

Abstract
Age-related diastolic dysfunction is a major factor in the epidemic of heart failure. In patients hospitalized with heart failure, diastolic heart failure is now as common as systolic heart failure. We now have many successful treatments for HFrEF, while specific treatment options for HFpEF patients remain elusive. The lack of treatments for HFpEF reflects our very incomplete understanding of this constellation of diseases. There are many pathophysiological factors in HFpEF, but aging appears to play an important role. Here we propose that aging of the myocardium is itself a specific pathophysiological process. New insights into the aging heart, including hormonal controls and specific molecular pathways such as microRNAs, are pointing to myocardial aging as a potentially reversible process. While the overall process of aging remains mysterious, understanding the molecular pathways of myocardial aging has never been more important. Unraveling these pathways could lead to new therapies for the enormous and growing problem of HFpEF.
Keywords: Aging, diastolic dysfunction, diastolic heart failure, cardiac hypertrophy
I. THE SIGNIFICANCE OF HFpEF AND AGING
For thousands of years, if people were lucky enough to survive childhood illnesses and reach adulthood, they had a good chance of living into their 50’s and 60’s.1 However, routine survival into the 80’s and 90’s is a truly new event in human life. As the world’s population ages, the prevalence of age-related diseases is growing dramatically. In the United States, for example, one in every eight Americans is now above age 65, and by 2030 the proportion of Americans over 65 will reach 19%.2 Heart failure affects ~1% of individuals over 50 and increases progressively with age.3 Thus, with the ongoing steep rise in the world population of elderly individuals4, age-related heart failure is certain to become an increasingly prevalent health condition and a leading cause of mortality in the elderly.5,6 Although heart failure traditionally is associated with reduced contractile function of the myocardium, dilation of the left ventricle, and reduced ejection fraction, there is a growing epidemic of heart failure accompanied by preserved ejection fraction (HFpEF).7 This form of heart failure usually has a normal sized left ventricle, often but not always with hypertrophy and is characterized by a global impairment of depressed cardiovascular function.8 Approximately 50% of patients hospitalized for heart failure have HFpEF, and the mortality risk for these patients is equivalent to those with heart failure accompanied by reduced ejection fraction (~50% die within 3 years9–11).
Recent studies have defined aging as one factor in the HFpEF epidemic.12, 13 Echocardiographic studies often reveal normal or near normal ejection fraction in elderly patients with heart failure, with abnormal diastolic relaxation and left ventricular filling.14–16 As observed by Borlaug et al, LV stiffness increases progressively with age despite reduction in arterial load.16 Abnormal diastolic filling is associated with female gender, obesity, age >65 years, hypertension, renal disease and diabetes, suggesting that distinct risk factors and pathological mechanisms underlie these conditions.6, 12
An overview of HFpEF with particular emphasis on the clinical aspects will be provided in the accompanying review by Kass and colleagues.17 Here we specifically review molecular insights into age-related contributors to changes in myocardial function, an area of paramount importance, which may also affect globally cardiovascular reserve. Understanding the molecular events in myocardial aging will be essential to developing treatments for the types of heart failure we will see in this century.
II. DISSECTING THE COMPONENTS OF MYOCARDIAL REMODELING IN AGE-RELATED HFpEF
Aging is an evolutionarily conserved yet poorly understood process that leads to deterioration of many physiological functions over the lifespan of an organism.18 Studies have suggested that aging may contribute independently to deterioration of diastolic function.19 Normal cardiac aging is characterized by structural and functional changes. Increased cardiomyocyte size, increased apoptosis with decreased myocyte number, increased collagen deposition and also functional changes at the cellular level may all contribute to abnormal diastolic function with normal aging20. The consequences of these changes are an increase in left ventricular diastolic stiffness with aging.19, 21 In both the Baltimore Longitudinal Study on Aging and the Framingham Heart Study, left ventricular hypertrophy increased with age, while systolic function was maintained.22 However, while cardiomyocyte hypertrophy occurs in aging human hearts, co-morbid diseases such as hypertension also are much more common in the elderly, complicating our understanding of the effects of aging.23 Left ventricular diastolic filling rate deteriorates with progressive age24 and this decline is observed as early as age 20 years in humans.25 By 80 years of age, the reduction in early diastolic filling is as profound as 50%.25 Adult mammalian heart replenishes its cardiomyocyte pool both during physiological aging and in response to injury 26–28; cardiomyocyte refreshment, which occurs at a low rate (~1%/year) in youth, appears to slow even further with advancing age.26 Aging does not appear to affect the low rate of cardiomyocyte apoptosis in normal human hearts.29 In the myocardial extracellular matrix, aging leads to increased deposition of extracellular matrix components, principally collagen, with increased fibril diameter and collagen cross-linking, an increase in the ratio of type I to type III collagen, decreased elastin content, and an increase in fibronectin.13, 30–37 These changes may contribute to exercise intolerance with advancing age, though skeletal muscle function declines with age, as well.15, 20 .
Models of cardiac aging
Cardiac aging in rodents recapitulates many changes observed in humans, with age-dependent increases in left ventricular mass index as well as impaired left ventricular filling.22, 38 Studies have documented an age-dependent increase in cardiomyocyte size in wild-type mice,39–41 and age-dependent cardiomyocyte hypertrophy also occurs in rats.42 Studies of rodents suggest multiple potential mechanisms of cardiomyopathy in aging. For example, cardiac angiotensin II levels are higher in senescent animals.43 Adrenergic and cholinergic signaling may also play a role, as mice with disruption of the type 5 adenyl cyclase are protected from age-related cardiac hypertrophy and fibrosis,40 and mice with reduced function of the choline transporter exhibit age-dependent decreases in fractional shortening and increases in ventricular size and fibrosis.44 Mitochondrial dysfunction likely also contributes, as the aging myocardium exhibits increased mitochondrial protein oxidation and increased mitochondrial DNA mutations.22 In addition to cardiomyocyte hypertrophy, aging mice develop progressive myocardial fibrosis, associated with molecular signatures of immune cell and inflammatory dysregulation.45 Furthermore, cardiomyocyte stability and intercellular mechanical and functional coupling may be perturbed in aged animals due to disruption of junctional adhesion proteins.46
III. AGE RELATED DETERMINANTS OF DIASTOLIC DYSFUNCTION
The development of heart failure symptoms in patients with diastolic dysfunction has profound implications, as it carries a 60% 5-year mortality prognosis.5 What tips patients into the symptomatic phase is unknown, as patients are known to have impaired diastolic filling before symptoms emerge.5, 13, 14 Aging may play a fundamental role in modifying both the passive stiffness of the myocardium as well as the active diastolic relaxation properties of the myocytes.
Myocardial Interstitial Fibrosis
Interstitial fibrosis is a hallmark of cardiac aging and a major contributor to myocardial stiffness.47, 48 The myocardial extracellular matrix (ECM) is not just a passive scaffold for tissue architecture but also a dynamic participant in cellular signaling.20, 49 Studies have shown more than doubling of extracellular matrix content in the myocardium of senescent rats.41, 47, 48 The collagenous weave is not only thicker but increased cross-linking among the collagen filaments confers greater rigidity to the myocardium.20 Fibroblasts are the principal cells secreting extracellular matrix components, including collagen, fibronectin, and laminin. In aging as well as under a wide range of hypertrophic stimuli, fibroblasts undergo activation and phenotypic transformation to myofibroblasts, which are characterized by expression of the contractile protein alpha-SMA.49–51 Myofibroblasts control ECM composition by regulating the secretion and activity of proteolytic enzymes, including members of the family of matrix metalloproteinases (MMPs) and their inhibitors plasminogen activator inhibitor 1 (PAI-1) and tissue inhibitors of matrix metalloproteinases (TIMPs).51 Fibroblasts are under the active control of multiple signaling hormones and cytokines, 49, 51 and the pro-fibrotic neurohormonal cascades relevant to cardiac aging are discussed below.
TGF-β Signaling Pathway
TGFβ is one of the most extensively studied fibroblast-activating growth factors. It mediates myofibroblast transformation as well as transcriptional suppression of the MMPs, thus tipping the proteolytic balance towards net matrix accumulation.48, 52–55 Brooks et al studied heterozygous TGFβ(+/−) deficient mice which at 24 months of age exhibited decreased myocardial fibrosis with a total of 4% interstitial collagen as opposed to 10% observed in wild type mice.55 The loss of one TGFβ allele also contributed to improved myocardial compliance and performance. Blocking TGFβ activity through administration of a neutralizing antibody attenuates diastolic dysfunction in a pressure overload model of cardiac hypertrophy.54, 55 TGFβ expression is also upregulated by angiotensin II signaling through the AT1 receptor.56, 57 Angiotensin II increases TGFβ1 mRNA and protein expression levels in both cardiomyocytes and fibroblasts.56–62 Administration of ACE inhibitors or AT1 receptor blockers ameliorates cardiac hypertrophy and decreases TGFβ1 levels, implicating TGFβ as a mediator of ATII effects.61, 62 Furthermore, Schultz et al have demonstrated that TGF-β1 −/− mice (bred on immunocompromised Rag1 −/− background to protect from lethality of complete TGFβ1 loss) are protected from the hypertrophic effects of Angiotensin II stimulation.63 Angiotensin II also activates cardiac fibroblast function through signaling via endothelin-1 (ET-1), IL-6 and periostin.48, 56, 64
Downstream TGFβ signaling pathways have been extensively studied. TGFβ binds to the constitutively active type II receptor (TβRII) at the surface of the target cell and subsequently recruits and phosphorylates a type I receptor (TβRI), also known as ALK5.59 Downstream, Smad2 and Smad3 are activated through phosphorylation by ALK5, and form a complex with Smad4 that translocates to the nucleus and affects gene expression.48, 59 Non-Smad signaling pathways mediated by TGFβ include Erk, JNK, p38 MAPK.59, 60 Although these downstream signaling pathways have not been explored extensively in the context of cardiac aging, their role in pro-fibrotic signaling has been shown in a number of animal models.48, 52
Oxidative Balance
Another important contributor to activation of pro-fibrotic signaling pathways in the aging myocardium is the presence of a positive oxidative balance.41, 65 Increased levels of reactive oxygen species (ROS) in the senescent myocardium can cause direct TGFβ activation resulting in upregulation of its downstream effector connective tissue growth factor (CTGF).59, 65 Indeed, NADPH oxidase 4 dependent generation of hydrogen peroxide is required for TGF-β1-induced conversion of cardiac fibroblasts to myofibroblasts.65 Furthermore, scavenging of reactive oxygen species through mitochondrially-targeted catalase expression ameliorates the age-related myocardial fibrosis.41 Other TGFβ activators include thrombospondin-1 as well as enzymes such as MMP-2, MMP-9 and plasmin which through their proteolytic activity also regulate the kinetics of TGFβ release from the ECM.59, 66, 67
Matrix Metalloproteinases
Many of the matrix metalloproteinases family of enzymes as well as their inhibitors have been explored in the context of cardiac aging. Epidemiological studies have shown correlations between increasing age and regulation of these enzymes.68, 69 Aging may produce a shift in the balance between matrix metalloproteinases and tissue inhibitors of matrix metalloproteinases that ultimately translates in increased matrix accumulation. In a study of healthy subjects with no prior diagnoses of cardiovascular disease, Bonnema et al showed that age correlates with an increase in MMP-2, MMP-7, TIMP-1, -2 and -4 levels, as well as with a decrease in MMP-9 plasma levels.68 Analysis of Framingham subjects has also shown an age dependent increase in TIMP-1 plasma levels that was related to major cardiovascular risk factors and to indices of LV hypertrophy.70 Direct genetic evidence from animal models for the specific role of these enzymes in aging-related ventricular remodeling are lacking. Chiao et al conducted a genetic study in which MMP-9 null mice of all age groups showed no variation in ventricular filling, unlike wild-type senescent mice where impaired diastolic filling occurs with aging.71 This functional preservation in MMP-9 deficient mice correlates with attenuation of the fibrotic remodeling observed with age.
