

Abstract
Heart failure (HF) causes a tremendous burden on the worldwide healthcare system, affecting more than 23 million people. There are many cardiovascular disorders that contribute to the development of HF and multiple risk factors that accelerate its occurrence, but regardless of its underlying cause, HF is characterized by a marked decrease in myocardial contractility and loss of pump function. One biomarker molecule consistently shown to be upregulated in human HF and several animal models is G protein-coupled receptor (GPCR) kinase 2 (GRK2), a kinase originally discovered to be involved in GPCR desensitization, especially β-adrenergic receptors (βARs). Indeed, higher levels of GRK2 can impair βAR-mediated inotropic reserve and its inhibition or molecular reduction has shown to improve pump function in several animal models including a pre-clinical pig model of HF. Recently, non-classical roles for GRK2 in cardiovascular disease have been described, including negative regulation of insulin signaling, a role in myocyte cell survival and apoptotic signaling, and it has been shown to be localized in/on mitochondria. These new roles of GRK2 suggest that GRK2 may be a nodal link in the myocyte, influencing both cardiac contractile function and cell metabolism and survival and contributing to HF independent of its canonical role on GPCR desensitization. In this review, classical and non-classical roles for GRK2 will be discussed, focusing on recently discovered roles for GRK2 in cardiomyocyte metabolism and the effects that these roles may have on myocardial contractile function and HF development.
Keywords: heart failure, GRK2, metabolism, cardiomyocytes
INTRODUCTION
Heart failure (HF) is the leading cause of death in developed countries and is caused by many different disease conditions, including coronary artery disease, high blood pressure and diabetes. At its root, HF is the inability of the heart to adequately pump blood and meet the oxygen demands of the body. One of the first molecular events associated with HF is an increase in the G protein-coupled receptor (GPCR) kinase-2 (GRK2), a ubiquitously expressed kinase first discovered to regulate β-adrenergic receptors (βARs) and a prototypic member of a kinase family that phosphorylates and desensitizes agonist-occupied GPCRs1. In the heart, this kinase is particularly important for triggering deactivation and down-regulation of βARs, impairing the myocyte’s ability to contract. The upregulation of GRK2 occurs initially after cardiac injury or stress and is necessary to shutdown over-activated βARs that occur as a result of compensated increases in catecholamines to drive the impaired heart from the activation of the sympathetic nervous system. This is the start of a vicious cycle of adrenergic signaling impairment where excess catecholamines (norepinephrine and epinephrine) are produced to compensate for decreased βAR signaling that ultimately keeps contractility diminished. Two decades of research have overwhelmingly uncovered that inhibition of GRK2 is beneficial for restoration of inotropic reserve and surprisingly, lowering GRK2 levels and activity in the hearts of several animal models has led to the prevention or reversal of HF2. This is due, in part, to the normalization of βAR levels that leads to a neurohormonal feedback that acts as a sympatholytic to undo the vicious adrenergic pathological cycle3. Further, targeting GRK2 appears complementary to βAR blockade as both can resensitize the receptor system over time and GRK2 inhibition also has extra-βAR effects that appear to contribute to its therapeutic benefit in HF3.
Recently, there has been increased interest in novel, non-canonical roles of GRK2 where this kinase is a central molecule in a complex ‘interactome’ that influences a wide variety of signaling networks and cellular functions4. For example, GRK2 has been found to play a role in insulin signaling, and also to be important for apoptosis induction in the injured heart. Contradicting the idea that GRK2 is/was primarily a cytosolic molecule, it was recently discovered that GRK2 also locates to the mitochondria and this localization is significantly increased following myocardial ischemic and oxidative stress. Below, we discuss in detail the influences that GRK2 has on glucose metabolism, ischemic injury and mitochondrial health and function and how these diverse actions can affect myocardial contractility and HF development.
GRK2 Function, Regulation and Role in the Heart and HF
GPCRs are a large superfamily of cell surface receptor proteins that are important for modulating a wide variety of physiological functions; together they constitute the most pharmacologically targeted protein family. Upon ligand binding, the receptors undergo conformational changes that ultimately result in the release of heterotrimeric G proteins that act as downstream signaling effectors of multiple pathways. GRKs are a small group of serine/threonine kinases that recognize only agonist-activated GPCRs, phosphorylating them and triggering the process of desensitization, which is marked by the loss of G protein coupling5. In general, these kinases work in concert with β-arrestins to desensitize, internalize and ultimately down-regulate GPCRs (Figure 1). Additionally, β-arrestins can also act as platforms to induce signaling outcomes in cells that can be independent of G protein activation6 (Figure 1).
Figure 1.
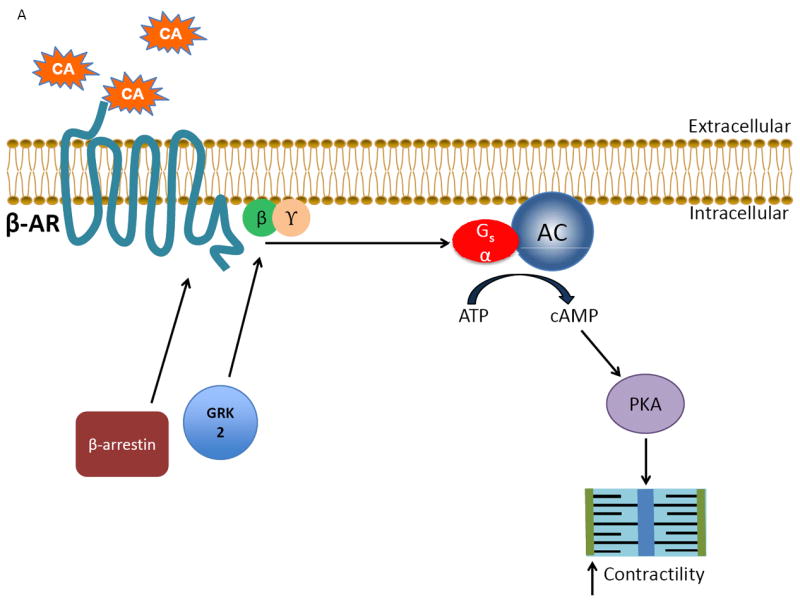
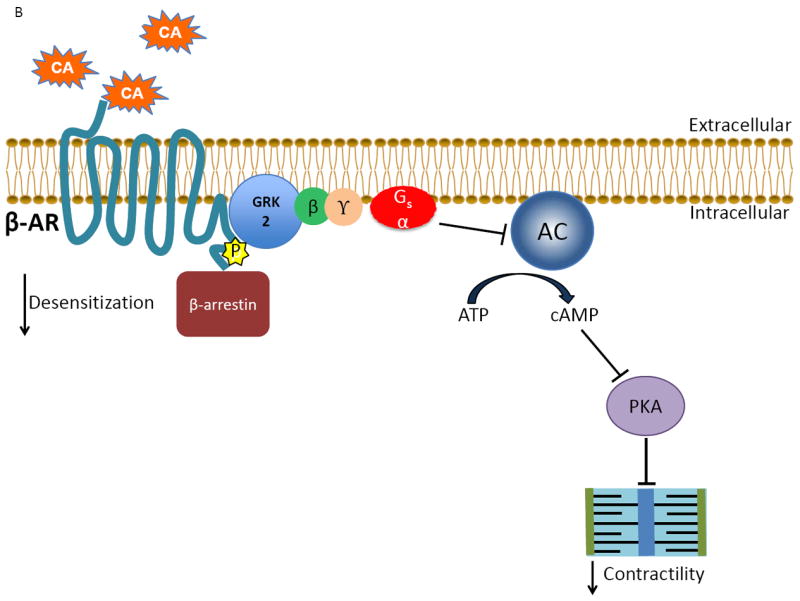
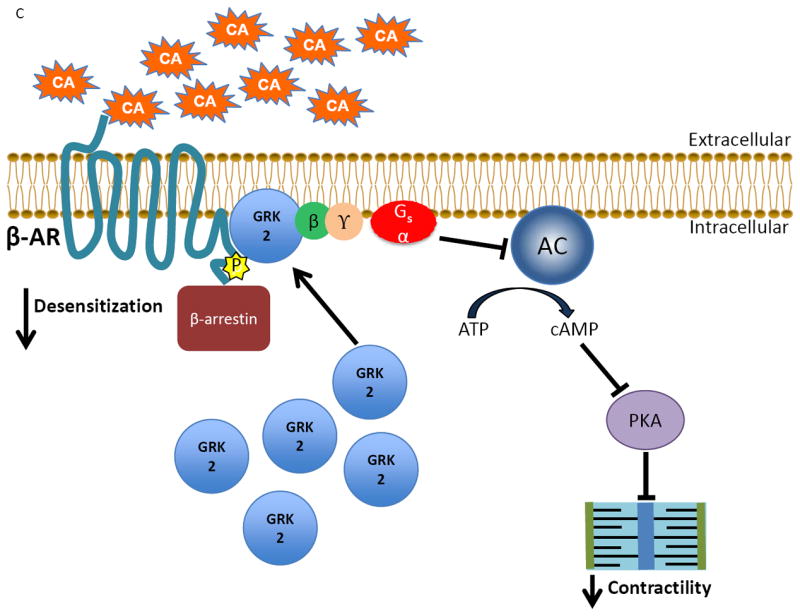
The classical actions of GRK2 on the cardiac βARs during physiological conditions and disease states. A) Under normal conditions, catecholamines (CA) activate the βAR, stimulating downstream signaling through Gαs that ultimately leads to increased contractility by activating adenylate cyclase (AC) and protein kinase A (PKA). B) As receptor activation occurs the desensitization of the signal is triggered simultaneously when the normally cytosolic GRK2 translocates and anchors to the membrane by binding to Gβγ via its C-terminus where it is able to interact with the agonist-occupied receptor and phosphorylate it, which begins the G protein uncoupling process. G protein activation is blocked by recruitment of β-arrestin molecules to the phosphorylated receptor. β-arrestins also start the receptor internalization and downregulation process as well as initiate novel signaling events. C) Following cardiac injury, GRK2 levels are increased due to enhanced βAR activation following stress-induced increases in catecholamines. The increase in GRK2 helps to prevent over-stimulation of the βAR initially, but over time a vicious cycle of chronically increased βAR desensitization occurs as well as increased pathological effects of GRK2, contributing to the progression of heart failure.
