

Abstract
Non-alcoholic fatty liver disease (NAFLD) is the most common chronic liver disease in the Western world, and it persists at a high prevalence. NAFLD is characterised by the accumulation of triglycerides in the liver and includes a spectrum of histopathological findings, ranging from simple fatty liver through non-alcoholic steatohepatitis (NASH) to fibrosis and ultimately cirrhosis, which may progress to hepatocellular carcinoma. The pathogenesis of NAFLD is closely related to the metabolic syndrome and insulin resistance. Understanding the pathophysiology and treatment of NAFLD in humans has currently been limited by the lack of satisfactory animal models. The ideal animal model for NAFLD should reflect all aspects of the intricate etiopathogenesis of human NAFLD and the typical histological findings of its different stages. Within the past several years, great emphasis has been placed on the development of an appropriate model for human NASH. This paper reviews the widely used experimental models of NAFLD in rats. We discuss nutritional, genetic and combined models of NAFLD and their pros and cons. The choice of a suitable animal model for this disease while respecting its limitations may help to improve the understanding of its complex pathogenesis and to discover appropriate therapeutic strategies. Considering the legislative, ethical, economical and health factors of NAFLD, animal models are essential tools for the research of this disease.
Keywords: Animal model, High-fat diet, Methionine- and choline-deficient diet, Non-alcoholic fatty liver disease, Non-alcoholic steatohepatitis, Otsuka-Long-Evans-Tokushima fatty rats, Zucker rats
Core tip: Non-alcoholic fatty liver disease (NAFLD) is the most common chronic liver disease in the Western world, but its pathogenesis is still not fully understood. This paper reviews the commonly used experimental models of NAFLD in rats. We discuss nutritional, genetic and combined models of NAFLD, and we highlight their pros and cons with special emphasis on nutritional models. To date, there is not an ideal model that reflects all aspects of the complex etiopathogenesis of human NAFLD and the typical histological features of its different stages.
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) is the most common chronic liver disease in the Western world, where its prevalence is estimated to be approximately 20%-30% of the adult population[1]. A steep increase in the prevalence of NAFLD correlates to increased obesity in the world. NAFLD is characterised by the accumulation of triglycerides in the liver and includes a spectrum of histopathological findings, including simple fatty liver, non-alcoholic steatohepatitis (NASH), fibrosis and ultimately cirrhosis, which may progress to hepatocellular carcinoma. The pathogenesis of NAFLD is closely related to metabolic syndrome and insulin resistance. NAFLD is thought to be the hepatic manifestation of metabolic syndrome[2]. Despite intensive research over the past decades, the pathogenesis of NAFLD is not completely understood, and treatment of the disease has not been fully defined.
PATHOGENESIS
NAFLD develops over years as a result of the accumulation of different risk factors involving varying degrees of genetic susceptibility. Nutritional habits, amount and composition of diet, physical inactivity, chronic stress and lifestyle are the major non-genetic factors determining the manifestation and severity of the disease. Similar to the pathogenesis of metabolic syndrome, of which NAFLD is a constituent[2], there is presumed to be a polygenic genetic background to this disease[3]. The pathogenesis of NAFLD in humans is very complex, and there are many players in the field, each of which may interact with one another. Moreover, some risk factors may be more or less pronounced and may arise or disappear over time. Currently, it is not fully understood why some patients with similar risk factors do not develop fatty accumulation, while others develop steatosis and steatohepatitis. The pathogenesis of NAFLD appears to be unique in each patient, but the risk factors are likely to be the same.
The natural history of NAFLD has been not fully clarified and appears to be slow in most patients. The progress from simple steatosis to advanced stages of the disease often takes years. It is not clear whether all patients with simple steatosis are at risk of steatohepatitis or advanced fibrosis. In contrast to patients with simple steatosis, those with NASH have a higher risk of death from cardiovascular and liver-related diseases[4]. NAFLD with elevated liver enzymes is associated with a clinically significant risk of developing end-stage liver disease, and most NAFLD patients will develop diabetes or impaired glucose tolerance in the long term[4]. In the United States, an estimated one-third of the population has NAFLD, and approximately 2%-5% have NASH[1].
The accumulation of lipids in the liver results from an imbalance among hepatic lipid intake, synthesis, degradation and secretion[5]. Fatty acids for the synthesis of triglycerides are acquired from different sources, including de novo formation in the liver, from portal blood as free fatty acids (FFAs) and from circulating lipoproteins, mainly from chylomicrons. Insulin resistance plays a key role in the hepatic accumulation of fats[5]. Serum FFAs are elevated by accelerated lipolysis in the peripheral adipose tissue and visceral fat in the state of insulin resistance. Hyperglycaemia caused by peripheral insulin resistance also promotes the synthesis of fatty acids in the liver. The elevation of chylomicrons is typical after intake of food containing high amounts of fats. Peripheral insulin resistance is often accompanied by increased insulin levels, and hyperinsulinaemia is known to inhibit the formation and secretion of very low-density lipoproteins (VLDL) from the liver in NASH patients[6]. Another effect of hyperinsulinaemia and hyperglycaemia is the inhibition β-oxidation of fatty acids and the promotion of lipogenesis in the liver[7]. Insulin is the primary stimulator of hepatic lipogenesis through activation of the sterol regulatory element-binding protein-1c (SREBP-1c) transcription factor[8]. Insulin resistance is found in the majority of patients with primary NAFLD and is more severe in NASH[9].
Some patients with simple steatosis progress to NASH. According to the “two-hit” theory[10], liver steatosis sensitises hepatocytes to the second hits, leading to hepatocyte damage, inflammation and fibrosis. These second insults may be increased oxidative stress (induction of microsomal cytochrome P450) and lipid peroxidation, mitochondrial dysfunction, cytokine/adipokine imbalance [tumour necrosis factor α (TNF-α), interleukin 6 (IL-6), adiponectin, leptin, resistin, etc.], lipotoxicity of FFAs, hepatic accumulation of cholesterol, lipopolysaccharide derived from GUT and the activation of innate immunity[10,11]. Exceeding defence and self-reparative mechanisms of hepatocytes lead to cell death. The importance of particular factors and their interactions are still not fully understood. The main difference between simple steatosis and NASH is in the degrees of hepatocyte injury and apoptosis[12]. Increased apoptosis is thought by some authors to be the third hit leading to advanced fibrosis and cirrhosis[13]. Overall chronic liver injury with a subsequent chronic regenerative response in the terrain of NASH leads to the activation of hepatic stellate cells and a fibrogenic response. With regards to fibrogenesis in NAFLD, special emphasis should be placed on chronic inflammation, oxidative stress/lipid peroxidation, leptin, adiponectin, platelet-derived growth factor, transforming growth factor β1 (TGF-β1), renin-angiotensin system and hepatocyte death[11]. The possible progression of human NAFLD is briefly depicted in Figure 1. Recently, a more realistic concept of the progression to NASH was introduced, which takes into account cumulative small pathogenic hits[14].
Figure 1.
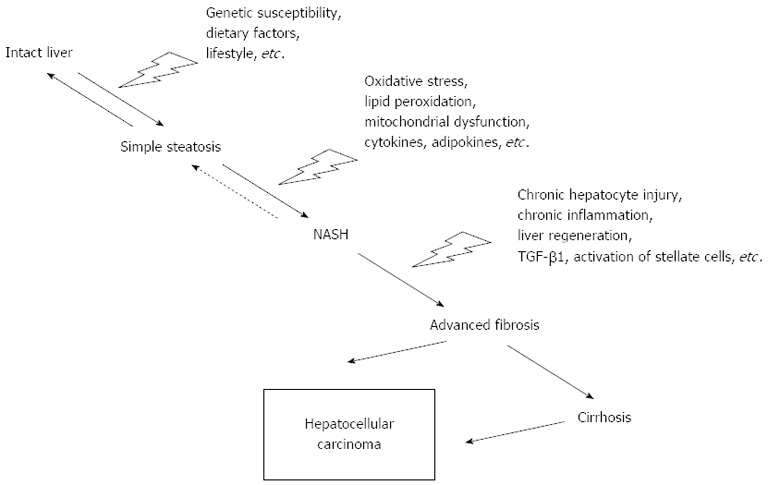
Schematic diagram of the possible progression of non-alcoholic fatty liver disease. TGF-β1: Transforming growth factor β1; NASH: Non-alcoholic steatohepatitis.
HISTOLOGY
NAFLD is histologically defined by the presence of liver steatosis exceeding 5% of hepatocytes, regardless of whether it is macrovesicular, mixed or microvesicular[15]. In adults, NAFLD is characterised predominantly by macrovesicular steatosis that is usually present in a zone 3 or panacinar distribution[16]. NASH is defined by the presence of hepatocellular ballooning (most frequently in zone 3), lobular or portal inflammation (lymphocytes, monocytes, eosinophils, and macrophages) and eventually fibrosis in the terrain of steatotic liver[17]. The presence of a characteristic ballooning injury is the key feature to the diagnosis of NASH. Hepatocyte apoptosis is often observed in NASH, and the severity of steatohepatitis correlates with the degree of apoptosis[18]. Examination of histological samples from liver biopsies remains the gold standard for accurately assessing the degree of steatosis, necroinflammatory changes and fibrosis, and these may distinguish NASH from simple steatosis[19].
ANIMAL MODELS OF NAFLD
Understanding the pathophysiology and treatment of NAFLD in human beings has been limited by the lack of satisfactory animal models. The ideal animal model for NAFLD should reflect all aspects of the intricate etiopathogenesis of human NAFLD and the typical histological findings of its different stages. Until now, there was no such animal model. Thus, researchers have attempted to introduce a suitable model of NAFLD, imitating at least the most important pathogenic and histological features of NAFLD. Animal models should be reproducible, reliable, simple, affordable and technically available with minimal disadvantages. Mice and rats have mostly been used as experimental animals of NAFLD[20]. Most models have been genetic models, high-fat diets or choline-methionine-deficient diets. The development of hepatic steatosis is common, and some also develop histological features of steatohepatitis; only a minority progress to fibrosis[20].
