

Summary
The primary cilium is a nexus of cell signaling, and ciliary dysfunction is associated with polycystic kidney disease, retinal degeneration, polydactyly, neural tube defects, and obesity (ciliopathies) [1]. Signaling molecules for cilium-associated pathways are concentrated in the cilium, and this is essential for efficient signaling. Cilia are nucleated from centrioles, and aberrant centriole numbers are seen in many cancers and in some ciliopathies [2]. We tested the effect of supernumerary centrioles on cilium function, and found that cells with extra centrioles often formed more than one cilium, had reduced ciliary concentration of Smoothened in response to Sonic hedgehog stimulation, and reduced Shh pathway transcriptional activation. This ciliary dilution phenotype was also observed with the serotonin receptor Htr6, fibrocystin PKHD1, and Arl13b. The presence of extra centrioles and cilia disrupted epithelial organization in 3-D spheroid culture. Cells mutant for the tuberous sclerosis gene Tsc2 also had extra cilia and diluted ciliary protein. In most cells extra cilia were clustered and shared the same ciliary pocket, suggesting that the ciliary pocket is the rate-limiting structure for trafficking of ciliary proteins. Thus, extra centrioles and cilia disrupt signaling, and may contribute to disease phenotypes.
Results and Discussion
The primary cilium grows from the older of the two centrioles in quiescent cells. Centriole number is maintained by a duplication and segregation mechanism linked to the cell cycle [3], and centriole amplification is a common characteristic of many cancers [4]. Centriole amplification has also been observed in renal tissues from mouse models and human patients bearing mutations in a subset of ciliopathy genes, including cells from PKD1 deletion mice and human autosomal dominant polycystic kidney disease patients [5]; fibroblasts and mesenchymal cells from PKD2 deletion mice [6]; cells from tuberous sclerosis (TSC1 deletion) mice [7]; and cells from Mks1 or Mks3 mouse and human patients with Meckel syndrome [8]. It is unclear what phenotypes are conferred upon cells having extra centrosomes that might lead to cilia-related diseases. Here we consider the possibility that extra centrosomes might result in more than one primary cilium, and thus affect normal ciliary signaling.
Supernumerary centrioles cause super-ciliated cells defective in Shh signaling
To investigate the effects of multiple cilia, we manipulated the number of cilia independent of genome ploidy. Overexpression of the kinase Plk4 induces the formation of multiple centrioles in a diversity of cell types, thus we tested whether Plk4-induced centriole amplification would result in the formation of extra cilia. Plk4 was overexpressed by two methods: transient transfection of a Plk4-expressing plasmid, and transient induction of a tet-inducible Plk4 stable cell line. Since Plk4 is an unstable protein, both methods resulted in a transient increase in Plk4 levels, and both gave similar results, causing the formation of extra centrioles as described [9].
To test whether the extra centrioles created by Plk4 overexpression can make cilia, cells overexpressing Plk4 were held in S-phase for 16 h by thymidine treatment, then released from S-phase for two subsequent rounds of cell division to allow the newly-assembled centrioles to mature and potentially become competent for cilium formation [10]. We found that in many cells with extra centrioles, some of those centrioles formed a cilium, resulting in super-ciliated cells containing two to six primary cilia per cell (Figure 1A–B and Supplementary Table 1). As expected, the fraction of ciliated cells with more than one cilium increased over time due to maturation of centrioles by passage through the cell cycle (Supplementary Table 1). Quantification of cilium length in cells with one, two or three primary cilia showed that, on average, the length of each cilium in these cells was similar (~ 4 μm, Figure 1C).
Figure 1. Super-ciliated cells are defective in Shh signaling.