There are seven mammalian GRKs that have been characterized to date and they are divided into three subfamilies based on function, receptor specificity and tissue distribution: GRK1 and GRK7, GRK2 and 3 (also known as βARK1 and 2), and GRKs 4,5 and 6. GRK1 and GRK7 are restricted to the retinal rods and cones; however, several of the other GRKs are ubiquitously expressed, including GRK2, which (along with GRK5) is the most highly expressed GRK in the heart. Despite their differences in tissue specificity all GRKs share a similar overall structure, consisting of a central highly conserved catalytic domain similar to other AGC kinases flanked by variable N- and C-termini7. These terminal domains in GRK2 contain binding sites for multiple proteins, including PI3K/Akt, clathrin and α-actinin, which are not present in GRK5. The C-terminus of GRK2 also contains a pleckstrin homology domain that facilitates binding to the dissociated Gβγ subunit of an activated GPCR. This is how the primarily cytosolic GRK2 (and the homologous GRK3) localize to the sarcolemmal membrane where it can interact with and phosphorylate an activated GPCR. The C-terminus of GRK2 anchors to membrane phospholipids while the N-terminus interacts with receptors, putting targeted Ser/Thr residues of the cytoplasmic tail of the GPCR in contact with the central GRK catalytic domain. This regulation of GRK2 ensures the fidelity of the activation/deactivation process since Gβγ is not free to bind to and facilitate the membrane translocation of GRK2 unless a receptor system is activated8 (Figure 1). Peptides from this C-terminal domain of GRK2 can keep the kinase from localizing to the membrane and blocking GPCR desensitization: the entire C-tail of GRK2 (known as the βARKct) has been used as an effective in vitro and in vivo GRK2 inhibitor9.
During HF, GRK2 is increased 3-4 fold in the myocardium. GRK2 up-regulation appears to be one of the first molecular alterations in the myocyte after cardiac injury/stress and this has been shown to precede other βAR and functional abnormalities. In fact, human studies have shown that GRK2 up-regulation in failing myocardium may have novel diagnostic and prognostic value as its levels in the heart are mirrored by levels in white blood cells and so it can be measured peripherally10-15. Levels in failing myocardium correlate with cardiac dysfunction and improved cardiac function in HF is associated with lower GRK2 levels11,13,14,16. These aspects of human heart disease are also mirrored in animal models of HF, where GRK2 levels are increased early in pathological cardiac hypertrophy and myocardial ischemia, which appears instrumental in HF development17. While initially adaptive to compensate for increased catecholamine stimulation, over time, the excess GRK2 causes dysregulation of the βAR system, leading to a loss of inotropic reserve and contributing to HF.
The overall importance of GRK2 to the heart has been demonstrated through studies with genetically engineered mice. The global loss of GRK2 by homologous recombination led to embryonic lethality with cardiac malformations and dysplagia18 that doesn’t appear to be cardiomyocyte autonomous19. Heterozygous GRK2 knockout (KO) mice with 50% less GRK2 in all tissues have increased cardiac function20 and have been shown to have a favorable phenotype in other aspects of cardiac and metabolic regulation4 which will be discussed in more detail below. Further, cardiac-specific GRK2 KO mice where GRK2 is ablated after birth leads to improved cardiac function and prevention of HF development after a myocardial infarction (MI)21. Moreover, when KO of GRK2 is induced in the cardiomyocyte after HF development, there was active reverse remodeling and improved cardiac function21. Both of these lines of GRK2 KO mice also lead to significant cardioprotection acutely after MI22. In a manner fairly identical to loss of myocyte GRK2, transgenic expression of the βARKct as a GRK2 inhibitor led to rescue of several mouse models of HF9,23,24 and also was cardioprotective25. Of interest, when GRK2 is ablated in myocytes early in development there were some remaining abnormalities into adulthood: mice exhibited super-sensitivity to the negative effects of chronic catecholamine toxicity19.
This developmental importance of GRK2 may extend to the endothelium: deleting GRK2 in endothelial cells using a Tie-2-Cre mouse resulted in some abnormalities and changes in vasculogenesis as GRK2 is deleted prenatally in this murine model26,27. Interestingly, the increased inflammatory and oxidative stress seen in the endothelium of Tie-2-GRK2 KO mice is in contrast to vascular results found in heterozygous global GRK2 KO mice with similar decreases in GRK2 expression in endothelium, as these mice have improved endothelium function and also prevent the oxidative stress and dysfunction caused by angiotensin II (AngII) treatment28. Further, studies using GRK2 inhibitors, albeit not as specific, also demonstrate prevention of endothelial dysfunction in diabetic and obese mice29,30. More studies are needed to determine why endothelium specific loss of GRK2 is detrimental, while simultaneous decrease of GRK2 in the endothelium and other tissues produces an opposite phenotype.
The Relationship Between Cardiac Function, HF and Metabolism
The idea that changes in the energetics of the heart play an important role in the pathogenesis of HF is not novel. The heart expends more energy than any other organ and in order to provide enough fuel to carry out its functions, it must use energy derived from glucose and fatty acids to generate the force required for muscle contraction. As a result, metabolism and heart function are not mutually exclusive, but intimately linked. There are three different components to energy metabolism: substrate utilization, oxidative phosphorylation and ATP transfer and utilization. It is well established that after cardiac trauma there is a change in the relative amounts of substrate utilization to cover cardiac energy demands. In the healthy heart, fatty acid oxidation accounts for 65%, while glucose accounts for 30% of energy utilization. Nevertheless, the use of metabolic substrates can modify according to different physiological and pathological conditions and also to O2 availability. Healthy exercise, for example, increases glucose utilization according to its intensity, and if it is prevalently aerobic or anaerobic. In pathological conditions such as acute ischemia, the reduced O2 availability increases glucose utilization in order to reduce oxygen wastage while accumulation of free fatty acids is toxic and induces damage to the membrane and death of the cell31-33. This metabolic flexibility is instead lost during a chronic condition such as HF34-35 as observed in PET studies where fatty acid utilization during cardiomyopathy is dramatically increased while glucose extraction is decreased in the injured heart36. Insulin signaling is the major regulator of cardiac glucose extraction and use. Cardiac insulin signaling is also important for the healthy and injured heart independent of its ability to promote glucose extraction and utilization. Activation of the Insulin Receptor/IRS1/PI3K/AKT/mTOR pathway is indeed fundamental for the cardiac adaptive response to stress; where the short-term activation of this pathway promotes physiological hypertrophy and protection from myocardial injury37; its long-term activation, however, causes pathological hypertrophy and heart failure38-39. The correct balance of this signaling is therefore pivotal for the correct geometry and remodeling of the injured heart. Since glucose metabolism is so crucial in the failing heart, the role GRK2 can play in myocyte insulin signaling, discussed below, becomes significant.