In the past several years, great emphases have been placed on the development of an appropriate model for human NASH. Most models successfully simulate the histological patterns of the disease, but they often lack the important metabolic or genetic context of human NASH (i.e., obesity, insulin resistance, hyperglycaemia, hyperinsulinaemia, dyslipidaemia, cytokine/adipokine imbalance)[14].
It should also be mentioned that, according to the natural history and pathogenesis of human NAFLD, there cannot be an ideal model. The progress from simple steatosis to NASH likely lasts years, and only a minority of patients with simple fatty liver progress to steatohepatitis. Thus, the models of steatohepatitis do not reflect this aspect even if the shorter lifespan of rats is taken into account. From this point of view, models of chronic overnutrition with a spontaneous progression of steatosis to steatohepatitis may be more valid than other models[20]. Moreover, these models exhibit a more complex interplay between metabolic abnormalities and liver injury[20].
In contrast to humans, data obtained from animal experiments are quite homogenous due to different sampling, even in wild type strains. Animal studies are generally performed on well-defined populations (species, strain, sex, weight or age, laboratory conditions, type of diet, etc.). In contrast, there is considerable heterogeneity in the human population, even if the study is defined by sex, place, age, ethnicity, and weight. There remains high interindividual variability concerning genetic background, comorbidities, physical activity, medication, diet composition, life style, etc. Thus, the interpretability of the data to humans is limited. Moreover, rodents have unique characteristics that affect the development of NAFLD and metabolic syndrome, which further hinder simple interpretations of the data. Crucial findings in animal models must be verified by clinical research when possible.
This review attempts to describe and briefly summarise the existing experimental models of NAFLD in rats. The paper is mainly focused on commonly used rat models of NAFLD in vivo. We discuss nutritional, genetic and combined models of the disease. The basic characteristics of the most widely employed models described in the text are summarised in Table 1. We do not review pharmacologic or toxic models of liver steatosis (dexamethasone, tamoxifen, orotic acid-diet, etc.), which are distinct from the natural etiopathogenesis of primary human NAFLD. Suitable animal models may help us to understand the complex pathogenesis of NAFLD and to discover new non-pharmacological or drug interventions for the treatment of disease. It can also further reveal risk factors of the progression of disease and uncover therapeutic strategies against the progression of NAFLD.
Table 1.
Characteristics of the most frequently used models of non-alcoholic fatty liver disease in rats
| Model | Obesity | Insulin resistance | Plasma cytokines/adipokines | Dyslipidaemia | Elevation of transaminases | Liver histology | |||
| Steatosis | Inflammation | Hepatocyte ballooning | Fibrosis | ||||||
| Nutritional models | |||||||||
| High-fat diets1 | Yes[27,28,34,35]/no[29,36]/not describe[37,38] | Yes[27-29,34,35]/not described[36-38] | ↑TNF-α[27,34]↓adiponectin[28,34,35] | Yes[27,28]/no[36]/not described[29,34,35,37,38] | Yes[27,28,34,38]/no[23,29,35,36] | Yes[23,27-29,34-38]/no[26] | Yes[27-29,34,37,38]/no[23,35,36] | Yes[34,37]/not described[23,27-29,35,36,38] | Yes[28,34]/no[23,35,36]/not described[27,37,38] |
| Atherogenic diet[41-43] (10% w/w lard, 2% w/w cholesterol) | Yes | Yes | ↑TNF-α | Yes | Yes | Yes | Yes | Yes | Yes |
| High-fructose/sucrose diet[45-47] | No | Yes | ↑IL-1β, IL-2, IL-6 | Yes | No | Yes | Yes[46]/No[45] | Not described | Yes[46]/not described[45] |
| High-fructose, high-fat diet[48] | Yes | Yes | ↑leptin | Yes | Yes | Yes | Yes | Not described | Yes |
| Cafeteria diet[49] | Yes | Yes | Not described | Not described | Not described | Yes | Yes | Not described | Not described |
| Methionine- and choline-deficient diet [52,54] | Weight loss! | No | ↑IL-1β, IL-6, TNF-α | ↓triglycerides, ↓cholesterol | Yes | Yes | Yes | Not described | Yes |
| Choline-deficient diet[52-53] | Yes | Yes | Not described | Yes | No or mild | Yes | Yes[52] | Not described | Mild after 90 d[53] |
| Genetic models | |||||||||
| Zucker fatty rats[72-74,77,78] | Yes | Yes | ↑adiponectin, leptin, TNF-α, | Yes | Yes | Yes | No | Not described | No |
| Zucker fatty rats + high-fat diet[78] | Yes | Yes | ↑TNF-α | Not described | Yes | Yes | Yes | Yes | Yes |
| Otsuka Long-Evans Tokushima fatty rats[98-100] | Yes | Yes | ↑leptin, IL-6↓adiponectin | Yes | Yes (AST) | Yes | Yes | Yes | No |
Characteristics of high-fat models depend mainly on the rat strain, amount and composition of fat, duration of feeding, experimental design, etc. AST: Aspartate aminotransferase; IL: Interleukin; TNF: Tumour necrosis factor.
NUTRITIONAL MODELS
Nutritional models of NAFLD can be classified according to the mechanism by which steatosis and other changes are induced[21]. The first group includes models with increased lipid import or synthesis in the liver (high-fat diets, high-fructose/sucrose diets, combined diets). The second group is defined by reduced lipid export or catabolism (methionine- and choline-deficient diet; choline-deficient, L-amino acid-defined diet). Because few NAFLD patients exhibit evident genetic defects, the use of dietary models of NAFLD is more relevant to human disease than genetic models[20]. Currently, the most common rat model of NAFLD uses high-fat diets. Additionally, methionine- and choline-deficient diets and high-fructose diets are widely employed.
High-fat diets
First, we should define what a high-fat diet (HFD) means for rats. Standard rat diets contain lower amounts of fat than the recommended human diet. In contrast to approximately 30% of total energy intake from fats in humans, the standard rat diets contain less than 10% of kcal fat, whereas high-fat diets and very high-fat diets contain 30%-50% and more than 50% of kcal fat, respectively[22]. Through literature review, we found that the use of high-fat diets is very popular, but there are many variations of high-fat diets with different compositions and experimental designs; thus, the results obtained from these studies are inconsistent. In some experiments, HFD induced hepatic steatosis of varying degrees and patterns[23-25], while in others, HFD did not lead to the development of fat accumulation in the liver[26]. Moreover, HFD led to histopathological hepatic changes similar to human NASH in several other experiments[27-29]. Studies also differ in regards to the development of insulin resistance, obesity and dyslipidaemia, and other known pathogenic factors of human NAFLD. These studies vary in the time of feeding; rat strain; sex and age of rats; the amount of energy derived from fats; the origin of fats; the ratio of saturated (SFA), monounsaturated (MUFA) and polyunsaturated (PUFA) fatty acids; and the proportion of ω-3 and ω-6 PUFA. Regarding the composition of fats, fatty acids considerably influence the expression of many genes, particularly in the liver[30]. Diets also vary in the composition of non-fat nutrients (the percentage of proteins and saccharides; the ratio of mono-, oligo- and polysaccharides). Most diets are fed ad libitum, but in some papers, controlled feeding via intragastric cannula was used[27]. All laboratory rodent species are susceptible to the induction of metabolic alterations upon HFD feeding[31]. HFDs are not only used for the induction of fatty liver changes[32] but are also employed as models of metabolic syndrome, dyslipidaemia, obesity and insulin resistance[22]. Because insulin resistance is one of the most important pathogenic factors of NAFLD[2], feeding rats with a HFD is legitimate and more closely resembles the pathogenesis of human NAFLD than genetic models and nutrient-deficient diets.
There are interstrain differences in the susceptibility of rats to HFD. Examination of 3 strains of male rats fed a HFD (71% of kcal fat) for 3 wk showed that all three strains developed steatosis affecting > 66% of liver cells, but the histological patterns were different[24]. Microvesicular, mixed, and macrovesicular steatosis were found in Lewis, Wistar, and Sprague-Dawley rats, respectively. Moreover, the highest degree of fibrosis, hepatocyte damage, and reduced blood flow velocity in central veins were observed in the Sprague-Dawley strain[24]. Another study compared Lewis and Sprague-Dawley rats of both genders fed a HFD for 3 wk. The livers of all experimental groups on a HFD demonstrated evidence of steatosis, but similarly to the previous article, Lewis rats developed microvesicular steatosis, whereas Sprague-Dawley rats presented macrovesicular steatosis accompanied by pronounced fibrosis[25]. This experiment also confirmed the higher susceptibility of male rats (of both strains) to the development of steatosis[25], which corresponds to observations in humans, where NAFLD is preferentially found in men or postmenopausal women[33]. Our previous observations showed that HFD (71% of kcal fat) feeding does not differ in the degree and pattern of steatosis and liver triglyceride content in male Sprague-Dawley and Wistar rats, respectively[23]. It was also confirmed that the time of feeding and the amount of dietary fats corresponded to the degree of liver pathology and other markers of liver tissue injury[28,34].
In 2006, Buettner et al[35] compared the metabolic and molecular effects of different HFDs. They fed male Wistar rats with a HFD containing 42% of energy in fats. They used four different sources of fats-lard (comparable quantities of SFA and MUFA), olive oil (mainly MUFA), coconut fat (mainly SFA) and fish oil (mainly ω-3 PUFA). Histopathological samples of the livers of rats fed by diets containing lard, olive oil and coconut fat exhibited mainly microvesicular steatosis without inflammation or fibrosis. These diets also up-regulated key genes of lipid synthesis. In contrast, the major enzymes of fatty acid oxidation were down-regulated in these groups. SREBP-1c was up-regulated in all HFDs, but peroxisome proliferator activated receptor-α (PPAR-α), the major regulator of fatty acid oxidation, was up-regulated only in the liver of rats fed a diet containing ω-3 PUFA. The diets based on lard and olive oil induced more pronounced obesity and insulin resistance and the highest accumulation of fats in the liver. In another study, a beneficial effect of ω-3 PUFA was shown[34]. Giving ω-3 PUFA reduced the histological severity of HFD-induced NAFLD in Sprague-Dawley rats.
