

Abstract
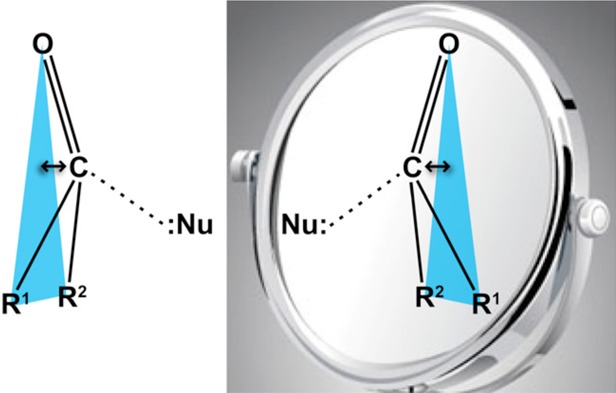
An n→π* interaction stems from the delocalization of the electron pair (n) of a donor group into the antibonding orbital (π*) of a carbonyl group. Crystallographic analyses of five pairs of diastereoisomers demonstrate that an n→π* interaction can induce chirality in an otherwise planar, prochiral carbonyl group. Thus, a subtle delocalization of electrons can have stereochemical consequences.
“Noncovalent” interactions can involve electron delocalization and thus partial covalency. For example, the partial covalency in a hydrogen bond stems from delocalization of the electron lone pair (n) of the hydrogen-bond acceptor into the antibonding orbital (σ*) of the hydrogen bond donor.1 Similarly, the partial covalency in an n→π* interaction stems from the delocalization of the electron pair (n) of a donor group into the antibonding orbital (π*) of a carbonyl group.2 This interaction is reminiscent of the Bürgi–Dunitz trajectory for nucleophilic addition to carbonyl groups.3 In common protein secondary structures, the electron-pair donor is a proximal amide oxygen in the backbone.2g,2i The partial covalency in these interactions induces an intimate association of the interacting donor and acceptor groups, wherein their van der Waals surfaces interpenetrate.
Recently, we proposed that an n→π* interaction can engender experimentally detectable pyramidalization of a planar carbonyl group.2a In this pyramidalization, the acceptor carbonyl carbon would be displaced toward the donor and away from the plane formed by the three atoms linked to the carbonyl carbon (Figure 1). Such pyramidalization would transform an otherwise prochiral carbonyl group into a chiral entity.4,5 Although analogous distortions have been noted for reactive carbonyl groups (e.g., ketones) proximal to potent nucleophiles (e.g., amines),3 it is less clear if they can result from an n→π* interaction involving weak nucleophiles, such as the oxygen of a carbonyl group. If pyramidalization were to result from such a weak force, its magnitude would be small,2y,6 requiring that crystal structures be solved by direct methods rather than by experimental phasing or molecular replacement. Even so, any observed pyramidalization could be attributable to the packing of the crystal lattice or other forces.7
Figure 1.
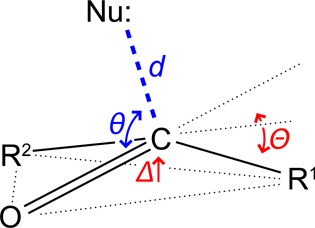
Parameters describing carbonyl group pyramidalization due to an n→π* interaction.
To overcome such artifacts, we sought to determine the crystal structures of diastereoisomers that differ in the position of one of two putative electron-pair donors with respect to an acceptor carbonyl group in the same molecule. We reasoned that if the displacement of the carbonyl carbon correlated with the position of the donor, then an n→π* interaction is responsible for the carbonyl pyramidalization. Conversely, the lack of such a correlation would implicate other forces.
Previously, we showed that installation of a fluoro group at Cγ of AcProOMe enlists a gauche effect.8 In the R configuration, a fluoro group enforces a Cγ-exo ring pucker, which brings the donor and acceptor carbonyl groups together for a strong “frontal” n→π* interaction.9 Conversely, in the S configuration, a fluoro group enforces a Cγ-endo ring pucker, which allows for only a weak n→π* interaction between the more distal donor and acceptor carbonyl groups.9 The endo pucker does, however, place the S-configured fluoro group in close proximity to the other “rear” face of the carbonyl group. We envisioned that a transannular n→π* interaction10 can be effected by this “rear” face of the carbonyl if the S-configured fluoro group is replaced with a good electron-pair donor.11 The direction of carbonyl carbon displacement induced by a “rear” n→π* interaction would be opposite to that effected by the “frontal” n→π* interaction.