Recently, novel insights in the connection between myocardium and global metabolism have been revealed. Interestingly, Grueter et al. demonstrated the importance of the heart in regulating systemic energy consumption40. Mice with cardiac-specific overexpression of mediator complex subunit 13 (MED13), a protein that controls transcription by nuclear hormone receptors, were resistant to high fat diet (HFD)-induced obesity and demonstrated improved insulin sensitivity and glucose tolerance40. These mice also exhibited higher levels of oxygen consumption, CO2 production and suppression of thyroid hormone receptor transcriptional activation, suggesting a hyper-metabolic state due to overexpression of MED13 exclusively in the heart40. The regulation of MED13 was found to be controlled by a cardiac-specific micro-RNA (miRNA)40. Therefore, there is now a precedent for cardiomyocytes having some regulatory control over global metabolism. GRK2 activity may be an important link in this process as discussed below.
GRK2 and insulin resistance: implications for linking cardiac metabolism and function
Although a relationship between insulin resistance and β-adrenergic overstimulation has been observed for many years, the relationship between the two was not completely understood until the involvement and importance of GRK2 was elucidated. In addition to the classical role of GRK2 in cardiac βAR signaling regulation through the manipulation of down-stream signaling which can have an indirect effect on insulin signaling, GRK2 can also directly regulate insulin signaling via different levels down-stream of βAR signaling. Additionally GRK2 can influence insulin signaling independent of adrenergic signaling; GRK2 can regulate insulin receptor signaling in response to insulin itself as well41. Novel aspects of GRK2 in its regulation of insulin signaling and myocyte glucose metabolism are highlighted in the next sections.
Neurohormonal Activation and Insulin Resistance
Crosstalk between insulin and the sympathetic nervous system (SNS) was noted several years ago. Insulin can affect a wide variety of cellular events; these include glucose transport, intermediary metabolism, DNA, RNA and protein synthesis rates, gene transcription, cell growth and cellular differentiation. In the heart, neurohormonal overactivation of both the SNS and renin-angiotensin-aldosterone (RAAS) systems, which is a common feature of chronic HF, is accompanied by insulin resistance (IR). Increased catecholamines confer myocardial damage and significant oxygen wasting by increasing the levels of ATP consumption42. Norepinephrine also increases levels of plasma free fatty acids (FFA)43 and promotes coronary vasoconstriction44, which further increases oxygen consumption45. Thus, central and peripheral activation of the SNS and also the RAAS system46, both directly and through amplified plasma FFA, correlates with systemic and cardiac IR47. Consequently, IR in HF is associated with markers of SNS activation, both in patients and in large animal models of HF43,47. In this scenario, therapies aimed to counteract neurohormonal activation would reduce IR and improve cardiac metabolism. Importantly, current front line therapy for HF targeting overactive SNS and RAAS activities do not clearly reduce IR in the failing heart. Indeed, a relative contraindication for βAR blockers in diabetic patients is still present. Importantly, RAAS antagonism with ACE inhibitors, AT1 blockers and aldosterone antagonists can reduce peripheral IR caused by Ang II and aldosterone, particularly in the case of Type II Diabetes and obesity.
It is possible that neurohormonal activation causes several different cellular modifications, with particular consideration to the expression or activity of molecules involved in key pathways that regulate cell physiology and cardiac metabolism. Therefore, relative “non-specific” receptor antagonism may not be sufficient means to normalize some specific cellular modifications induced by persistent hormonal stimulation and could be alternatively treated using therapies that focus on one particular molecule. As detailed below, one molecule that is emerging as such a target is GRK2. In fact, a recent study has shown that GRK2 in brown fat suppresses thermogenic gene expression and its loss increases energy expenditure, which may be responsible for the prevention of obesity and metabolic syndrome in the heterozygous GRK2 KO mice48. There are other molecules implicated in insulin resistance during sympathetic stimulation that are beyond the scope of this review; Akt, a downstream effector of insulin signaling whose phosphorylation/activation is decreased in severe dialated cardiomyopathy and also PTEN, a phosphatase that is increased in cardiomyopathy and is known to prevent Akt phosphorylation49. However, the fact that GRK2 is such a powerful regulator of myocardial function through GPCR-dependent mechanisms and its elucidation as having a key influence on cardiac metabolism appears to link contractility and metabolism via this kinase.
GRK2 and cardiac insulin signaling
Our group and others have explored the role of GRK2 in cardiovascular disease and have described its role in the pathophysiology of HF2. As previously mentioned, GRK2 upregulation brought about by catecholamine stimulation is a hallmark of failing myocardium; this phenomenon has also been observed during conditions with chronic insulin stimulation. The connection between GRK2 and insulin signaling has been convincingly demonstrated by experiments showing that treatment with insulin can swiftly increase the cellular content of GRK2, within a time frame of 15 to 30 minutes, suggesting that increased transcription or translation is not the causal mechanism50. Interestingly, studies have shown that insulin-like growth factor-1 (IGF1) can induce cellular accumulation of GRK2 by inhibition of mdm2, an ubiquitin ligase responsible for GRK2 ubiquitination and degradation51. IGF-1 stimulation also recruits GRK2 to the membrane where it appears that GRK2 can desensitize this receptor tyrosine kinase (RTK)52. This is certainly plausible since earlier studies have shown that the IGF-1 receptor can activate G proteins and especially Gβγ-dependent signaling that can bind the C-terminus of GRK253.These data with IGF-1 and mdm2 suggests that this could also be the mechanism for how GRK2 levels increase after chronic insulin treatment. This has significant pathophysiological significance since, as described below, GRK2 is a negative regulator of insulin signaling and alone can induce IR.
Several lines of evidence lead to the conclusion that GRK2 is a crucial modulator of IR, both systemically and in the heart54,55. Early data implicated GRK2 increases after chronic catecholamine exposure as being responsible for the well-known βAR-mediated IR50. Recently, GRK2 has been directly implicated in pathologies associated with metabolic disorders with characteristics of elevated insulin levels and IR including aging and obesity56. Of note, the expression of GRK2 is increased in important tissues, including myocytes, in different experimental models of IR56. Interestingly, lowering GRK2 systemically by fifty percent protects mice against TNFα, aging or HFD-stimulated negative modifications in glucose homeostasis and insulin signaling56. These novel findings were shown by using the heterozygous global GRK2 KO mice and indicate a critical role for GRK2 in the alterations of insulin sensitivity in physiological and disease conditions56. Exercise has also been shown to lower GRK2 levels, which improves insulin sensitivity in spontaneously hypertensive rats; this was also accomplished by targeted GRK2 silencing in endothelial cells57. Moreover, utilizing small peptides to inhibit GRK2 restored glucose tolerance in animal models of IR50,58. Therefore, GRK2 promotes IR, reduced glucose metabolism and metabolic syndrome, which are conditions associated with lower cardiac contractile function and add to the potential mechanisms for the apparent therapeutic benefit in the heart with GRK2 inhibition. It appears that targeting GRK2 will ameliorate both detrimental pathways, although since both are closely associated and linked, the improvement of one could influence the other.
Data from our lab has recently emerged showing how GRK2 can directly promote IR and lowered glucose metabolism in the cardiomyocyte, directly implicating the kinase activity of GRK2 in this process. Data had existed in other cell types that GRK2’s influence as a negative regulator of insulin signaling was due to non-kinase actions of GRK2 through protein-protein interactions. First, in liver cells and adipocytes, GRK2, through a Regulator of G Protein Signaling (RGS) domain present within its N-terminus, was shown to block Gαq/11 signaling downstream of insulin stimulation and its RTK activation59-61. GRK2 has also been shown to bind to and inhibit Akt, which would have a negative impact on glucose transporter 4 (GLUT4) translocation to the membrane to increase glucose uptake into cells62. However, neither one of these occurs in myocytes as insulin treatment induces the loss of downstream insulin signaling (Akt activation and GLUT4 membrane translocation) due to the direct interaction with insulin receptor substrate-1 (IRS1)41. In fact, when GRK2 is elevated to levels seen in human HF, there is a significant defect in myocardial glucose uptake and impaired insulin signaling in myocytes41. GRK2 directly phosphorylates IRS1 at the inhibitory Ser307 residue, promoting dissociation of the insulin receptor signaling complex and attenuating signaling to downstream effectors such as Akt and GLUT441 (Figure 2).
Figure 2.