Hunting for a new high-fat animal model of NASH
In 2004, Lieber et al[29] introduced a new high-fat model of NASH in rats. Feeding Sprague-Dawley rats a liquid HFD (71% of kcal fat) for 3 wk incurred liver steatosis, inflammation and mitochondrial abnormalities without a significant elevation of serum transaminases. The histological observations were accompanied by increased hepatic TNF-α and TNF-α mRNA, collagen type 1 and procollagen mRNA, induction of cytochrome P450 2E1 and elevated markers of lipid peroxidation. Although there was no difference in final bodyweight, plasma insulin concentrations were increased. In this work, a control group received a diet containing 35% of kcal fat without significant liver changes[29], but a diet containing 35% of kcal fat is thought to be a high-fat diet for rats[22]. Our study showed that Wistar rats fed a diet with 35% of kcal fat exhibited considerable microvesicular steatosis[23]. Significant steatosis was not found in Sprague-Dawley rats, although the liver triglyceride content was significantly increased (3-fold)[23]. Using similar or equivalent diets in Sprague-Dawley rats, we[23] and others[36] failed to reproduce the histological picture of NASH, even when the time of feeding was extended to 5 or 6 wk.
In another study, feeding male Sprague-Dawley rats a high-fat emulsion by gavage for 6 wk successfully induced obesity, insulin resistance, hyperlipidaemia, hepatic steatosis, pericentral necrosis, inflammation and mitochondrial lesions[27]. These findings were accompanied by lipid peroxidation, elevated expression of cytochrome P450 2E1, reduced PPAR-α in the liver, and high aminotransferase activity and serum TNF-α levels[27]. This model reproduces several characteristics of human NASH but also introduces several limitations, such as forced overfeeding, high content of polyunsaturated fatty acids, technical requirements and skills.
Another attempt to establish a rat model of NASH used total enteral nutrition to overfeed male Sprague-Dawley rats a HFD (5%-70% of kcal corn oil) for 21-65 d[28]. The hepatic triglyceride content was dependent on the percentage of corn oil in the diet. Histopathological findings of NASH (steatosis, macrophage infiltration, apoptosis, and focal necrosis) were present in the 70% corn oil group. In this group, markers of lipid peroxidation, TNF-α expression, expression of cytochrome P450 2E1 in the liver and serum alanine aminotransferase (ALT) were elevated. Although the intragastric infusion of HFD results in pathology that is clinically similar to NASH, the method of feeding does not reflect human NAFLD.
During the last few years, there has been an upswing of HFD models of NASH[28,34,37,38]. These models successfully induce histopathological patterns of human NASH and the main metabolic perturbations of metabolic syndrome. The histological picture shows liver steatosis, lobular inflammatory infiltrates, hepatocyte necrosis and apoptosis, hepatocyte ballooning, and zone 3 fibrosis. Other characteristic features of NAFLD/metabolic syndrome are also observed (visceral obesity, insulin resistance, dyslipidaemia, elevated hepatic cytochrome P450 2E1 and TNF-α expression, oxidative stress and lipid peroxidation, increased serum ALT, and decreased adiponectin and PPAR-α expression).
The majority of nutritional HFD models of NASH use fat with high amounts of corn oil, which mainly contains PUFA, of which > 98% are ω-6 PUFA. In contrast, saturated or monounsaturated fats are primarily found in the human diet. Thus, the use of such diets does not correlate with the dietary aetiology of NAFLD in humans. Moreover, ω-6 PUFAs exert proinflammatory characteristics in the body and enhance lipid peroxidation[39]. When male Wistar rats were fed ad libitum with a HFD containing mainly SFA (67% of energy from fats, coconut fat or butter) for 14 wk, neither liver steatosis nor increased liver triglyceride content were induced[26].
In conclusion, there is a high variability in the different diets and experimental protocols. Choosing suitable diet compositions, durations of feeding, strains and sexes of rats are crucial events in the experimental design. The Sprague-Dawley strain and male gender appear more susceptible to the development of NAFLD by HFD feeding. The specific mechanisms of interstrain differences have not been fully clarified. Using a HFD with high amounts of ω-6 PUFA can induce histopathological changes similar to NASH with some features of metabolic syndrome, but liver inflammation may be at least partially caused by the proinflammatory effect of ω-6 PUFA. Another limiting factor may be the lower reproducibility of nutritional models of NASH based on HFD in different laboratories.
High-cholesterol and high-fat, high-cholesterol diets (atherogenic diets)
Diets with supplemented cholesterol (up to 3% w/w) were originally used for the study of atherosclerosis pathogenesis. It was observed that diets containing cholesterol, particularly with the combination of higher fat and cholic acid in the chow, induced fatty liver changes. Dietary cholesterol is an important risk factor for the progression of simple steatosis to NASH in a mouse model[40]. Thus, the employment of high-fat, high-cholesterol diets has become popular for the induction of NAFLD in rats. Atherogenic diets may be easily prepared by the addition of extra fat and cholesterol (i.e., 10% w/w lard and 2% w/w cholesterol) to standard pellet chow, and they induce liver steatosis, necrosis, hepatocyte ballooning, steatohepatitis and fibrosis[41-43]. Furthermore, obesity, dyslipidaemia, insulin resistance and elevated serum aminotransferases accompany these liver changes.
High-fructose/sucrose diet
A shift in the eating habits of Western countries in recent decades has been characterised by an increase in mono- and disaccharides, primarily sucrose (50% fructose) and fructose (i.e., candy, soft drinks). Fructose and other saccharides in excess are known to promote de novo lipogenesis. In humans, higher amounts of fructose in the diet are associated with metabolic syndrome, obesity and NAFLD[44]. Fructose promotes protein fructosylation and the formation of reactive oxygen species in the liver[44]. For rat models of metabolic syndrome or NAFLD, fructose or sucrose can be added into the diet or drinking water. A fructose-enriched diet induces steatosis and, in some experiments, inflammation and periportal fibrosis[45-47]. The histopathological findings were accompanied by insulin resistance, dyslipidaemia, elevated proinflammatory cytokines and markers of hepatic lipid peroxidation.
Combined diets
Various “fast food” diets containing higher amounts of fat and fructose are used to induce NAFLD in rats. Moreover, these diets can be supplemented with cholesterol or trans-fatty acids. Feeding male Wistar rats a high-fructose, high-fat diet together with fructose in the drinking water for 16 wk resulted in marked obesity, impaired glucose tolerance, dyslipidaemia, hyperinsulinaemia, increased plasma leptin and liver steatosis, inflammation, and fibrosis[48]. The cardiovascular system (increased systolic blood pressure, endothelial dysfunction, inflammation, fibrosis and hypertrophy of the left ventricle), kidneys (inflammation and fibrosis) and pancreas were also affected.
The cafeteria diet is a special combined diet that more accurately reflects the variety of highly palatable, energy dense, unhealthy foods in Western countries. Feeding a cafeteria diet is a better model for human obesity and metabolic syndrome compared to a simple HFD[49]. Male Wistar rats fed standard chow with concurrently offered cafeteria food (cookies, cereals, cheese, processed meats, crackers, etc.) ad libitum for 15 wk developed hyperphagia, resulting in severe obesity and prediabetes (glucose and insulin intolerance). This diet induced panlobular microvesicular steatosis, steatohepatitis and chronic inflammation in white and brown adipose tissues. Liver histopathological and metabolic alterations were more pronounced in the cafeteria diet compared to the lard-based HFD[49]. Voluntary feeding a cafeteria diet closely reflects the etiopathogenesis of human NAFLD.
According to the point of view of changing eating behaviour in Western populations, combined diets (high-fructose, high-fat diet or cafeteria diet) are the most relevant model for metabolic syndrome and NAFLD in rats.
Methionine- and choline-deficient diet
One of the most commonly used dietary models of NAFLD and NASH is the methionine- and choline-deficient diet (MCDD). Choline is an essential substance that is involved in many metabolic reactions in rats (e.g., methylation or transport of lipids). It is used for the formation of many biologically important compounds, such as phosphatidylcholine, sphingomyelin and acetylcholine. In humans, choline deficiency can be found in malnutrition, alcohol abuse or during pregnancy and lactation, and it may lead to the development of hepatic steatosis[50]. When choline is lacking in the diet, methionine can be used for the synthesis of choline. The absence of both choline and methionine substantially disturbs the formation of phosphatidylcholine. Phosphatidylcholine is essential for the normal formation of VLDL and its secretion from the liver[51]. Feeding rats a MCDD results in a rapid accumulation of triglycerides in the liver with the subsequent development of steatohepatitis and fibrogenesis[52]. In contrast to MCDD, a choline-deficient diet with sufficient supplementation of methionine does not result in the blockage of VLDL secretion, thereby leading to steatosis with a lower degree of inflammation in the rat liver[52,53].
In 2006, Veteläinen et al[52] described in a detail course of histological changes in an MCDD model in Wistar rats. At 1 wk, feeding MCDD induced mainly microvesicular steatosis with a macrovesicular component, predominantly in the acinar zone 3. At 3 wk, the pattern of steatosis changed to macrovesicular and scattered foci of inflammatory cells. At 5 wk, diffuse and extensive macrovesicular steatosis with foci of mononuclear inflammatory cells together with the occasional spotty necrosis occurred. At 7 wk, extensive macrovesicular steatosis, accumulation of cellular debris and numerous clusters of inflammatory cells developed. At this time, increased perivenular fibrosis with occasional centrilobular fibrosis was reported. Biochemically confirmed hepatic steatosis was accompanied by the accumulation of cholesterol in the liver[52].