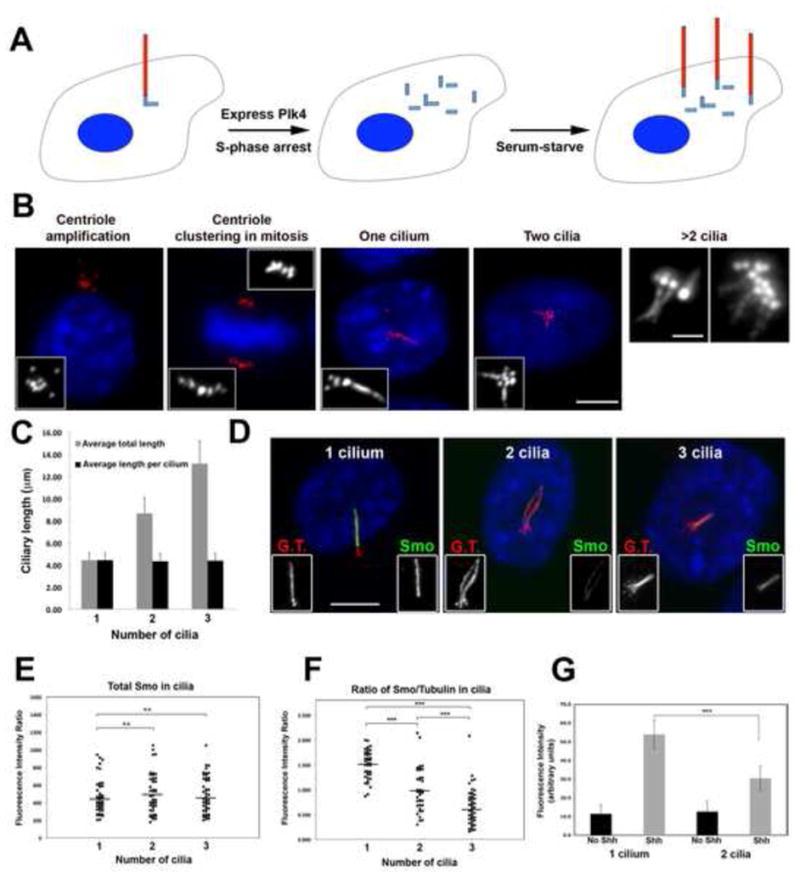
(A) Schematic depicting generation of super-ciliated cells. (B) Examples of NIH3T3 cells with amplified centrioles and cilia, stained for glutamylated tubulin (centrioles and cilia; red) and DNA (blue). Scale bars, 10 μm and 2 μm (right panel). (C) Average lengths of cilia in mono- and super-ciliated cells. (D) Endogenous Smo localization (green) in mono- and super-ciliated cells. Cilia were co-stained for glutamylated tubulin (G.T., red) and DNA (blue). Scale bar, 10 μm. (E) Distribution of total Smo signal intensity in cilia of mono-, bi- and tri-ciliated cells. (F) Ratio of Smo/tubulin levels in cilia of mono-, bi- and tri-ciliated cells. (G) Graph of GFP signal intensity in mono- and bi-ciliated cells before and after Shh treatment. For all graphs, N = 50 for each sample (from 2 independent experiments; *** indicates P < 0.0001).
We considered that an increased number of primary cilia might cause changes in signaling pathways that rely on cilium function. We focused first on the transmembrane protein Smoothened (Smo), which translocates into the cilium in response to Sonic Hedgehog (Shh) ligand [11]; this translocation is essential in activating downstream signaling in mammalian cells [12]. Plk4 was expressed in mouse embryonic fibroblasts to generate super-ciliated cells, which were treated with Shh, and stained for Smo (Figure 1D; [12]). We used quantitative single-cell fluorescence to measure the total ciliary amounts of Smo in cells with one, two, or three primary cilia (Figure 1D). Surprisingly, the total amount of ciliary Smo was similar in mono-, bi- and tri-ciliated cells (Figure 1E). Since the total cilium length increased linearly with cilium number (Figure 1C), this was most likely due to reduced concentration of Smo in each cilium in superciliated cells. To confirm this, the amount of Smo per unit length of cilium was determined by measuring the ratio of Smo signal intensity with respect to ciliary axoneme tubulin staining. The amount of Smo per unit length was reduced in bi-ciliated cells compared to mono-ciliated cells, and further reduced in tri-ciliated cells (Figure 1F), indicating that the Smo protein is diluted in the cilia of super-ciliated cells.
We tested whether the dilution of ciliary Smo in super-ciliated cells perturbed the response of the pathway. Hedgehog response was assayed using an NIH3T3 reporter cell line that expresses GFP under the control of Gli response elements [13], [14]. NIH3T3::Gli-GFP cells were transfected with Plk4 to generate super-ciliated cells and treated with Shh, inducing expression of Gli-GFP. Quantification of GFP levels in mono- versus bi-ciliated cells revealed a significant reduction in the latter (Figure 1G), demonstrating that dilution of Smo in cilia of super-ciliated cells results in defective activation of the Shh pathway.