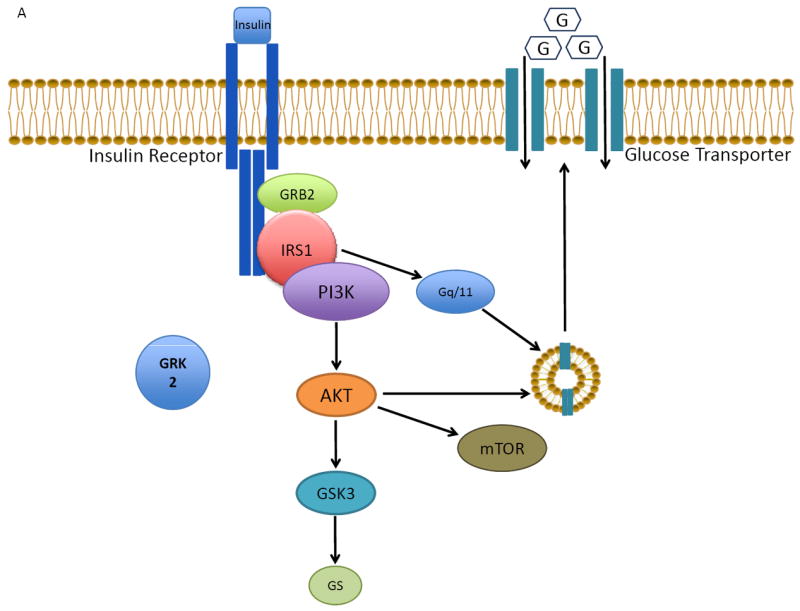
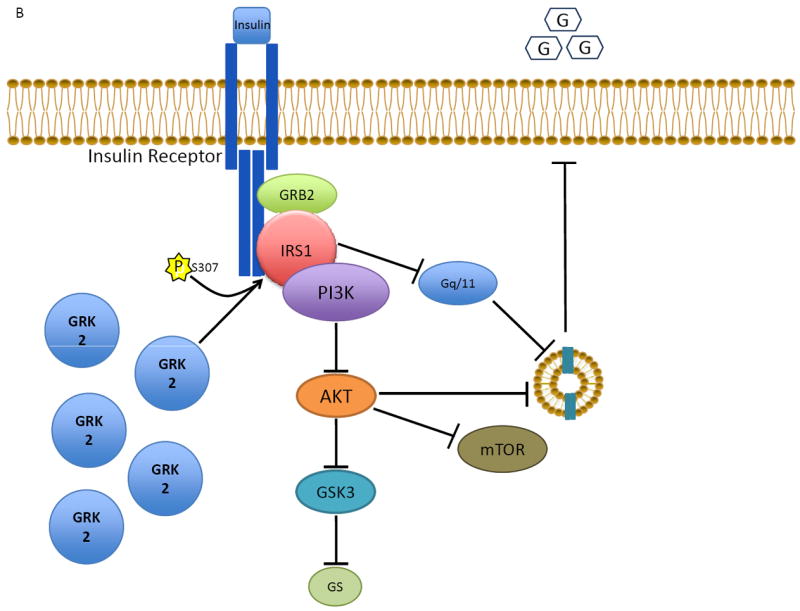
The role of GRK2 in cardiac insulin receptor signaling and myocyte glucose uptake. A) Normally, insulin binding to its receptor activates a downstream signaling process involving IRS1 that ultimately results in the movement of GLUT4 from the cytosol to the membrane, allowing for increased glucose uptake. B) Excess GRK2, as in heart failure, attenuates the insulin response via phosphorylation of IRS1 and inhibition of GLUT4 membrane translocation. The net result of GRK2’s action on the insulin signaling pathway in the myocyte is insulin resistance and glucose intolerance.
Importantly, elevated myocardial GRK2 levels enhanced negative cardiac glucose metabolism after ischemic injury and these effects precede GRK2-mediated ventricular contractile dysfunction41. This was the first report to link adrenergic control of contractility and metabolism; GRK2 appears to be this nodal link and the metabolic dysfunction that occurs first after cardiac injury may contribute to pump failure. Importantly, to further strengthen this nodal link of GRK2, cardiac-specific GRK2 KO mice have more myocardial glucose uptake that is maintained even after ischemia and insulin signaling remains intact with significantly decreased IRS1 phosphorylation41. This improved glucose metabolism in the face of decreased GRK2 remains normalized out to at least 8 weeks after MI in mice. It is accompanied by HF prevention because cardiac contractility is not adversely affected as it is in wild-type mice or GRK2 overexpressing mice post-MI41.
Interestingly, the interaction between GRK2 and IRS1 is dependent on an intact C-terminus of GRK2 since introduction of the βARKct peptide inhibits insulin-mediated GRK2-dependent IRS1 phosphorylation while βARKct expression improves Akt activation and GLUT4 membrane translocation in response to insulin41 (Figure 2). Moreover, βARKct gene delivery to the hearts of rats using adeno-associated virus serotype 6 (AAV6) prior to ischemic injury prevented insulin resistance and myocardial glucose uptake remained high41. These results could mean the direct interaction between GRK2 and IRS1 takes place within the C-tail of GRK2 or that activation of the insulin receptor stimulates a pool of G proteins, like the IGF-1 receptor, and the Gβγ released recruits GRK2 to the membrane where it can interact with IRS1 (Figure 2). Overall, given the higher efficiency of glucose in ATP production and the lower impact in oxidative stress with respect to other substrates, these data argue strongly that the role of GRK2 in the pathogenesis of HF is due at least in part by negative alterations in cardiac metabolism.
GRK2 and Mitochondrial Function – Relevance to Cardiac Metabolism and Myocyte Survival
GRK2 Localizes to Mitochondria
An interesting idea that arises from these data is that GRK2 may be a master regulator of cellular metabolism by controlling signal transduction from multiple receptors (GPCRs and RTKs) in addition to the cellular production and expenditure of energy through the regulation of βAR-mediated contractile function. In an interesting finding that can support this hypothesis, GRK2 has been found to localize in/on mitochondria. This was first observed in the brain63 and more recently in other cells including cardiac myocytes64,65. During basal states or resting conditions, the substrates, binding partners or functions of GRK2 within myocardial mitochondria are not known, however a study in non-cardiomyocytes did show GRK2 to be in involved in ATP production64.
What is known for myocytes is that when levels of GRK2 are elevated 3-4 fold as is found in human HF, there is more GRK2 associated with mitochondria as well, leading to reduced calcium tolerance of the mitochondrial permeability transition pore (MPTP) basally65, cytochrome C release and increased apoptotic signaling after ischemic stress22 (Figure 3). As discussed more below, cardiac-specific GRK2 KO mice had reduced ischemic injury due to lower cytochrome C release from mitochondria and decreased apoptotic signaling, consistent with the hypothesis that GRK2 mediates these events22.
Figure 3.
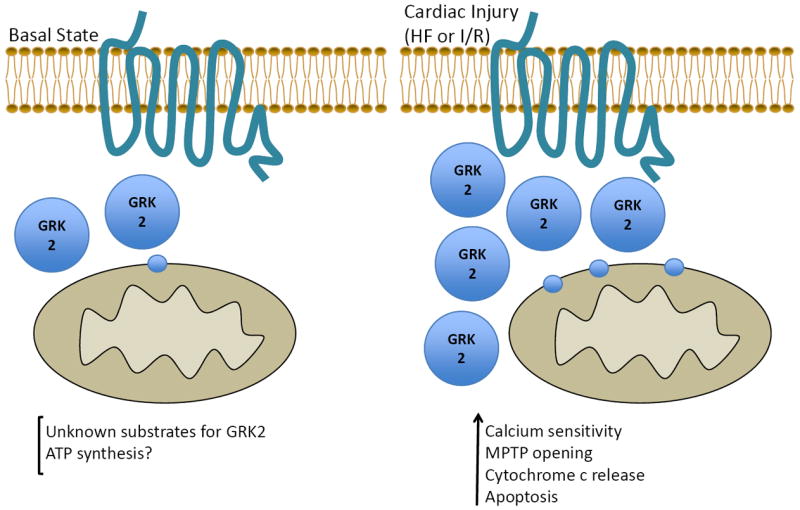
GRK2 at the mitochondria during basal and injured conditions. During healthy conditions, GRK2 may play a role in ATP synthesis or other functions as its binding partners or substrates are as yet unknown. Following cardiac injury, as GRK2 expression increases there is higher amounts associated with mitochondria, which have detrimental effects including increased calcium sensitivity to mitochondrial permeability transition pore (MPTP) opening, release of cytochrome C and an increase in apoptotic signaling, contributing to heart failure progression.
As detailed in the next section, it is clear that GRK2 is a pro-death kinase and the activity of this kinase is dependent, at least in part, on its mitochondrial localization. Importantly, in the setting of chronic HF development, the increase in cellular and mitochondrial GRK2 has negative effects on βAR and insulin signaling, resulting in impaired cellular metabolism and survival that eventually leads to contractile dysfunction. More studies are needed to determine the GRK2 targets in mitochondria, if any, that may affect basal metabolic functions.