Feeding rats with MCDD results in malnutrition; thus, rats lose weight, and the weight of the liver proportionally decreases[52]. Liver injury induced by MCDD is accompanied by elevated serum transaminases and proinflammatory cytokines, such as IL-1β, IL-6 and TNF-α[54]. Due to hepatic blockage of VLDL, serum levels of triglycerides and cholesterol are reduced[52]. In contrast to the pathogenesis of human NAFLD, MCDD does not induce insulin resistance in rats[52,55]. Feeding MCDD increases lipid peroxidation and lowers the hepatic content of reduced glutathione (GSH)[52,56]. The lack of methionine may be partially responsible for the decreased glutathione synthesis. An increase in oxidative stress and lipid peroxidation precedes the onset of steatohepatitis in MCDD-fed rats, and lipid peroxidation corresponds to hepatocellular damage and increased hepatic TNF-α[52]. By contrast, liver regeneration following a 2/3 partial hepatectomy in Wistar rats is not significantly impaired by MCDD feeding[57]. Liver mitochondria from MCDD rats show higher oxidative activity but a lower efficiency of oxidative phosphorylation; thus, mitochondrial ATP synthesis efficiency was decreased[58]. Liver mitochondria isolated from MCDD-fed rats exhibit higher hydrogen peroxide production, decreased respiratory control index of complex I, up-regulation of uncoupling protein-2, increased permeability of the inner mitochondrial membrane for H+ and, as a consequence, a reduction in mitochondrial membrane potential[56,59,60].
In a study by Kirsch et al[61], species (rat and mouse), strain (Wistar, Long-Evans and Sprague-Dawley rats, and C57/BL6 mice) and sex differences were investigated in an MCDD model of NASH. The degrees of steatosis, hepatic lipid content and serum ALT activity were significantly higher in male rats than in female rats. Wistar rats had higher liver lipid levels, serum ALT levels, and liver mass/body mass ratios than Long-Evans and Sprague-Dawley rats. The mechanisms underlying these strain differences are uncertain. Feeding rats with MCDD for 4 wk induced only minor necrosis and inflammation, and fibrosis was absent[61]. In contrast to mice, rats fed MCDD develop steatohepatitis more slowly[21,61].
The choline-deficient, L-amino acid-defined (CDAA) diet has similar characteristics as the MCDD. The CDAA diet contains a low amount of methionine; thus the mechanism by which this diet induces typical hepatic changes is similar to MCDD. Feeding a CDAA diet induces liver steatosis, which is followed by more severe steatosis with inflammation, increased oxidative stress and fibrosis, cirrhosis, preneoplastic lesions and hepatocellular carcinoma[62-64]. Similar to MCDD, rats fed a CDAA diet do not develop typical characteristics of metabolic syndrome. This model may be employed for the study of hepatocarcinogenesis from steatohepatitis.
Although the MCDD model is one of the best defined rat models of NASH and is widely used to study NASH, it does not reflect several causative features of human NASH[65]. In contrast to humans, rats fed MCDD lose weight, have low serum triglyceride and cholesterol levels and have normal insulin sensitivity. This model induces changes in the liver by nutritional deficiency and does not reflect the metabolic profile and aetiology of NAFLD in humans. MCDD imitates several histopathological features of human NAFLD, but the distribution of steatosis may vary. Depending on the time of feeding, MCDD induces hepatic steatosis, steatohepatitis or fibrosing steatohepatitis and thus simulates the histological picture of NAFLD. This model is more suitable for studying the consequences of fat accumulation, inflammation, oxidative stress and fibrogenesis in the liver than the complex pathogenesis of human NAFLD. In contrast to most other NAFLD models, MCDD induces reproducible histological changes with significant inflammation and fibrogenesis. In conclusion, the MCDD model mimics only histopathological features but lacks the main aetiopathogenic factors of human disease.
Choline-deficient diet
Choline-deficient diet (CDD) induces hepatic steatosis, and the degree of steatosis is dependent upon the time of feeding[52]. In contrast to MCDD, CDD-fed rats do not develop severe fibrosis, and the inflammation is of a lower extent[52]. The accumulation of fats in the liver is accompanied by non-significant changes or mild elevations in serum transaminases[52,53]. Feeding CDD for 90 d leads to the development of moderate fibrotic alterations in the liver of male Wistar rats[53]. Liver steatosis in this model is characterised by the development of obesity, dyslipidaemia (increase in plasma concentrations of triglycerides and cholesterol) and insulin resistance after 7 wk[52]. Hepatic GSH and lipid peroxidation were reported to be unaltered[52]. In contrast, Vendemiale et al[66] and Hensley et al[67] showed a greater mitochondrial content of oxidised lipids and proteins, lower concentration of glutathione and increased mitochondrial ROS formation. The activity of mitochondrial complex I, mitochondrial ATP synthase activity and hepatic ATP levels were significantly reduced in the fatty livers of CDD-fed rats[66,67].
In summary, the CDD and MCDD are two distinct models in rats, which differ not only in their histological findings but also in the metabolic and other characteristics of rats (insulin resistance, inflammatory response, oxidative stress)[52]. CDD induces fatty liver accompanied by metabolic alterations typical of metabolic syndrome.
GENETIC MODELS
Genetic models can be further subdivided into naturally occurring genetic models and models of induced genetic mutations. There is a variety of genetic models of NAFLD in mice[68]. In view of the polygenic genetic background of this disease[3], monogenic mouse models of NAFLD do not fully replicate the etiopathogenesis of human NAFLD, but they may provide valuable results concerning particular events in the pathogenesis of NAFLD. Moreover, genetically modified mice are less available and more expensive. In contrast to mice, there are very few genetically modified rats.
The Zucker fatty rats
One of the most commonly used models of NAFLD in rats is a genetic model of the obese Zucker rats (the Zucker fatty rats, fa/fa rats, ZFR). ZFR have a natural mutation in the leptin receptor (fa allele), which decreases the affinity of the receptor for leptin and alters signal transduction[69]. ZFR are homozygotes for the fa allele, while heterozygotes for the fa allele (lean Zucker rats) serve as controls. Leptin is a peptide hormone secreted from adipose tissue, which participates in the regulation of feeding behaviour and energy expenditure[70]. This hormone is also involved in the modulation of cell death and fibrogenesis[71]. Thus, the mutation in the leptin receptor leads to leptin resistance. ZFR are widely used as an animal model of genetic obesity and metabolic syndrome[22]. ZFR develop severe obesity and are hyperleptinaemic, hyperphagic, inactive, obese and insulin-resistant (hyperinsulinaemia, mild hyperglycaemia, hyperlipidaemia)[72,73]. ZFR rats are also used as a model of insulin resistance. Hyperlipidaemia in ZFR is characterised by increased VLDL and high-density lipoproteins (HDL) without significant changes in low-density lipoprotein (LDL) cholesterol but with reduced expression of the hepatic LDL receptor[74,75]. It was also observed that the systolic arterial blood pressure increased (at 28 wk)[76], and plasma adiponectin levels were higher[73] in ZFR than in lean controls.
Macrovesicular or microvesicular steatosis is present in ZFR without signs of progression to steatohepatitis[77]. The accumulation of fat in the liver of Zucker obese rats is localised to the periportal area[78]. The liver of obese Zucker rats have lower amounts of GSH and vitamin E and decreased activity of catalase[79]. The regenerative capacity of the liver of fa/fa rats is significantly decreased after 2/3 partial hepatectomy[80]. As a consequence of leptin resistance, increased expressions of SREBP-1c and the carbohydrate response element binding protein (ChREBP) were reported[81,82]. The elevations of SREBP-1c mRNA was accompanied by increased expression of lipogenic enzymes and the accumulation of triglycerides in the liver[81].
As in ob/ob mice, ZFR do not spontaneously develop steatohepatitis, and they require a “second hit” for the progression of steatosis to steatohepatitis. A second hit could be exposure to lipopolysaccharide[77], a combination of diet rich in disaccharide and low-dose lipopolysaccharide[83], a high-fat diet[78] or the combination of MCDD with a high fat diet[84].
Because the dietary models (except MCDD) more closely resemble the pathogenesis of human NAFLD compared to the genetic models, the use of dietary interventions for genetic models has become more popular. After feeding fa/fa rats a high-saturated fat diet (60% of energy of diet derived from lard) for 8 wk, more severe micro- and macrovesicular steatosis and steatohepatitis developed[78]. Liver injury was accompanied by fasting hyperglycaemia and increased serum activity of ALT, TNF-α and TGF-β, increased collagen deposition, and greater activation of hepatic stellate cells. Markers of oxidative stress, such as lipid peroxidation, protein carbonyls, glutathione, and antioxidant enzymes, were significantly aggravated in ZFR fed a high-fat diet.
A special genetic model of obesity with type 2 diabetes, the Zucker diabetic fatty (ZDF) rats, was derived from ZFR by the selective breeding of hyperglycaemic ZFR. ZDF male rats typically develop hyperinsulinaemia, peripheral insulin resistance, hyperglycaemia and non-insulin dependent diabetes by 12 wk of age[85,86]. ZDF females are more resistant to the spontaneous development of diabetes, and the induction of type II diabetes in female ZDF rats is successful only when they are fed a high-fat diet[87].
ZFR partially simulates human metabolic syndrome (obesity, insulin resistance, dyslipidaemia, hyperinsulinaemia, steatosis), but they have several disadvantages. Because leptin or leptin receptor mutations are rare in humans[88], ZFR do not reflect the multifactorial etiopathogenesis of human NAFLD. Moreover, ZFR do not spontaneously develop steatohepatitis and are resistant to liver fibrosis. ZFR require further interventions for the progression from simple steatosis to steatohepatitis.