Titin and Myocyte Stiffness
Titin has been identified as a major molecular determinant of myocyte stiffness.72–79 As the largest molecular component of the myocyte structure spanning from Z disk to the M band of the sarcomere, it has been shown to modulate cardiomyocyte passive stiffness through its I-band region, which has spring-like properties that regulate early diastolic recoil and late diastolic resistance to stretch.78, 79 Titin modulates its stiffness through post-translational modifications, including phosphorylation by protein kinases A, C-alpha and G, which decrease its compliance.78–84
In HFpEF patients, titin is largely hypophosphorylated.80 A proposed mechanism by the Paulus group postulates that the increased oxidative stress in diastolic dysfunction depletes the nitric oxide (NO) reserve, thus lowering PKG activity.73, 74 Furthermore, the abundant ROS generate disulfide bridges that shorten titin’s N2B segment, increasing its stiffness.83 Relative hypophosphorylation of the stiff N2B titin isoform ultimately increases resting tension of cardiomyocytes that contributes to the high diastolic LV stiffness observed in failing human hearts and restoring phosphorylation of the titin N2B segment in elderly hypertensive dogs lowered diastolic LV stiffness.76 In addition, multiple transgenic mouse models have been created showing that the absence of either the N2B or PEVK segment, or shortening the tandem immunoglobulin segment, is sufficient to increase myocardial stiffness and cause impaired diastolic filling.77, 79, 83 Thus, titin is emerging as an important mediator of diastolic dysfunction, but the effects of aging on titin are incompletely described.
Calcium Signaling and Active Diastolic Relaxation
In addition to the above discussed changes in passive myocardial stiffness, impairment in active diastolic relaxation has been well documented in diastolic dysfunction.85–88 Cardiac relaxation occurs when Ca2+ reuptake into the sarcoplasmic reticulum occurs through sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA) or extruded extracellularly through the sarcolemal Na+/Ca2+ exchanger (NCX). Regulation of SERCA channel activity occurs through interaction with the regulatory protein phospholamban (PLN). In its unphosphorylated state, PLN interacts with SERCA2a and decreases its affinity for Ca2+. Phosphorylation of PLN by PKA and Ca2+/calmodulin-dependent protein kinase (CaMK) disrupts this inhibitory interaction and augments the SERCA pump activity.89
Myocardial preparations from experimental models of impaired diastolic function have revealed a decreased rate of intracellular Ca2+ decay resulting in prolonged action potential and impaired diastolic filling.87, 88 Studies have been equivocal correlating expression levels of calcium handling proteins and aging-related HFpEF. Brian et al and others have shown that human SERCA2a levels correlate inversely with age, while Babušíková et al and others have shown no changes in SERCA2a protein or mRNA expression levels in senescent murine myocardium compared to young control animals.90–95 When SERCA levels are reported together with PLN levels, the evidence has suggested decreased SERCA2a/PLN ratio with age.92, 95, 96 Restoring the SERCA/PLN balance through gene transfer of the SERCA2a protein in senescent rat myocardium improves age-related diastolic dysfunction.97, 98
Conflicting results have been reported regarding protein and mRNA expression levels of RyR, L-type Ca2+ channels, and NCX.98–102 However, more recent evidence suggests the role of post-translational modification of the Ca2+-handling proteins in modifying their activity level. Specifically, in multiple separate studies it has been shown that the increased oxidative stress observed in senescent myocardium leads to oxidative damage of the SERCA pump, thus decreasing its Ca2+ sequestering activity and prolonging diastolic relaxation.91–93 Phosphorylation is another mechanism of post-translational signaling control in Ca2+ homeostasis.91 The aged myocardium has lower responsiveness to beta-adrenergic stimulation, and this has been shown to translate into reduced PKA and CaMK mediated phosphorylation of PLN and RyR, thus decreasing Ca2+ handling rate.102
Aging-Related Diastolic Dysfunction and Mitochondria
Both animal and human studies have shown that with aging, mitochondrial DNA accumulates mutations with reported increases as much as 16-fold, and the components of the mitochondrial apparatus function at a decreased level.103–105 Vermulst et al showed that mitochondrial DNA deletions play much greater role than point mutations in premature cardiac aging.104, 105 In a genetic murine model, mice with homozygous mutation of mitochondrial polymerase γ (Polgm/m) exhibit accelerated aging including cardiac senescence features such as cardiac hypertrophy and dysfunction.103 The loss of mitochondrial polymerase proofreading capacity leads to deficiently functioning components of the cellular energetics machinery that contribute directly to increased oxidative stress. The specific role of mitochondria in myocyte oxidative stress is supported by the finding that overexpression of catalase targeted to mitochondria (mCAT) but not targeted to peroxisomes (pCAT) protects against cardiac hypertrophy, fibrosis, and failure.106, 107 Dai et al show that mCAT mice exhibit significantly attenuated cardiac aging as demonstrated by increased left ventricular myocardial index, myocardial performance index and E/A ratio on echocardiography.41, 107 On a histopathological level, mice with overexpressed mitochondrial catalase exhibit decreased myocardial hypertrophy and interstitial fibrosis, as well as about 20% lifespan extension.
Dysfunctional mitochondria are eliminated by mitophagy, a specialized form of macroautophagy 108, 109 This process is particularly important in cells like cardiomyocytes where autophagy is the major determinant in protein and organelle turnover.110 Macroautophagy efficiency progressively declines in cardiomyocytes during aging.111, 112 As a result, dysfunctional mitochondria that are more prone to release ROS accumulate within aging cardiomyocytes contributing to increase oxidative stress.113
The renin-angiotensin aldosterone system is a driver of mitochondrial dysfunction.114, 115 Dai et al have shown elevated myocardial levels of angiotensin II in aging hearts.114 Angiotensin II signals through binding to the angiotensin receptor-1 (ATR1), a Gαq coupled receptor, which has been shown to stimulate the NOX4 isoform of NADPH oxidase on the mitochondrial membrane.114, 115 Reactive oxygen species generated by NADPH oxidase sets off a ROS-mediated-ROS generation propagating a vicious cycle of oxidative stress damaging mitochondrial components further exacerbating the above cycle.116, 117 Thus, activation of angiotensin II signaling may promote myocardial aging.
IV. MOLECULAR PATHWAYS IN AGE-RELATED DIASTOLIC DYSFUNCTION
The NO-cGMP-PKG signaling axis/Inflammation
Aging is associated with a systemic proinflammatory state, so called ‘inflamm-aging’, that may lead to a functional decline in multiple organs even in absence of a specific disease118 and can activate signaling cascades leading to myocardial structural and functional remodeling.119–126 Indeed, multiple cross-sectional studies show that increasing age is associated with elevated circulating levels of inflammatory markers, including TNFα, IL-6, IL-18, MCP-1, soluble ST2, and pentraxin 3 (Figure 1).119, 121, 124 An emerging theory for the pathogenesis of HFpEF proposes that a systemic proinflammatory state produced by comorbidities, including aging, causes coronary microvascular endothelial inflammation. This inflammation ultimately results in increased interstitial fibrosis and cardiomyocytes stiffness that contributes to high diastolic left ventricular stiffness and heart failure development.119 Moreover, the presence of circulating pro-inflammatory cytokines is predictive of the development of heart failure symptoms.124
Figure 1. Aging itself leads to molecular changes that contribute to diastolic impairment.
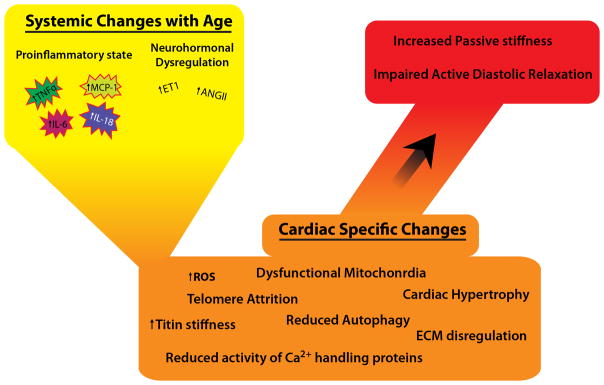
Age is associated with systemic changes and myocardial molecular dysfunction that translate into structural changes believed to contribute to HFpEF.