Myocyte survival and the role of mitochondrial localized GRK2
Studies using a model of in vivo I/R injury have shown unequivocally that increased GRK2 expression in the heart enhances cardiac injury while inhibition with βARKct expression is robustly cardioprotective25. In addition to the above potential effects of mitochondrial GRK2 in cardiac metabolism, we have found a definitive mitochondria-dependent mode of pro-death kinase action in the myocyte following ischemic injury and the pursuing oxidative stress via Reactive Oxygen Species (ROS)65. Interestingly, like Akt and PKC mitochondrial translocation, GRK2 movement from the cytosol to mitochondria in response to ROS is mediated by the chaperone protein Hsp9065. This protein-protein interaction is mediated by the phosphorylation of GRK2 itself at Ser670 by MAP kinases (presumably ERK) (Figure 4). This residue resides in the C-tail and is also present in the GRK2 inhibitor, βARKct; ROS results in the phosphorylation of the βARKct peptide when it is expressed in myocytes and the binding of phosphorylated βARKct prevents endogenous GRK2 from binding to Hsp90 and translocating to the mitochondria (Figure 4). Moreover, expression of a Ser670Ala mutant of βARKct can no longer prevent myocyte death after oxidative stress, as it cannot compete with GRK2 for binding to Hsp9065. We have previously found the βARKct to be cardioprotective in the ischemic heart and Chen et al., determined that this mechanism of protection involves preventing the localization of GRK2 to the mitochondria65.
Figure 4.
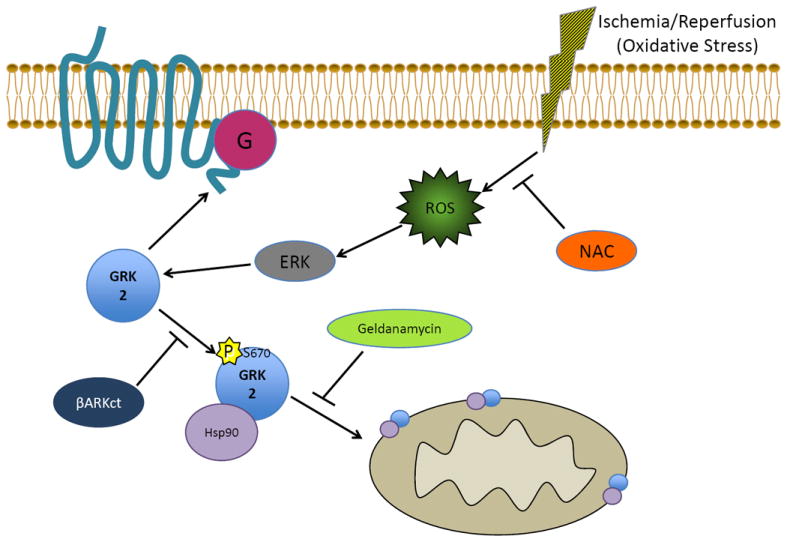
Mechanism for how GRK2 moves to the mitochondria after cardiac injury. Reactive oxygen species (ROS) produced as a result of ischemia/reperfusion injury or oxidative stress can activate ERK MAP kinase, which is able to phosphorylate GRK2 at Serine 670, facilitating the interaction of GRK2 with the chaperone protein Hsp90. Hsp90 aids in the movement of GRK2 to the mitochondria where it induces its pro-death actions in myocytes. The change in GRK2 localization can be prevented in multiple ways including expression of βARKct (through competition with Hsp90 binding as the βARKct has Ser670), the Hsp90 inhibitor Geldanamycin and the anti-oxidant N-acetyl-cysteine (NAC).
Recent studies in cardiomyocyte-specific GRK2 KO mice have also hinted at the prodeath actions of GRK2 at the level of mitochondria as loss of myocyte GRK2 after I/R injury decreased cytochrome C release and increased levels of anti-apoptotic Bcl-2 proteins22. Lowering GRK2 is cardioprotective and it resembles βARKct-mediated protection as both molecular manipulations decrease mitochondrial levels of GRK2. Prevention of the pro-death actions of GRK2 at the level of the mitochondria provides a novel mechanism to add to the therapeutic potential of GRK2 inhibition as a clinical therapy for HF. There are still many questions to be answered regarding the role of GRK2 in the mitochondria during both healthy and disease states; including the identification of potential substrates for GRK2 in the mitochondria, the precise function of mitochondia-localized GRK2 under basal conditions and lastly, if GRK2 either directly or indirectly hinders pro-survival functions of Hsp90.
Other Cell Survival Roles for GRK2 – Regulation of Cellular REDOX States through eNOS
In addition to the above mitochondrial-dependent mechanism of GRK2 mediated myocyte apoptosis, we have uncovered another key mechanism of GRK2 in relation to the REDOX state of the myocyte that appears to have crucial importance in regulating cardiac injury after ischemia. In the original study demonstrating the pro-death actions of elevated myocardial GRK2 and the cardioprotective effects of βARKct expression after I/R, we found that inhibition of GRK2 in the myocyte was accompanied by increased Akt and endothelial nitric oxide synthase (eNOS) activity, which led to increased nitric oxide production in the ischemic heart25. Consistent with this, GRK2 overexpression decreased eNOS-NO25. Myocyte death is a consequence of I/R injury and has detrimental effects on left ventricular function, contributing to the development of HF. Protective pathways, like PI3K/Akt and increased levels of nitric oxide allow for the preservation of myocardial contractility adding to the importance of GRK2 in not only influencing contractility directly through βAR regulation but by other means including cardiac metabolism and cell survival.
Recently, we have uncovered that this is due to a direct interaction between GRK2 and eNOS as we were able to co-immunoprecipitate (co-IP) GRK2 and eNOS from the mouse heart as well as myocytes; ischemia or oxidative stress increased the interaction66. Interestingly, within this interaction, GRK2 and eNOS negatively regulate one another. The negative regulation of GRK2 by eNOS occurs via the direct S-nitrosylation of GRK2 at Cys340 as previously found67. Thus, when NO bioavailability is high GRK2 is inhibited and cardiac contractile function and cell survival is enhanced. Indeed, studies have shown that increased Snitrosylation of GRK2 at this site enhances βAR-mediated cardiac contractility67. GRK2 inhibits eNOS activation by a yet unknown mechanism, however Ser1177 is involved (although this doesn’t appear to be the target of direct GRK2 kinase activity)66. Therefore, in HF or following myocardial injury, the increased expression of GRK2 suppresses eNOS activity preventing increased NO that ultimately feedbacks to less GRK2 inhibition through S-nitrosylation, creating a vicious cycle of the negative actions of this kinase. Further studies using a novel GRK2-Cys340Ser knock-in mouse demonstrates that the lack of dynamic S-nitrosylation control of GRK2 can directly influence cardiac ischemic injury as these mice have more GRK2 activity and greater injury after I/R66. Interestingly, we also found that the full cardioprotection effect of βARKct expression is dependent on eNOS activity66, which suggests that the GRK2-eNOS interaction may also influence GRK2 mitochondrial localization and this is the subject of ongoing investigations. Nitrates and β-blockers are some of the most commonly prescribed drugs for HF and the molecular mechanisms underlying their effectiveness is not clearly understood; elucidating the reciprocal relationship between GRK2 and eNOS lends insight into this and underscores the pathological role of GRK2, implicating that direct inhibition may be a more effective treatment in HF.
Conclusion
Overwhelming evidence revealed over the course of the last two decades has convincingly demonstrated the benefits of preventing excessive GRK2 expression activity in the cardiomyocyte after cardiac injury and during HF. While initially this therapeutic benefit of GRK2 inhibition was attributed to increases in contractility resulting from enhanced or restored βAR signaling, recent data has now come to light focusing on the harmful adrenergic-independent activities of GRK2 displayed during cardiac injury including cardiac metabolism and cell survival. These latter effects of GRK2 are not only βAR independent but are GPCR–independent and also include a novel cellular localization of GRK2 in/on mitochondria and novel binding partners (i.e. eNOS). Although specific targets and potential substrates still need to be elucidated, the mitochondrial localization of GRK2 points towards this kinase being a nodal link between cellular metabolism, cell survival and cardiac contractility. Based on the vast amount of research demonstrating the potential therapeutic effects of GRK2 inhibition during HF in animal models, it is interesting to speculate whether the benefits are due to the classical effects of GRK2 activity or the novel actions of GRK2. It is clear that the altered βAR signaling alterations found in the failing heart with GRK2 inhibition support and improve cardiac contractile function, however recent data regarding cardiac metabolic defects (i.e., insulin resistance) being improved with GRK2 inhibition lends a novel perspective to the overall therapeutic landscape41. Moreover, given the importance of the mitochondria as the cellular powerhouse and the initiator of apoptosis during basal and disease states, the significance of this localization, and its inhibition, could be considerably important in the pathological development of heart failure. Additionally, the interaction with and inhibition of eNOS, the catalyst of NO production, adds another dimension to the importance of regulating GRK2 levels to maintain NO levels, as NO is important to maintain vascular tone and cardiac function. Therefore it is probable that inhibition of both classical and novel aspects of GRK2 localization and function contribute to the beneficial effects seen in the heart after injury/stress. Keeping these critical cellular events in mind, these novel discoveries have placed even more emphasis on the importance of inhibiting GRK2 in multiple disease models, including HF and conditions associated with metabolic syndromes, hypertension and Type 2 diabetes. More studies are needed that specifically target the different roles of GRK2 in the failing heart in order to discern alterations in GPCR desensitization from these novel and exciting roles of GRK2 in the cardiomyocyte.
