation is that a recombination event occurred between a portion of a pFiD188-like plasmid and the chromosome of A25f. Consistent with previously reported results, A25f delineates a small portion of the linear plasmid as necessary for phytopathogenicity [19].
In A21d2, only 11 of the 28 best hit CDSs in the 18 kb scaffold are co-linear to the U1 region, starting at RFA21d2_02316 (attR) and unexpectedly and abruptly ending after RFA21d2_02306, an AraC-like encoding CDS (fasR; Fig. 3B). We used thermal asymmetric interlaced (TAIL) PCR to determine flanking sequences. The resultant sequences were nearly 1 kb long, partially similar in sequence to each other, and mapped to separate single locations that coincided to a region between RFA21d2_03323 and RFA21d2_03324 within a 169 kb long scaffold, node_3. Consistent with previous searches, no fas operon was present in node_3 and no functional replacement of the Fas proteins could be predicted from the translated sequences of node_3. The A21d2 homolog of pFi_057 was not found in this block of co-linear CDSs and is instead located over 1 Mb away.
The fas operon of A21d2 is replaced by a gene fusion encoding IPT and LOG domains
The absence of the fas operon from A21d2 was not expected. The locus is replaced by a novel cluster of three overlapping CDSs (Fig. 6A). The outer two CDSs, RFA21d2_02305 and _02303, are annotated as containing a single Radical SAM domain and an Abhydrolase_6 domain, respectively. Neither of the domains are present in the Fas proteins of pFiD188 and the single Radical SAM domain is different from those predicted for the translated sequences of mtr1 and the homologous mtr2, both of which are located between fasR and the fas operon in pFiD188 [19]. RFA21d2_02305 and _02303 were not expressed in cells grown in any of the three media tested and not predicted to be involved in cytokinin metabolism (Fig. 6A).
Figure 6. RFA21d2_02304 is a gene fusion unique to A21d2.

(A) Structure and predicted functional domains for RFA21d2_02303-02305. Functional domains were identified using PFAM (PFxxxx) and mapped to each of the sequences. Sequence logos were generated and shown for the isopentyltransferase and phosphoribohydrolase domains. The best matching residues of RFA21d2_02304 are shown and mapped to their corresponding locations in the diagram. Amino acids are colored according to class. The “*” highlights conserved residues; the single “R” at positions 166 and 378 are not part of the sequence logos but are included to show their conservation. Small arrows show the locations of the binding sequences for oligonucleotides used in RT-PCR (see inset: L = LB; I = minimal media+leafy gall extracts; M = minimal media). RT-PCRs with oligonucleotides that span both overlapping regions were negative and are not shown. (B) Unrooted phylogenetic tree for IPTs. Scale bar = number of amino acid substitutions per site. (C) Unrooted phylogenetic tree for phosphoribohydrolases. Scale bar = number of amino acid substitutions per site. One branch was split for sizing purposes.
Remarkably, RFA21d2_02304 is a chimera between fasD- and fasF-like sequences and the possibility of an assembly mistake was dismissed based on PCR and Sanger sequencing that confirmed the sequence of the locus (Fig. 6A; Table S2). Both IPT and cytokinin phosphoribohydrolase domains are present in RFA21d2_02304. Three residues necessary for IPT function and conserved in all IPT sequences previously examined by others are present in the N-terminal portion of RFA21d2_02304 [28]. There are also three conserved and predicted catalytic residues in the C-terminal portion of RFA21d2_02304 that are found in similar positions relative to other bona fide LOG proteins [29]. Unlike fasD of D188 and the CDSs flanking RFA21d2_02304, the chimera is expressed under what is considered non-inducing conditions (Fig. 6A; Fig. S3). These results indicated the three CDSs are expressed as independent monocistronic messages.
The two domains of RFA21d2_02304 formed branches distinct from the 14 other FasD and FasF homologs (Figs. 6B and 6C). The IPT region is more similar to FasD than the LOG region is to FasF, and as previously shown, IPTs from phytopathogenic Rhodococcus cluster separately from those of plants and other bacteria [21], [30]. The exceptions are IPTs of Streptomyces spp., which also encode the fas operon [31]. The LOG region of RFA21d2_02304 and FasF are more similar to LOG of plants than to either of the two homologs present in Rhodococcus. In all, the data suggested that RFA21d2_02304 was acquired horizontally from another source, as opposed to being derived from a series of recombination events within a pFiD188-like plasmid.
Isopentenyladenine is the only cytokinin that specifically accumulates in extracts of D188 grown in culture and under inducing conditions
The absence of fasA from A21d2 is not consistent with the model that predicts the necessity for a mixture of cytokinin types (isopentenyladenine (iP), tZ, ciz-zeatin (cZ), and the methylthio derivatives of the latter two) for virulence [18], [20]. The 20 Rhodococcus isolates encode homologs of all the enzymes necessary for the methylerythritol phosphate (MEP) pathway, including a homolog of isopentenyl diphosphate (IPP) isomerase (IDI1) that catalyzes the isomerization of IPP to dimethylallyl diphosphate (DMADP; Fig. 1; Table S3). The MEP pathway synthesizes DMADP and the intermediate, (E)-4-hydroxy-3-methyl-but-2-enyl diphosphate (HMBDP), both of which are isoprenoid side chain donors that can be used by IPTs to synthesize the isopentenyladenine ribotide (iPAMP) and tZ ribotide (tZRMP; [32]–[35]). In contrast, homologs for most of the enzymes of the mevalonate (MVA) pathway that synthesizes DMADP were not identified (Table S3).
Because DMADP and HMBDP are potentially available, it is conceivable that the activities of FasD and FasF could enable the synthesis of both iP and tZ by Rhodococcus in culture. To test this, we profiled cytokinins from genetic variants of D188 grown in minimal media supplemented with extracts of leafy galls, uninfected plants, or H2O as a mock treatment. Of the 32 cytokinins that were profiled, only eight could be detected: iP, cZ, 2-methylthio-cis-zeatin (2MeScZ), their ribosides, as well as iPAMP and tZRMP; tZ was not detected (Table S4). iP was the only active cytokinin type that significantly accumulated specifically in preparations from bacteria grown in leafy gall extracts and in a fasD/pFiD188-dependent manner (Fig. 7). In contrast, none of the detectable Z-type cytokinins accumulated to significant levels in a treatment-dependent manner. All detectable cytokinin types started to accumulate at later time points in the ΔfasD mutant, independent of treatment (Fig. S4). Expression profiles of attE and fasD showed induced expression specific to bacterial cells grown in extracts of leafy galls, as previously reported (Fig. S2; [16]. Maximum expression was observed at 6 hours (10,000X and 100X more expression for attE and fasD, respectively, relative to expression in cells grown in extracts of uninfected plants) and rapidly declined to basal levels by 12 hours after growth. Data indicated that phytopathogenic Rhodococcus synthesizes only one active type of cytokinin in a fasD-and leafy gall-dependent manner.
Figure 7. Isopentenyladenine is the only active cytokinin that accumulates in a fasD and leafy gall extract-dependent manner.

Wild type D188 (blue), ΔfasD (green) and ΔpFiD188 (red), were grown in media augmented with extracts from leafy galls, uninfected plants, and water as a mock. Of 32 cytokinin types that were profiled, only eight were detected and the six most abundant types are shown. Change in concentration (y-axis) is presented for only the first four time points (x-axis). Colored “*” indicates significant difference in cytokinin accumulation in corresponding mutant genotype relative to wild type and corresponding time point (p-value threshold = 0.05). Experiments were repeated twice with similar results.
The fasR CDSs of A25f and A21d2 are substantially more polymorphic than other family members
Putative AraC-like encoding CDSs are located between the att operon and fas operon or fas-like CDSs in both A25f and A21d2 (RFA25f_04127 and RFA21d2_02306, respectively; Fig. 3). RFA25f_04127 and RFA21d2_02306 are predicted to be functional and clustered with fasR in a phylogenetic tree, supporting their membership to this family (bootstrap percentage of 100%; Fig. S5). However, the sequences of fasR are highly polymorphic (Fig. 8A). The translated sequence of the A25f CDS shares 97% identity with FasR of D188. In RFA21d2_02306, there are 343 SNPs (32% of all possible sites), of which 89, or 26% of the SNPs, are non-synonymous substitutions.
Figure 8. RFA25f_04833 and RFA21d2_02306 are polymorphic in sequence.