Inflamed coronary microvascular endothelial cells, as evidenced by the upregulated expression of endothelial adhesion molecules including vascular cell adhesion molecule (VCAM-1) and E-selectin in myocardial samples from HFpEF patients,119, 127, 128 produce reactive oxygen species.129–135 Increases in reactive oxygen species can cause reduction in bioavailability of nitric oxide (NO) for adjacent cardiomyocytes. Reduced NO causes decreases in cGMP levels, which in turn decreases protein kinase G (PKG) activity in cardiomyocytes. The inflamed endothelium enables binding and translocation of inflammatory cells, further propagating an inflammatory state within myocardium.119 Low PKG activity translates into cardiomyocyte hypertrophy and hypophosphorylation of titin, thereby increasing stiffness. Stiff cardiomyocytes and increased collagen deposition by myofibroblasts cause diastolic LV impairment.119
Neurohormonal pro-fibrotic signaling through angiotensin II and endothelin is also facilitated by the presence of endothelial inflammation.47, 119, 131 Counterbalancing the effect of the oxidative stress on cGMP levels with long-term use of sildenafil has shown benefit for diastolic LV function.131 However, in the largest trial studying PDE5 inhibitors, the RELAX study (Evaluating the Effectiveness of Sildenafil at Improving Health Outcomes and Exercise Ability in People With Diastolic Heart Failure), sildenafil showed no effect on exercise tolerance in the enrolled subjects with HFpEF.136
V. METABOLISM AND CARDIAC AGING
Reducing calorie intake in many species leads to increased longevity and slows the aging effects in key organ systems.137, 138 In fact, Doppler studies in humans practicing caloric restriction reveal no changes in systolic function but improved diastolic function.139 In rats, caloric restriction improves calcium handling and diastolic function.140 It was long thought that lower calorie intake leads to diminished metabolism-associated “wear and tear” by decreasing generation of harmful metabolic byproducts, such as reactive oxygen species. However, further studies have shown the activation of “longevity pathways” that control genes protective against apoptosis and maladaptive remodeling.141, 142
For example, the sirtuins (silent information regulators, SIRTs) as well as the IGF-1/Akt pathways have been identified as key nutrient sensors involved in cardiac and organismal longevity. Conserved from lower organisms, such as yeast, flies and worms, up to humans, the 7 members of the sirtuins class are diversely positioned in the nucleus and cytosol responding to the cellular energy balance through their NAD+ cofactor.142–149 The characterization of the role of the first member of this class, SIrt1, in cardiac aging has yielded equivocal results. Alcendor et al have shown that low (2.5-fold) to moderate (7.5-fold) cardiac-specific overexpression of Sirt1 in a transgenic mouse model attenuated age-dependent cardiac remodeling and dysfunction, as well as served a cardioprotective role in the presence of oxidative stress (such as paraquat administration) through FOXO-dependent signaling.145 In contrast, high expression levels (12.5-fold) of Sirt1 increased baseline myocyte oxidative stress, apoptosis and cellular hypertrophy that reflected in a functional deterioration of the heart.145 In addition, Sirt1 haploinsufficiency ameliorates the extent of cardiac hypertrophy in the presence of a pressure overload stimulus.149 Sundaresan et al demonstrated that the pro-hypertrophic properties of Sirt1 are mediated through crosstalk with the Akt pathway, which participates in cell survival, protein synthesis and metabolism. Sirt1 deacetylates Akt, thus freeing it to bind and activate PIP3.150, 151 Sirt1 also deacetylates PDK1 allowing Akt phosphorylation by this phosphokinase, thus augmenting Akt activity by as much as ~1000-fold. Sirt1, therefore, plays a dual function in cardiac aging.150–152
Another member of the sirtuin family, Sirt3, is a NAD+ dependent histone deacetylase primarily localized to the mitochondrial membrane that has been shown to regulate pathways in energy metabolism, apoptosis and reactive oxygen species synthesis.153–157 Sirt3 knockout mice do not exhibit grossly different phenotype but microscopic examination reveals premature aging including mitochondrial swelling, cellular hypertrophy and fibrosis as early as 13 months of age.153–155 Sirt3 −/− mice also demonstrate greater susceptibility to stress such as transverse aortic banding, to which they respond with an exaggerated hypertrophy and fibrosis.153–155 On the contrary, Sirt3 overexpression is cardioprotective in the setting of hypertrophy induced by stimulation with angiotensin II or isoproterenol.154 Hafner et al proposed a mechanism of Sirt3 to involve prevention of mitochondrial dysfunction by way of decreasing the activity of the mitochondrial permeability transition pore (mPTP) through deacetylation of its regulatory component cyclophilin D.154 Furthermore, Sirt3 activates FOXO3a-mediated expression of the anti-oxidant enzymes superoxide dismutase and catalase.158
Recent studies have shed light on the crosstalk between Sirt6 and the IGF-1/Akt pathway and its cardiac effects.150, 157 Sirt6 −/− mice exhibit the most progerian phenotype of all sirtuin knockout animal models.150 These transgenic mice are severely hypoglycemic and die within 1 month of age. Sundaresan et al showed that myocardial samples from humans with heart failure as well as from mouse models of cardiac hypertrophy induced by transverse aortic constriction, angiotensin II or isoproterenol showed markedly reduced Sirt6 levels.157 Both single and double Sirt6 knock-out mice develop significant cardiac hypertrophy and dysfunction.157 The myocardium of these animals exhibits fibrosis, myocyte hypertrophy, apoptotic and fetal gene expression. Sirt6 overexpression, on the other hand, protects remodeling and dysfunction in the presence of hypertrophic stimuli.157 Sirt6 is, thus, a major regulator on the crossroads of two nutrient-sensing pathways.
Sirt7 is another sirtuin family member and similar to Sirt6, its downregulation through double gene knock-out in a mouse model leads to myocardial hypertrophy and decreased lifespan.159 However, much less extensive evidence exists for Sirt 7 and the rest of the sirtuin enzymes on their roles on cardiac aging. The Sirt proteins through their complex interactions with multiple signaling pathways involved in nutrient responses and mitochondrial function are emerging as important therapeutic targets in the context of cardiac aging.
VI. NOVEL DISCOVERIES IN CARDIAC AGING
SMP30
Senescence marker protein 30 (SMP30), a 34-kDa protein, ubiquitously expressed in human organs and preserved across species, has recently been identified as another player in cardioprotection in the setting of aging-associated myocardial changes.160, 161 Misaka et al demonstrated that SMP30 is a marker of cardiac senescence as levels of this protein decrease in the murine myocardium with aging by as much as 40%.162, 163 SMP30 has been shown to play a role in multi-organ senescence, including brain, lungs and kidneys.162 Misaka’s laboratory created an Ang II-induced model of cardiac hypertrophy and showed that SMP30 knock-out mice exhibit greater myocardial remodeling when exposed to angiotensin II.162, 163 They also demonstrated that SMP30 deficiency leads to increased myocardial oxidative stress concomitant with increased NADPH oxidase activity. SMP30, therefore, may tip the redox balance in the aging heart.
GDF11
The quest for a humoral factor that can rejuvenate aging phenotypes has been long-standing. Recently, growth differentiation factor 11 (GDF11), a member of the activin/TGF-β superfamily, has been identified as a factor that carries the potential to reverse aging-related cardiac remodeling.164 Heterochronic parabiosis, an experimental procedure whereby two animals of different ages are joined together, identified GDF11 as a candidate hormone that controls the aging myocardial phenotype. Further elucidation of how GDF11 fits in the multiple signaling cascades identified to play a role in cardiac aging is pending.
miRNA Signaling
MicroRNAs (miRNAs) are endogenous small noncoding RNAs, 20–23-nucleotides in length, which have emerged as important post-translational regulators of numerous cardiovascular processes, from myocardial infarction to cardiac aging.165–167 They are characterized by target promiscuity, as a single miRNA is known to target the expression of up to 100 of genes by hybridization with complementary sequences on mRNAs and thus triggering their degradation or translational inhibition.165 Expressional survey of the 17–92 miRNA cluster in the aging heart has implicated miRNA 18 and miRNA 19 as potential regulators of aging cardiomyopathy through their targeting of pro-fibrotic pathways involving TGF-beta and TSP-1 signaling.168 Another study conducted in vitro has also shown that miRNA-22 is upregulated with age in mouse fibroblast isolates.169 The authors identified mimecan (osteoglycan, described together with TGF-beta1 and beta2) as its target under inverse regulation relationship.169
More recently, exciting studies have revealed that miR-34a has been implicated in cardiac aging.170–173 Predominantly expressed by cardiomyocytes, miR-34a is upregulated in aging mouse hearts as well as in human heart biopsies (~2-fold).170 The role of miR-34a in cardiac aging has been linked to regulation of apoptosis. Using microRNA target prediction tools, Boon et al identified Ppp1r10 (PNUTS) as a downstream target which is downregulated by miR-34a (luciferase assays have shown direct targeting of mi-34a to the 3′UTR of PNUTS).170 PNUTS has known antiapoptotic effects - it reduces telomere attrition in vitro and DNA damage through the DNA damage response pathways involving CHK2 activation in the presence of TRF2.170–173 miRNAs are opening new potential therapeutic frontiers in cardiac aging but additional studies delineating their biology in aging are needed.
Telomere attrition
Telomere shortening may contribute to functional decline in different tissues, including myocardial tissue.174 Telomere shortening has been recently identified as a biomarker of lifetime stress, as early as in childhood175 and this stress-related telomere shortening could be responsible for accelerated biological aging.176 Telomere dysfunction-induced cellular phenotypes characterized by proliferative arrest, apoptosis, and senescence may be less relevant considering that genesis of cardiomyocytes occurs at a low rate by the division of pre-existing cardiomyocytes during normal aging.177 Interestingly, however, telomeres may regulate functional changes in cardiomyocytes. p53 activation induced by telomere dysfunction may directly affect function of mitochondria and metabolism in cardiomyocytes by repressing peroxisome proliferator-activated receptor gamma coactivator (PGC)-1α and PGC-1β thus contributing to the development of an aging dysfunctional phenotype.178
VII. FUTURE DIRECTIONS AND THERAPEUTIC IMPLICATIONS
In the twenty-first century we are seeing our rapidly aging population afflicted with a cardiovascular syndrome that we are ill prepared to face therapeutically, as we simply don’t understand the disease. It is common practice to use HFpEF as a term covering a syndrome that is likely a set of diseases of diverse pathophysiologies. Careful dissection of the various pathophysiological factors at play in this clinical syndrome is important in order to design effective therapeutics (Figure 2).
Figure 2. New molecular pathways in aging biology could lead to new treatments of age-related diastolic heart failure.

Existing therapies for systolic heart failure have been unsuccessful at treating HFpEF. Novel therapeutic pathways including microRNAs, metabolic factors and age-dependent circulating hormones offer the new opportunities for developing successful treatments.
Systemic inflammation could be a common pathway toward diastolic dysfunction. However, recent discoveries of new molecular pathways in aging suggest that we are at the very beginning of understanding the specific role that aging plays in the myocardium. As these new pathways are explored, it raises the exciting possibility that some effects of myocardial aging are potentially reversible.
Supplementary Material
Acknowledgments
SOURCES OF FUNDING:
This work was supported in part by grants from NIH (R01AG040019, R01HL117986).
Nonstandard Abbreviations and Acronyms
- MMPs
matrix metalloproteinases
- TIMPs
tissue inhibitors of matrix metalloproteinases
- CTGF
connective tissue growth factor
Footnotes
In April 2014, the average time from submission to first decision for all original research papers submitted to Circulation Research was 14.38 days.
DISCLOSURES:
None
References
- 1.Kaplan H, Hill K, Lancaster J, Hurtado AM. A theory of human life history evolution: Diet, intelligence, and longevity. Evolutionary Anthropology: Issues, News, and Reviews. 2000;9:156–185. [Google Scholar]
- 2.Aging Ao. Aging statistics.
- 3.Mosterd A, Hoes AW. Clinical epidemiology of heart failure. Heart. 2007;93:1137–1146. doi: 10.1136/hrt.2003.025270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.U.S. Census Bureau USIP, by Age S, Race and Hispanic Origin.