Generality of the ciliary dilution phenotype in super-ciliated cells
We next addressed the generality and nature of the dilution phenotype in super-ciliated cells by examining the ciliary concentrations of additional proteins that localize to cilia. We found that the serotonin 6 (Htr6) receptor, the fibrocystin protein, and the GTPase Arl13b all exhibited a dilution phenotype similar to that of Smo in super-ciliated cells (Supplemental Data and Supplementary Figures S1 and S2). The ciliary dilution phenotype was observed even under conditions in which the total concentration of the transported protein was not limiting (Supplemental Data and Supplementary Figures S1). Since super-ciliated cells assembled cilia of similar length to those in mono-ciliated cells, we reasoned that components of the ciliary machinery might not display the ciliary dilution phenotype. Consistent with this hypothesis, the ciliary concentration of IFT88, a component of intraflagellar transport, was equal in mono- and bi-ciliated cells (Supplementary Figure S2C). Lastly, we found that the ciliary dilution phenotype was dependent on number of cilia rather than number of centrioles (Supplementary Figure S2B), and was independent of nuclear ploidy (Supplementary Figure S2D), suggesting that the mechanism determining ciliary protein levels assesses the number of cilia per cell, rather than cilia per unit genome.
Disease-related defects associated with presence of extra cilia
We sought to determine the functional consequences of having extra cilia in a cell/tissue system that relies on cilium function. Ciliary signaling is essential in organizing the architecture of epithelial cells; components of both the canonical and non-canonical (planar cell polarity) Wnt signaling pathways localize to cilia, and depend on proper cilia function to establish cell polarity [15]. Growth of epithelial cells in a 3D gel matrix allows cells to organize into polarized, spheroid structures that resemble in vivo epithelium architecture [16]. Spheroid systems have been used to study the activities of oncogenes with regard to their ability to disrupt epithelial architecture early during tumor formation [16], and have recently been used to assay cilia dysfunction related to mutations implicated in ciliopathies [17], [18]. IMCD-3 cells grown in Matrigel for four days formed spheroids with a prominent lumen, apically located cilia projecting into the lumen, and well defined apical and basolateral junctions (Figure 2A). In contrast, 84% of super-ciliated IMCD-3 cells made by transient expression of Plk4 failed to develop spheroid structures, instead forming clusters of cells with irregular or non-existent lumens, disorganized apical and basolateral junctions, and randomly oriented cilia (Figure 2A and 2B). Note that it is not possible to distinguish between the contribution of the extra centrioles versus the extra cilia in this assay, since perturbing ciliary structure and function (even in cells with the normal complement of centrioles) leads to defective spheroid formation [17], [18]. Thus, the presence of extra centrosomes and/or cilia disrupts epithelial organization in this in vitro tissue model.
Figure 2. Disease-related defects associated with presence of extra cilia.
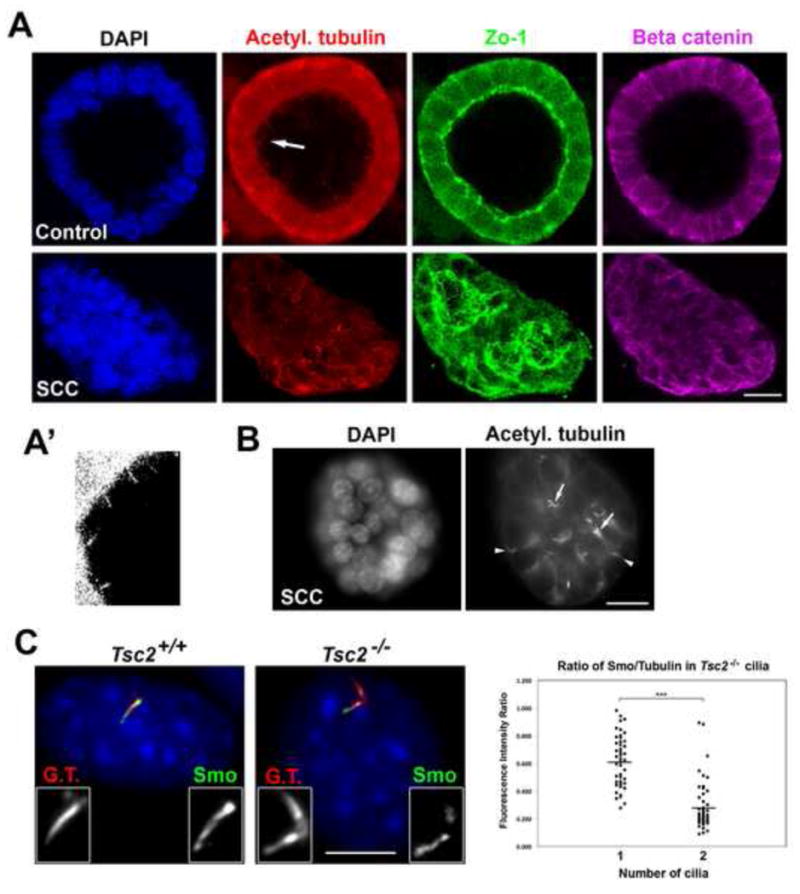