(A) Predicted structure of the 348 amino acid FasR sequence. Functional domains are indicated along the top with arrows and barrels representing predicted β-sheets and α-helices, respectively. Boxed region = a region of fasR from D188 that is absent from the reported sequence. Synonymous (red) and non-synonymous substitutions (blue) and INDELs (green arrow heads with number of nucleotide differences indicated) are plotted according to their location in the sequences. Clusters of non-synonymous substitutions in fasR of A21d2 are denoted with, “*”. Substitutions for the other 12 FasR sequences (Other 12) represent the sum total of all sites with substitutions. (B) Sliding-window analysis of synonymous and non-synonymous substitutions in fasR of A21d2 paired with fasR of D188. (C) Sliding-window analysis of codon adaptation index in fasR of A21d2 and D188. The x-axis is based on coordinates of the translated sequence that are aligned to predicted structure shown in panel A.
RFA21d2_02306 is nevertheless predicted to be under purifying selection (Ka/Ks = 0.327; p-value = 9.3×10−20; paired with allele from D188). RFA21d2_02306 is also not optimized for translation (CAI = 0.221; 59 ribosomal protein encoding genes = 0.667; genome average = 0.530). This was not surprising since fasR includes an unusually high number of rare TTA leucine codons [16]. In attempts to model the patterns of substitutions, we mapped them onto the predicted secondary structure of FasR (Fig. 8A). Both domains of the predicted structure are similar to other AraC-like regulators [36]–[39]. One region within the putative ligand binding domain was identified as having a high Ka/Ks and was significant in a permutation test (p-value = 0.033; Fig. 8B; [40]). The Ks values started trending upward thereafter within the putative DNA binding domain and culminated in a region with very high values (p-value = 0.04). This region overlaps two helix-turn-helix motifs predicted to be involved in binding DNA, based on alignments to AraC. In addition, a larger portion of this region correlated with above average CAI values in RFA21d2_02306 relative to FasR of D188 (Fig. 8C). Therefore, despite evidence for purifying selection and a high density of synonymous substitutions in the putative DNA binding domain, diversifying selection was identified in a putative ligand binding region.
Discussion
Phytopathogenic Rhodococcus are unlike most plant pathogens. These bacteria are Gram-positive Actinobacteria with members capable of causing growth deformities and persisting, often through the life of the host plant. To gain insights into the evolution of Rhodococcus virulence and develop resources for understanding the mechanisms of phytopathogenesis, we determined and characterized the genome sequences for 20 isolates. Analyses suggested that the acquisition of just four functions is sufficient for diverse isolates of Rhodococcus to gain the trait of phytopathogenicity. Additionally, the discovery of a novel gene chimera and profiling of the exemplar isolate challenged the cytokinin mixture model of Rhodococcus virulence.
Rhodococcus requires four horizontally acquired functions to be phytopathogenic
Virulence evolution of phytopathogenic Rhodococcus species can be modeled by the co-option mechanism proposed for R. equi [10], [24]. It has been previously reported that the chromosomal locus vicA, which encodes malate synthase that functions in the glyoxylate shunt pathway of the Krebs cycle, is necessary for full symptom development [41]. Additionally, the predicted dependency of the Fas proteins on the MEP pathway for the necessary substrate for cytokinin biosynthesis is another example of co-option (Fig. 1). Few other loci could be identified that had evidence for HGT and implicated in virulence. Only a miniscule fraction of the CDSs associated with regions with signatures of HGT were found conserved in pathogenic isolates and all of these CDSs have homologs encoded on a pFiD188-like plasmid (Fig. 4). Analysis of functional categories of CDSs associated with regions acquired via HGT also failed to support HGT as having a large-scale contribution in virulence evolution (Fig. S1). Similar findings were described for the COG categories enriched in the “flexible” genome (Fig. S2). The results are consistent with the linear plasmid being the key acquisition for virulence and the majority of horizontally acquired functions contributing to the adaptation of Rhodococcus to their outside-host environments. Indeed, R. fascians is considered a soil bacterium and closely related isolates have been isolated from extreme environments, suggesting an ability of these organisms to use a wide range of substrates [42]–[44].
The structures and sequences of virulence loci of A25f and A21d2 are novel
We suggest that the horizontal acquisition of just four functions involved in secondary metabolism (att), gene transcription (fasR), cytokinin biosynthesis (fasD), and cytokinin activation (fasF) is sufficient for Rhodococcus to infect plants. Isolates A25f and A21d2 are unique among the sequenced samples. In A25f a recombination event likely occurred that introduced a block of CDSs from a linear plasmid into the chromosome. Gene fusions are generally predicted to occur by insertions and deletions between juxtaposed genes, such as those within an operon, and disseminated via HGT [45], [46]. However, for A21d2 a fusion would have had to occur between two CDSs separated by fasE. Stronger evidence against a direct fusion event comes from the observation that the IPT and LOG domains of RFA21d2_02304 formed branches clearly distinct from their homologs and that RFA21d2_02304 overlaps two CDSs absent from other sequenced isolates of Rhodococcus (Fig. 6). We therefore conclude that RFA21d2_02304 was acquired from another organism in what could be a non-orthologous displacement of the fas operon.
Integration of virulence loci into the chromosome is expected to confer a greater state of stability. However, the rarity of the virulence loci of A25f and A21d2 among the sequenced isolates is not consistent with this expectation. A potential explanation is that because the pFiD188-like linear plasmids are conjugative, the mobility of these vehicles may compensate for any lowered stability by enabling rapid dissemination throughout the population. This is highly plausible especially considering that acquisition of the four functions, most frequently vectored by a plasmid, is sufficient for genetically divergent Rhodococcus isolates to be phytopathogenic (Fig. 2). Alternatively, the events that led to the changes in A25f and A21d2 could have occurred relatively recently or the 20 isolates that were sequenced do not adequately reflect the diversity of the population. We also cannot ignore the likelihood for trade-offs that occur in non-obligatory pathogens that persist in both within and outside-host environments.
The att locus is conserved in the phytopathogenic Rhodococcus isolates
The att locus is the most conserved in sequence and structure across all phytopathogenic isolates examined and consistently clustered with the fas locus (Fig. 3). Its product is also the only known target of host resistance [47]. Together, these data suggest the product of att is an important virulence determinant in Rhodococcus despite the attenuated phenotype of the Δatt mutant [17]. The infection methods used in the laboratory may possibly circumvent att dependence, since the Δatt mutant can cause disease if the host is wounded prior to infection. It has been suggested that att is important for the early transition of R. fascians from epiphyte to endophyte and its ability to ingress into host tissue [17]. Indeed, attE expression peaks to extremely high levels within six hours of growth in inducing media and rapidly decays thereafter (Fig. S3). Seemingly counter to the importance of att is its absence from Streptomyces turgidiscabies, which does have the fas operon and can form galls on plants ([31]; PRJNA185785). However, att may not be necessary because S. turgidiscabies does not ingress but rather, it primarily enters its host through wounds and natural openings.
A new model for Rhodococcus virulence
The data generally supported the model identifying pFiD188 as necessary for R. fascians and the role of cytokinins in the pathology of plant-associated Rhodococcus. However, several lines of evidence presented in this study are consistent with a mechanism of virulence where only the iP cytokinin type, not a mixture of cytokinins, is synthesized and necessary for Rhodococcus phytopathogenicity (Figs. 1, 3, 6, and 7). We and others could not detect tZ and/or demonstrate its FasD-dependent synthesis in culture (Fig. 7; [18], [48]). The other two free base types that could be detected, cZ and 2MeScZ, accumulated independently of fas gene induction, similar to previous reports (Fig. 7; Fig. S4; [20], [21], [49]–[53]). The generation of tZ from cZ by the action of a cis-trans isomerase is not well substantiated, so it is unlikely that this could be a source of tZ [21].
Evidence also argues against FasD being able to directly produce significant levels of tZ even though bacterial IPTs can use HMBDP as a prenyl donor without the activity of a P450 monooxygenase [28], [32]. The Tzs IPT of A. tumefaciens has a hydrophilic region in the substrate-binding cavity that contributes to its ability to use both DMADP and HMBDP as substrates [28]. A substitution mutation that affected the hydrophilic region increased the specificity of A. tumefaciens Tzs for DMADP by ∼100-fold [28]. FasD of D188 has an alanine at a position that corresponds to one of the key hydrophilic residues of Tzs and the Kcat/Km value for FasD is 10X higher when DMADP is provided as a substrate, relative to HMBDP [18]. Therefore, while it is possible that FasD could use HMBDP to generate Tz, data suggests otherwise. Additionally, the novel gene chimera of A21d2 indicates that only the IPT and LOG functions are necessary for virulence (Figs. 3 and 6). The IPT domain of RFA21d2_02304 also has amino acid substitutions that affect the predicted hydrophilic region that contribute to the increased specificity of Tzs for HMBDP [28]. RFA21d2_02304 is thus not predicted to efficiently synthesize tZ.