There are several other rat models utilising altered leptin pathways in which hepatic steatosis develops. These are spontaneously hypertensive obese rats (Koletsky’s rats), stroke-prone spontaneously hypertensive rats (SHRSP5/Dmcr) or corpulent JCR:LA-cp rats, which have a recessive mutation in the leptin receptor gene that blocks expression of the protein[89]. These rats exhibit several similar phenotypic characteristics to ZFR. They are hyperphagic, obese, hyperlipidaemic and insulin resistant[90,91]. Although they develop simple fatty liver[91,92], these models have not been widely employed for the study of NAFLD. The exposure of stroke-prone spontaneously hypertensive rats to a high-fat, high-cholesterol diet results in steatosis, necrosis with lymphocyte infiltrations, hepatocyte ballooning with Mallory-Denk bodies and fibrosis[93].
Transgenic spontaneously hypertensive rats overexpressing SREBP-1a
Recently, a new transgenic model of SREBP-1a overexpression was introduced in the spontaneously hypertensive rat[94]. SREBP-1 gene polymorphisms are associated with obesity, severe insulin resistance and type 2 diabetes in humans[95]. Transgenic spontaneously hypertensive rats (SHR) overexpressing SREBP-1a rats are non-obese, but they spontaneously develop hypertension, fatty liver, and several important characteristics of metabolic syndrome (hyperinsulinaemia, hyperglycaemia, hypertriglyceridaemia, and hypercholesterolaemia)[94]. Biochemical examination confirmed the accumulation of triglycerides in the liver and found elevated hepatic levels of cholesterol[94]. Transgenic SHR overexpressing SREBP-1a rats fed with a high-fructose diet exhibit significantly increased oxidative stress in the liver (enhanced lipid peroxidation, decreased activities of antioxidative enzymes and lower glutathione), which was more pronounced in older rats[96]. Similarly, the progression of histopathological findings was age-dependent. Centrilobular steatosis without inflammation was observed at 2 mo of age, whereas prominent fatty changes accompanied by focal liver cell necrosis and inflammatory infiltrate similar to human NASH were found after 16 mo[96]. Transgenic SHR overexpressing SREBP-1a rats may provide a tool for studying the pathogenic mechanisms and treatment of metabolic syndrome associated with NAFLD[94].
Otsuka Long-Evans Tokushima fatty rats
Otsuka Long-Evans Tokushima fatty rats (OLETF) were obtained in 1984 by the selective breeding of an outbred colony of Long-Evans rats exhibiting a diabetic phenotype (polyuria, polydipsia, obesity). OLETF rats have a deletion in the promoter region of the gene encoding the cholecystokinin-1 (CCK-1) receptor and in the first and second exons of this gene, resulting in low or absent expression of CCK-1 receptor mRNA[97]. A control strain, the LETO rats, which originated from the same colony of rats, expresses the CCK-1 receptor and does not exhibit the phenotype of OLETF rats. CCK-1 participates in satiety control in the hypothalamus; thus, OLETF rats exhibit hyperphagia, and they are mildly obese, hyperlipidaemic, hyperglycaemic (after the age of 18 wk), hyperinsulinaemic, insulin resistant and have diabetes mellitus[55,98-100]. Serum concentrations of leptin and IL-6 are elevated, while adiponectin is lower in these rats[98]. OLETF rats are used as a model for metabolic syndrome with diabetes[22]. OLETF rats spontaneously develop liver steatosis when fed a standard diet ad libitum[100]. At 22-38 wk of age, micro- and macrovesicular steatosis (with a maximum at approximately 30 wk) and hepatocyte ballooning without collagen deposition or fibrosis were observed, but the steatosis disappeared after 42 wk[100]. mRNA expressions of lipogenic genes, such as SREBP-1c and ChREBP, were significantly elevated in the liver of OLETF rats prior to the onset of hepatic steatosis (10 wk) and then gradually decreased to control values (50 wk)[100]. Contrarily, PPAR-α is down-regulated in younger OLETF rats (12 and 28 wk)[101].
For the induction of steatohepatitis in OLETF rats, MCDD or a combined diet with choline and methionine deficiency and high-fat content can be used[55,102]. In the OLETF rats, MCDD induces hepatic lobular inflammation and perivenular and pericellular fibrosis in zone 3 with increased expression of TGF-β1 and activation of stellate cells[102]. The advantage of a combination of MCDD and HFD in OLETF rats is the development of steatohepatitis with enhanced insulin resistance, more pronounced inflammation and fibrosis (pre-cirrhosis), and increased mRNA expression of lipogenic genes[55]. In contrast to the finding of Ota et al[55], Song et al[100] described hepatocyte ballooning in OLETF (and LETO) rats only when a combined diet (MCDD + HFD) was used.
Similar to the Zucker fatty rats, OLETF rats do not reflect the etiopathogenesis of human NAFLD (mutation in CCK-1 receptor). OLETF rats are recommended at the age of 22-38 wk as animal models of hepatic steatosis, but they are not a suitable model for NASH without an additional dietary hit.
Prague hereditary hypercholesterolaemic rats
Prague hereditary hypercholesterolaemic rats were crossbred from Wistar rats as a model for polygenic hypercholesterolaemia induced by dietary cholesterol (2% w/w cholesterol diet). Hypercholesterolaemia is caused by the elevation of cholesterol in VLDL, intermediate-density lipoproteins, and LDL but not in HDL[103]. When fed standard chow, serum cholesterol is only mildly elevated[103]. In contrast to 5% dietary fat alone (standard chow), a high cholesterol diet in these rats induces the development of fatty liver and the accumulation of cholesterol[103]. Thus, the presence of cholesterol in the diet is necessary for the development of fatty liver. This combined model of fatty liver is also not widely used.
CONCLUSION
Although there is not an ideal model for human non-alcoholic fatty liver disease that reflects all the clinical, histological, aetiological and pathogenic aspects of human disease, choosing an appropriate model for studying particular events of NAFLD while respecting its limitations has contributed greatly to the understanding of this disease, its progression and treatment. Considering the legislative, ethical, economical and health factors of NAFLD, animal models are and will continue to be an essential tool for further research of this disease.
Footnotes
Supported by Programme PRVOUK P37/02
P- Reviewers: Gwak GY, Mueller-Wieland D S- Editor: Qi Y L- Editor: A E- Editor: Ma S
References
- 1.Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther. 2011;34:274–285. doi: 10.1111/j.1365-2036.2011.04724.x. [DOI] [PubMed] [Google Scholar]
- 2.Yki-Järvinen H. Nutritional modulation of nonalcoholic fatty liver disease and insulin resistance: human data. Curr Opin Clin Nutr Metab Care. 2010;13:709–714. doi: 10.1097/MCO.0b013e32833f4b34. [DOI] [PubMed] [Google Scholar]
- 3.Anstee QM, Day CP. The genetics of NAFLD. Nat Rev Gastroenterol Hepatol. 2013;10:645–655. doi: 10.1038/nrgastro.2013.182. [DOI] [PubMed] [Google Scholar]
- 4.Ekstedt M, Franzén LE, Mathiesen UL, Thorelius L, Holmqvist M, Bodemar G, Kechagias S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology. 2006;44:865–873. doi: 10.1002/hep.21327. [DOI] [PubMed] [Google Scholar]
- 5.Tessari P, Coracina A, Cosma A, Tiengo A. Hepatic lipid metabolism and non-alcoholic fatty liver disease. Nutr Metab Cardiovasc Dis. 2009;19:291–302. doi: 10.1016/j.numecd.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 6.Charlton M, Sreekumar R, Rasmussen D, Lindor K, Nair KS. Apolipoprotein synthesis in nonalcoholic steatohepatitis. Hepatology. 2002;35:898–904. doi: 10.1053/jhep.2002.32527. [DOI] [PubMed] [Google Scholar]
- 7.Samuel VT, Liu ZX, Qu X, Elder BD, Bilz S, Befroy D, Romanelli AJ, Shulman GI. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem. 2004;279:32345–32353. doi: 10.1074/jbc.M313478200. [DOI] [PubMed] [Google Scholar]
- 8.Ferré P, Foufelle F. SREBP-1c transcription factor and lipid homeostasis: clinical perspective. Horm Res. 2007;68:72–82. doi: 10.1159/000100426. [DOI] [PubMed] [Google Scholar]
- 9.Valenti L, Fracanzani AL, Dongiovanni P, Santorelli G, Branchi A, Taioli E, Fiorelli G, Fargion S. Tumor necrosis factor alpha promoter polymorphisms and insulin resistance in nonalcoholic fatty liver disease. Gastroenterology. 2002;122:274–280. doi: 10.1053/gast.2002.31065. [DOI] [PubMed] [Google Scholar]