Acknowledgments
WJK is the William Wikoff Smith Chair in Cardiovascular Medicine.
Funding Sources:
WJK is supported by the following NIH grants: R37 HL061690, R01 HL085503, P01 HLHL091799, P01 HL075443 (Project 2) and P01 HL108806 (Project 3).
Non-Standard Abbreviations and Acronyms
- βAR
β-adrenergic receptor
- βARKct
carboxyl terminus of β-adrenergic receptor kinase
- eNOS
endothelial nitric oxide synthase
- ERK
extracellular signal-regulated kinase
- FFA
free fatty acid
- GPCR
G protein-coupled receptor
- GRK2
G protein-coupled receptor kinase 2
- HF
heart failure
- HFD
high fat diet
- Hsp90
heat shock protein 90
- IGF1
insulin-like growth factor-1
- IR
insulin resistance
- I/R
ischemia-reperfusion
- IRS1
insulin receptor substrate-1
- MI
myocardial infarction
- MPTP
mitochondrial permeability transition pore
- mTOR
mammalian target of rapamycin
- RAAS
renin-angiotensin-aldosterone systems
- ROS
reactive oxygen species
- RTK
receptor tyrosine kinase
- SNS
sympathetic nervous system
Footnotes
Disclosures:
None.
References
- 1.Maurice JP, Shah AS, Kypson AP, Hata JA, White DC, Glower DD, Koch WJ. Molecular β-adrenergic signaling abnormalities in failing rabbit hearts after infarction. Am J Physiol. 1999;276:H1853–H1860. doi: 10.1152/ajpheart.1999.276.6.H1853. [DOI] [PubMed] [Google Scholar]
- 2.Huang ZM, Gold JI, Koch WJ. G protein-coupled receptor kinases in normal and failing myocardium. Front Biosci (Landmark Ed) 2011;16:3047–60. doi: 10.2741/3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rengo G, Lymperopoulos A, Koch WJ. Myocardial adeno-associated virus serotype 6-βARKct gene therapy improves cardiac function and normalizes the neurohormonal axis in chronic heart failure. Circulation. 2009;119:89–98. doi: 10.1161/CIRCULATIONAHA.108.803999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Penela P, Murga C, Ribas C, Lafarga V, Mayor F. The complex G protein-coupled receptor kinase 2 (GRK2) interactome unveils new physiopathological targets. Br J Pharmacol. 2010;160:821–832. doi: 10.1111/j.1476-5381.2010.00727.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Freedman NJ, Liggett SB, Drachman DE, Pei G, Caron MG, Lefkowitz RJ. Phosphorylation and desensitization of the human β1-adrenergic receptor: Involvement of G protein-coupled receptor kinases and cAMP-dependent protein kinase. J Biol Chem. 1995;270:17953–17961. doi: 10.1074/jbc.270.30.17953. [DOI] [PubMed] [Google Scholar]
- 6.Premont RT, Gainetdinov RR. Physiological roles of G protein-coupled receptor kinases and arrestins. Annu Rev Physiol. 2007;69:511–534. doi: 10.1146/annurev.physiol.69.022405.154731. [DOI] [PubMed] [Google Scholar]
- 7.Lyon AM, Taylor VG, Tesmer JJ. Strike a pose: Gαq complexes at the membrane. Trends Pharmacol Sci. 2014;35:23–30. doi: 10.1016/j.tips.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pitcher JA, Freedman NJ, Lefkowitz RJ. G protein-coupled receptor kinases. Ann Rev Biochem. 1998;67:653–692. doi: 10.1146/annurev.biochem.67.1.653. [DOI] [PubMed] [Google Scholar]
- 9.Rockman HA, Chien KR, Choi DJ, Iaccarino G, Hunter JJ, Ross J, Jr, Lefkowitz RJ, Koch WJ. Expression of a β-adrenergic receptor kinase 1 inhibitor prevents the development of myocardial failure in gene-targeted mice. Proc Natl Acad Sci USA. 1998;95:7000–5. doi: 10.1073/pnas.95.12.7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iaccarino G, Barbato E, Cipolletta E, De Amicis V, Margulies KB, Leosco D, Trimarco B, Koch WJ. Elevated myocardial and lymphocyte GRK2 expression and activity in human heart failure. Eur Heart J. 2005;26:1752–8. doi: 10.1093/eurheartj/ehi429. [DOI] [PubMed] [Google Scholar]
- 11.Hata JA, Williams ML, Schroder JN, Lina B, Keys JR, Blaxall BC, Petrofski JA, Jakoi A, Milano CA, Koch WJ. Lymphocyte levels of GRK2 (βARK1) mirror changes in the LVAD-supported failing human heart: lower GRK2 associated with improved β-adrenergic signaling after mechanical unloading. J Card Fail. 2006;12:360–8. doi: 10.1016/j.cardfail.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 12.Bonita RE, Raake PW, Otis NJ, Chuprun JK, Spivack T, Dasgupta A, Whellan DJ, Mather PJ, Koch WJ. Dynamic changes in lymphocyte GRK2 levels in cardiac transplant patients: a biomarker for left ventricular function. Clin Transl Sci. 2010;3:14–8. doi: 10.1111/j.1752-8062.2010.00176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akhter SA, D’Souza KM, Malhotra R, Staron ML, Valeroso TB, Fedson SE, Anderson AS, Raman J, Jeevanandam V. Reversal of impaired myocardial β-adrenergic receptor signaling by continuous-flow left ventricular assist device support. J Heart Lung Transplant. 2010;29:603–9. doi: 10.1016/j.healun.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rengo G, Galasso G, Femminella GD, Parisi V, Zincarelli C, Pagano G, Lucia CD, Cannavo A, Liccardo D, Marciano C, Vigorito C, Giallauria F, Ferrara N, Furgi G, Filardi PP, Koch WJ, Leosco D. Reduction of lymphocyte G protein-coupled receptor kinase-2 (GRK2) after exercise training predicts survival in patients with heart failure. Eur J Prev Cardiol. 2014;21:4–11. doi: 10.1177/2047487313491656. [DOI] [PubMed] [Google Scholar]
- 15.Leosco D, Fortunato F, Rengo G, Iaccarino G, Sanzari E, Golina L, Zincarelli C, Canonico V, Marchese M, Koch WJ, Rengo F. Lymphocyte G-protein-coupled receptor kinase-2 is upregulated in patients with Alzheimer’s disease. Neurosci Lett. 2007;415:279–82. doi: 10.1016/j.neulet.2007.01.034. [DOI] [PubMed] [Google Scholar]
- 16.Leosco D, Parisi V, Femminella GD, Formisano R, Petraglia L, Allocca E, Bonaduce D. Effects of exercise training on cardiovascular adrenergic system. Front Physiol. 2013;28:348. doi: 10.3389/fphys.2013.00348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lymperopoulos A, Rengo G, Koch WJ. GRK2 inhibition in heart failure: something old, something new. Curr Pharm Des. 2012;18:186–91. doi: 10.2174/138161212799040510. [DOI] [PubMed] [Google Scholar]
- 18.Jaber M, Koch WJ, Rockman H, Smith B, Bond RA, Sulik KK, Ross J, Lefkowitz RJ, Caron MG, Giros B. Essential role of ß-adrenergic receptor kinase 1 in cardiac development and function. Proc Natl Acad Sci USA. 1996;93:12974–12979. doi: 10.1073/pnas.93.23.12974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matkovich SJ, Diwan A, Klanke JL, Hammer DJ, Marreez Y, Odley AM, Brunskill EW, Koch WJ, Schwartz RJ, Dorn GW., 2nd Cardiac-specific ablation of G-protein receptor kinase 2 redefines its roles in heart development and β-adrenergic signaling. Circ Res. 2006;99:996–1003. doi: 10.1161/01.RES.0000247932.71270.2c. [DOI] [PubMed] [Google Scholar]
- 20.Rockman HA, Choi DJ, Akhter SA, Jaber M, Giros B, Lefkowitz RJ, Caron MG, Koch WJ. Control of myocardial contractile function by the level of β-adrenergic receptor kinase 1 in genetargeted mice. J Biol Chem. 1998;273:18180–4. doi: 10.1074/jbc.273.29.18180. [DOI] [PubMed] [Google Scholar]