- 5.Alagiakrishnan K, Banach M, Jones LG, Datta S, Ahmed A, Aronow WS. Update on diastolic heart failure or heart failure with preserved ejection fraction in the older adults. Ann Med. 2013;45:37–50. doi: 10.3109/07853890.2012.660493. [DOI] [PubMed] [Google Scholar]
- 6.Shioi T, Inuzuka Y. Aging as a substrate of heart failure. J Cardiol. 2012;60:423–428. doi: 10.1016/j.jjcc.2012.07.015. [DOI] [PubMed] [Google Scholar]
- 7.Desai A, Fang JC. Heart failure with preserved ejection fraction: Hypertension, diabetes, obesity/sleep apnea, and hypertrophic and infiltrative cardiomyopathy. Heart Fail Clin. 2008;4:87–97. doi: 10.1016/j.hfc.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 8.Borlaug BA, Olson TP, Lam CS, Flood KS, Lerman A, Johnson BD, Redfield MM. Global cardiovascular reserve dysfunction in heart failure with preserved ejection fraction. Journal of the American College of Cardiology. 2010;56:845–854. doi: 10.1016/j.jacc.2010.03.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ouzounian M, Lee DS, Liu PP. Diastolic heart failure: Mechanisms and controversies. Nat Clin Pract Cardiovasc Med. 2008;5:375–386. doi: 10.1038/ncpcardio1245. [DOI] [PubMed] [Google Scholar]
- 10.Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med. 2006;355:251–259. doi: 10.1056/NEJMoa052256. [DOI] [PubMed] [Google Scholar]
- 11.Bhatia RS, Tu JV, Lee DS, Austin PC, Fang J, Haouzi A, Gong Y, Liu PP. Outcome of heart failure with preserved ejection fraction in a population-based study. N Engl J Med. 2006;355:260–269. doi: 10.1056/NEJMoa051530. [DOI] [PubMed] [Google Scholar]
- 12.Gottdiener JS, Bartz T, DeFilippi C, Kop W, Kitzman D, Barasch E, Seliger S, Lloyd-Jones D. Echocardiographic and biomarker phenotype of heart failure with preserved ejection fraction (hfpef) in older individuals in comparison to hypertension without heart failure (htn), elderly with risk factors, and healthy aging. Importance of myocyte injury, fibrosis, lv hypertrophy, and diastolic load. Journal of the American College of Cardiology. 2012;59:E852–E852. [Google Scholar]
- 13.Borlaug BA, Paulus WJ. Heart failure with preserved ejection fraction: Pathophysiology, diagnosis, and treatment. Eur Heart J. 2011;32:670–679. doi: 10.1093/eurheartj/ehq426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benjamin EJ, Levy D, Anderson KM, Wolf PA, Plehn JF, Evans JC, Comai K, Fuller DL, Sutton MS. Determinants of doppler indexes of left ventricular diastolic function in normal subjects (the framingham heart study) Am J Cardiol. 1992;70:508–515. doi: 10.1016/0002-9149(92)91199-e. [DOI] [PubMed] [Google Scholar]
- 15.Swinne CJ, Shapiro EP, Jamart J, Fleg JL. Age-associated changes in left ventricular outflow tract geometry in normal subjects. Am J Cardiol. 1996;78:1070–1073. doi: 10.1016/s0002-9149(96)00542-5. [DOI] [PubMed] [Google Scholar]
- 16.Borlaug BA, Redfield MM, Melenovsky V, Kane GC, Karon BL, Jacobsen SJ, Rodeheffer RJ. Longitudinal changes in left ventricular stiffness: A community-based study. Circ Heart Fail. 2013;6:944–952. doi: 10.1161/CIRCHEARTFAILURE.113.000383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sharma K, Kass D. Heart failure with preserved ejection fraction. Circulation research. 2014 doi: 10.1161/CIRCRESAHA.115.302922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ho AD, Wagner W, Mahlknecht U. Stem cells and ageing. The potential of stem cells to overcome age-related deteriorations of the body in regenerative medicine. EMBO Rep. 2005;6(Spec No):S35–38. doi: 10.1038/sj.embor.7400436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Redfield MM, Jacobsen SJ, Borlaug BA, Rodeheffer RJ, Kass DA. Age- and gender-related ventricular-vascular stiffening: A community-based study. Circulation. 2005;112:2254–2262. doi: 10.1161/CIRCULATIONAHA.105.541078. [DOI] [PubMed] [Google Scholar]
- 20.Lakatta EG, Levy D. Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises: Part ii: The aging heart in health: Links to heart disease. Circulation. 2003;107:346–354. doi: 10.1161/01.cir.0000048893.62841.f7. [DOI] [PubMed] [Google Scholar]
- 21.Chen CH, Nakayama M, Nevo E, Fetics BJ, Maughan WL, Kass DA. Coupled systolic-ventricular and vascular stiffening with age: Implications for pressure regulation and cardiac reserve in the elderly. Journal of the American College of Cardiology. 1998;32:1221–1227. doi: 10.1016/s0735-1097(98)00374-x. [DOI] [PubMed] [Google Scholar]
- 22.Dai DF, Rabinovitch PS. Cardiac aging in mice and humans: The role of mitochondrial oxidative stress. Trends Cardiovasc Med. 2009;19:213–220. doi: 10.1016/j.tcm.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pugh KG, Wei JY. Clinical implications of physiological changes in the aging heart. Drugs Aging. 2001;18:263–276. doi: 10.2165/00002512-200118040-00004. [DOI] [PubMed] [Google Scholar]
- 24.Bursi F, Weston SA, Redfield MM, Jacobsen SJ, Pakhomov S, Nkomo VT, Meverden RA, Roger VL. Systolic and diastolic heart failure in the community. JAMA. 2006;296:2209–2216. doi: 10.1001/jama.296.18.2209. [DOI] [PubMed] [Google Scholar]
- 25.Dannenberg AL, Levy D, Garrison RJ. Impact of age on echocardiographic left ventricular mass in a healthy population (the framingham study) Am J Cardiol. 1989;64:1066–1068. doi: 10.1016/0002-9149(89)90816-3. [DOI] [PubMed] [Google Scholar]
- 26.Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnable-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S, Frisen J. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hsieh PC, Segers VF, Davis ME, MacGillivray C, Gannon J, Molkentin JD, Robbins J, Lee RT. Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nat Med. 2007;13:970–974. doi: 10.1038/nm1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McPherron AC, Huynh TV, Lee SJ. Redundancy of myostatin and growth/differentiation factor 11 function. BMC Dev Biol. 2009;9:24. doi: 10.1186/1471-213X-9-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mallat Z, Fornes P, Costagliola R, Esposito B, Belmin J, Lecomte D, Tedgui A. Age and gender effects on cardiomyocyte apoptosis in the normal human heart. J Gerontol A Biol Sci Med Sci. 2001;56:M719–723. doi: 10.1093/gerona/56.11.m719. [DOI] [PubMed] [Google Scholar]
- 30.Olivetti G, Melissari M, Capasso JM, Anversa P. Cardiomyopathy of the aging human heart. Myocyte loss and reactive cellular hypertrophy. Circulation research. 1991;68:1560–1568. doi: 10.1161/01.res.68.6.1560. [DOI] [PubMed] [Google Scholar]
- 31.Martos R, Baugh J, Ledwidge M, O’Loughlin C, Conlon C, Patle A, Donnelly SC, McDonald K. Diastolic heart failure: Evidence of increased myocardial collagen turnover linked to diastolic dysfunction. Circulation. 2007;115:888–895. doi: 10.1161/CIRCULATIONAHA.106.638569. [DOI] [PubMed] [Google Scholar]
- 32.Kitzman DW. Diastolic heart failure in the elderly. Heart Fail Rev. 2002;7:17–27. doi: 10.1023/a:1013745705318. [DOI] [PubMed] [Google Scholar]
- 33.Olivetti G, Giordano G, Corradi D, Melissari M, Lagrasta C, Gambert SR, Anversa P. Gender differences and aging: Effects on the human heart. Journal of the American College of Cardiology. 1995;26:1068–1079. doi: 10.1016/0735-1097(95)00282-8. [DOI] [PubMed] [Google Scholar]
- 34.Olivetti G, Melissari M, Balbi T, Quaini F, Sonnenblick EH, Anversa P. Myocyte nuclear and possible cellular hyperplasia contribute to ventricular remodeling in the hypertrophic senescent heart in humans. Journal of the American College of Cardiology. 1994;24:140–149. doi: 10.1016/0735-1097(94)90554-1. [DOI] [PubMed] [Google Scholar]
- 35.Gazoti Debessa CR, Mesiano Maifrino LB, Rodrigues de Souza R. Age related changes of the collagen network of the human heart. Mech Ageing Dev. 2001;122:1049–1058. doi: 10.1016/s0047-6374(01)00238-x. [DOI] [PubMed] [Google Scholar]
- 36.Eghbali M, Eghbali M, Robinson TF, Seifter S, Blumenfeld OO. Collagen accumulation in heart ventricles as a function of growth and aging. Cardiovasc Res. 1989;23:723–729. doi: 10.1093/cvr/23.8.723. [DOI] [PubMed] [Google Scholar]
- 37.Barasch E, Gottdiener JS, Aurigemma G, Kitzman DW, Han J, Kop WJ, Tracy RP. The relationship between serum markers of collagen turnover and cardiovascular outcome in the elderly: The cardiovascular health study. Circ Heart Fail. 2011;4:733–739. doi: 10.1161/CIRCHEARTFAILURE.111.962027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lakatta EG. Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises: Part iii: Cellular and molecular clues to heart and arterial aging. Circulation. 2003;107:490–497. doi: 10.1161/01.cir.0000048894.99865.02. [DOI] [PubMed] [Google Scholar]
- 39.Treuting PM, Linford NJ, Knoblaugh SE, Emond MJ, Morton JF, Martin GM, Rabinovitch PS, Ladiges WC. Reduction of age-associated pathology in old mice by overexpression of catalase in mitochondria. J Gerontol A Biol Sci Med Sci. 2008;63:813–822. doi: 10.1093/gerona/63.8.813. [DOI] [PubMed] [Google Scholar]
- 40.Yan L, Vatner DE, O’Connor JP, Ivessa A, Ge H, Chen W, Hirotani S, Ishikawa Y, Sadoshima J, Vatner SF. Type 5 adenylyl cyclase disruption increases longevity and protects against stress. Cell. 2007;130:247–258. doi: 10.1016/j.cell.2007.05.038. [DOI] [PubMed] [Google Scholar]
- 41.Dai DF, Santana LF, Vermulst M, Tomazela DM, Emond MJ, MacCoss MJ, Gollahon K, Martin GM, Loeb LA, Ladiges WC, Rabinovitch PS. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation. 2009;119:2789–2797. doi: 10.1161/CIRCULATIONAHA.108.822403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang M, Zhang J, Walker SJ, Dworakowski R, Lakatta EG, Shah AM. Involvement of nadph oxidase in age-associated cardiac remodeling. Journal of molecular and cellular cardiology. 2010;48:765–772. doi: 10.1016/j.yjmcc.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Groban L, Pailes NA, Bennett CD, Carter CS, Chappell MC, Kitzman DW, Sonntag WE. Growth hormone replacement attenuates diastolic dysfunction and cardiac angiotensin ii expression in senescent rats. J Gerontol A Biol Sci Med Sci. 2006;61:28–35. doi: 10.1093/gerona/61.1.28. [DOI] [PubMed] [Google Scholar]