(A–B) Immunofluorescence images of mono-ciliated (Control) and super-ciliated (SCC) IMCD-3 cells grown in 3D culture. Most mono-ciliated cells formed spheroids with a prominent lumen (93%, N = 400), whereas most super-ciliated cells failed to develop spheroids (84%, N = 400); P < 0.0001. Arrow in (A) denotes the region that is magnified 3-fold and displayed in (A′). Arrows in (B) mark cells in an SCC cluster with multiple cilia; arrowheads mark mis-oriented cilia. Scale bars, 10 μm. (C) Immunofluorescence images of endogenous Smo (green) in mono-ciliated Tsc2+/+ and bi-ciliated Tsc2−/− MEFs, co-stained with glutamylated tubulin (G.T., red) and DNA (blue). Scale bar, 10 μm. Graph (right panel) shows ratio of Smo/glutamylated tubulin levels. N = 50 for each sample (from 2 independent experiments; P < 0.0001).
We next examined the effect of extra cilia caused by a naturally-occurring disease mutation. Tuberous sclerosis is a multi-systemic disease in which patients develop benign tumors in the brain, retina, kidney, heart and skin [19]. Causative mutations occur in two tumor suppressor genes (TSC1 and TSC2), the gene products of which function as a complex to inhibit mTOR [20]. Interestingly, mutations in TSC1 and TSC2 have recently been linked to ciliary dysfunction. MEFs isolated from Tsc1−/− and Tsc2−/− mice show enhanced ciliary formation, and many Tsc2−/− MEFs have extra centrosomes and cilia [21]. To test whether Tsc2−/− super-ciliated MEFs showed the ciliary dilution phenotype Tsc2−/− and control Tsc2+/+ MEFs were treated with Shh and the amount of Smo per unit cilium length was determined. Ciliary Smo concentration was reduced in bi-ciliated Tsc2−/− cells compared to mono-ciliated cells of either Tsc2−/− or Tsc2+/+ genotype (Figure 2C). This phenotype might be relevant to tuberous sclerosis disease, given the similarity between the disease phenotype at the tissue level and the epithelial disorganization we observed in spheroid cultures.
Finally, to address potential mechanisms underlying the ciliary dilution effect, we examined the ultrastructure of the cilia in super-ciliated cells. In the experiments above we noted that in most super-ciliated cells (>95%) the centrioles were clustered, and that the primary cilia were situated next to each other. This raised the possibility that the multiple cilia in super-ciliated cells shared the same membrane compartment at the surface of the cell. The proximal portion of the primary cilium is part of a specialized membrane compartment, the ciliary pocket [22], an invagination of the plasma membrane in which the primary cilium is rooted. The ciliary, or flagellar, pocket is important for trafficking of ciliary components in trypanosomatids and is the unique site of endocytosis and exocytosis [23], and may be performing a similar role in mammalian cells [24]. We tested whether the multiple cilia in our super-ciliated cells might occupy the same ciliary pocket, limiting the amount of ciliary signaling proteins available to each cilium. To test this hypothesis, we used transmission electron microscopy to examine the ciliary pocket in super-ciliated RPE-1 cells. Longitudinal sections through the ciliary region of control mono-ciliated cells showed the typical ciliary pocket structure (Figure 3A). Remarkably, serial sections of super-ciliated cells revealed that in each cell in which multiple cilia could be identified (n = 6 cells), those cilia occupied the same ciliary pocket (Figure 3A). To test whether the ciliary dilution phenotype might be due to this clustering of cilia, we compared the concentration of ciliary Arl13b in cells where the cilia were clustered to that in the small fraction of cells (<5%) in which the cilia were unclustered, and presumably residing in separate ciliary pockets (Figure 3B). We found that the concentration of Arl13b in unclustered cilia was similar to that in cells with one cilium (Figure 3C), supporting the hypothesis that the ciliary pocket is the rate-limiting structure for trafficking of ciliary proteins.
Figure 3. Architecture of the ciliary pocket in super-ciliated cells.