- 21.Raake P, Vinge LE, Gao E, Boucher M, Chen X, Kerkela R, DeGeorge BR, Jr, Matkovich S, Houser SR, Most P, Eckhart AD, Dorn GW, II, Koch WJ. G protein-coupled receptor kinase 2 ablation in cardiac myocytes before or after myocardial infarction prevents heart failure. Circ Res. 2008;103:413–422. doi: 10.1161/CIRCRESAHA.107.168336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fan Q, Chen M, Zuo L, Shang X, Huang ZM, Ciccarelli M, Raake P, Brinks H, Chuprun JK, Dorn GW, II, Koch WJ, Gao E. Myocardial ablation of G protein-coupled receptor kinase 2 (GRK2) decreases ischemia/reperfusion injury through an anti-intrinsic apoptotic pathway. PLOS One. 2013;8:e66234. doi: 10.1371/journal.pone.0066234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Freeman K, Lerman I, Kranias E, Bohlmeyer T, Bristow MR, Lefkowitz RJ, Iaccarino G, Koch WJ, Leinwand LA. Alterations in cardiac adrenergic signaling and calcium cycling differentially affect the progression of cardiomyopathy. J Clin Invest. 2001;107:967–74. doi: 10.1172/JCI12083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harding V, Jones L, Lefkowitz RJ, Koch WJ, Rockman HA. Cardiac βARK1 inhibition prolongs survival and augments β blocker therapy in a mouse model of severe heart failure. Proc Natl Acad Sci USA. 2001;98:5809–5814. doi: 10.1073/pnas.091102398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brinks H, Boucher M, Gao E, Chuprun JK, Pesant S, Raake PW, Huang ZM, Xang X, Qiu G, Gumpert A, Harris DM, Eckhart AD, Most P, Koch WJ. Level of G protein-coupled receptor kinase-2 determines myocardial ischemia/reperfusion injury via pro- and anti-apoptotic mechanisms. Circ Res. 2010;107:1140–1149. doi: 10.1161/CIRCRESAHA.110.221010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ciccarelli M, Sorriento D, Franco A, Fusco A, Del Guidice C, Annunziata R, Cipolletta E, Monti MG, Dorn GW, II, Trimarco B, Iaccarino G. Endothelial G protein-coupled receptor kinase 2 regulates vascular homeostasis through the control of free radical oxygen species. Arterioscler Thromb Vasc Biol. 2013;33:2415–24. doi: 10.1161/ATVBAHA.113.302262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rivas V, Carmona R, Muñoz-Chápuli R, Mendiola M, Nogués L, Reglero C, Miguel-Martin M, García-Escudero, Dorn GW, II, Hardisson D, Mayor F, Jr, Penela P. Developmental and tumoral vascularization is regulated by G protein-coupled receptor kinase 2. J Clin Invest. 2013;123:4714–30. doi: 10.1172/JCI67333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Avendaño MS, Lucas E, Jurado-Pueyo M, Martínez-Revelles S, Vila-Bedmar R, Mayor F, Jr, Salaices M, Biones AM, Murga C. Increased nitric oxide bioavailability in adult GRK2 hemizygous mice protects against Angiotensin II-induced hypertension. Hypertension. 2014;63:369–75. doi: 10.1161/HYPERTENSIONAHA.113.01991. [DOI] [PubMed] [Google Scholar]
- 29.Taguchi K, Matsumoto T, Kamata K, Kobayashi T. Inhibitor of G protein-coupled receptor kinase 2 normalizes vascular endothelial function in type 2 diabetic mice by improving β-arrestin 2 translocation and ameliorating Akt/eNOS signal dysfunction. Endocrinology. 2012;153:2985–96. doi: 10.1210/en.2012-1101. [DOI] [PubMed] [Google Scholar]
- 30.Taguchi K, Matsumoto T, Kamata K, Kobayashi T. G protein-coupled receptor kinase 2, with β-arrestin 2, impairs insulin-induced Akt/endothelial nitric oxide synthase signaling in ob/ob mouse aorta. Diabetes. 2012;61:1978–85. doi: 10.2337/db11-1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang HX, Zang YM, Huo JH, Liang SJ, Zhang HF, Wang YM, Fan Q, Guo WY, Wang HC, Gao FJ. Physiologically tolerable insulin reduces myocardial injury and improves cardiac functional recovery in myocardial ischemic/reperfused dogs. Cardiovasc Pharmacol. 2006;48:306–13. doi: 10.1097/01.fjc.0000249873.73197.c3. [DOI] [PubMed] [Google Scholar]
- 32.Shiojima I, Walsh K. Regulation of cardiac growth and coronary angiogenesis by the Akt/PKB signaling pathway. Genes Dev. 2006;20:3347–65. doi: 10.1101/gad.1492806. [DOI] [PubMed] [Google Scholar]
- 33.Diaz R, Goyal A, Mehta SR, Afzal R, Xavier D, Pais P, Chrolavicius S, Zhu J, Kazmi K, Liu L, Budaj A, Zubaid M, Avezum A, Ruda M, Yusuf S. Glucose-insulin-potassium therapy in patients with ST-segment elevation myocardial infarction. JAMA. 2007;298:2399–2405. doi: 10.1001/jama.298.20.2399. [DOI] [PubMed] [Google Scholar]
- 34.Kolwicz SC, Jr, Purohit S, Tian R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res. 2013;113:603–16. doi: 10.1161/CIRCRESAHA.113.302095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neubauer S. The failing heart--an engine out of fuel. N Engl J Med. 2007;356:1140–51. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 36.Taylor M, Wallhaus TR, Degrado TR, Russel DC, Stanko P, NIckles RJ, Stone CK. An evaluation of myocardial fatty acid and glucose uptake using PET with [18F]fluoro-6-thiaheptadecanoic acid and [18F]FDG in patients with congestive heart failure. J Nucl Med. 2001;42:55–62. [PubMed] [Google Scholar]
- 37.Shiojima I, Yefremashvili M, Luo Z, Kureishi Y, Takahashi A, Tao J, Rosenzweig A, Kahn CR, Abel ED, Walsh KJ. Akt signaling mediates postnatal heart growth in response to insulin and nutritional status. Biol Chem. 2002;277:37670–7. doi: 10.1074/jbc.M204572200. [DOI] [PubMed] [Google Scholar]
- 38.Kemi OJ, Ceci M, Wisloff U, Grimaldi S, Gallo P, Smith GL, Condorelli G, Ellingsen OJ. Activation or inactivation of cardiac Akt/mTOR signaling diverges physiological from pathological hypertrophy. Cell Physiol. 2008;214:316–21. doi: 10.1002/jcp.21197. [DOI] [PubMed] [Google Scholar]
- 39.Matsui T, Nagoshi T, Rosenzweig A. Akt and PI 3-kinase signaling in cardiomyocyte hypertrophy and survival. Cell Cycle. 2003;2:220–3. [PubMed] [Google Scholar]
- 40.Grueter CE, van Rooji E, Johnson BA, DeLeon SM, Sutherland LB, Qi X, Gautron L, Elmquist JK, Bassal-Duby R, Olson EN. A cardiac microRNA governs systemic energy homeostasis by regulation of MED13. Cell. 2012;149:671–83. doi: 10.1016/j.cell.2012.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ciccarelli M, Chuprun JK, Rengo G, Gao E, Wei Z, Peroutka RJ, Gold JI, Gumpert A, Chen M, Otis NJ, Dorn GW, 2nd, Trimarco B, Iaccarino G, Koch WJ. G protein-coupled receptor kinase 2 activity impairs cardiac glucose uptake and promotes insulin resistance after myocardial ischemia. Circulation. 2011;123:1953–62. doi: 10.1161/CIRCULATIONAHA.110.988642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Opie LH, Thandroyen FT, Muller C, Bricknell OL. Adrenaline-induced “oxygen-wastage” and enzyme release from working rat heart. Effects of calcium antagonism, beta-blockade, nicotinic acid and coronary artery ligation. J Mol Cell Cardiol. 1979;11:1073–94. doi: 10.1016/0022-2828(79)90395-x. [DOI] [PubMed] [Google Scholar]
- 43.Paolisso G, De Riu S, Marrazzo G, Verza M, Varricchio M, D’Onofrio F. Insulin resistance and hyperinsulinemia in patients with chronic congestive heart failure. Metabolism. 1991;40:972–7. doi: 10.1016/0026-0495(91)90075-8. [DOI] [PubMed] [Google Scholar]