- 44.English BA, Appalsamy M, Diedrich A, Ruggiero AM, Lund D, Wright J, Keller NR, Louderback KM, Robertson D, Blakely RD. Tachycardia, reduced vagal capacity, and age-dependent ventricular dysfunction arising from diminished expression of the presynaptic choline transporter. Am J Physiol Heart Circ Physiol. 2010;299:H799–810. doi: 10.1152/ajpheart.00170.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cieslik KA, Taffet GE, Carlson S, Hermosillo J, Trial J, Entman ML. Immune-inflammatory dysregulation modulates the incidence of progressive fibrosis and diastolic stiffness in the aging heart. Journal of molecular and cellular cardiology. 2010 doi: 10.1016/j.yjmcc.2010.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boengler K, Konietzka I, Buechert A, Heinen Y, Garcia-Dorado D, Heusch G, Schulz R. Loss of ischemic preconditioning’s cardioprotection in aged mouse hearts is associated with reduced gap junctional and mitochondrial levels of connexin 43. Am J Physiol Heart Circ Physiol. 2007;292:H1764–1769. doi: 10.1152/ajpheart.01071.2006. [DOI] [PubMed] [Google Scholar]
- 47.Biernacka A, Frangogiannis NG. Aging and cardiac fibrosis. Aging and disease. 2011;2:158–173. [PMC free article] [PubMed] [Google Scholar]
- 48.Dobaczewski M, Chen W, Frangogiannis NG. Transforming growth factor (tgf)-beta signaling in cardiac remodeling. Journal of molecular and cellular cardiology. 2011;51:600–606. doi: 10.1016/j.yjmcc.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cieslik KA, Taffet GE, Carlson S, Hermosillo J, Trial J, Entman ML. Immune-inflammatory dysregulation modulates the incidence of progressive fibrosis and diastolic stiffness in the aging heart. Journal of molecular and cellular cardiology. 2011;50:248–256. doi: 10.1016/j.yjmcc.2010.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen W, Frangogiannis NG. The role of inflammatory and fibrogenic pathways in heart failure associated with aging. Heart Fail Rev. 2010;15:415–422. doi: 10.1007/s10741-010-9161-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ghosh AK, Bradham WS, Gleaves LA, De Taeye B, Murphy SB, Covington JW, Vaughan DE. Genetic deficiency of plasminogen activator inhibitor-1 promotes cardiac fibrosis in aged mice: Involvement of constitutive transforming growth factor-beta signaling and endothelial-to-mesenchymal transition. Circulation. 2010;122:1200–1209. doi: 10.1161/CIRCULATIONAHA.110.955245. [DOI] [PubMed] [Google Scholar]
- 52.Bujak M, Frangogiannis NG. The role of tgf-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. 2007;74:184–195. doi: 10.1016/j.cardiores.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seeland U, Haeuseler C, Hinrichs R, Rosenkranz S, Pfitzner T, Scharffetter-Kochanek K, Bohm M. Myocardial fibrosis in transforming growth factor-beta(1) (tgf-beta(1)) transgenic mice is associated with inhibition of interstitial collagenase. Eur J Clin Invest. 2002;32:295–303. doi: 10.1046/j.1365-2362.2002.00985.x. [DOI] [PubMed] [Google Scholar]
- 54.Kuwahara F, Kai H, Tokuda K, Kai M, Takeshita A, Egashira K, Imaizumi T. Transforming growth factor-beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation. 2002;106:130–135. doi: 10.1161/01.cir.0000020689.12472.e0. [DOI] [PubMed] [Google Scholar]
- 55.Brooks WW, Conrad CH. Myocardial fibrosis in transforming growth factor beta(1)heterozygous mice. Journal of molecular and cellular cardiology. 2000;32:187–195. doi: 10.1006/jmcc.1999.1065. [DOI] [PubMed] [Google Scholar]
- 56.Gray MO, Long CS, Kalinyak JE, Li HT, Karliner JS. Angiotensin ii stimulates cardiac myocyte hypertrophy via paracrine release of tgf-beta 1 and endothelin-1 from fibroblasts. Cardiovasc Res. 1998;40:352–363. doi: 10.1016/s0008-6363(98)00121-7. [DOI] [PubMed] [Google Scholar]
- 57.Chen K, Mehta JL, Li D, Joseph L, Joseph J. Transforming growth factor beta receptor endoglin is expressed in cardiac fibroblasts and modulates profibrogenic actions of angiotensin ii. Circulation research. 2004;95:1167–1173. doi: 10.1161/01.RES.0000150369.68826.2f. [DOI] [PubMed] [Google Scholar]
- 58.Billet S, Bardin S, Verp S, Baudrie V, Michaud A, Conchon S, Muffat-Joly M, Escoubet B, Souil E, Hamard G, Bernstein KE, Gasc JM, Elghozi JL, Corvol P, Clauser E. Gain-of-function mutant of angiotensin ii receptor, type 1a, causes hypertension and cardiovascular fibrosis in mice. J Clin Invest. 2007;117:1914–1925. doi: 10.1172/JCI28764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shi Y, Massague J. Mechanisms of tgf-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 60.Rosenkranz S. Tgf-beta1 and angiotensin networking in cardiac remodeling. Cardiovasc Res. 2004;63:423–432. doi: 10.1016/j.cardiores.2004.04.030. [DOI] [PubMed] [Google Scholar]
- 61.Basso N, Cini R, Pietrelli A, Ferder L, Terragno NA, Inserra F. Protective effect of long-term angiotensin ii inhibition. Am J Physiol Heart Circ Physiol. 2007;293:H1351–1358. doi: 10.1152/ajpheart.00393.2007. [DOI] [PubMed] [Google Scholar]
- 62.Benigni A, Corna D, Zoja C, Sonzogni A, Latini R, Salio M, Conti S, Rottoli D, Longaretti L, Cassis P, Morigi M, Coffman TM, Remuzzi G. Disruption of the ang ii type 1 receptor promotes longevity in mice. J Clin Invest. 2009;119:524–530. doi: 10.1172/JCI36703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schultz Jel J, Witt SA, Glascock BJ, Nieman ML, Reiser PJ, Nix SL, Kimball TR, Doetschman T. Tgf-beta1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin ii. J Clin Invest. 2002;109:787–796. doi: 10.1172/JCI14190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li L, Fan D, Wang C, Wang JY, Cui XB, Wu D, Zhou Y, Wu LL. Angiotensin ii increases periostin expression via ras/p38 mapk/creb and erk1/2/tgf-beta1 pathways in cardiac fibroblasts. Cardiovasc Res. 2011;91:80–89. doi: 10.1093/cvr/cvr067. [DOI] [PubMed] [Google Scholar]
- 65.Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, Sorescu D. Nad(p)h oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circulation research. 2005;97:900–907. doi: 10.1161/01.RES.0000187457.24338.3D. [DOI] [PubMed] [Google Scholar]
- 66.Eghbali M, Tomek R, Sukhatme VP, Woods C, Bhambi B. Differential effects of transforming growth factor-beta 1 and phorbol myristate acetate on cardiac fibroblasts. Regulation of fibrillar collagen mrnas and expression of early transcription factors. Circulation research. 1991;69:483–490. doi: 10.1161/01.res.69.2.483. [DOI] [PubMed] [Google Scholar]
- 67.Chua CC, Chua BH, Zhao ZY, Krebs C, Diglio C, Perrin E. Effect of growth factors on collagen metabolism in cultured human heart fibroblasts. Connect Tissue Res. 1991;26:271–281. doi: 10.3109/03008209109152444. [DOI] [PubMed] [Google Scholar]
- 68.Bonnema DD, Webb CS, Pennington WR, Stroud RE, Leonardi AE, Clark LL, McClure CD, Finklea L, Spinale FG, Zile MR. Effects of age on plasma matrix metalloproteinases (mmps) and tissue inhibitor of metalloproteinases (timps) J Card Fail. 2007;13:530–540. doi: 10.1016/j.cardfail.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lindsey ML, Goshorn DK, Squires CE, Escobar GP, Hendrick JW, Mingoia JT, Sweterlitsch SE, Spinale FG. Age-dependent changes in myocardial matrix metalloproteinase/tissue inhibitor of metalloproteinase profiles and fibroblast function. Cardiovasc Res. 2005;66:410–419. doi: 10.1016/j.cardiores.2004.11.029. [DOI] [PubMed] [Google Scholar]
- 70.Sundstrom J, Evans JC, Benjamin EJ, Levy D, Larson MG, Sawyer DB, Siwik DA, Colucci WS, Wilson PW, Vasan RS. Relations of plasma total timp-1 levels to cardiovascular risk factors and echocardiographic measures: The framingham heart study. Eur Heart J. 2004;25:1509–1516. doi: 10.1016/j.ehj.2004.05.029. [DOI] [PubMed] [Google Scholar]
- 71.Chiao YA, Dai Q, Zhang J, Lin J, Lopez EF, Ahuja SS, Chou YM, Lindsey ML, Jin YF. Multi-analyte profiling reveals matrix metalloproteinase-9 and monocyte chemotactic protein-1 as plasma biomarkers of cardiac aging. Circ Cardiovasc Genet. 2011;4:455–462. doi: 10.1161/CIRCGENETICS.111.959981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Borbely A, van der Velden J, Papp Z, Bronzwaer JG, Edes I, Stienen GJ, Paulus WJ. Cardiomyocyte stiffness in diastolic heart failure. Circulation. 2005;111:774–781. doi: 10.1161/01.CIR.0000155257.33485.6D. [DOI] [PubMed] [Google Scholar]
- 73.Borbely A, Falcao-Pires I, van Heerebeek L, Hamdani N, Edes I, Gavina C, Leite-Moreira AF, Bronzwaer JG, Papp Z, van der Velden J, Stienen GJ, Paulus WJ. Hypophosphorylation of the stiff n2b titin isoform raises cardiomyocyte resting tension in failing human myocardium. Circulation research. 2009;104:780–786. doi: 10.1161/CIRCRESAHA.108.193326. [DOI] [PubMed] [Google Scholar]