The original model for R. fascians phytopathogenicity is predicated on the necessity of fasA for virulence [15]. However, the original fasA mutant was generated via insertion of a plasmid and the sufficiency of fasA in complementing the mutant was not tested, opening the possibility for polar effects. In addition, FasA has not been confirmed as having P450 monooxygenase activity and the absence of detectable levels of tZ indicate fasA may be dispensable. FasB and FasC, like deoxyxylulose 5-phosphate (DXP) synthase, the first enzyme of the MEP pathway, are members of thiamine pyrophosphate enzyme superfamilies [54]. We speculate that the possible redundancy in function could explain the dispensability of fasB and fasC. The superfluousness of fasE (cytokinin oxidase/dehydrogenase) for virulence solves the conundrum of why a pathogen has an operon that encodes an enzyme with a function that diametrically opposes those that synthesize and activate cytokinins as virulence factors. There are two explanations as to why functions predicted to be dispensable are still in the population of phytopathogenic Rhodococcus clades. The enzymatic functions of the FasA-C, and -E may be unnecessary but the proteins may be required for scaffolding and forming a protein complex for efficient cytokinin biosynthesis and activation. Alternatively or additionally, the CDSs may have not been purged because membership in the fas operon could confer a level of immunity to the effects of mutation or are important in contributing to the regulation of gene expression [55].
New insights into cytokinin metabolism
The characterization of pathogen-encoded virulence proteins and molecules has provided many new insights into host growth, development, and signaling. Specifically, bacterial-encoded IPTs have contributed much to our understanding in cytokinin metabolism in plants [21]. The association of fasF with fasD in the fas operon was key to recognizing that LOG of plants is involved in cytokinin metabolism and provided evidence for the existence of the direct activation pathway of cytokinins [22]. The discovery of RFA21d2_02304 in this study raises the possibility that the phosphoribohydrolase complexes with IPT in other phytopathogenic Rhodococcus and also in plants. Gene fusions are important for increasing functional complexity through the generation of multi-domain proteins, often by uniting domains from those that physically interact and function in a common pathway. The low in vitro turnover rate of FasD led to the speculation of product inhibition [18]. The possibility of a complex with FasF indicates the potential for a direct activation of cytokinins and alleviation of inhibition. In A21d2, the fusion could be more efficient at coupling these two reactions, a possibility that will be addressed in future studies. It will also be interesting to determine if, and which, members of the IPT and LOG families form complexes in plants [56], [57].
Materials and Methods
Nucleic acid preparations, Sanger sequencing, and PCRs
Genomic DNA was extracted from cells grown directly from stocks. The Wizard Genomic DNA Purification Kit was used, according to the instructions of the manufacturer, to extract genomic DNA (Promega Corporation, Madison, WI, USA). Except for D188, isolates had undergone a limited number of passages.
Genomic DNA isolated from appropriate isolates was used as templates in PCRs with primers specific to the CDS (Table S5). Thermal Asymmetric Interlaced PCRs (TAIL) were done using nested oligonucleotides designed to Contig_21 and genomic DNA from A21d2, according to methods previously described (Table S5; [58]). Eight arbitrary degenerative primer pools were used in the amplification steps and successfully amplified products were sequenced and aligned to the A21d2 genome sequence.
For Sanger sequencing, products were treated with 2.5 U of Exonuclease I and 0.25 U of Shrimp Alkaline Phosphatase (SAP) for 40 minutes at 37°C and heat killed at 80°C for 20 minutes. Products were sequenced on an ABI 3730 DNA Analyzer in the Center for Genome Research and Biocomputing (CGRB) at Oregon State University.
Trizol Reagent (Life Technologies, Carlsbad, CA, USA) and ∼200 µl of 500 nm glass beads were added to Rhodococcus cells and disrupted using a FastPrep-24 homogenizer (MP Biomedical, Santa Ana, CA, USA) at 6.0 m/s for 1 minute. Samples were treated with DNase I (NEB, Ipswich, MA, USA), quantified and analyzed using a BioAnalyzer (Agilent Technologies, Santa Clara, CA, USA). First-strand cDNA was synthesized using 100 ng of total RNA and the Reverse Transcription System (Promega Corporation). One µl of the reverse transcription reaction was mixed with 10 µl LightCycler 480 SYBR Green I Master, 2.0 µl primer mix (250 nM each) and water for a final volume of 20 µl. Quantitative PCR was done on a LightCycler 480 System (Roche Applied Science, Germany). Gene expression was calculated using the deltadeltaCt method relative to the expression of the 16S gene and corresponding genes in the mock treatment [59]. For RT-PCR, first-strand cDNA was synthesized from 1.0 µg total RNA using Quantas (Life Technologies, Carlsbad, CA, USA). cDNA equivalent to 50 ng of starting RNA template was used in PCRs. Samples lacking reverse transcriptase were included as controls. Oligonucleotide sequences are available in Table S5.
Next-generation sequencing, assembly, and annotation
Library construction and sequencing were done in the CGRB (Illumina GAIIx, HiSeq, and MiSeq, 454 Jr.), DNAVision SA (Illumina HiSeq of D188), or HTSF@UNC (Mate-pair sequencing of A44a).
Genomes were assembled using Velvet, with hash lengths of 75 [60]. Insert sizes were independently determined based on estimated fragment sizes from each library preparation. A44a was assembled using Velvet 0.7.55, D188 was assembled using Velvet 1.1.05, and the other 18 isolates were assembled using Velvet_1.2.08. For each genome, multiple assemblies were done, in which coverage cutoff, expected coverage, and hash length parameters were changed. The highest quality assembly was identified based on number of contigs and having a sum total size between 5–6 Mb. Contigs from isolate A44a were reordered with Mauve, using the genome sequence of Rhodococcus jostii RHA1 as a reference [61], [62]. All other assemblies were directly or indirectly re-ordered based on the genome sequence of A44a. Prokka was used to annotate genomes. Prokka identifies tRNAs, rRNAs, and CDSs using Aragaron, rnammer, and prodigal, respectively (Prokka: Prokaryotic Genome Annotation System - http://vicbioinformatics.com/). CDSs were annotated based on BLAST analysis to a database of genomes core to the Rhodococcus genus, including all whole-genome assemblies, followed by BLAST analysis to the UniProt database [63], [64]. The remaining CDSs were then used in searches against the HMM databases using HMMER3 [65].
Phylogenetic analyses
Publically available sequences were gathered from the NCBI nr, nt, or wgs databases (http://www.ncbi.nlm.nih.gov/). Lists of sequences were manually curated. Sequences were aligned using L-INS-i algorithm in MAFFT and the most appropriate models of substitution were selected using the BIC selection method implemented in ProtTest 3 [66], [67]. Trees were generated using RAxML, with the -f a setting, and 1000 bootstrap replicates, unless otherwise noted [68]. Alignments were visualized and trimmed using Belvu [69]. Gblocks was used to trim the MLSA alignments with half gapped positions allowed [70]. Images were generated using the iTOL [71]. Only bootstrap values equal to or greater than 50 were shown.
For IPT-encoding genes, sequences were gathered by using RFD188_04926 (fasD) from D188 as the query in tBLASTn searches against both the nt and wgs NCBI databases. A total of 50 and 25 translated sequences, from the nt and wgs databases respectively, all IPT-encoding genes from Arabidopsis thaliana and Oryza sativa, and fasD from the Rhodococcus strains sequenced in this study were used to construct the tree. A similar approach was used for LOG-encoding genes with the exception that RFD188_04928 (fasF), RFD188_01289 (LOG-1), and RFD188_01290 (LOG-2) were used as query sequences. The top 50 hits for each query, 28 LOG family genes in the UniProtKB database and those found in the Rhodococcus strains sequenced in this study were used to construct the tree1. A total of 308 AraC-type sequences were identified, using BLASTP analysis, from the translated CDSs of all 20 Rhodococcus isolates (exceeded an e-value threshold of 1×10−5). Seventy of the sequences were used to construct the tree.
Bioinformatic analyses
InterProScan was used to analyze the translated sequences of RFA21d2_02303-_2305 for domain analysis and generation of sequence logos [72]. Sequences from IPR005269 (LOG family, 9681 sequences) and IPR002648 (IPT domain, 144 sequences) were downloaded from the InterPro database and aligned using MAFFT–auto and L-INS-i, respectively [67], [73]. Sequences without the conserved domains were removed, leaving 9648 and 75 sequences, respectively. Logos were generated using WebLogo 3 (http://weblogo.threeplusone.com/; [74]).