To test this hypothesis, we synthesized and examined by X-ray crystallography structures of five pairs of diastereoisomers (1–5) with “frontal” or “rear” n→π* interactions arising from the R and S diastereoisomers, respectively (Figure 2). We quantified the extent of pyramidalization by using the parameters Δ and Θ (Figure 1). In accordance with our hypothesis, we found that the displacement of the carbonyl carbon is always toward the electron-pair donor that is proximal to the acceptor carbonyl carbon (Figure 2 and Table 1).
Figure 2.
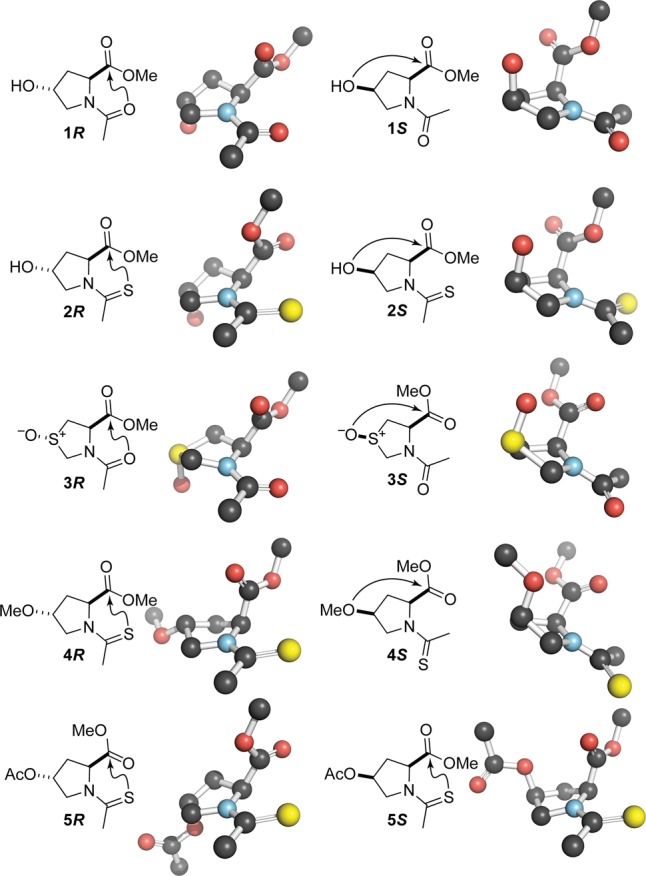
n→π* Interactions that elicit carbonyl group pyramidalization in five pairs of diastereoisomers (1–5). Structures show non-hydrogen atoms as determined by X-ray crystallography. For computational analyses of n→π* interactions in crystalline 1–5, see Figure S7 and Table S37 in the Supporting Information.
Table 1. Structural Parameters for Crystalline Compounds 1–5.
| compd | donor atom | C′i=Oi face | d (Å) | θ (deg) | Δ (Å) | Θ (deg) |
|---|---|---|---|---|---|---|
| 1Ra | O | re | 2.83 | 91.8 | 0.017 | 2.1 |
| 1Sb | O | re | 2.95 | 100.1 | 0.027 | 3.3 |
| 2R | S | re | 3.05 | 105.7 | 0.041 | 5.0 |
| 2S | O | re | 2.86 | 106.0 | 0.013 | 1.6 |
| 3Rc | O | re | 3.01 | 91.9 | 0.025 | 2.9 |
| 3Sc | O | si | 2.93 | 98.9 | 0.054 | 6.3 |
| 4R | S | re | 3.31 | 103.1 | 0.026 | 3.0 |
| 4S | O | si | 3.02 | 100.3 | 0.025 | 3.0 |
| 5R | S | si | 3.03 | 106.5 | 0.053 | 6.3 |
| 5S | S | re | 3.37 | 92.3 | 0.007 | 0.8 |
Compounds 1 and 5 offer particular insight. In crystalline 1R and 1S, the re face of the carbonyl group is exposed to an n→π* interaction from two distinct donors (Figure 2). In crystalline 5R and 5S, the si or re face, respectively, of the carbonyl group is exposed to an n→π* interaction from the same donor. Despite these distinctions, the displacement of the carbonyl carbon in all four of these compounds is always toward the donor (Table 1).
These analyses indicate that carbonyl pyramidalization does indeed result from n→π* electronic delocalization. The induction of a stereogenic center is thus a signature of the n→π* interaction. Integrating the solid-state data herein with solution-state data from infrared spectroscopy2j,2l,9a,13 indicates that, despite being relatively weak, an n→π* interaction can have important consequences for carbonyl groups.