- 74.LeWinter MM, Granzier H. Cardiac titin: A multifunctional giant. Circulation. 2010;121:2137–2145. doi: 10.1161/CIRCULATIONAHA.109.860171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fukuda N, Wu Y, Nair P, Granzier HL. Phosphorylation of titin modulates passive stiffness of cardiac muscle in a titin isoform-dependent manner. J Gen Physiol. 2005;125:257–271. doi: 10.1085/jgp.200409177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bishu K, Hamdani N, Mohammed SF, Kruger M, Ohtani T, Ogut O, Brozovich FV, Burnett JC, Jr, Linke WA, Redfield MM. Sildenafil and b-type natriuretic peptide acutely phosphorylate titin and improve diastolic distensibility in vivo. Circulation. 2011;124:2882–2891. doi: 10.1161/CIRCULATIONAHA.111.048520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chung CS, Hutchinson KR, Methawasin M, Saripalli C, Smith JE, 3rd, Hidalgo CG, Luo X, Labeit S, Guo C, Granzier HL. Shortening of the elastic tandem immunoglobulin segment of titin leads to diastolic dysfunction. Circulation. 2013;128:19–28. doi: 10.1161/CIRCULATIONAHA.112.001268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kruger M, Kotter S, Grutzner A, Lang P, Andresen C, Redfield MM, Butt E, dos Remedios CG, Linke WA. Protein kinase g modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circulation research. 2009;104:87–94. doi: 10.1161/CIRCRESAHA.108.184408. [DOI] [PubMed] [Google Scholar]
- 79.Hidalgo C, Hudson B, Bogomolovas J, Zhu Y, Anderson B, Greaser M, Labeit S, Granzier H. Pkc phosphorylation of titin’s pevk element: A novel and conserved pathway for modulating myocardial stiffness. Circulation research. 2009;105:631–638. 617. doi: 10.1161/CIRCRESAHA.109.198465. p following 638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hamdani N, Bishu KG, von Frieling-Salewsky M, Redfield MM, Linke WA. Deranged myofilament phosphorylation and function in experimental heart failure with preserved ejection fraction. Cardiovasc Res. 2013;97:464–471. doi: 10.1093/cvr/cvs353. [DOI] [PubMed] [Google Scholar]
- 81.Raskin A, Lange S, Banares K, Lyon RC, Zieseniss A, Lee LK, Yamazaki KG, Granzier HL, Gregorio CC, McCulloch AD, Omens JH, Sheikh F. A novel mechanism involving four-and-a-half lim domain protein-1 and extracellular signal-regulated kinase-2 regulates titin phosphorylation and mechanics. J Biol Chem. 2012;287:29273–29284. doi: 10.1074/jbc.M112.372839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hamdani N, Krysiak J, Kreusser MM, Neef S, Dos Remedios CG, Maier LS, Kruger M, Backs J, Linke WA. Crucial role for ca2(+)/calmodulin-dependent protein kinase-ii in regulating diastolic stress of normal and failing hearts via titin phosphorylation. Circulation research. 2013;112:664–674. doi: 10.1161/CIRCRESAHA.111.300105. [DOI] [PubMed] [Google Scholar]
- 83.Grutzner A, Garcia-Manyes S, Kotter S, Badilla CL, Fernandez JM, Linke WA. Modulation of titin-based stiffness by disulfide bonding in the cardiac titin n2-b unique sequence. Biophys J. 2009;97:825–834. doi: 10.1016/j.bpj.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hudson B, Hidalgo C, Saripalli C, Granzier H. Hyperphosphorylation of mouse cardiac titin contributes to transverse aortic constriction-induced diastolic dysfunction. Circulation research. 2011;109:858–866. doi: 10.1161/CIRCRESAHA.111.246819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Movsesian MA, Karimi M, Green K, Jones LR. Ca(2+)-transporting atpase, phospholamban, and calsequestrin levels in nonfailing and failing human myocardium. Circulation. 1994;90:653–657. doi: 10.1161/01.cir.90.2.653. [DOI] [PubMed] [Google Scholar]
- 86.Freeman K, Lerman I, Kranias EG, Bohlmeyer T, Bristow MR, Lefkowitz RJ, Iaccarino G, Koch WJ, Leinwand LA. Alterations in cardiac adrenergic signaling and calcium cycling differentially affect the progression of cardiomyopathy. J Clin Invest. 2001;107:967–974. doi: 10.1172/JCI12083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Janczewski AM, Lakatta EG. Modulation of sarcoplasmic reticulum ca(2+) cycling in systolic and diastolic heart failure associated with aging. Heart Fail Rev. 2010;15:431–445. doi: 10.1007/s10741-010-9167-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hohendanner F, Ljubojevic S, MacQuaide N, Sacherer M, Sedej S, Biesmans L, Wakula P, Platzer D, Sokolow S, Herchuelz A, Antoons G, Sipido K, Pieske B, Heinzel FR. Intracellular dyssynchrony of diastolic cytosolic [ca(2)(+)] decay in ventricular cardiomyocytes in cardiac remodeling and human heart failure. Circulation research. 2013;113:527–538. doi: 10.1161/CIRCRESAHA.113.300895. [DOI] [PubMed] [Google Scholar]
- 89.MacLennan DH, Kranias EG. Phospholamban: A crucial regulator of cardiac contractility. Nat Rev Mol Cell Biol. 2003;4:566–577. doi: 10.1038/nrm1151. [DOI] [PubMed] [Google Scholar]
- 90.Cain BS, Meldrum DR, Joo KS, Wang JF, Meng X, Cleveland JC, Jr, Banerjee A, Harken AH. Human serca2a levels correlate inversely with age in senescent human myocardium. Journal of the American College of Cardiology. 1998;32:458–467. doi: 10.1016/s0735-1097(98)00233-2. [DOI] [PubMed] [Google Scholar]
- 91.Babusikova E, Lehotsky J, Dobrota D, Racay P, Kaplan P. Age-associated changes in ca(2+)-atpase and oxidative damage in sarcoplasmic reticulum of rat heart. Physiol Res. 2012;61:453–460. doi: 10.33549/physiolres.932320. [DOI] [PubMed] [Google Scholar]
- 92.Lancel S, Qin F, Lennon SL, Zhang J, Tong X, Mazzini MJ, Kang YJ, Siwik DA, Cohen RA, Colucci WS. Oxidative posttranslational modifications mediate decreased serca activity and myocyte dysfunction in galphaq-overexpressing mice. Circulation research. 2010;107:228–232. doi: 10.1161/CIRCRESAHA.110.217570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Qin F, Siwik DA, Lancel S, Zhang J, Kuster GM, Luptak I, Wang L, Tong X, Kang YJ, Cohen RA, Colucci WS. Hydrogen peroxide-mediated serca cysteine 674 oxidation contributes to impaired cardiac myocyte relaxation in senescent mouse heart. J Am Heart Assoc. 2013;2:e000184. doi: 10.1161/JAHA.113.000184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vangheluwe P, Tjwa M, Van Den Bergh A, Louch WE, Beullens M, Dode L, Carmeliet P, Kranias E, Herijgers P, Sipido KR, Raeymaekers L, Wuytack F. A serca2 pump with an increased ca2+ affinity can lead to severe cardiac hypertrophy, stress intolerance and reduced life span. Journal of molecular and cellular cardiology. 2006;41:308–317. doi: 10.1016/j.yjmcc.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 95.Schwinger RH, Bohm M, Schmidt U, Karczewski P, Bavendiek U, Flesch M, Krause EG, Erdmann E. Unchanged protein levels of serca ii and phospholamban but reduced ca2+ uptake and ca(2+)-atpase activity of cardiac sarcoplasmic reticulum from dilated cardiomyopathy patients compared with patients with nonfailing hearts. Circulation. 1995;92:3220–3228. doi: 10.1161/01.cir.92.11.3220. [DOI] [PubMed] [Google Scholar]
- 96.Xu A, Narayanan N. Effects of aging on sarcoplasmic reticulum ca2+-cycling proteins and their phosphorylation in rat myocardium. The American journal of physiology. 1998;275:H2087–2094. doi: 10.1152/ajpheart.1998.275.6.H2087. [DOI] [PubMed] [Google Scholar]
- 97.Meyer M, Dillmann WH. Sarcoplasmic reticulum ca(2+)-atpase overexpression by adenovirus mediated gene transfer and in transgenic mice. Cardiovasc Res. 1998;37:360–366. doi: 10.1016/s0008-6363(97)00270-8. [DOI] [PubMed] [Google Scholar]
- 98.Schmidt U, del Monte F, Miyamoto MI, Matsui T, Gwathmey JK, Rosenzweig A, Hajjar RJ. Restoration of diastolic function in senescent rat hearts through adenoviral gene transfer of sarcoplasmic reticulum ca(2+)-atpase. Circulation. 2000;101:790–796. doi: 10.1161/01.cir.101.7.790. [DOI] [PubMed] [Google Scholar]
- 99.Shanmugam M, Gao S, Hong C, Fefelova N, Nowycky MC, Xie LH, Periasamy M, Babu GJ. Ablation of phospholamban and sarcolipin results in cardiac hypertrophy and decreased cardiac contractility. Cardiovasc Res. 2011;89:353–361. doi: 10.1093/cvr/cvq294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Louch WE, Vangheluwe P, Bito V, Raeymaekers L, Wuytack F, Sipido KR. Phospholamban ablation in hearts expressing the high affinity serca2b isoform normalizes global ca(2)(+) homeostasis but not ca(2)(+)-dependent hypertrophic signaling. Am J Physiol Heart Circ Physiol. 2012;302:H2574–2582. doi: 10.1152/ajpheart.01166.2011. [DOI] [PubMed] [Google Scholar]
- 101.Schmidt AG, Zhai J, Carr AN, Gerst MJ, Lorenz JN, Pollesello P, Annila A, Hoit BD, Kranias EG. Structural and functional implications of the phospholamban hinge domain: Impaired sr ca2+ uptake as a primary cause of heart failure. Cardiovasc Res. 2002;56:248–259. doi: 10.1016/s0008-6363(02)00541-2. [DOI] [PubMed] [Google Scholar]
- 102.Chu G, Lester JW, Young KB, Luo W, Zhai J, Kranias EG. A single site (ser16) phosphorylation in phospholamban is sufficient in mediating its maximal cardiac responses to beta -agonists. J Biol Chem. 2000;275:38938–38943. doi: 10.1074/jbc.M004079200. [DOI] [PubMed] [Google Scholar]
- 103.Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, Tornell J, Jacobs HT, Larsson NG. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 104.Vermulst M, Wanagat J, Kujoth GC, Bielas JH, Rabinovitch PS, Prolla TA, Loeb LA. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat Genet. 2008;40:392–394. doi: 10.1038/ng.95. [DOI] [PubMed] [Google Scholar]
- 105.Vermulst M, Bielas JH, Kujoth GC, Ladiges WC, Rabinovitch PS, Prolla TA, Loeb LA. Mitochondrial point mutations do not limit the natural lifespan of mice. Nat Genet. 2007;39:540–543. doi: 10.1038/ng1988. [DOI] [PubMed] [Google Scholar]
- 106.Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 107.Dai DF, Chen T, Wanagat J, Laflamme M, Marcinek DJ, Emond MJ, Ngo CP, Prolla TA, Rabinovitch PS. Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell. 2010;9:536–544. doi: 10.1111/j.1474-9726.2010.00581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang K, Klionsky DJ. Mitochondria removal by autophagy. Autophagy. 2011;7:297–300. doi: 10.4161/auto.7.3.14502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dutta D, Calvani R, Bernabei R, Leeuwenburgh C, Marzetti E. Contribution of impaired mitochondrial autophagy to cardiac aging: Mechanisms and therapeutic opportunities. Circulation research. 2012;110:1125–1138. doi: 10.1161/CIRCRESAHA.111.246108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146:682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 111.Inuzuka Y, Okuda J, Kawashima T, Kato T, Niizuma S, Tamaki Y, Iwanaga Y, Yoshida Y, Kosugi R, Watanabe-Maeda K, Machida Y, Tsuji S, Aburatani H, Izumi T, Kita T, Shioi T. Suppression of phosphoinositide 3-kinase prevents cardiac aging in mice. Circulation. 2009;120:1695–1703. doi: 10.1161/CIRCULATIONAHA.109.871137. [DOI] [PubMed] [Google Scholar]