For reciprocal best-hit BLAST analysis, the translated sequences of all CDSs of pFiD188 were used as queries in BLASTP searches of the A25f and A21d2 assemblies. We used the pFiD188 sequence from D188 generated in this study because of the discrepancies between it and the deposited sequence (JN093097; [19]). Top hitting homologs (p-value threshold = 1×10−5) were used as queries in reciprocal BLASTP analysis of the D188 assembly. Results were confirmed with tBLASTn searches.
The core and flexible genomes were identified using reciprocal best-hit BLASTP analysis of translated CDSs. Translated sequences were iteratively added into groups of orthologs, starting with strain D188. Threshold cut-offs of 35% identity and at least 50% gene coverage of the subject by the query were used.
Secondary structure predictions were made using jpred and visualized using jalview [75], [76]. ParaAT was used to generate alignments as input for Ka/Ks calculator [77], [78]. A 120-nucleotide window was slid along the sequence in 12 nucleotide increments for analysis of Ka/Ks and codon usage. For the permutation test, the ParaAT-aligned sequences were split based on aligned codons and the order of the codons was randomly permuted 1000 times. Values were calculated for each of the permuted sequences, using the same sliding window approach. We enumerated the number of times four (Ka and Ka/Ks) and six (Ks) consecutive windows were found in the permutations. Threshold values for window peaks were set at 1.5 * (standard deviation) from the mean for Ka and 2 * (standard deviation) from the mean for Ks and Ka/Ks, in relation to the initial aligned fasR sequences.
The SuperGenome based on the 13 pFiD188-like sequences was aligned with Mauve then generated and visualized using GenomeRing [62], [79].
Alien hunter (default settings) was used to predict regions acquired by horizontal gene transfer [26]. A custom BLAST database was generated using the translated CDSs from Streptomyces coelicolor A3, Nocardia farcinica, Corynebacterium diphtheriae NCTC 13129, Gordonia KTR9, Mycobacterium tuberculosis H37Rv, Rhodococcus jostii RHA1, Rhodococcus opacus B4, Rhodococcus equi 103S, and Rhodococcus erythropolis PR4 [24], [61], [80]–[86]. Homology was determined based on BLASTP top-hit analysis (threshold cut-offs = >50% in length and ≥35% identity).
WebMGA (default settings) and Revigo (Allowed similarity = 0.9) were used to assign genes to Cluster of Orthologous Groups and Gene Ontology (GO) categories, respectively [87], [88]. Revigo was also used to analyze GO categories. Fisher’s exact test was used to determine statistically significant differences in COG assignments (p-value≤0.01).
Graphs were generated in R [89].
Inkscape was used to design line drawings and figures (inkscape.org/).
Bacterial strains and growth conditions
The 20 isolates of Rhodococcus (Table 1) were grown in Luria-Bertani (LB) media at 28°C. When appropriate, media were amended with 30 µg/ml kanamycin.
Plant growth conditions and plant infection
For root inhibition assays, N. benthamiana was germinated on MS agar plates (half-strength MS, 0.5 M MES). Three-day-old germinated seedlings were inoculated with suspensions of Rhodococcus (OD600 = 0.5; 10 mM MgCl2 buffer) or mock inoculated (buffer only) and grown vertically for 1 week at room temperature with 16 hours of light. Plantlets were photographed and ImageJ was used to measure root lengths [90]. Comparisons for each Rhodococcus isolate were made relative to average root length of corresponding seedlings inoculated with ΔpFiD188 in the same experimental replicate. Tukey’s HSD test was used to determine significance.
Leafy galls were induced in 3–4-week-old N. benthamiana plants, as previously described [14]. The apical meristems of plants were pinched using forceps and inoculated with 10 µl of Rhodococcus (OD600 = 0.5; 10 mM MgCl2 buffer). Plants were grown under conditions described above and assessed weekly. Leafy galls were photographed at 4 weeks post-inoculation. For leafy gall extracts, 4 week-old leafy galls were excised from plants, ground in sterile water using a mortar and pestle, and filter sterilized (22 µm filter). Extracts were frozen at −80°C until use. Induction was done as previously described [91].
Cytokinin profiling
Cytokinins were extracted from R. fascians D188 and its variants and quantified, using previously published methods [91], [92]. All supernatants were spiked with deutered cytokinin standards, including [2H6]iP, [2H6][9R]iP, [2H6](9G)iP, [2H6](7G)iP, [2H5]DHZ, [2H5][9R]DHZ, [2H5](9G)DHZ, [2H5](7G)DHZ, [2H5](OG)DHZ, [2H5](OG)[9R]DHZ, [2H5]2MeScZ, [2H5]2MeScZR, [2H6]MS-iP and [2H6]MS-iPA (Table S5; Olchemlm Ltd., Olomouc, Czech Republic). The pH of the samples was adjusted to pH 7 and samples were purified on a DEAE-Sephadex column (2 ml, HCO3 −) with a RP-C18 column coupled underneath. After washing with ddH2O, the fraction containing the cytokinin free bases, ribosides, and glucosides was eluted with 80% methanol. Eluates were dried under vacuum, diluted with PBS and immunoaffinity purified using isoprenoid cytokinin IAC columns (Olchemlm Ltd.). After elution with ice-cold 100% methanol, eluates were dried in vacuum and stored at −20°C until further analysis. Cytokinin-phosphates (retained in the DEAE column) were eluted with NH4HCO3 and concentrated on a RP-C18 column before elution with 80% methanol. After vacuum concentration and dissolution with 0.01M Tris-HCl (pH 9.6), the cytokinin-phosphates fraction was treated with alkaline phosphatase and further immunoaffinity purified as described above.
Cytokinins were quantified using an electrospray ACQUITY TQP UPLC-MS/MS device (Waters, Micromass Ltd., United Kingdom). Samples (10 µl) were injected into an ACQUITY TQP UPLC BEH C18 column (1.7 µm×2.1 mm×50 mm, Waters) and eluted with ammonium acetate (1.0 mM) in 10% methanol (A) and 100% methanol (B). The UPLC gradient was as follows: linear gradient of 100% A to 55.6% A and 44.4% B in 8 minutes, followed by a wash with 100% B and an equilibration to initial conditions with 100% A at a flow rate of 0.3 ml/min. The effluent was introduced into the electrospray source at a temperature of 150°C. Quantitative analysis was carried out using the internal standard ratio methods and the deuterated isotopes. The ESI(+)-MRM (multiple reactant monitoring) mode was used for quantification based on specific diagnostic transitions for the different compounds analyzed (Table S6). Chromatograms were processed using the Masslynx software (Waters) and the concentrations were calculated following the principles of isotope dilution.
Two-way analysis of variance followed by Dunnett’s multiple comparison test were used to determine statistical significance. The two-way analysis of variance examined the influence of both time and bacterial strain on cytokinin levels assuming equal variance across both groups. Dunnett's test was used to compare cytokinin levels in ΔfasD and ΔpFiD188 to D188, within each time point, assuming equal variance among the treatment strains.
Supporting Information
Analysis of functional categories associated with coding sequences in regions acquired via HGT (A) Analysis of clusters of orthologous groups (COGs). COGs of CDSs exclusive to pathogenic isolates were compared to those common among all 20 isolates. CDSs putatively acquired by HGT were categorized as exclusive to two or more pathogenic isolates (dark red) and those present in two or more isolates regardless of phytopathogenicity trait (blue). CDSs were assigned to COG functional groups (x-axis) and displayed as the percent of COGs per category relative to the total number of COGs assigned (y-axis). (B) Scatterplot of GO functional categories. GO terms were assigned to all coding sequences identified in pathogenic isolates and in regions potentially acquired via HGT. The semantic similarities were calculated, GO terms representative of clusters were generated, and plotted in a scatterplot. Clusters (circles) are placed based on semantically similarities, with more similar clusters placed closer to each other. Plot sizes represent the frequency of GO terms of a cluster relative to the total number of terms in the GO database. Heatmap represents semantic uniqueness, calculated relative to all terms in the list. High-level functions are indicated for clusters of the more frequently observed terms.
(EPS)
Analysis of functional categories associated with the core and flexible genomes COGs of CDSs that were core (blue) to all 24 isolates were compared to those in the flexible (red) genome. CDSs were assigned to COG functional groups (x-axis) and displayed as the percent of COGs per category relative to the total number of COGs assigned (y-axis).
(EPS)
Marker genes of the att and fas operon were induced in the presence of leafy gall extract. D188 was grown in media augmented with extracts from leafy galls (red), uninfected plants (blue), or water (mock). Samples were taken and RNA was extracted at the times indicated. Expression of attE and fasD was determined using qRT-PCR and calculated as expression relative to 16S and corresponding genes in D188 grown in a mock-treated culture.