- 10.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 11.Marra F, Gastaldelli A, Svegliati Baroni G, Tell G, Tiribelli C. Molecular basis and mechanisms of progression of non-alcoholic steatohepatitis. Trends Mol Med. 2008;14:72–81. doi: 10.1016/j.molmed.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 12.Wieckowska A, Zein NN, Yerian LM, Lopez AR, McCullough AJ, Feldstein AE. In vivo assessment of liver cell apoptosis as a novel biomarker of disease severity in nonalcoholic fatty liver disease. Hepatology. 2006;44:27–33. doi: 10.1002/hep.21223. [DOI] [PubMed] [Google Scholar]
- 13.Jou J, Choi SS, Diehl AM. Mechanisms of disease progression in nonalcoholic fatty liver disease. Semin Liver Dis. 2008;28:370–379. doi: 10.1055/s-0028-1091981. [DOI] [PubMed] [Google Scholar]
- 14.Larter CZ, Yeh MM. Animal models of NASH: getting both pathology and metabolic context right. J Gastroenterol Hepatol. 2008;23:1635–1648. doi: 10.1111/j.1440-1746.2008.05543.x. [DOI] [PubMed] [Google Scholar]
- 15.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 16.Kleiner DE, Brunt EM. Nonalcoholic fatty liver disease: pathologic patterns and biopsy evaluation in clinical research. Semin Liver Dis. 2012;32:3–13. doi: 10.1055/s-0032-1306421. [DOI] [PubMed] [Google Scholar]
- 17.Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, Charlton M, Sanyal AJ. The diagnosis and management of non-alcoholic fatty liver disease: practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology. 2012;55:2005–2023. doi: 10.1002/hep.25762. [DOI] [PubMed] [Google Scholar]
- 18.Ribeiro PS, Cortez-Pinto H, Solá S, Castro RE, Ramalho RM, Baptista A, Moura MC, Camilo ME, Rodrigues CM. Hepatocyte apoptosis, expression of death receptors, and activation of NF-kappaB in the liver of nonalcoholic and alcoholic steatohepatitis patients. Am J Gastroenterol. 2004;99:1708–1717. doi: 10.1111/j.1572-0241.2004.40009.x. [DOI] [PubMed] [Google Scholar]
- 19.Wieckowska A, Feldstein AE. Diagnosis of nonalcoholic fatty liver disease: invasive versus noninvasive. Semin Liver Dis. 2008;28:386–395. doi: 10.1055/s-0028-1091983. [DOI] [PubMed] [Google Scholar]
- 20.Schattenberg JM, Galle PR. Animal models of non-alcoholic steatohepatitis: of mice and man. Dig Dis. 2010;28:247–254. doi: 10.1159/000282097. [DOI] [PubMed] [Google Scholar]
- 21.London RM, George J. Pathogenesis of NASH: animal models. Clin Liver Dis. 2007;11:55–74, viii. doi: 10.1016/j.cld.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 22.Fellmann L, Nascimento AR, Tibiriça E, Bousquet P. Murine models for pharmacological studies of the metabolic syndrome. Pharmacol Ther. 2013;137:331–340. doi: 10.1016/j.pharmthera.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 23.Kučera O, Garnol T, Lotková H, Staňková P, Mazurová Y, Hroch M, Bolehovská R, Roušar T, Červinková Z. The effect of rat strain, diet composition and feeding period on the development of a nutritional model of non-alcoholic fatty liver disease in rats. Physiol Res. 2011;60:317–328. doi: 10.33549/physiolres.932022. [DOI] [PubMed] [Google Scholar]
- 24.Rosenstengel S, Stoeppeler S, Bahde R, Spiegel HU, Palmes D. Type of steatosis influences microcirculation and fibrogenesis in different rat strains. J Invest Surg. 2011;24:273–282. doi: 10.3109/08941939.2011.586094. [DOI] [PubMed] [Google Scholar]
- 25.Stöppeler S, Palmes D, Fehr M, Hölzen JP, Zibert A, Siaj R, Schmidt HH, Spiegel HU, Bahde R. Gender and strain-specific differences in the development of steatosis in rats. Lab Anim. 2013;47:43–52. doi: 10.1177/0023677212473717. [DOI] [PubMed] [Google Scholar]
- 26.Romestaing C, Piquet MA, Bedu E, Rouleau V, Dautresme M, Hourmand-Ollivier I, Filippi C, Duchamp C, Sibille B. Long term highly saturated fat diet does not induce NASH in Wistar rats. Nutr Metab (Lond) 2007;4:4. doi: 10.1186/1743-7075-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zou Y, Li J, Lu C, Wang J, Ge J, Huang Y, Zhang L, Wang Y. High-fat emulsion-induced rat model of nonalcoholic steatohepatitis. Life Sci. 2006;79:1100–1107. doi: 10.1016/j.lfs.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 28.Baumgardner JN, Shankar K, Hennings L, Badger TM, Ronis MJ. A new model for nonalcoholic steatohepatitis in the rat utilizing total enteral nutrition to overfeed a high-polyunsaturated fat diet. Am J Physiol Gastrointest Liver Physiol. 2008;294:G27–G38. doi: 10.1152/ajpgi.00296.2007. [DOI] [PubMed] [Google Scholar]
- 29.Lieber CS, Leo MA, Mak KM, Xu Y, Cao Q, Ren C, Ponomarenko A, DeCarli LM. Model of nonalcoholic steatohepatitis. Am J Clin Nutr. 2004;79:502–509. doi: 10.1093/ajcn/79.3.502. [DOI] [PubMed] [Google Scholar]
- 30.Jump DB, Tripathy S, Depner CM. Fatty acid-regulated transcription factors in the liver. Annu Rev Nutr. 2013;33:249–269. doi: 10.1146/annurev-nutr-071812-161139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tschöp M, Heiman ML. Rodent obesity models: an overview. Exp Clin Endocrinol Diabetes. 2001;109:307–319. doi: 10.1055/s-2001-17297. [DOI] [PubMed] [Google Scholar]
- 32.Takahashi Y, Soejima Y, Fukusato T. Animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J Gastroenterol. 2012;18:2300–2308. doi: 10.3748/wjg.v18.i19.2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hamaguchi M, Kojima T, Ohbora A, Takeda N, Fukui M, Kato T. Aging is a risk factor of nonalcoholic fatty liver disease in premenopausal women. World J Gastroenterol. 2012;18:237–243. doi: 10.3748/wjg.v18.i3.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Svegliati-Baroni G, Candelaresi C, Saccomanno S, Ferretti G, Bachetti T, Marzioni M, De Minicis S, Nobili L, Salzano R, Omenetti A, et al. A model of insulin resistance and nonalcoholic steatohepatitis in rats: role of peroxisome proliferator-activated receptor-alpha and n-3 polyunsaturated fatty acid treatment on liver injury. Am J Pathol. 2006;169:846–860. doi: 10.2353/ajpath.2006.050953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buettner R, Parhofer KG, Woenckhaus M, Wrede CE, Kunz-Schughart LA, Schölmerich J, Bollheimer LC. Defining high-fat-diet rat models: metabolic and molecular effects of different fat types. J Mol Endocrinol. 2006;36:485–501. doi: 10.1677/jme.1.01909. [DOI] [PubMed] [Google Scholar]
- 36.Ahmed U, Redgrave TG, Oates PS. Effect of dietary fat to produce non-alcoholic fatty liver in the rat. J Gastroenterol Hepatol. 2009;24:1463–1471. doi: 10.1111/j.1440-1746.2009.05870.x. [DOI] [PubMed] [Google Scholar]
- 37.Wang Y, Ausman LM, Russell RM, Greenberg AS, Wang XD. Increased apoptosis in high-fat diet-induced nonalcoholic steatohepatitis in rats is associated with c-Jun NH2-terminal kinase activation and elevated proapoptotic Bax. J Nutr. 2008;138:1866–1871. doi: 10.1093/jn/138.10.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Y, Hai J, Li L, Chen X, Peng H, Cao M, Zhang Q. Administration of ghrelin improves inflammation, oxidative stress, and apoptosis during and after non-alcoholic fatty liver disease development. Endocrine. 2013;43:376–386. doi: 10.1007/s12020-012-9761-5. [DOI] [PubMed] [Google Scholar]
- 39.Simopoulos AP. Evolutionary aspects of diet, the omega-6/omega-3 ratio and genetic variation: nutritional implications for chronic diseases. Biomed Pharmacother. 2006;60:502–507. doi: 10.1016/j.biopha.2006.07.080. [DOI] [PubMed] [Google Scholar]
- 40.Wouters K, van Gorp PJ, Bieghs V, Gijbels MJ, Duimel H, Lütjohann D, Kerksiek A, van Kruchten R, Maeda N, Staels B, et al. Dietary cholesterol, rather than liver steatosis, leads to hepatic inflammation in hyperlipidemic mouse models of nonalcoholic steatohepatitis. Hepatology. 2008;48:474–486. doi: 10.1002/hep.22363. [DOI] [PubMed] [Google Scholar]
- 41.Wang W, Zhao C, Zhou J, Zhen Z, Wang Y, Shen C. Simvastatin ameliorates liver fibrosis via mediating nitric oxide synthase in rats with non-alcoholic steatohepatitis-related liver fibrosis. PLoS One. 2013;8:e76538. doi: 10.1371/journal.pone.0076538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu ZJ, Fan JG, Ding XD, Qiao L, Wang GL. Characterization of high-fat, diet-induced, non-alcoholic steatohepatitis with fibrosis in rats. Dig Dis Sci. 2010;55:931–940. doi: 10.1007/s10620-009-0815-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang W, Wang LW, Wang LK, Li X, Zhang H, Luo LP, Song JC, Gong ZJ. Betaine protects against high-fat-diet-induced liver injury by inhibition of high-mobility group box 1 and Toll-like receptor 4 expression in rats. Dig Dis Sci. 2013;58:3198–3206. doi: 10.1007/s10620-013-2775-x. [DOI] [PubMed] [Google Scholar]