The ten crystal structures have other notable features. Six of the diastereoisomers adopt a ψ dihedral angle (Ni–Cαi–C′i–Ni+1 but with an oxygen replacing Ni+1) close to that in a right-handed α-helix (ψ ≈ −45°; 1R, 3R, 3S, 4R, 4S, and 5S), whereas four adopt a ψ dihedral angle close to that in a left-handed polyproline II helix (ψ ≈ 150°; 1S, 2R, 2S, and 5R). Evidently, either of these rotamers can gain stability from a “frontal” or “rear” n→π* interaction. In addition, all five of the R diastereoisomers have a trans (Z) amide bond, whereas three of the S diastereoisomers have a cis (E) amide bond (1S, 3S, and 4S). These data are consistent with the trans amide bond being stabilized by a “frontal” n→π* interaction,14 which is weaker in the presence of a “rear” n→π* interaction.2v,11 The bridging acetoxy oxygen of 5S is the weakest transannular donor in 1S–5S and alone fails to manifest a significant “rear” n→π* interaction.
Our data provide insight, in particular, on oxygen versus sulfur as a donor for an n→π* interaction. The pyramidalization effected by the thioamide donor in 2R is much greater than that by the isosteric amide donor in 1R (Table 1). This observation is consistent with a quantum mechanical basis for the n→π* interaction, as we have described elsewhere.2a,2u An oxygen can, however, be a strong donor, as in 3S. Although a sulfoxide is not a true isostere of a carbonyl group, its use to enhance an n→π* interaction merits consideration.
Our findings have general implications. Carbonyl pyramidalization has been reported in protein structures determined at high resolution.2y,6 These deformations could stem from an n→π* interaction. As the nitrogen of peptide bonds can be pyramidal,15 our data suggest that every backbone atom in a protein (except Cα of glycine residues) can be a stereogenic center. Moreover, pyramidalization of a carbonyl carbon could be accompanied by increased electron density on the carbonyl oxygen,16 making that oxygen a better hydrogen-bond acceptor. Such enhanced chirality and hydrogen-bonding ability might play unappreciated roles in biomolecular recognition processes. Finally, we note that n→π* interactions have the potential to relay chirality through a polypeptide or other organic polymer.
Acknowledgments
We are grateful to I. A. Guzei for assistance with X-ray crystallography and to C. N. Bradford, B. R. Caes, and M. D. Shoulders (University of Wisconsin–Madison), and B. Paul (New York University) for contributive discussions. R.W.N. was supported by Predoctoral Training Grant T32 GM008349 (NIH). This work was supported by Grant R01 AR044276 (NIH).
Supporting Information Available
Procedures for the synthesis and crystallographic analysis of compounds 2, 4, and 5, and results from computational analyses of n→π* interactions in crystalline 1–5. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Pimentel G. C., McClellan A. L.. The Hydrogen Bond; W. H. Freeman: San Francisco, CA, 1960. [Google Scholar]; b Reed A. E.; Curtiss L. A.; Weinhold F. Chem. Rev. 1988, 88, 899–926. [Google Scholar]; c Isaacs E. D.; Shukla A.; Platzman P. M.; Barbiellini D. R.; Tulk C. A. Phys. Rev. Lett. 1999, 82, 600–603. [Google Scholar]; d Weinhold F. In Advances in Protein Chemistry: Peptide Solvation and H-Bonds; Elsevier Academic Press: San Diego, CA, 2006; Vol. 72, pp 121–155. [Google Scholar]; e Khaliullin R. Z.; Cobar E. A.; Lochan R. C.; Bell A. T.; Head-Gordon M. J. Phys. Chem. A 2007, 111, 8753–8765. [DOI] [PubMed] [Google Scholar]; f Weinhold F.; Klein R. A.. Chem. Educ. Res. Pract.2014, 15, DOI: 10.1039/C4RP00030G. [DOI]