(EPS)
Complete cytokinin profiling data. Three genotypes of D188, wild type (blue), ΔfasD (green) and ΔpFiD188 (red), were grown in media augmented with extracts from leaf galls, uninfected plants, and water as a mock. Of 32 cytokinin types that were profiled, only eight were detected and the six most abundant types are shown (iP: isopentenyladenine; cZ: cis-zeatin; 2MeScZ: 2-methylthio-cis-zeatin; and their ribosides). Change in concentration (y-axis) is presented for all time points (x-axis). Colored “*” indicates significant difference in cytokinin accumulation in corresponding mutant genotype relative to wild type and corresponding time point (p-value threshold = 0.05). Experiments were repeated twice with similar results.
(EPS)
RFA25f_04833 and RFA21d2_02306 are members of the fasR family. The alignment of the translated sequences for predicted AraC-type transcriptional regulators was used to generate a phylogenetic tree. Only candidates from the original 20 isolates were included. A single candidate AraC-type transcriptional regulator from Rhodococcus jostii RHA1 was used as the root. Genes are labeled by locus tag or with GI number (R. jostii RHA1). The FasR sequences are in bold. Node values indicate bootstrap percentages out of 1000 iterations. The scale bar represents the mean number of amino acid substitutions per site.
(EPS)
Predicted CDSs of the thirteen pFiD188-like sequences. *Regions were designated based on previously published determinations; §Functions were based on the annotations of the query sequences described in this study. The query sequences were derived from the CDS of the first isolate in which it appears in the table. Homologs were identified using reciprocal best BLAST hit analysis.
(XLSX)
Reciprocal best BLAST hit analysis of pFiD188 against the genome assemblies of A25f and A21d2. *Locus identifiers from previously published pFID188 linear plasmid sequence; §Based on this study.
(PDF)
Homologs of enzymes of the methylerythritol phosphate pathway are present in all 20 isolates of Rhodococcus . *Locus; % identity; BLAST e-value. Column abbreviations: Deoxyxylulose 5-phosphate synthase (DXPS); DXP reductiosomerase (DXPR); isopentenyl diphosphate isomerase (IDI1); 4-diphosphocytidyl-2C-methyl-D-erythirol synthase (CMS); 4-(cytidine 5′-diphospho)-2-C-methyl-D-erythritol kinase (CMK); 2-C-methyl-D-erythritol 2,4-cyclodiphosphate synthase (MCS); hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate synthase (HDS); hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate reductase (HDR); No homologs of the MEP pathway were identified: 3-hydroxy-3-methylglutaryl-CoA synthase (HMGS; N. farcinia; YP_118423); 3-hydroxy-3-methylglutaryl-CoA reductase (HMGR; class I; N. farcinia; YP_118422); Mevalonate Kinase (MVK; N. farcinia; YP_118418); Phosphomevalonate kinase (PMK; N. farcinia; YP_118420); Mevalonate-5-decarboxylase (MDC; N. farcinia; YP_118419). YP_118418 identified homologs in isolates of sub-clades i and ii but e-values ranged from 6e-10 ∼ 3e-10 and CDSs are annotated as galactokinase. ∧Genbank ID number of sequence used for BLAST searches.
(PDF)
Cytokinin types that were profiled from D188.
(PDF)
Sequences of oligonucleotides used in this study.
(PDF)
Parent ions and diagnostic transitions used in multiple reactant monitoring (MRM) for the analysis of cytokinins by ACQUITY TQP UPLC-MS/MS.
(PDF)
Acknowledgments
We would like to thank Alex Buchanan, Ethan Chang, Skylar Fuller, Tyler Horton, and Arthur To of the Chang lab for their assistance; Mark Desenko, Matthew Peterson, and Chris Sullivan of the CGRB, Jean-François Laes and Patrice Godard of DNAVision SA (Belgium), and Drs. Corbin Jones and Piotr Mieczkowski of the HTSF@UNC for sequencing and data processing; Danny Vereecke (Department of Plant Production, University College Ghent, Ghent University, Belgium) for providing the D188 mutant strains; Vanya I. Miteva for providing the isolates obtained from a Greenland ice core; and the Saturday Academy and the Apprenticeships in Science & Engineering for their support and assistance in mentoring E. Hu.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. The 20 whole Genome Shotgun projects are linked to BioProject PRJNA233522. The Whole Genome Shotgun projects have been deposited at DDBJ/EMBL/GenBank under the accessions JMEM00000000 (LMG3625), JMEN00000000 (LMG3623), JMEO00000000 (LMG3616), JMEP00000000 (LMG3605), JMEQ00000000 (LMG3602), JMER00000000 (GIC36), JMES00000000 (GIC26), JMET00000000 (D188), JMEU00000000 (A78), JMEV00000000 (A76), JMEW00000000 (A73a), JMEX00000000 (A44A), JMEY00000000 (A3b), JMEZ00000000 (A25f), JMFA00000000 (A21d2), JMFB00000000 (05-561-1), JMFC00000000 (05-339-1), JMFD00000000 (04-516), JMFE00000000 (02-816c), JMFF00000000 (02-815). The versions described in this paper are versions XXXX010000000. The sequences of the ordered contigs, Genbank-formatted files, and nucleotide as well amino acid sequences are available for download at: http://dx.doi.org/10.7267/N9PN93H8.
Funding Statement
This work is supported by a grant from the Agricultural Research Foundation awarded to JHC. EAS is supported by USDA (United States Department of Agriculture) NIFA (National Institute of Food and Agriculture) post-doctoral fellowship #2013-67012-21139. EWD is supported by a Provost’s Distinguished Graduate Fellowship awarded by Oregon State University. This material is based upon work supported by the National Science Foundation Graduate Research Fellowship under Grant No. DGE-1314109 to EWD. Any opinion, findings, and conclusions or recommendations expressed in this material are those of the authors(s) and do not necessarily reflect the views of the National Science Foundation. This work is also supported in part by the Society of American Florists and a cooperative agreement with USDA-ARS (Agricultural Research Service) awarded to MLP. OMV is a Post-doctoral Researcher of the FRS-FNRS (Fonds de la Recherche Scientifique, Belgium) and supported by grants from FRS-FNRS and the Fonds David et Alice Van Buuren (Belgium). MB is a Senior Research Associate of the FRS-FNRS (Fonds de la Recherche Scientifique, Belgium). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Win J, Chaparro-Garcia A, Belhaj K, Saunders DGO, Yoshida K, et al. (2012) Effector biology of plant-associated organisms: concepts and perspectives. Cold Spring Harb Symp Quant Biol 77: 235–247 10.1101/sqb.2012.77.015933 [DOI] [PubMed] [Google Scholar]
- 2. Xin X-F, He SY (2013) Pseudomonas syringae pv. tomato DC3000: a model pathogen for probing disease susceptibility and hormone signaling in plants. Annu Rev Phytopathol 51: 473–498 10.1146/annurev-phyto-082712-102321 [DOI] [PubMed] [Google Scholar]
- 3. Brown JKM, Tellier A (2011) Plant-parasite coevolution: bridging the gap between genetics and ecology. Annu Rev Phytopathol 49: 345–367 10.1146/annurev-phyto-072910-095301 [DOI] [PubMed] [Google Scholar]
- 4. Juhas M, van der MeerJR, Gaillard M, Harding RM, Hood DW, et al. (2009) Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol Rev 33: 376–393 10.1111/j.1574-6976.2008.00136.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Day WA, Fernández RE, Maurelli AT (2001) Pathoadaptive mutations that enhance virulence: genetic organization of the cadA regions of Shigella spp. Infect Immun 69: 7471–7480 10.1128/IAI.69.12.7471-7480.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ma W, Dong FFT, Stavrinides J, Guttman DS (2006) Type III effector diversification via both pathoadaptation and horizontal transfer in response to a coevolutionary arms race. PLoS Genet 2: e209 10.1371/journal.pgen.0020209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gürtler V, Mayall BC, Seviour R (2004) Can whole genome analysis refine the taxonomy of the genus Rhodococcus? FEMS Microbiol Rev 28: 377–403. [DOI] [PubMed] [Google Scholar]
- 8. Larkin MJ, Kulakov LA, Allen CCR (2005) Biodegradation and Rhodococcus–masters of catabolic versatility. Curr Opin Biotechnol 16: 282–290 10.1016/j.copbio.2005.04.007 [DOI] [PubMed] [Google Scholar]
- 9. Francis I, Holsters M, Vereecke D (2010) The Gram-positive side of plant-microbe interactions. Environ Microbiol 12: 1–12 10.1111/j.1462-2920.2009.01989.x [DOI] [PubMed] [Google Scholar]
- 10. Stes E, Vandeputte OM, Jaziri El M, Holsters M, Vereecke D (2011) A successful bacterial coup d’état: how Rhodococcus fascians redirects plant development. Annu Rev Phytopathol 49: 69–86 10.1146/annurev-phyto-072910-095217 [DOI] [PubMed] [Google Scholar]