- 44.Lim JS, Mietus-Snyder M, Valente A, Schwarz JM, Lustig RH. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat Rev Gastroenterol Hepatol. 2010;7:251–264. doi: 10.1038/nrgastro.2010.41. [DOI] [PubMed] [Google Scholar]
- 45.Armutcu F, Coskun O, Gürel A, Kanter M, Can M, Ucar F, Unalacak M. Thymosin alpha 1 attenuates lipid peroxidation and improves fructose-induced steatohepatitis in rats. Clin Biochem. 2005;38:540–547. doi: 10.1016/j.clinbiochem.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 46.Kawasaki T, Igarashi K, Koeda T, Sugimoto K, Nakagawa K, Hayashi S, Yamaji R, Inui H, Fukusato T, Yamanouchi T. Rats fed fructose-enriched diets have characteristics of nonalcoholic hepatic steatosis. J Nutr. 2009;139:2067–2071. doi: 10.3945/jn.109.105858. [DOI] [PubMed] [Google Scholar]
- 47.Tian YF, He CT, Chen YT, Hsieh PS. Lipoic acid suppresses portal endotoxemia-induced steatohepatitis and pancreatic inflammation in rats. World J Gastroenterol. 2013;19:2761–2771. doi: 10.3748/wjg.v19.i18.2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Panchal SK, Poudyal H, Iyer A, Nazer R, Alam MA, Diwan V, Kauter K, Sernia C, Campbell F, Ward L, et al. High-carbohydrate, high-fat diet-induced metabolic syndrome and cardiovascular remodeling in rats. J Cardiovasc Pharmacol. 2011;57:611–624. doi: 10.1097/FJC.0b013e31821b1379. [DOI] [PubMed] [Google Scholar]
- 49.Sampey BP, Vanhoose AM, Winfield HM, Freemerman AJ, Muehlbauer MJ, Fueger PT, Newgard CB, Makowski L. Cafeteria diet is a robust model of human metabolic syndrome with liver and adipose inflammation: comparison to high-fat diet. Obesity (Silver Spring) 2011;19:1109–1117. doi: 10.1038/oby.2011.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lombardi B, Pani P, Schlunk FF. Choline-deficiency fatty liver: impaired release of hepatic triglycerides. J Lipid Res. 1968;9:437–446. [PubMed] [Google Scholar]
- 51.Ghoshal AK. New insight into the biochemical pathology of liver in choline deficiency. Crit Rev Biochem Mol Biol. 1995;30:263–273. doi: 10.3109/10409239509083487. [DOI] [PubMed] [Google Scholar]
- 52.Veteläinen R, van Vliet A, van Gulik TM. Essential pathogenic and metabolic differences in steatosis induced by choline or methione-choline deficient diets in a rat model. J Gastroenterol Hepatol. 2007;22:1526–1533. doi: 10.1111/j.1440-1746.2006.04701.x. [DOI] [PubMed] [Google Scholar]
- 53.Al-Humadi H, Theocharis S, Dontas I, Stolakis V, Zarros A, Kyriakaki A, Al-Saigh R, Liapi C. Hepatic injury due to combined choline-deprivation and thioacetamide administration: an experimental approach to liver diseases. Dig Dis Sci. 2012;57:3168–3177. doi: 10.1007/s10620-012-2299-9. [DOI] [PubMed] [Google Scholar]
- 54.Tahan V, Eren F, Avsar E, Yavuz D, Yuksel M, Emekli E, Imeryuz N, Celikel C, Uzun H, Haklar G, et al. Rosiglitazone attenuates liver inflammation in a rat model of nonalcoholic steatohepatitis. Dig Dis Sci. 2007;52:3465–3472. doi: 10.1007/s10620-007-9756-x. [DOI] [PubMed] [Google Scholar]
- 55.Ota T, Takamura T, Kurita S, Matsuzawa N, Kita Y, Uno M, Akahori H, Misu H, Sakurai M, Zen Y, et al. Insulin resistance accelerates a dietary rat model of nonalcoholic steatohepatitis. Gastroenterology. 2007;132:282–293. doi: 10.1053/j.gastro.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 56.Serviddio G, Bellanti F, Giudetti AM, Gnoni GV, Petrella A, Tamborra R, Romano AD, Rollo T, Vendemiale G, Altomare E. A silybin-phospholipid complex prevents mitochondrial dysfunction in a rodent model of nonalcoholic steatohepatitis. J Pharmacol Exp Ther. 2010;332:922–932. doi: 10.1124/jpet.109.161612. [DOI] [PubMed] [Google Scholar]
- 57.Zhang BH, Weltman M, Farrell GC. Does steatohepatitis impair liver regeneration? A study in a dietary model of non-alcoholic steatohepatitis in rats. J Gastroenterol Hepatol. 1999;14:133–137. doi: 10.1046/j.1440-1746.1999.01822.x. [DOI] [PubMed] [Google Scholar]
- 58.Romestaing C, Piquet MA, Letexier D, Rey B, Mourier A, Servais S, Belouze M, Rouleau V, Dautresme M, Ollivier I, et al. Mitochondrial adaptations to steatohepatitis induced by a methionine- and choline-deficient diet. Am J Physiol Endocrinol Metab. 2008;294:E110–E119. doi: 10.1152/ajpendo.00407.2007. [DOI] [PubMed] [Google Scholar]
- 59.Serviddio G, Bellanti F, Tamborra R, Rollo T, Capitanio N, Romano AD, Sastre J, Vendemiale G, Altomare E. Uncoupling protein-2 (UCP2) induces mitochondrial proton leak and increases susceptibility of non-alcoholic steatohepatitis (NASH) liver to ischaemia-reperfusion injury. Gut. 2008;57:957–965. doi: 10.1136/gut.2007.147496. [DOI] [PubMed] [Google Scholar]
- 60.Stärkel P, Sempoux C, Leclercq I, Herin M, Deby C, Desager JP, Horsmans Y. Oxidative stress, KLF6 and transforming growth factor-beta up-regulation differentiate non-alcoholic steatohepatitis progressing to fibrosis from uncomplicated steatosis in rats. J Hepatol. 2003;39:538–546. doi: 10.1016/s0168-8278(03)00360-x. [DOI] [PubMed] [Google Scholar]
- 61.Kirsch R, Clarkson V, Shephard EG, Marais DA, Jaffer MA, Woodburne VE, Kirsch RE, Hall Pde L. Rodent nutritional model of non-alcoholic steatohepatitis: species, strain and sex difference studies. J Gastroenterol Hepatol. 2003;18:1272–1282. doi: 10.1046/j.1440-1746.2003.03198.x. [DOI] [PubMed] [Google Scholar]
- 62.Kawaratani H, Tsujimoto T, Kitazawa T, Kitade M, Yoshiji H, Uemura M, Fukui H. Innate immune reactivity of the liver in rats fed a choline-deficient L-amino-acid-defined diet. World J Gastroenterol. 2008;14:6655–6661. doi: 10.3748/wjg.14.6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nakae D, Mizumoto Y, Andoh N, Tamura K, Horiguchi K, Endoh T, Kobayashi E, Tsujiuchi T, Denda A, Lombardi B. Comparative changes in the liver of female Fischer-344 rats after short-term feeding of a semipurified or a semisynthetic L-amino acid-defined choline-deficient diet. Toxicol Pathol. 1995;23:583–590. doi: 10.1177/019262339502300504. [DOI] [PubMed] [Google Scholar]
- 64.Uto H, Nakanishi C, Ido A, Hasuike S, Kusumoto K, Abe H, Numata M, Nagata K, Hayashi K, Tsubouchi H. The peroxisome proliferator-activated receptor-gamma agonist, pioglitazone, inhibits fat accumulation and fibrosis in the livers of rats fed a choline-deficient, l-amino acid-defined diet. Hepatol Res. 2005;32:235–242. doi: 10.1016/j.hepres.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 65.Larter CZ. Not all models of fatty liver are created equal: understanding mechanisms of steatosis development is important. J Gastroenterol Hepatol. 2007;22:1353–1354. doi: 10.1111/j.1440-1746.2007.05004.x. [DOI] [PubMed] [Google Scholar]
- 66.Vendemiale G, Grattagliano I, Caraceni P, Caraccio G, Domenicali M, Dall’Agata M, Trevisani F, Guerrieri F, Bernardi M, Altomare E. Mitochondrial oxidative injury and energy metabolism alteration in rat fatty liver: effect of the nutritional status. Hepatology. 2001;33:808–815. doi: 10.1053/jhep.2001.23060. [DOI] [PubMed] [Google Scholar]
- 67.Hensley K, Kotake Y, Sang H, Pye QN, Wallis GL, Kolker LM, Tabatabaie T, Stewart CA, Konishi Y, Nakae D, et al. Dietary choline restriction causes complex I dysfunction and increased H(2)O(2) generation in liver mitochondria. Carcinogenesis. 2000;21:983–989. doi: 10.1093/carcin/21.5.983. [DOI] [PubMed] [Google Scholar]
- 68.Nagarajan P, Mahesh Kumar MJ, Venkatesan R, Majundar SS, Juyal RC. Genetically modified mouse models for the study of nonalcoholic fatty liver disease. World J Gastroenterol. 2012;18:1141–1153. doi: 10.3748/wjg.v18.i11.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yamashita T, Murakami T, Iida M, Kuwajima M, Shima K. Leptin receptor of Zucker fatty rat performs reduced signal transduction. Diabetes. 1997;46:1077–1080. doi: 10.2337/diab.46.6.1077. [DOI] [PubMed] [Google Scholar]
- 70.Ahima RS, Flier JS. Leptin. Annu Rev Physiol. 2000;62:413–437. doi: 10.1146/annurev.physiol.62.1.413. [DOI] [PubMed] [Google Scholar]
- 71.Qamar A, Sheikh SZ, Masud A, Jhandier MN, Inayat IB, Hakim W, Mehal WZ. In vitro and in vivo protection of stellate cells from apoptosis by leptin. Dig Dis Sci. 2006;51:1697–1705. doi: 10.1007/s10620-006-9244-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Godbole V, York DA. Lipogenesis in situ in the genetically obese Zucker fatty rat (fa/fa): role of hyperphagia and hyperinsulinaemia. Diabetologia. 1978;14:191–197. doi: 10.1007/BF00429780. [DOI] [PubMed] [Google Scholar]