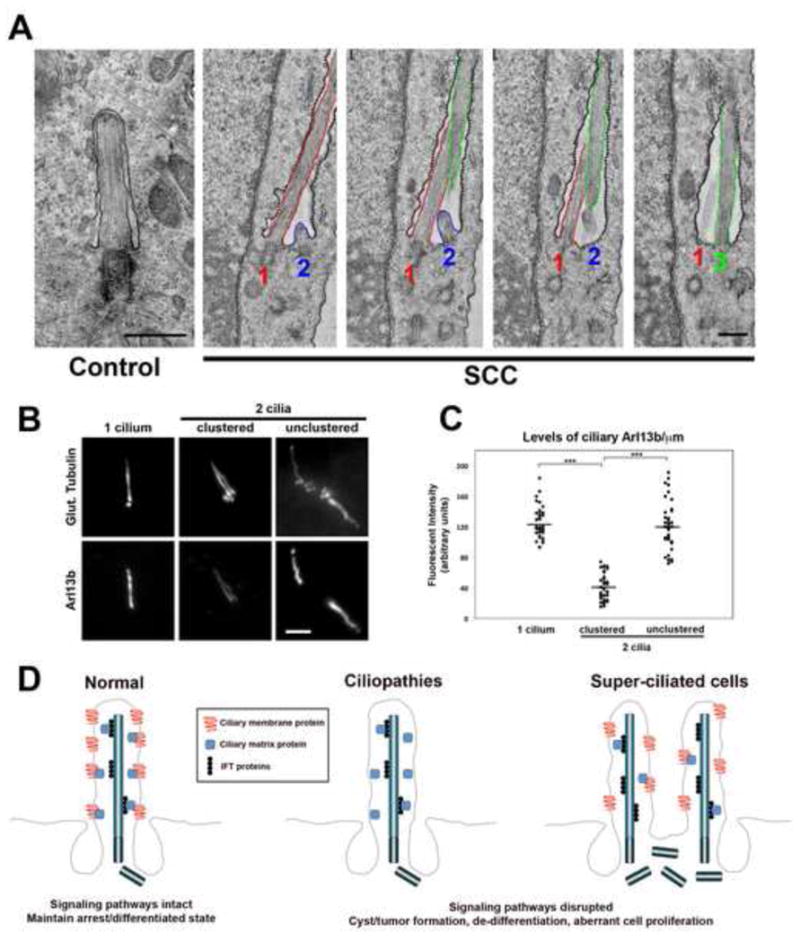
(A) Transmission electron microscopy of wild-type (Control) and super-ciliated (SCC) RPE-1 cells. Images show a set of four serial sections through the ciliary pocket of a super-ciliated cell. In this cell there are three centrioles with associated cilia (numbered 1–3), as well as other centrioles not associated with cilia, below the pocket. The membrane of the ciliary pocket is indicated with black stippling, and the membranes of the three cilia are colored red, blue and green, as indicated. Cilia 1 and 3 are oriented mostly in the plane of the sections, whereas cilium 2 is oriented obliquely to the section plane and is thus only partially visible. Scale bars, 0.5 μm. (B) Immunofluorescence images of endogenous Arl13b in RPE-1 cells, containing either 1, 2-culstered or 2-unclustered cilia. Cells were co-stained with glutamylated tubulin. Scale bar, 2 μm. (C) Level of endogenous Arl13b per unit length, in mono-ciliated, bi-ciliated clustered and unclustered cells. N = 40 for each sample (from 2 independent experiments; P < 0.0001).(D) Model depicting ciliary dysfunction caused by mutation of a ciliopathy protein, resulting in loss of a cilium signaling component, or by dilution of such components in super-ciliated cells where the cilia are residing in a single ciliary pocket.
Conclusion
We show that extra centrioles often result in extra cilia, and that presence of extra cilia results in reduced ciliary concentration of signaling molecules in cilia. This dilution phenotype is likely a direct effect of extra cilia because the same effect was observed in super-ciliated cells generated by transient Plk4 expression or by a cytokinesis block, as well as in Tsc2−/− null MEFs. That the presence of extra cilia can lead to altered cilium structure and function is supported by results demonstrating a reduction in canonical Wnt pathway activation in bi-ciliated cells [25]. How might the presence of extra cilia result in a signaling defect? In the case of the Shh pathway, we propose that decreased pathway activation results from decreased ciliary concentration of Smo, as well as other components of that pathway, several of which are known to be localized to the cilium [12]. This would result in reduced interaction between these components in the ciliary compartment, and compromised signaling (Figure 3D). This contrasts with the mechanism of ciliary dysfunction caused by mutations in canonical ciliopathy genes, in which the molecular defect appears to be the absence of particular ciliary proteins (Figure 3D). Although the mechanisms of ciliary dysfunction are different in these cases, we posit that ciliary dilution due to supernumerary centrosomes and cilia might lead to the same phenotypic output.