- 112.Taneike M, Yamaguchi O, Nakai A, Hikoso S, Takeda T, Mizote I, Oka T, Tamai T, Oyabu J, Murakawa T, Nishida K, Shimizu T, Hori M, Komuro I, Takuji Shirasawa TS, Mizushima N, Otsu K. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy. 2010;6:600–606. doi: 10.4161/auto.6.5.11947. [DOI] [PubMed] [Google Scholar]
- 113.Terman A, Kurz T, Navratil M, Arriaga EA, Brunk UT. Mitochondrial turnover and aging of long-lived postmitotic cells: The mitochondrial-lysosomal axis theory of aging. Antioxidants & redox signaling. 2010;12:503–535. doi: 10.1089/ars.2009.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dai DF, Johnson SC, Villarin JJ, Chin MT, Nieves-Cintron M, Chen T, Marcinek DJ, Dorn GW, 2nd, Kang YJ, Prolla TA, Santana LF, Rabinovitch PS. Mitochondrial oxidative stress mediates angiotensin ii-induced cardiac hypertrophy and galphaq overexpression-induced heart failure. Circulation research. 2011;108:837–846. doi: 10.1161/CIRCRESAHA.110.232306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dai DF, Chen T, Szeto H, Nieves-Cintron M, Kutyavin V, Santana LF, Rabinovitch PS. Mitochondrial targeted antioxidant peptide ameliorates hypertensive cardiomyopathy. Journal of the American College of Cardiology. 2011;58:73–82. doi: 10.1016/j.jacc.2010.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mollnau H, Wendt M, Szocs K, Lassegue B, Schulz E, Oelze M, Li H, Bodenschatz M, August M, Kleschyov AL, Tsilimingas N, Walter U, Forstermann U, Meinertz T, Griendling K, Munzel T. Effects of angiotensin ii infusion on the expression and function of nad(p)h oxidase and components of nitric oxide/cgmp signaling. Circulation research. 2002;90:E58–65. doi: 10.1161/01.res.0000012569.55432.02. [DOI] [PubMed] [Google Scholar]
- 117.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ros)-induced ros release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192:1001–1014. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Franceschi C, Bonafe M, Valensin S, Olivieri F, De Luca M, Ottaviani E, De Benedictis G. Inflamm-aging. An evolutionary perspective on immunosenescence. Annals of the New York Academy of Sciences. 2000;908:244–254. doi: 10.1111/j.1749-6632.2000.tb06651.x. [DOI] [PubMed] [Google Scholar]
- 119.Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. Journal of the American College of Cardiology. 2013;62:263–271. doi: 10.1016/j.jacc.2013.02.092. [DOI] [PubMed] [Google Scholar]
- 120.Puntmann VO, Taylor PC, Mayr M. Coupling vascular and myocardial inflammatory injury into a common phenotype of cardiovascular dysfunction: Systemic inflammation and aging - a mini-review. Gerontology. 2011;57:295–303. doi: 10.1159/000316577. [DOI] [PubMed] [Google Scholar]
- 121.Miles EA, Rees D, Banerjee T, Cazzola R, Lewis S, Wood R, Oates R, Tallant A, Cestaro B, Yaqoob P, Wahle KW, Calder PC. Age-related increases in circulating inflammatory markers in men are independent of bmi, blood pressure and blood lipid concentrations. Atherosclerosis. 2008;196:298–305. doi: 10.1016/j.atherosclerosis.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 122.Bruunsgaard H, Skinhoj P, Pedersen AN, Schroll M, Pedersen BK. Ageing, tumour necrosis factor-alpha (tnf-alpha) and atherosclerosis. Clin Exp Immunol. 2000;121:255–260. doi: 10.1046/j.1365-2249.2000.01281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kalogeropoulos A, Georgiopoulou V, Psaty BM, Rodondi N, Smith AL, Harrison DG, Liu Y, Hoffmann U, Bauer DC, Newman AB, Kritchevsky SB, Harris TB, Butler J Health ABCSI. Inflammatory markers and incident heart failure risk in older adults: The health abc (health, aging, and body composition) study. Journal of the American College of Cardiology. 2010;55:2129–2137. doi: 10.1016/j.jacc.2009.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Collier P, Watson CJ, Voon V, Phelan D, Jan A, Mak G, Martos R, Baugh JA, Ledwidge MT, McDonald KM. Can emerging biomarkers of myocardial remodelling identify asymptomatic hypertensive patients at risk for diastolic dysfunction and diastolic heart failure? Eur J Heart Fail. 2011;13:1087–1095. doi: 10.1093/eurjhf/hfr079. [DOI] [PubMed] [Google Scholar]
- 125.Ather S, Chan W, Bozkurt B, Aguilar D, Ramasubbu K, Zachariah AA, Wehrens XH, Deswal A. Impact of noncardiac comorbidities on morbidity and mortality in a predominantly male population with heart failure and preserved versus reduced ejection fraction. Journal of the American College of Cardiology. 2012;59:998–1005. doi: 10.1016/j.jacc.2011.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lam CS, Lyass A, Kraigher-Krainer E, Massaro JM, Lee DS, Ho JE, Levy D, Redfield MM, Pieske BM, Benjamin EJ, Vasan RS. Cardiac dysfunction and noncardiac dysfunction as precursors of heart failure with reduced and preserved ejection fraction in the community. Circulation. 2011;124:24–30. doi: 10.1161/CIRCULATIONAHA.110.979203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Csiszar A, Ungvari Z, Koller A, Edwards JG, Kaley G. Proinflammatory phenotype of coronary arteries promotes endothelial apoptosis in aging. Physiol Genomics. 2004;17:21–30. doi: 10.1152/physiolgenomics.00136.2003. [DOI] [PubMed] [Google Scholar]
- 128.Csiszar A, Labinskyy N, Smith K, Rivera A, Orosz Z, Ungvari Z. Vasculoprotective effects of anti-tumor necrosis factor-alpha treatment in aging. Am J Pathol. 2007;170:388–398. doi: 10.2353/ajpath.2007.060708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chiossi G, Costantine MM, Tamayo E, Orise P, Hankins GD, Saade GR, Longo M. Effect of age and gender on the progression of adult vascular dysfunction in a mouse model of fetal programming lacking endothelial nitric oxide synthase. Am J Physiol Heart Circ Physiol. 2011;301:H297–305. doi: 10.1152/ajpheart.01284.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Maruyama Y. Aging and arterial-cardiac interactions in the elderly. Int J Cardiol. 2012;155:14–19. doi: 10.1016/j.ijcard.2011.01.087. [DOI] [PubMed] [Google Scholar]
- 131.Hammond J, Balligand JL. Nitric oxide synthase and cyclic gmp signaling in cardiac myocytes: From contractility to remodeling. Journal of molecular and cellular cardiology. 2012;52:330–340. doi: 10.1016/j.yjmcc.2011.07.029. [DOI] [PubMed] [Google Scholar]
- 132.Lam CS, Brutsaert DL. Endothelial dysfunction: A pathophysiologic factor in heart failure with preserved ejection fraction. Journal of the American College of Cardiology. 2012;60:1787–1789. doi: 10.1016/j.jacc.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 133.Calderone A, Thaik CM, Takahashi N, Chang DL, Colucci WS. Nitric oxide, atrial natriuretic peptide, and cyclic gmp inhibit the growth-promoting effects of norepinephrine in cardiac myocytes and fibroblasts. J Clin Invest. 1998;101:812–818. doi: 10.1172/JCI119883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Griendling KK, Sorescu D, Ushio-Fukai M. Nad(p)h oxidase: Role in cardiovascular biology and disease. Circulation research. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 135.Schulz E, Jansen T, Wenzel P, Daiber A, Munzel T. Nitric oxide, tetrahydrobiopterin, oxidative stress, and endothelial dysfunction in hypertension. Antioxidants & redox signaling. 2008;10:1115–1126. doi: 10.1089/ars.2007.1989. [DOI] [PubMed] [Google Scholar]
- 136.Redfield MM, Chen HH, Borlaug BA, Semigran MJ, Lee KL, Lewis G, LeWinter MM, Rouleau JL, Bull DA, Mann DL, Deswal A, Stevenson LW, Givertz MM, Ofili EO, O’Connor CM, Felker GM, Goldsmith SR, Bart BA, McNulty SE, Ibarra JC, Lin G, Oh JK, Patel MR, Kim RJ, Tracy RP, Velazquez EJ, Anstrom KJ, Hernandez AF, Mascette AM, Braunwald E, Trial R. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: A randomized clinical trial. JAMA. 2013;309:1268–1277. doi: 10.1001/jama.2013.2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Weiss EP, Fontana L. Caloric restriction: Powerful protection for the aging heart and vasculature. Am J Physiol Heart Circ Physiol. 2011;301:H1205–1219. doi: 10.1152/ajpheart.00685.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Fontana L. Calorie restriction and cardiometabolic health. Eur J Cardiovasc Prev Rehabil. 2008;15:3–9. doi: 10.1097/HJR.0b013e3282f17bd4. [DOI] [PubMed] [Google Scholar]
- 139.Meyer TE, Kovacs SJ, Ehsani AA, Klein S, Holloszy JO, Fontana L. Long-term caloric restriction ameliorates the decline in diastolic function in humans. Journal of the American College of Cardiology. 2006;47:398–402. doi: 10.1016/j.jacc.2005.08.069. [DOI] [PubMed] [Google Scholar]
- 140.Shinmura K, Tamaki K, Sano M, Murata M, Yamakawa H, Ishida H, Fukuda K. Impact of long-term caloric restriction on cardiac senescence: Caloric restriction ameliorates cardiac diastolic dysfunction associated with aging. Journal of molecular and cellular cardiology. 2011;50:117–127. doi: 10.1016/j.yjmcc.2010.10.018. [DOI] [PubMed] [Google Scholar]
- 141.Ungvari Z, Parrado-Fernandez C, Csiszar A, de Cabo R. Mechanisms underlying caloric restriction and lifespan regulation: Implications for vascular aging. Circulation research. 2008;102:519–528. doi: 10.1161/CIRCRESAHA.107.168369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Crow MT. Sir-viving cardiac stress: Cardioprotection mediated by a longevity gene. Circulation research. 2004;95:953–956. doi: 10.1161/01.RES.0000148666.20729.1c. [DOI] [PubMed] [Google Scholar]