- 44.Kostis JB, Sanders M. The association of heart failure with insulin resistance and the development of type 2 diabetes. Am J Hypertens. 2005;18:731–7. doi: 10.1016/j.amjhyper.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 45.Sasaoka T, Wada T, Tsuneki H. Lipid phosphatases as a possible therapeutic target in cases of type 2 diabetes and obesity. Pharmacol Ther. 2006;112:799–809. doi: 10.1016/j.pharmthera.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 46.Zucker IH. Novel mechanisms of sympathetic regulation in chronic heart failure. Hypertension. 2006;48:1005–11. doi: 10.1161/01.HYP.0000246614.47231.25. [DOI] [PubMed] [Google Scholar]
- 47.Opie LH. The metabolic syndrome, does it exist? In: Opie LH, Kasuga M, Yellon DM, editors. Diabetes at the Limits. Vol. 2. Cape Town, South Africa: University of Cape Town Press; 2006. pp. 95–110. [Google Scholar]
- 48.Vila-Bedmar R, Garcia-Guerra L, Nieto-Vazquez I, Mayor F, Jr, Lorenzo M, Murga C, Fernández-Veledo S. GRK2 contribution to the regulation of energy expenditure and brown fat function. FASEB J. 2012;26:3503–14. doi: 10.1096/fj.11-202267. [DOI] [PubMed] [Google Scholar]
- 49.Ashrafian H, Frenneaux MP, Opie LH. Metabolic mechanisms in heart failure. Circulation. 2007;116:434–48. doi: 10.1161/CIRCULATIONAHA.107.702795. [DOI] [PubMed] [Google Scholar]
- 50.Cipolletta E, Campanile A, Santulli G, Sanzari E, Leosco D, Campiglia P, Trimarco B, Iaccarino G. The G protein coupled receptor kinase 2 plays an essential role in β-adrenergic receptor-induced insulin resistance. Cardiovasc Res. 2009;84:407–15. doi: 10.1093/cvr/cvp252. [DOI] [PubMed] [Google Scholar]
- 51.Salcedo A, Mayor F, Jr, Penela P. Mdm2 is involved in the ubiquitination and degradation of G-protein-coupled receptor kinase 2. EMBO J. 2006;25:4752–62. doi: 10.1038/sj.emboj.7601351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zheng H, Worrall C, Shem H, Issad T, Seregard S, Girnita A, Girnita L. Selective recruitment of G protein-coupled receptor kinases (GRKs) controls signaling of the insulin-like growth factor 1 receptor. Proc Natl Acad Sci U S A. 2012;109:7055–60. doi: 10.1073/pnas.1118359109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Luttrell LM, van Biesen T, Hawes BE, Koch WJ, Touhara K, Lefkowitz RJ. Gβγ subunits mediate mitogen-activated protein kinase activation by the tyrosine kinase insulin-like growth factor 1 receptor. J Biol Chem. 1995;270:16495–8. doi: 10.1074/jbc.270.28.16495. [DOI] [PubMed] [Google Scholar]
- 54.Mayor F, Jr, Lucas E, Jurado-Peuyo M, Garcia-Guerra L, Nieto-Vazquez I, Vila-Bedmar R, Fernandez-Veledo S, Murga C. G protein-coupled receptor kinase 2 (GRK2): A novel modulator of insulin resistance. Arch Physiol Biochem. 2011;117:125–30. doi: 10.3109/13813455.2011.584693. [DOI] [PubMed] [Google Scholar]
- 55.Ciccarelli M, Cipolletta E, Iaccarino G. GRK2 at the control shaft of cellular metabolism. Curr Pharm Des. 2012;18:121–7. doi: 10.2174/138161212799040493. [DOI] [PubMed] [Google Scholar]
- 56.Garcia-Guerra L, Nieto-Vazquez I, Vila-Bedmar R, Jurado-Pueyo M, Zalba G, Diez J, Murga C, Fernandez-Veledo S, Mayor F, Jr, Lorenzo M. G protein-coupled receptor kinase 2 plays a relevant role in insulin resistance and obesity. Diabetes. 2010;59:2407–17. doi: 10.2337/db10-0771. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 57.Xing W, Li Y, Zhang H, Mi C, Hou Z, Quon MJ, Gao F. Improvement of vascular insulin sensitivity by downregulation of GRK2 mediates exercise-induced alleviation of hypertension in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2013;305:H1111–9. doi: 10.1152/ajpheart.00290.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Anis Y, Leshem O, Reuveni H, Wexler I, Ben Sasson R, Yahalom B, Laster M, Raz I, Ben Sasson S, Shafrir E, Ziv E. Antidiabetic effect of novel modulating peptides of G-protein-coupled kinase in experimental models of diabetes. Diabetologia. 2004;47:1232–44. doi: 10.1007/s00125-004-1444-1. [DOI] [PubMed] [Google Scholar]
- 59.Usui H, Nishiyama M, Moroi K, Shibasaki T, Zhou J, Ishida J, Fukamizu A, Haga T, Sekiya S, Kimura S. RGS domain in the amino-terminus of G protein-coupled receptor kinase inhibits Gq-mediated signaling. Int J Mol Med. 2000;5:335–40. doi: 10.3892/ijmm.5.4.335. [DOI] [PubMed] [Google Scholar]
- 60.Usui I, Imamura T, Satoh H, Huang J, Babendure JL, Hupfeld CJ, Olefsky JM. GRK2 is an endogenous protein inhibitor of the insulin signaling pathway for glucose transport stimulation. EMBO J. 2004;23:2821–9. doi: 10.1038/sj.emboj.7600297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Usui I, Imamura T, Babendure JL, Satoh H, Lu JC, Hupfeld CJ, Olefsky JM. G protein-coupled receptor kinase 2 mediates endothelin-1-induced insulin resistance via the inhibition of both Gαq/11 and insulin receptor substrate-1 pathways in 3T3-L1 adipocytes. Mol Endocrinol. 2005;19:2760–8. doi: 10.1210/me.2004-0429. [DOI] [PubMed] [Google Scholar]
- 62.Liu S, Premont RT, Kontos CD, Zhu S, Rockey DC. A crucial role for GRK2 in regulation of endothelial cell nitric oxide synthase function in portal hypertension. Nat Med. 2005;11:952–8. doi: 10.1038/nm1289. [DOI] [PubMed] [Google Scholar]
- 63.Obrenovich ME, Smith MA, Siedlak SL, Chen SG, de la Torre JC, Perry G, Aliev G. Overexpression of GRK2 in Alzheimer disease and in a chronic hypoperfusion rat model is an early marker of brain mitochondrial lesions. Neurotox Res. 2006;10:43–56. doi: 10.1007/BF03033333. [DOI] [PubMed] [Google Scholar]
- 64.Fusco A, Santulli G, Sorriento D, Cipolletta E, Garbi C, Dorn GW, 2nd, Trimarco B, Feliciello A, Iaccarino G. Mitochondrial localization unveils a novel role for GRK2 in organelle biogenesis. Cell Signal. 2012;24:468–75. doi: 10.1016/j.cellsig.2011.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen M, Sato PY, Chuprun JK, Peroutka RJ, Otis NJ, Ibetti J, Pan S, Sheu S, Gao E, Koch WJ. Prodeath signaling of G protein-coupled receptor kinase 2 in cardiac myocytes after ischemic stress occurs via extracellular signal-regulated kinase-dependent heat shock protein 90-mediated mitochondrial targeting. Circ Res. 2013;112:1121–34. doi: 10.1161/CIRCRESAHA.112.300754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huang ZM, Gao E, Fonseca F, Hayashi H, Shang Xiying, Hoffman NE, Chuprun JK, Tian X, Madesh M, Lefer DJ, Stamler JS, Koch WJ. Convergence of G-protein coupled receptor and nitric oxide pathways determines the outcome to cardiac ischemic injury. Science Signaling. 2013;6:ra95. doi: 10.1126/scisignal.2004225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Whalen EJ, Foster MW, Matsumoto A, Ozawa K, Violin JD, Que LG, Nelson CD, Benhar M, Keys JR, Rockman HA, Koch WJ, Daaka Y, Lefkowitz RJ, Stamler JS. Regulation of β-adrenergic receptor signaling by S-nitrosylation of G-protein-coupled receptor kinase 2. Cell. 2007;129:511–522. doi: 10.1016/j.cell.2007.02.046. [DOI] [PubMed] [Google Scholar]