- 11. Cornelis K, Ritsema T, Nijsse J, Holsters M, Goethals K, et al. (2001) The plant pathogen Rhodococcus fascians colonizes the exterior and interior of the aerial parts of plants. Molecular Plant-Microbe Interactions 14: 599–608 10.1094/MPMI.2001.14.5.599 [DOI] [PubMed] [Google Scholar]
- 12. Putnam ML, Miller ML (2007) Rhodococcus fascians in herbaceous perennials. Plant Dis 91: 1064–1076 10.1094/PDIS-91-9-1064 [DOI] [PubMed] [Google Scholar]
- 13. Vereecke D, Burssens S, Simón-Mateo C, Inzé D, Van Montagu M, et al. (2000) The Rhodococcus fascians-plant interaction: morphological traits and biotechnological applications. Planta 210: 241–251. [DOI] [PubMed] [Google Scholar]
- 14. Crespi M, Messens E, Caplan AB, Van Montagu M, Desomer J (1992) Fasciation induction by the phytopathogen Rhodococcus fascians depends upon a linear plasmid encoding a cytokinin synthase gene. EMBO J 11: 795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Crespi M, Vereecke D, Temmerman W, Van Montagu M, Desomer J (1994) The fas operon of Rhodococcus fascians encodes new genes required for efficient fasciation of host plants. J Bacteriol 176: 2492–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Temmerman W, Vereecke D, Dreesen R, Van Montagu M, Holsters M, et al. (2000) Leafy gall formation is controlled by fasR, an AraC-type regulatory gene in Rhodococcus fascians . J Bacteriol 182: 5832–5840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maes T, Vereecke D, Ritsema T, Cornelis K, Thu HN, et al. (2001) The att locus of Rhodococcus fascians strain D188 is essential for full virulence on tobacco through the production of an autoregulatory compound. Mol Microbiol 42: 13–28. [DOI] [PubMed] [Google Scholar]
- 18. Pertry I, Václavíková K, Gemrotová M, Spíchal L, Galuszka P, et al. (2010) Rhodococcus fascians impacts plant development through the dynamic fas-mediated production of a cytokinin mix. Molecular Plant-Microbe Interactions 23: 1164–1174 10.1094/MPMI-23-9-1164 [DOI] [PubMed] [Google Scholar]
- 19. Francis I, De Keyser A, De Backer P, Simón-Mateo C, Kalkus J, et al. (2012) pFiD188, the Linear Virulence Plasmid of Rhodococcus fascians D188. Molecular Plant-Microbe Interactions 25: 637–647 10.1094/MPMI-08-11-0215 [DOI] [PubMed] [Google Scholar]
- 20. Pertry I, Václavíková K, Depuydt S, Galuszka P, Spíchal L, et al. (2009) Identification of Rhodococcus fascians cytokinins and their modus operandi to reshape the plant. Proc Natl Acad Sci USA 106: 929–934 10.1073/pnas.0811683106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Frébort I, Kowalska M, Hluska T, Frébortová J, Galuszka P (2011) Evolution of cytokinin biosynthesis and degradation. J Exp Bot 62: 2431–2452 10.1093/jxb/err004 [DOI] [PubMed] [Google Scholar]
- 22. Kurakawa T, Ueda N, Maekawa M, Kobayashi K, Kojima M, et al. (2007) Direct control of shoot meristem activity by a cytokinin-activating enzyme. Nature 445: 652–655 10.1038/nature05504 [DOI] [PubMed] [Google Scholar]
- 23. Takei K, Yamaya T, Sakakibara H (2004) Arabidopsis CYP735A1 and CYP735A2 encode cytokinin hydroxylases that catalyze the biosynthesis of trans-Zeatin. J Biol Chem 279: 41866–41872 10.1074/jbc.M406337200 [DOI] [PubMed] [Google Scholar]
- 24. Letek M, González P, Macarthur I, Rodríguez H, Freeman TC, et al. (2010) The genome of a pathogenic Rhodococcus: cooptive virulence underpinned by key gene acquisitions. PLoS Genet 6: e1001145 10.1371/journal.pgen.1001145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bargen von K, Haas A (2009) Molecular and infection biology of the horse pathogen Rhodococcus equi . FEMS Microbiol Rev 33: 870–891 10.1111/j.1574-6976.2009.00181.x [DOI] [PubMed] [Google Scholar]
- 26. Vernikos GS, Parkhill J (2006) Interpolated variable order motifs for identification of horizontally acquired DNA: revisiting the Salmonella pathogenicity islands. Bioinformatics (Oxford, England) 22: 2196–2203 10.1093/bioinformatics/btl369 [DOI] [PubMed] [Google Scholar]
- 27. Hjerde E, Pierechod MM, Williamson AK, Bjerga GEK, Willassen NP, et al. (2013) Draft Genome Sequence of the Actinomycete Rhodococcus sp. Strain AW25M09, Isolated from the Hadsel Fjord, Northern Norway. Genome Announc 1: e0005513 10.1128/genomeA.00055-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sugawara H, Ueda N, Kojima M, Makita N, Yamaya T, et al. (2008) Structural insight into the reaction mechanism and evolution of cytokinin biosynthesis. Proc Natl Acad Sci USA 105: 2734–2739 10.1073/pnas.0707374105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jeon WB, Allard STM, Bingman CA, Bitto E, Han BW, et al. (2006) X-ray crystal structures of the conserved hypothetical proteins from Arabidopsis thaliana gene loci At5g11950 and AT2g37210. Proteins 65: 1051–1054 10.1002/prot.21166 [DOI] [PubMed] [Google Scholar]
- 30. Kakimoto T (2003) Biosynthesis of cytokinins. J Plant Res 116: 233–239 10.1007/s10265-003-0095-5 [DOI] [PubMed] [Google Scholar]
- 31. Joshi MV, Loria R (2007) Streptomyces turgidiscabies possesses a functional cytokinin biosynthetic pathway and produces leafy galls. Molecular Plant-Microbe Interactions 20: 751–758 10.1094/MPMI-20-7-0751 [DOI] [PubMed] [Google Scholar]
- 32. Krall L, Raschke M, Zenk MH, Baron C (2002) The Tzs protein from Agrobacterium tumefaciens C58 produces zeatin riboside 5′-phosphate from 4-hydroxy-3-methyl-2-(E)-butenyl diphosphate and AMP. FEBS letters 527: 315–318. [DOI] [PubMed] [Google Scholar]
- 33. Rodríguez-Concepción M, Boronat A (2002) Elucidation of the methylerythritol phosphate pathway for isoprenoid biosynthesis in bacteria and plastids. A metabolic milestone achieved through genomics. Plant Physiology 130: 1079–1089 10.1104/pp.007138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sakakibara H, Kasahara H, Ueda N, Kojima M, Takei K, et al. (2005) Agrobacterium tumefaciens increases cytokinin production in plastids by modifying the biosynthetic pathway in the host plant. Proc Natl Acad Sci USA 102: 9972–9977 10.1073/pnas.0500793102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lombard J, Moreira D (2011) Origins and early evolution of the mevalonate pathway of isoprenoid biosynthesis in the three domains of life. Mol Biol Evol 28: 87–99 10.1093/molbev/msq177 [DOI] [PubMed] [Google Scholar]
- 36. Gallegos MT, Schleif R, Bairoch A, Hofmann K, Ramos JL (1997) Arac/XylS family of transcriptional regulators. Microbiol Mol Biol Rev 61: 393–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Soisson SM, MacDougall-Shackleton B, Schleif R, Wolberger C (1997) Structural basis for ligand-regulated oligomerization of AraC. Science 276: 421–425. [DOI] [PubMed] [Google Scholar]
- 38. Kwon HJ, Bennik MH, Demple B, Ellenberger T (2000) Crystal structure of the Escherichia coli Rob transcription factor in complex with DNA. Nat Struct Biol 7: 424–430 10.1038/75213 [DOI] [PubMed] [Google Scholar]
- 39. Lowden MJ, Skorupski K, Pellegrini M, Chiorazzo MG, Taylor RK, et al. (2010) Structure of Vibrio cholerae ToxT reveals a mechanism for fatty acid regulation of virulence genes. Proc Natl Acad Sci USA 107: 2860–2865 10.1073/pnas.0915021107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Parmley JL, Hurst LD (2007) How common are intragene windows with KA>KS owing to purifying selection on synonymous mutations? J Mol Evol 64: 646–655 10.1007/s00239-006-0207-7 [DOI] [PubMed] [Google Scholar]
- 41. Vereecke D, Cornelis K, Temmerman W, Jaziri M, Van Montagu M, et al. (2002) Chromosomal locus that affects pathogenicity of Rhodococcus fascians . J Bacteriol 184: 1112–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bell KS, Philp JC, Aw DW, Christofi N (1998) The genus Rhodococcus . J Appl Microbiol 85: 195–210. [DOI] [PubMed] [Google Scholar]
- 43. La Duc MT, Dekas A, Osman S, Moissl C, Newcombe D, et al. (2007) Isolation and characterization of bacteria capable of tolerating the extreme conditions of clean room environments. Appl Environ Microbiol 73: 2600–2611 10.1128/AEM.03007-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Konishi M, Nishi S, Fukuoka T, Kitamoto D, Watsuji T-O, et al. (2014) Deep-sea Rhodococcus sp. BS-15, Lacking the Phytopathogenic fas Genes, Produces a Novel Glucotriose Lipid Biosurfactant. Mar Biotechnol. doi:10.1007/s10126-014-9568-x. [DOI] [PubMed]
- 45. Yanai I, Wolf YI, Koonin EV (2002) Evolution of gene fusions: horizontal transfer versus independent events. Genome Biol 3: research0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pasek S, Risler J-L, Brézellec P (2006) Gene fusion/fission is a major contributor to evolution of multi-domain bacterial proteins. Bioinformatics 22: 1418–1423 10.1093/bioinformatics/btl135 [DOI] [PubMed] [Google Scholar]