Centrosome aberrations are associated with renal cystic disease [2], but a causative role for centrosome defects in cystogenesis has not been established. We speculate that the ciliary signaling defects in super-ciliated cells might play a role in the initiation of cyst formation. Interestingly, cells in kidney tissue from a patient bearing a mutation in MKS3 were found to have extra centrioles and cilia in vivo [8], and kidney cysts in patients with mutations in PKD1 have super-ciliated cells in vivo (our unpublished results). Thus, defects caused by extra cilia might be an important aspect of a set of cilia-related diseases.
How might the concentration of ciliary proteins be affected by extra cilia? Localization to the cilium is controlled at several levels, including a barrier to diffusion of cytoplasmic and membrane proteins into the cilium [26], [27], specific membrane-targeting pathways for ciliary membrane proteins [28] and a cilium-specific transport mechanism (intraflagellar transport [29]). We propose that these pathways act at the level of the ciliary pocket, the unique membrane structure at the base of the cilium. We find that more than one cilium can occupy the ciliary pocket in super-ciliated cells, suggesting that the ciliary pocket is the rate-limiting structure for trafficking of ciliary proteins, and that the ciliary dilution phenotype derives from the distribution of trafficked proteins among the cilia sharing that pocket. Further work on the factors involved in centriole clustering in interphase and on the assembly of the ciliary pocket will help to establish the molecular mechanism of the ciliary dilution phenotype.
Experimental Procedures
Generating super-ciliated cells
Extra centrioles were generated by transfecting cells with a construct expressing Plk4 using Lipofectamine 2000 (Invitrogen), according to the manufacturer’s protocol. Cells were arrested in S-phase by treatment with 2 mM thymidine for 16 h to induce centriole amplification, then incubated with fresh medium and allowed to grow for 2 additional days to allow centriole maturation. Alternatively, tetracycline-inducible RPE-1::Tet-Plk4 were incubated with 1 μg/mL doxycycline and 2 mM thymidine for 16 h to induce expression of Plk4 while maintaining arrest in S-phase. Cells were then grown in fresh medium (without doxycycline) for 2 additional days to allow centriole maturation. To induce ciliogenesis, cells were incubated with low-serum (0.5%) medium for 24 h. Activation of the Shh pathway was achieved by treating cells (MEFs or NIH3T3) with Shh-conditioned medium for 16 h, as previously described.
Immunofluorescence microscopy
Samples were fixed in either 100% methanol at −20°C for 10 min, or with 4% paraformaldehyde in PBS at room temperature for 10 min, depending on antigen. Following fixation, cells were rinsed twice with PBS and briefly extracted with 0.1% Triton X-100 in PBS and blocked for 1 h at room temperature with 3% BSA (Sigma-Aldrich) in PBS. Samples were incubated with primary antibodies for either 1 h at room temperature or 4°C overnight. Primary antibodies used in this study were: Rabbit anti-Smo (1:2000; gift from Rajat Rohatgi, Stanford, CA), mouse anti-acetylated tubulin (clone 6-11b-1; 1:5000; Sigma), mouse anti-glutamylated tubulin (GT335; 1:2000; gift from C. Janke, Centre de Recherches de Biochimie Macromoléculaire, Montpellier, France), rabbit anti-GFP (generated in our lab), rabbit anti-IFT88 (1:1000; gift from Bradley Yoder, University of Alabama at Birmingham, AL), rat anti-Zo-1 (1:1000; American Research Products), and rabbit anti-β-catenin (1:1000; Santa Cruz Biotechnology). Alexa dye–conjugated secondary antibodies (Invitrogen) were used at a dilution of 1:500 at room temperature for 1 h. Samples were mounted using Mowiol mounting medium containing N-propyl gallate (Sigma-Aldrich). Images were captured using Openlab 4.0.4 (Improvision) controlling an Axiovert 200M microscope (Carl Zeiss MicroImaging, Inc.). Images of spheroids were captured on a Zeiss LSM 510 Confocal Laser Scanning microscope using a 25X oil-immersion objective and a scan zoom of 4. A series of digital optical sections consisting of 12 sections in the Z-axis (total of 25 μm) was captured, and three-dimensional image reconstructions were produced.