- 143.Cornwell GG, 3rd, Thomas BP, Snyder DL. Myocardial fibrosis in aging germ-free and conventional lobund-wistar rats: The protective effect of diet restriction. J Gerontol. 1991;46:B167–170. doi: 10.1093/geronj/46.5.b167. [DOI] [PubMed] [Google Scholar]
- 144.Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, Allison DB, Cruzen C, Simmons HA, Kemnitz JW, Weindruch R. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325:201–204. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Alcendor RR, Gao S, Zhai P, Zablocki D, Holle E, Yu X, Tian B, Wagner T, Vatner SF, Sadoshima J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circulation research. 2007;100:1512–1521. doi: 10.1161/01.RES.0000267723.65696.4a. [DOI] [PubMed] [Google Scholar]
- 146.Hsu CP, Odewale I, Alcendor RR, Sadoshima J. Sirt1 protects the heart from aging and stress. Biol Chem. 2008;389:221–231. doi: 10.1515/BC.2008.032. [DOI] [PubMed] [Google Scholar]
- 147.Sedding D, Haendeler J. Do we age on sirt1 expression? Circulation research. 2007;100:1396–1398. doi: 10.1161/01.RES.0000269326.37165.3d. [DOI] [PubMed] [Google Scholar]
- 148.Planavila A, Iglesias R, Giralt M, Villarroya F. Sirt1 acts in association with pparalpha to protect the heart from hypertrophy, metabolic dysregulation, and inflammation. Cardiovasc Res. 2011;90:276–284. doi: 10.1093/cvr/cvq376. [DOI] [PubMed] [Google Scholar]
- 149.Oka S, Alcendor R, Zhai P, Park JY, Shao D, Cho J, Yamamoto T, Tian B, Sadoshima J. Pparalpha-sirt1 complex mediates cardiac hypertrophy and failure through suppression of the err transcriptional pathway. Cell Metab. 2011;14:598–611. doi: 10.1016/j.cmet.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Pillai VB, Sundaresan NR, Gupta MP. Regulation of akt signaling by sirtuins: Its implication in cardiac hypertrophy and aging. Circulation research. 2014;114:368–378. doi: 10.1161/CIRCRESAHA.113.300536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sundaresan NR, Pillai VB, Wolfgeher D, Samant S, Vasudevan P, Parekh V, Raghuraman H, Cunningham JM, Gupta M, Gupta MP. The deacetylase sirt1 promotes membrane localization and activation of akt and pdk1 during tumorigenesis and cardiac hypertrophy. Sci Signal. 2011;4:ra46. doi: 10.1126/scisignal.2001465. [DOI] [PubMed] [Google Scholar]
- 152.Sundaresan NR, Pillai VB, Gupta MP. Emerging roles of sirt1 deacetylase in regulating cardiomyocyte survival and hypertrophy. Journal of molecular and cellular cardiology. 2011;51:614–618. doi: 10.1016/j.yjmcc.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, Gupta MP. Sirt3 blocks the cardiac hypertrophic response by augmenting foxo3a-dependent antioxidant defense mechanisms in mice. J Clin Invest. 2009;119:2758–2771. doi: 10.1172/JCI39162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hafner AV, Dai J, Gomes AP, Xiao CY, Palmeira CM, Rosenzweig A, Sinclair DA. Regulation of the mptp by sirt3-mediated deacetylation of cypd at lysine 166 suppresses age-related cardiac hypertrophy. Aging (Albany NY) 2010;2:914–923. doi: 10.18632/aging.100252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sadoshima J. Sirt3 targets mptp and prevents aging in the heart. Aging (Albany NY) 2011;3:12–13. doi: 10.18632/aging.100266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Liu Y, Zhang D, Chen D. Sirt3: Striking at the heart of aging. Aging (Albany NY) 2011;3:1–2. doi: 10.18632/aging.100256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Sundaresan NR, Vasudevan P, Zhong L, Kim G, Samant S, Parekh V, Pillai VB, Ravindra PV, Gupta M, Jeevanandam V, Cunningham JM, Deng CX, Lombard DB, Mostoslavsky R, Gupta MP. The sirtuin sirt6 blocks igf-akt signaling and development of cardiac hypertrophy by targeting c-jun. Nat Med. 2012;18:1643–1650. doi: 10.1038/nm.2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 159.Vakhrusheva O, Smolka C, Gajawada P, Kostin S, Boettger T, Kubin T, Braun T, Bober E. Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circulation research. 2008;102:703–710. doi: 10.1161/CIRCRESAHA.107.164558. [DOI] [PubMed] [Google Scholar]
- 160.Maruyama N, Ishigami A, Kondo Y. Pathophysiological significance of senescence marker protein-30. Geriatr Gerontol Int. 2010;10 (Suppl 1):S88–98. doi: 10.1111/j.1447-0594.2010.00586.x. [DOI] [PubMed] [Google Scholar]
- 161.Mizukami H, Saitoh S, Machii H, Yamada S, Hoshino Y, Misaka T, Ishigami A, Takeishi Y. Senescence marker protein-30 (smp30) deficiency impairs myocardium-induced dilation of coronary arterioles associated with reactive oxygen species. Int J Mol Sci. 2013;14:9408–9423. doi: 10.3390/ijms14059408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Misaka T, Suzuki S, Miyata M, Kobayashi A, Ishigami A, Shishido T, Saitoh S, Kubota I, Takeishi Y. Senescence marker protein 30 inhibits angiotensin ii-induced cardiac hypertrophy and diastolic dysfunction. Biochem Biophys Res Commun. 2013;439:142–147. doi: 10.1016/j.bbrc.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 163.Misaka T, Suzuki S, Miyata M, Kobayashi A, Shishido T, Ishigami A, Saitoh S, Hirose M, Kubota I, Takeishi Y. Deficiency of senescence marker protein 30 exacerbates angiotensin ii-induced cardiac remodelling. Cardiovasc Res. 2013;99:461–470. doi: 10.1093/cvr/cvt122. [DOI] [PubMed] [Google Scholar]
- 164.Loffredo FS, Steinhauser ML, Jay SM, Gannon J, Pancoast JR, Yalamanchi P, Sinha M, Dall’osso C, Khong D, Shadrach JL, Miller CM, Singer BS, Stewart A, Psychogios N, Gerszten RE, Hartigan AJ, Kim MJ, Serwold T, Wagers AJ, Lee RT. Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell. 2013;153:828–839. doi: 10.1016/j.cell.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Zamore PD, Haley B. Ribo-gnome: The big world of small rnas. Science. 2005;309:1519–1524. doi: 10.1126/science.1111444. [DOI] [PubMed] [Google Scholar]
- 166.Sayed D, Hong C, Chen IY, Lypowy J, Abdellatif M. Micrornas play an essential role in the development of cardiac hypertrophy. Circulation research. 2007;100:416–424. doi: 10.1161/01.RES.0000257913.42552.23. [DOI] [PubMed] [Google Scholar]
- 167.Kumarswamy R, Thum T. Non-coding rnas in cardiac remodeling and heart failure. Circulation research. 2013;113:676–689. doi: 10.1161/CIRCRESAHA.113.300226. [DOI] [PubMed] [Google Scholar]
- 168.van Almen GC, Verhesen W, van Leeuwen RE, van de Vrie M, Eurlings C, Schellings MW, Swinnen M, Cleutjens JP, van Zandvoort MA, Heymans S, Schroen B. Microrna-18 and microrna-19 regulate ctgf and tsp-1 expression in age-related heart failure. Aging Cell. 2011;10:769–779. doi: 10.1111/j.1474-9726.2011.00714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Jazbutyte V, Fiedler J, Kneitz S, Galuppo P, Just A, Holzmann A, Bauersachs J, Thum T. Microrna-22 increases senescence and activates cardiac fibroblasts in the aging heart. Age (Dordr) 2013;35:747–762. doi: 10.1007/s11357-012-9407-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Boon RA, Iekushi K, Lechner S, Seeger T, Fischer A, Heydt S, Kaluza D, Treguer K, Carmona G, Bonauer A, Horrevoets AJ, Didier N, Girmatsion Z, Biliczki P, Ehrlich JR, Katus HA, Muller OJ, Potente M, Zeiher AM, Hermeking H, Dimmeler S. Microrna-34a regulates cardiac ageing and function. Nature. 2013;495:107–110. doi: 10.1038/nature11919. [DOI] [PubMed] [Google Scholar]
- 171.Bernardo BC, Gao XM, Winbanks CE, Boey EJ, Tham YK, Kiriazis H, Gregorevic P, Obad S, Kauppinen S, Du XJ, Lin RC, McMullen JR. Therapeutic inhibition of the mir-34 family attenuates pathological cardiac remodeling and improves heart function. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:17615–17620. doi: 10.1073/pnas.1206432109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Emanueli C, Thum T. Mirnage-34 induces cardiac damage. Cell Res. 2013;23:866–867. doi: 10.1038/cr.2013.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Loffredo FS, Pancoast JR, Lee RT. Keep pnuts in your heart. Circulation research. 2013;113:97–99. doi: 10.1161/CIRCRESAHA.113.301933. [DOI] [PubMed] [Google Scholar]
- 174.Sahin E, Depinho RA. Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature. 2010;464:520–528. doi: 10.1038/nature08982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Mitchell C, Hobcraft J, McLanahan SS, Siegel SR, Berg A, Brooks-Gunn J, Garfinkel I, Notterman D. Social disadvantage, genetic sensitivity, and children’s telomere length. Proceedings of the National Academy of Sciences of the United States of America; 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Geronimus AT, Hicken MT, Pearson JA, Seashols SJ, Brown KL, Cruz TD. Do us black women experience stress-related accelerated biological aging?: A novel theory and first population-based test of black-white differences in telomere length. Human nature. 2010;21:19–38. doi: 10.1007/s12110-010-9078-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Senyo SE, Steinhauser ML, Pizzimenti CL, Yang VK, Cai L, Wang M, Wu TD, Guerquin-Kern JL, Lechene CP, Lee RT. Mammalian heart renewal by pre-existing cardiomyocytes. Nature. 2013;493:433–436. doi: 10.1038/nature11682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Sahin E, Colla S, Liesa M, Moslehi J, Muller FL, Guo M, Cooper M, Kotton D, Fabian AJ, Walkey C, Maser RS, Tonon G, Foerster F, Xiong R, Wang YA, Shukla SA, Jaskelioff M, Martin ES, Heffernan TP, Protopopov A, Ivanova E, Mahoney JE, Kost-Alimova M, Perry SR, Bronson R, Liao R, Mulligan R, Shirihai OS, Chin L, DePinho RA. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470:359–365. doi: 10.1038/nature09787. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.