- 47. Rajaonson S, Vandeputte OM, Vereecke D, Kiendrebeogo M, Ralambofetra E, et al. (2011) Virulence quenching with a prenylated isoflavanone renders the Malagasy legume Dalbergia pervillei resistant to Rhodococcus fascians . Environ Microbiol 13: 1236–1252 10.1111/j.1462-2920.2011.02424.x [DOI] [PubMed] [Google Scholar]
- 48. Akiyoshi DE, Regier DA, Gordon MP (1987) Cytokinin production by Agrobacterium and Pseudomonas spp. J Bacteriol 169: 4242–4248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Matsubara S, Armstrong DJ, Skoog F (1968) Cytokinins in tRNA of Corynebacterium fascians . Plant Physiology 43: 451–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rathbone MP, Hall RH (1972) Concerning the presence of the cytokinin, N6-(Δ2-isopentnyl) adenine, in cultures of Corynebacterium fascians . Planta 108: 93–102 10.1007/BF00386072 [DOI] [PubMed] [Google Scholar]
- 51. Einset JW, Skoog FK (1977) Isolation and identification of ribosyl–zeatin from transfer RNA of Corynebacterium fascians . Biochem Biophys Res Commun 79: 1117–1121 10.1016/0006-291X(77)91121-4 [DOI] [PubMed] [Google Scholar]
- 52. Miyawaki K, Tarkowski P, Matsumoto-Kitano M, Kato T, Sato S, et al. (2006) Roles of Arabidopsis ATP/ADP isopentenyltransferases and tRNA isopentenyltransferases in cytokinin biosynthesis. Proc Natl Acad Sci USA 103: 16598–16603 10.1073/pnas.0603522103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Agris PF, Vendeix FAP, Graham WD (2007) tRNA’s Wobble Decoding of the Genome: 40 Years of Modification. J Mol Biol 366: 1–13 10.1016/j.jmb.2006.11.046 [DOI] [PubMed] [Google Scholar]
- 54. Sprenger GA, Schörken U, Wiegert T, Grolle S, de Graaf AA, et al. (1997) Identification of a thiamin-dependent synthase in Escherichia coli required for the formation of the 1-deoxy-D-xylulose 5-phosphate precursor to isoprenoids, thiamin, and pyridoxol. Proc Natl Acad Sci USA 94: 12857–12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lynch M (2006) Streamlining and simplification of microbial genome architecture. Annu Rev Microbiol 60: 327–349 10.1146/annurev.micro.60.080805.142300 [DOI] [PubMed] [Google Scholar]
- 56. Takei K, Sakakibara H, Sugiyama T (2001) Identification of genes encoding adenylate isopentenyltransferase, a cytokinin biosynthesis enzyme, in Arabidopsis thaliana . J Biol Chem 276: 26405–26410 10.1074/jbc.M102130200 [DOI] [PubMed] [Google Scholar]
- 57. Kuroha T, Tokunaga H, Kojima M, Ueda N, Ishida T, et al. (2009) Functional analyses of LONELY GUY cytokinin-activating enzymes reveal the importance of the direct activation pathway in Arabidopsis. The Plant Cell 21: 3152–3169 10.1105/tpc.109.068676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Liu YG, Mitsukawa N, Oosumi T, Whittier RF (1995) Efficient isolation and mapping of Arabidopsis thaliana T-DNA insert junctions by thermal asymmetric interlaced PCR. Plant J 8: 457–463. [DOI] [PubMed] [Google Scholar]
- 59. Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 25: 402–408 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 60. Zerbino DR, Birney E (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18: 821–829 10.1101/gr.074492.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. McLeod MP, Warren RL, Hsiao WWL, Araki N, Myhre M, et al. (2006) The complete genome of Rhodococcus sp. RHA1 provides insights into a catabolic powerhouse. Proc Natl Acad Sci USA 103: 15582–15587 10.1073/pnas.0607048103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rissman AI, Mau B, Biehl BS, Darling AE, Glasner JD, et al. (2009) Reordering contigs of draft genomes using the Mauve aligner. Bioinformatics 25: 2071–2073 10.1093/bioinformatics/btp356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Magrane M, Consortium U (2011) UniProt Knowledgebase: a hub of integrated protein data. Database (Oxford) 2011: bar009 10.1093/database/bar009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Eddy SR (2011) Accelerated Profile HMM Searches. PLoS Comp Biol 7: e1002195 10.1371/journal.pcbi.1002195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Darriba D, Taboada GL, Doallo R, Posada D (2011) ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27: 1164–1165 10.1093/bioinformatics/btr088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Katoh K, Standley DM (2013) MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol Biol Evol 30: 772–780 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22: 2688–2690 10.1093/bioinformatics/btl446 [DOI] [PubMed] [Google Scholar]
- 69. Sonnhammer ELL, Hollich V (2005) Scoredist: a simple and robust protein sequence distance estimator. BMC Bioinformatics 6: 108 10.1186/147121056108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Castresana J (2000) Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17: 540–552. [DOI] [PubMed] [Google Scholar]
- 71. Letunic I, Bork P (2011) Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res 39: W475–W478 10.1093/nar/gkr201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, et al. (2005) InterProScan: protein domains identifier. Nucleic Acids Res 33: W116–W120 10.1093/nar/gki442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hunter S, Jones P, Mitchell A, Apweiler R, Attwood TK, et al. (2012) InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res 40: D306–D312 10.1093/nar/gkr948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Crooks GE, Hon G, Chandonia J-M, Brenner SE (2004) WebLogo: a sequence logo generator. Genome Res 14: 1188–1190 10.1101/gr.849004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cole C, Barber JD, Barton GJ (2008) The Jpred 3 secondary structure prediction server. Nucleic Acids Res 36: W197–W201 10.1093/nar/gkn238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ (2009) Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics 25: 1189–1191 10.1093/bioinformatics/btp033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zhang Z, Li J, Zhao X-Q, Wang J, Wong GK-S, et al. (2006) KaKs_Calculator: calculating Ka and Ks through model selection and model averaging. Genomics Proteomics Bioinformatics 4: 259–263 10.1016/S16720229(07)600072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zhang Z, Xiao J, Wu J, Zhang H, Liu G, et al. (2012) ParaAT: a parallel tool for constructing multiple protein-coding DNA alignments. Biochem Biophys Res Commun 419: 779–781 10.1016/j.bbrc.2012.02.101 [DOI] [PubMed] [Google Scholar]
- 79. Herbig A, Jager G, Battke F, Nieselt K (2012) GenomeRing: alignment visualization based on SuperGenome coordinates. Bioinformatics 28: i7–i15 10.1093/bioinformatics/bts217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, et al. (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393: 537–544 10.1038/31159 [DOI] [PubMed] [Google Scholar]
- 81. Bentley SD, Chater KF, Cerdeño-Tárraga A-M, Challis GL, Thomson NR, et al. (2002) Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417: 141–147. [DOI] [PubMed] [Google Scholar]
- 82. Cerdeño-Tárraga A-M, Efstratiou A, Dover LG, Holden MTG, Pallen M, et al. (2003) The complete genome sequence and analysis of Corynebacterium diphtheriae NCTC13129. Nucleic Acids Res 31: 6516–6523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ishikawa J, Yamashita A, Mikami Y, Hoshino Y, Kurita H, et al. (2004) The complete genomic sequence of Nocardia farcinica IFM 10152. Proc Natl Acad Sci USA 101: 14925–14930 10.1073/pnas.0406410101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Na K-S, Kuroda A, Takiguchi N, Ikeda T, Ohtake H, et al. (2005) Isolation and characterization of benzene-tolerant Rhodococcus opacus strains. Journal of Bioscience and Bioengineering 99: 378–382 10.1263/jbb.99.378 [DOI] [PubMed] [Google Scholar]
- 85. Sekine M, Tanikawa S, Omata S, Saito M, Fujisawa T, et al. (2006) Sequence analysis of three plasmids harboured in Rhodococcus erythropolis strain PR4. Environ Microbiol 8: 334–346 10.1111/j.1462-2920.2005.00899.x [DOI] [PubMed] [Google Scholar]
- 86. Chen H-P, Zhu S-H, Casabon I, Hallam SJ, Crocker FH, et al. (2012) Genomic and transcriptomic studies of an RDX (hexahydro-1,3,5-trinitro-1,3,5-triazine)-degrading actinobacterium. Appl Environ Microbiol 78: 7798–7800 10.1128/AEM.02120-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Supek F, Bošnjak M, Skunca N, Smuc T (2011) REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 6: e21800 10.1371/journal.pone.0021800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Wu S, Zhu Z, Fu L, Niu B, Li W (2011) WebMGA: a customizable web server for fast metagenomic sequence analysis. BMC Genomics 12: 444 10.1186/1471-2164-12-444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.R Core Team (2013) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. Available: http://www.R-project.org.