Quantification of signal intensity and statistical analysis
The ciliary fluorescence intensity was calculated using ImageJ software (http://rsbweb.nih.gov/ij/). Briefly, a region encompassing each cilium was selected, and the total fluorescence intensity for a particular protein was determined for each cilium. Subsequently, the total fluorescence intensity of glutamylated tubulin in each cilium was obtained using the same parameters. In both cases, a background subtraction was performed by quantifying the fluorescence intensity of a region of equal dimensions in the area neighboring the cilium. The ratio of protein/tubulin was determined and graphs were generated using Xcel software (Microsoft). For statistical analysis, unpaired two-tailed Student’s t-test was performed using Excel. N value for each experiment is noted in the respective figure legend.
Supplementary Material
Highghlights.
Supernumerary centrosomes nucleate cilia, resulting in super-ciliated cells (SCC)
Ciliary proteins are reduced in concentration in SCC, compromising signaling
Presence of extra cilia disrupts epithelial organization in 3-D spheroid culture
Extra cilia may contribute to disease phenotypes by disrupting signaling.
Acknowledgments
We thank R. Rohatgi for anti-Smo antibody, C. Janke for anti-glutamylated tubulin, B. Yoder for anti-IFT88, M. Nachury for IMCD-3::Htr6-GFP and IMCD-3:: CTSPKHD1-GFP cells, B. Tsou for RPE-1::Tet-Plk4 cells, E. Henske for Tsc2+/+ and Tsc2−/− MEFs, and J. Chen for NIH3T3:Gli-GFP reporter cells. We also thank John Perrino (Stanford Cell Sciences Imaging Facility) for help with electron microscopy. This work was supported by National Institutes of Health grant GM52022 to T. Stearns.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Waters AM, Beales PL. Ciliopathies: an expanding disease spectrum. Pediatr Nephrol. 2011;26:1039–1056. doi: 10.1007/s00467-010-1731-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee K, Battini L, Gusella GL. Cilium, centrosome and cell cycle regulation in polycystic kidney disease. Biochim Biophys Acta. 2011;1812:1263–1271. doi: 10.1016/j.bbadis.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hatch E, Stearns T. The life cycle of centrioles. Cold Spring Harb Symp Quant Biol. 2010;75:425–431. doi: 10.1101/sqb.2010.75.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Godinho SA, Kwon M, Pellman D. Centrosomes and cancer: how cancer cells divide with too many centrosomes. Cancer Metastasis Rev. 2009;28:85–98. doi: 10.1007/s10555-008-9163-6. [DOI] [PubMed] [Google Scholar]
- 5.Battini L, Macip S, Fedorova E, Dikman S, Somlo S, Montagna C, Gusella GL. Loss of polycystin-1 causes centrosome amplification and genomic instability. Hum Mol Genet. 2008;17:2819–2833. doi: 10.1093/hmg/ddn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burtey S, Riera M, Ribe E, Pennenkamp P, Rance R, Luciani J, Dworniczak B, Mattei MG, Fontes M. Centrosome overduplication and mitotic instability in PKD2 transgenic lines. Cell Biol Int. 2008;32:1193–1198. doi: 10.1016/j.cellbi.2008.07.021. [DOI] [PubMed] [Google Scholar]
- 7.Astrinidis A, Senapedis W, Henske EP. Hamartin, the tuberous sclerosis complex 1 gene product, interacts with polo-like kinase 1 in a phosphorylation-dependent manner. Hum Mol Genet. 2006;15:287–297. doi: 10.1093/hmg/ddi444. [DOI] [PubMed] [Google Scholar]
- 8.Tammachote R, Hommerding CJ, Sinders RM, Miller CA, Czarnecki PG, Leightner AC, Salisbury JL, Ward CJ, Torres VE, Gattone VH, 2nd, et al. Ciliary and centrosomal defects associated with mutation and depletion of the Meckel syndrome genes MKS1 and MKS3. Hum Mol Genet. 2009;18:3311–3323. doi: 10.1093/hmg/ddp272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Habedanck R, Stierhof YD, Wilkinson CJ, Nigg EA. The Polo kinase Plk4 functions in centriole duplication. Nat Cell Biol. 2005;7:1140–1146. doi: 10.1038/ncb1320. [DOI] [PubMed] [Google Scholar]