- 73.Oana F, Takeda H, Hayakawa K, Matsuzawa A, Akahane S, Isaji M, Akahane M. Physiological difference between obese (fa/fa) Zucker rats and lean Zucker rats concerning adiponectin. Metabolism. 2005;54:995–1001. doi: 10.1016/j.metabol.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 74.Liao W, Angelin B, Rudling M. Lipoprotein metabolism in the fat Zucker rat: reduced basal expression but normal regulation of hepatic low density lipoprotein receptors. Endocrinology. 1997;138:3276–3282. doi: 10.1210/endo.138.8.5337. [DOI] [PubMed] [Google Scholar]
- 75.Witztum JL, Schonfeld G. Lipoproteins in the plasma and hepatic perfusates of the Zucker fatty rat. Diabetes. 1979;28:509–516. doi: 10.2337/diab.28.5.509. [DOI] [PubMed] [Google Scholar]
- 76.Kurtz TW, Morris RC, Pershadsingh HA. The Zucker fatty rat as a genetic model of obesity and hypertension. Hypertension. 1989;13:896–901. doi: 10.1161/01.hyp.13.6.896. [DOI] [PubMed] [Google Scholar]
- 77.Yang SQ, Lin HZ, Lane MD, Clemens M, Diehl AM. Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proc Natl Acad Sci USA. 1997;94:2557–2562. doi: 10.1073/pnas.94.6.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Carmiel-Haggai M, Cederbaum AI, Nieto N. A high-fat diet leads to the progression of non-alcoholic fatty liver disease in obese rats. FASEB J. 2005;19:136–138. doi: 10.1096/fj.04-2291fje. [DOI] [PubMed] [Google Scholar]
- 79.Soltys K, Dikdan G, Koneru B. Oxidative stress in fatty livers of obese Zucker rats: rapid amelioration and improved tolerance to warm ischemia with tocopherol. Hepatology. 2001;34:13–18. doi: 10.1053/jhep.2001.25452. [DOI] [PubMed] [Google Scholar]
- 80.Selzner M, Clavien PA. Failure of regeneration of the steatotic rat liver: disruption at two different levels in the regeneration pathway. Hepatology. 2000;31:35–42. doi: 10.1002/hep.510310108. [DOI] [PubMed] [Google Scholar]
- 81.Kakuma T, Lee Y, Higa M, Wang Zw, Pan W, Shimomura I, Unger RH. Leptin, troglitazone, and the expression of sterol regulatory element binding proteins in liver and pancreatic islets. Proc Natl Acad Sci USA. 2000;97:8536–8541. doi: 10.1073/pnas.97.15.8536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Letexier D, Peroni O, Pinteur C, Beylot M. In vivo expression of carbohydrate responsive element binding protein in lean and obese rats. Diabetes Metab. 2005;31:558–566. doi: 10.1016/s1262-3636(07)70231-8. [DOI] [PubMed] [Google Scholar]
- 83.Fukunishi S, Nishio H, Fukuda A, Takeshita A, Hanafusa T, Higuchi K, Suzuki K. Development of Fibrosis in Nonalcoholic Steatosis through Combination of a Synthetic Diet Rich in Disaccharide and Low-Dose Lipopolysaccharides in the Livers of Zucker (fa/fa) Rats. J Clin Biochem Nutr. 2009;45:322–328. doi: 10.3164/jcbn.09-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yang YY, Tsai TH, Huang YT, Lee TY, Chan CC, Lee KC, Lin HC. Hepatic endothelin-1 and endocannabinoids-dependent effects of hyperleptinemia in nonalcoholic steatohepatitis-cirrhotic rats. Hepatology. 2012;55:1540–1550. doi: 10.1002/hep.25534. [DOI] [PubMed] [Google Scholar]
- 85.Clark JB, Palmer CJ, Shaw WN. The diabetic Zucker fatty rat. Proc Soc Exp Biol Med. 1983;173:68–75. doi: 10.3181/00379727-173-41611. [DOI] [PubMed] [Google Scholar]
- 86.Etgen GJ, Oldham BA. Profiling of Zucker diabetic fatty rats in their progression to the overt diabetic state. Metabolism. 2000;49:684–688. doi: 10.1016/s0026-0495(00)80049-9. [DOI] [PubMed] [Google Scholar]
- 87.Corsetti JP, Sparks JD, Peterson RG, Smith RL, Sparks CE. Effect of dietary fat on the development of non-insulin dependent diabetes mellitus in obese Zucker diabetic fatty male and female rats. Atherosclerosis. 2000;148:231–241. doi: 10.1016/s0021-9150(99)00265-8. [DOI] [PubMed] [Google Scholar]
- 88.Chalasani N, Crabb DW, Cummings OW, Kwo PY, Asghar A, Pandya PK, Considine RV. Does leptin play a role in the pathogenesis of human nonalcoholic steatohepatitis? Am J Gastroenterol. 2003;98:2771–2776. doi: 10.1111/j.1572-0241.2003.08767.x. [DOI] [PubMed] [Google Scholar]
- 89.Takaya K, Ogawa Y, Hiraoka J, Hosoda K, Yamori Y, Nakao K, Koletsky RJ. Nonsense mutation of leptin receptor in the obese spontaneously hypertensive Koletsky rat. Nat Genet. 1996;14:130–131. doi: 10.1038/ng1096-130. [DOI] [PubMed] [Google Scholar]
- 90.Koletsky S. Pathologic findings and laboratory data in a new strain of obese hypertensive rats. Am J Pathol. 1975;80:129–142. [PMC free article] [PubMed] [Google Scholar]
- 91.Elam MB, Wilcox HG, Cagen LM, Deng X, Raghow R, Kumar P, Heimberg M, Russell JC. Increased hepatic VLDL secretion, lipogenesis, and SREBP-1 expression in the corpulent JCR: LA-cp rat. J Lipid Res. 2001;42:2039–2048. [PubMed] [Google Scholar]
- 92.Wexler BC. Hyperlipidemia, hyperglycemia and hypertension in repeatedly bred parents of the obese spontaneously hypertensive rat (obese/SHR) unaccompanied by arteriosclerosis. Atherosclerosis. 1984;51:211–222. doi: 10.1016/0021-9150(84)90169-2. [DOI] [PubMed] [Google Scholar]
- 93.Kitamori K, Naito H, Tamada H, Kobayashi M, Miyazawa D, Yasui Y, Sonoda K, Tsuchikura S, Yasui N, Ikeda K, et al. Development of novel rat model for high-fat and high-cholesterol diet-induced steatohepatitis and severe fibrosis progression in SHRSP5/Dmcr. Environ Health Prev Med. 2012;17:173–182. doi: 10.1007/s12199-011-0235-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Qi NR, Wang J, Zidek V, Landa V, Mlejnek P, Kazdová L, Pravenec M, Kurtz TW. A new transgenic rat model of hepatic steatosis and the metabolic syndrome. Hypertension. 2005;45:1004–1011. doi: 10.1161/01.HYP.0000161995.64192.2b. [DOI] [PubMed] [Google Scholar]
- 95.Eberlé D, Clément K, Meyre D, Sahbatou M, Vaxillaire M, Le Gall A, Ferré P, Basdevant A, Froguel P, Foufelle F. SREBF-1 gene polymorphisms are associated with obesity and type 2 diabetes in French obese and diabetic cohorts. Diabetes. 2004;53:2153–2157. doi: 10.2337/diabetes.53.8.2153. [DOI] [PubMed] [Google Scholar]
- 96.Malínská H, Oliyarnyk O, Hubová M, Zídek V, Landa V, Simáková M, Mlejnek P, Kazdová L, Kurtz TW, Pravenec M. Increased liver oxidative stress and altered PUFA metabolism precede development of non-alcoholic steatohepatitis in SREBP-1a transgenic spontaneously hypertensive rats with genetic predisposition to hepatic steatosis. Mol Cell Biochem. 2010;335:119–125. doi: 10.1007/s11010-009-0248-5. [DOI] [PubMed] [Google Scholar]
- 97.Takiguchi S, Takata Y, Funakoshi A, Miyasaka K, Kataoka K, Fujimura Y, Goto T, Kono A. Disrupted cholecystokinin type-A receptor (CCKAR) gene in OLETF rats. Gene. 1997;197:169–175. doi: 10.1016/s0378-1119(97)00259-x. [DOI] [PubMed] [Google Scholar]
- 98.Jung TS, Kim SK, Shin HJ, Jeon BT, Hahm JR, Roh GS. α-lipoic acid prevents non-alcoholic fatty liver disease in OLETF rats. Liver Int. 2012;32:1565–1573. doi: 10.1111/j.1478-3231.2012.02857.x. [DOI] [PubMed] [Google Scholar]
- 99.Kawano K, Hirashima T, Mori S, Saitoh Y, Kurosumi M, Natori T. Spontaneous long-term hyperglycemic rat with diabetic complications. Otsuka Long-Evans Tokushima Fatty (OLETF) strain. Diabetes. 1992;41:1422–1428. doi: 10.2337/diab.41.11.1422. [DOI] [PubMed] [Google Scholar]
- 100.Song YS, Fang CH, So BI, Park JY, Lee Y, Shin JH, Jun DW, Kim H, Kim KS. Time course of the development of nonalcoholic Fatty liver disease in the Otsuka long-evans Tokushima Fatty rat. Gastroenterol Res Pract. 2013;2013:342648. doi: 10.1155/2013/342648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yeon JE, Choi KM, Baik SH, Kim KO, Lim HJ, Park KH, Kim JY, Park JJ, Kim JS, Bak YT, et al. Reduced expression of peroxisome proliferator-activated receptor-alpha may have an important role in the development of non-alcoholic fatty liver disease. J Gastroenterol Hepatol. 2004;19:799–804. doi: 10.1111/j.1440-1746.2004.03349.x. [DOI] [PubMed] [Google Scholar]
- 102.Uno M, Kurita S, Misu H, Ando H, Ota T, Matsuzawa-Nagata N, Kita Y, Nabemoto S, Akahori H, Zen Y, et al. Tranilast, an antifibrogenic agent, ameliorates a dietary rat model of nonalcoholic steatohepatitis. Hepatology. 2008;48:109–118. doi: 10.1002/hep.22338. [DOI] [PubMed] [Google Scholar]
- 103.Kovár J, Tonar Z, Heczková M, Poledne R. Prague hereditary hypercholesterolemic (PHHC) rat - a model of polygenic hypercholesterolemia. Physiol Res. 2009;58 Suppl 2:S95–S99. doi: 10.33549/physiolres.931916. [DOI] [PubMed] [Google Scholar]