- 90. Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Meth 9: 671–675 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Vandeputte O, Oden S, Mol A, Vereecke D, Goethals K, et al. (2005) Biosynthesis of auxin by the gram-positive phytopathogen Rhodococcus fascians is controlled by compounds specific to infected plant tissues. Appl Environ Microbiol 71: 1169–1177 10.1128/AEM.71.3.1169-1177.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Redig P, Schmülling T, Van Onckelen H (1996) Analysis of Cytokinin Metabolism in ipt Transgenic Tobacco by Liquid Chromatography-Tandem Mass Spectrometry. Plant Physiology 112: 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Analysis of functional categories associated with coding sequences in regions acquired via HGT (A) Analysis of clusters of orthologous groups (COGs). COGs of CDSs exclusive to pathogenic isolates were compared to those common among all 20 isolates. CDSs putatively acquired by HGT were categorized as exclusive to two or more pathogenic isolates (dark red) and those present in two or more isolates regardless of phytopathogenicity trait (blue). CDSs were assigned to COG functional groups (x-axis) and displayed as the percent of COGs per category relative to the total number of COGs assigned (y-axis). (B) Scatterplot of GO functional categories. GO terms were assigned to all coding sequences identified in pathogenic isolates and in regions potentially acquired via HGT. The semantic similarities were calculated, GO terms representative of clusters were generated, and plotted in a scatterplot. Clusters (circles) are placed based on semantically similarities, with more similar clusters placed closer to each other. Plot sizes represent the frequency of GO terms of a cluster relative to the total number of terms in the GO database. Heatmap represents semantic uniqueness, calculated relative to all terms in the list. High-level functions are indicated for clusters of the more frequently observed terms.
(EPS)
Analysis of functional categories associated with the core and flexible genomes COGs of CDSs that were core (blue) to all 24 isolates were compared to those in the flexible (red) genome. CDSs were assigned to COG functional groups (x-axis) and displayed as the percent of COGs per category relative to the total number of COGs assigned (y-axis).
(EPS)
Marker genes of the att and fas operon were induced in the presence of leafy gall extract. D188 was grown in media augmented with extracts from leafy galls (red), uninfected plants (blue), or water (mock). Samples were taken and RNA was extracted at the times indicated. Expression of attE and fasD was determined using qRT-PCR and calculated as expression relative to 16S and corresponding genes in D188 grown in a mock-treated culture.
(EPS)
Complete cytokinin profiling data. Three genotypes of D188, wild type (blue), ΔfasD (green) and ΔpFiD188 (red), were grown in media augmented with extracts from leaf galls, uninfected plants, and water as a mock. Of 32 cytokinin types that were profiled, only eight were detected and the six most abundant types are shown (iP: isopentenyladenine; cZ: cis-zeatin; 2MeScZ: 2-methylthio-cis-zeatin; and their ribosides). Change in concentration (y-axis) is presented for all time points (x-axis). Colored “*” indicates significant difference in cytokinin accumulation in corresponding mutant genotype relative to wild type and corresponding time point (p-value threshold = 0.05). Experiments were repeated twice with similar results.
(EPS)
RFA25f_04833 and RFA21d2_02306 are members of the fasR family. The alignment of the translated sequences for predicted AraC-type transcriptional regulators was used to generate a phylogenetic tree. Only candidates from the original 20 isolates were included. A single candidate AraC-type transcriptional regulator from Rhodococcus jostii RHA1 was used as the root. Genes are labeled by locus tag or with GI number (R. jostii RHA1). The FasR sequences are in bold. Node values indicate bootstrap percentages out of 1000 iterations. The scale bar represents the mean number of amino acid substitutions per site.
(EPS)
Predicted CDSs of the thirteen pFiD188-like sequences. *Regions were designated based on previously published determinations; §Functions were based on the annotations of the query sequences described in this study. The query sequences were derived from the CDS of the first isolate in which it appears in the table. Homologs were identified using reciprocal best BLAST hit analysis.
(XLSX)
Reciprocal best BLAST hit analysis of pFiD188 against the genome assemblies of A25f and A21d2. *Locus identifiers from previously published pFID188 linear plasmid sequence; §Based on this study.
(PDF)
Homologs of enzymes of the methylerythritol phosphate pathway are present in all 20 isolates of Rhodococcus . *Locus; % identity; BLAST e-value. Column abbreviations: Deoxyxylulose 5-phosphate synthase (DXPS); DXP reductiosomerase (DXPR); isopentenyl diphosphate isomerase (IDI1); 4-diphosphocytidyl-2C-methyl-D-erythirol synthase (CMS); 4-(cytidine 5′-diphospho)-2-C-methyl-D-erythritol kinase (CMK); 2-C-methyl-D-erythritol 2,4-cyclodiphosphate synthase (MCS); hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate synthase (HDS); hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate reductase (HDR); No homologs of the MEP pathway were identified: 3-hydroxy-3-methylglutaryl-CoA synthase (HMGS; N. farcinia; YP_118423); 3-hydroxy-3-methylglutaryl-CoA reductase (HMGR; class I; N. farcinia; YP_118422); Mevalonate Kinase (MVK; N. farcinia; YP_118418); Phosphomevalonate kinase (PMK; N. farcinia; YP_118420); Mevalonate-5-decarboxylase (MDC; N. farcinia; YP_118419). YP_118418 identified homologs in isolates of sub-clades i and ii but e-values ranged from 6e-10 ∼ 3e-10 and CDSs are annotated as galactokinase. ∧Genbank ID number of sequence used for BLAST searches.
(PDF)
Cytokinin types that were profiled from D188.
(PDF)
Sequences of oligonucleotides used in this study.
(PDF)
Parent ions and diagnostic transitions used in multiple reactant monitoring (MRM) for the analysis of cytokinins by ACQUITY TQP UPLC-MS/MS.
(PDF)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. The 20 whole Genome Shotgun projects are linked to BioProject PRJNA233522. The Whole Genome Shotgun projects have been deposited at DDBJ/EMBL/GenBank under the accessions JMEM00000000 (LMG3625), JMEN00000000 (LMG3623), JMEO00000000 (LMG3616), JMEP00000000 (LMG3605), JMEQ00000000 (LMG3602), JMER00000000 (GIC36), JMES00000000 (GIC26), JMET00000000 (D188), JMEU00000000 (A78), JMEV00000000 (A76), JMEW00000000 (A73a), JMEX00000000 (A44A), JMEY00000000 (A3b), JMEZ00000000 (A25f), JMFA00000000 (A21d2), JMFB00000000 (05-561-1), JMFC00000000 (05-339-1), JMFD00000000 (04-516), JMFE00000000 (02-816c), JMFF00000000 (02-815). The versions described in this paper are versions XXXX010000000. The sequences of the ordered contigs, Genbank-formatted files, and nucleotide as well amino acid sequences are available for download at: http://dx.doi.org/10.7267/N9PN93H8.