- 10.Hoyer-Fender S. Centriole maturation and transformation to basal body. Semin Cell Dev Biol. 2010;21:142–147. doi: 10.1016/j.semcdb.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 11.Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437:1018–1021. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- 12.Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317:372–376. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- 13.Cupido T, Rack PG, Firestone AJ, Hyman JM, Han K, Sinha S, Ocasio CA, Chen JK. The imidazopyridine derivative JK184 reveals dual roles for microtubules in Hedgehog signaling. Angew Chem Int Ed Engl. 2009;48:2321–2324. doi: 10.1002/anie.200805666. [DOI] [PubMed] [Google Scholar]
- 14.Hyman JM, Firestone AJ, Heine VM, Zhao Y, Ocasio CA, Han K, Sun M, Rack PG, Sinha S, Wu JJ, et al. Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proc Natl Acad Sci U S A. 2009;106:14132–14137. doi: 10.1073/pnas.0907134106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wallingford JB, Mitchell B. Strange as it may seem: the many links between Wnt signaling, planar cell polarity, and cilia. Genes Dev. 2011;25:201–213. doi: 10.1101/gad.2008011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 17.Sang L, Miller JJ, Corbit KC, Giles RH, Brauer MJ, Otto EA, Baye LM, Wen X, Scales SJ, Kwong M, et al. Mapping the NPHP-JBTS-MKS Protein Network Reveals Ciliopathy Disease Genes and Pathways. Cell. 2011;145:513–528. doi: 10.1016/j.cell.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Otto EA, Hurd TW, Airik R, Chaki M, Zhou W, Stoetzel C, Patil SB, Levy S, Ghosh AK, Murga-Zamalloa CA, et al. Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nat Genet. 2010;42:840–850. doi: 10.1038/ng.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Franz DN, Bissler JJ, McCormack FX. Tuberous sclerosis complex: neurological, renal and pulmonary manifestations. Neuropediatrics. 2010;41:199–208. doi: 10.1055/s-0030-1269906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Orlova KA, Crino PB. The tuberous sclerosis complex. Ann N Y Acad Sci. 2010;1184:87–105. doi: 10.1111/j.1749-6632.2009.05117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hartman TR, Liu D, Zilfou JT, Robb V, Morrison T, Watnick T, Henske EP. The tuberous sclerosis proteins regulate formation of the primary cilium via a rapamycin-insensitive and polycystin 1-independent pathway. Hum Mol Genet. 2009;18:151–163. doi: 10.1093/hmg/ddn325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghossoub R, Molla-Herman A, Bastin P, Benmerah A. The ciliary pocket: a once-forgotten membrane domain at the base of cilia. Biol Cell. 2011;103:131–144. doi: 10.1042/BC20100128. [DOI] [PubMed] [Google Scholar]
- 23.Field MC, Carrington M. The trypanosome flagellar pocket. Nat Rev Microbiol. 2009;7:775–786. doi: 10.1038/nrmicro2221. [DOI] [PubMed] [Google Scholar]
- 24.Molla-Herman A, Ghossoub R, Blisnick T, Meunier A, Serres C, Silbermann F, Emmerson C, Romeo K, Bourdoncle P, Schmitt A, et al. The ciliary pocket: an endocytic membrane domain at the base of primary and motile cilia. J Cell Sci. 2010;123:1785–1795. doi: 10.1242/jcs.059519. [DOI] [PubMed] [Google Scholar]
- 25.Lancaster MA, Schroth J, Gleeson JG. Subcellular spatial regulation of canonical Wnt signalling at the primary cilium. Nat Cell Biol. 2011;13:702–709. doi: 10.1038/ncb2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pazour GJ, Bloodgood RA. Targeting proteins to the ciliary membrane. Curr Top Dev Biol. 2008;85:115–149. doi: 10.1016/S0070-2153(08)00805-3. [DOI] [PubMed] [Google Scholar]
- 27.Hu Q, Milenkovic L, Jin H, Scott MP, Nachury MV, Spiliotis ET, Nelson WJ. A septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution. Science. 2010;329:436–439. doi: 10.1126/science.1191054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nachury MV, Seeley ES, Jin H. Trafficking to the ciliary membrane: how to get across the periciliary diffusion barrier? Annu Rev Cell Dev Biol. 2010;26:59–87. doi: 10.1146/annurev.cellbio.042308.113337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosenbaum JL, Witman GB. Intraflagellar transport. Nat Rev Mol Cell Biol. 2002;3:813–825. doi: 10.1038/nrm952. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.