- For recent literature, see:; a Choudhary A.; Gandla D.; Krow G. R.; Raines R. T. J. Am. Chem. Soc. 2009, 131, 7244–7246. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chiang Y.-C.; Lin Y.-J.; Horng J.-C. Protein Sci. 2009, 18, 1967–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Dai N.; Etzkorn F. A. J. Am. Chem. Soc. 2009, 131, 13728–13732. [DOI] [PubMed] [Google Scholar]; d Kuemin M.; Schweizer S.; Ochsenfeld C.; Wennemers H. J. Am. Chem. Soc. 2009, 131, 15474–15482. [DOI] [PubMed] [Google Scholar]; e Gorske B. C.; Stringer J. R.; Bastian B. L.; Fowler S. A.; Blackwell H. E. J. Am. Chem. Soc. 2009, 131, 16555–16567. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Choudhary A.; Fry C. G.; Raines R. T. ARKIVOC 2010, 251–262. [PMC free article] [PubMed] [Google Scholar]; g Bartlett G. J.; Choudhary A.; Raines R. T.; Woolfson D. N. Nat. Chem. Biol. 2010, 6, 615–620. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Choudhary A.; Kamer K. J.; Powner M. W.; Sutherland J. D.; Raines R. T. ACS Chem. Biol. 2010, 5, 655–657. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Fufezan C. Proteins 2010, 78, 2831–2838. [DOI] [PubMed] [Google Scholar]; j Kuemin M.; Nagel Y. A.; Schweizer S.; Monnard F. W.; Ochsenfeld C.; Wennemers H. Angew. Chem., Int. Ed. 2010, 49, 6324–6327. [DOI] [PubMed] [Google Scholar]; k Jakobsche C. E.; Choudhary A.; Raines R. T.; Miller S. J. J. Am. Chem. Soc. 2010, 132, 6651–6653. [DOI] [PMC free article] [PubMed] [Google Scholar]; l Shoulders M. D.; Kotch F. W.; Choudhary A.; Guzei I. A.; Raines R. T. J. Am. Chem. Soc. 2010, 132, 10857–10865. [DOI] [PMC free article] [PubMed] [Google Scholar]; m Choudhary A.; Pua K. H.; Raines R. T. Amino Acids 2011, 39, 181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]; n Choudhary A.; Raines R. T. Protein Sci. 2011, 20, 1077–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]; o Pollock S. B.; Kent S. B. H. Chem. Commun. 2011, 47, 2342–2344. [DOI] [PubMed] [Google Scholar]; p Choudhary A.; Kamer K. J.; Raines R. T. J. Org. Chem. 2011, 76, 7933–7937. [DOI] [PMC free article] [PubMed] [Google Scholar]; q Choudhary A.; Kamer K. J.; Raines R. T. Protein Sci. 2012, 21, 171–177. [DOI] [PMC free article] [PubMed] [Google Scholar]; r Erdmann R. S.; Wennemers H. J. Am. Chem. Soc. 2012, 134, 17117–17124. [DOI] [PubMed] [Google Scholar]; s Kamer K. J.; Choudhary A.; Raines R. T. J. Org. Chem. 2013, 78, 2099–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]; t Newberry R. W.; Raines R. T. Chem. Commun. 2013, 49, 7699–7701. [DOI] [PMC free article] [PubMed] [Google Scholar]; u Newberry R. W.; VanVeller B.; Guzei I. A.; Raines R. T. J. Am. Chem. Soc. 2013, 135, 7843–7846. [DOI] [PMC free article] [PubMed] [Google Scholar]; v Choudhary A.; Fry C. G.; Kamer K. J.; Raines R. T. Chem. Commun. 2013, 49, 8166–8168. [DOI] [PMC free article] [PubMed] [Google Scholar]; w Bartlett G. J.; Newberry R. W.; VanVeller B.; Raines R. T.; Woolfson D. N. J. Am. Chem. Soc. 2013, 135, 18682–18688. [DOI] [PMC free article] [PubMed] [Google Scholar]; x Guzei I. A.; Choudhary A.; Raines R. T. Acta Crystallogr. Sect. E 2013, 69, o805–o806. [DOI] [PMC free article] [PubMed] [Google Scholar]; y Newberry R. W.; Bartlett G. J.; VanVeller B.; Woolfson D. N.; Raines R. T. Protein Sci. 2014, 23, 284–288. [DOI] [PMC free article] [PubMed] [Google Scholar]; z Newberry R. W.; Raines R. T. ACS Chem. Biol. 2014, 9, 880–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bürgi H. B.; Dunitz J. D.; Shefter E. J. Am. Chem. Soc. 1973, 95, 5065–5067. [Google Scholar]; b Bürgi H. B.; Dunitz J. D.; Shefter E. Acta Crystallogr. Sect. B 1974, 30, 1517–1527. [Google Scholar]; c Bürgi H. B.; Dunitz J. D.; Lehn J. M.; Wipff G. Tetrahedron 1974, 30, 1563–1572. [Google Scholar]; d Bürgi H. B.; Dunitz J. D. Acc. Chem. Res. 1983, 16, 153–161. [Google Scholar]
- a LeBel J. A. Bulletin de la Société Chimique de Paris 1874, 22, 337–347. [Google Scholar]; b van’t Hoff J. H. Archives Néerlandaises des Sciences Exactes et Naturelles 1874, 9, 445–454. [Google Scholar]; c Richardson G. M., Ed. The Foundations of Stereo Chemistry: Memoirs by Pasteur, van’t Hoff, LeBel, and Wislicenus; American Book Co.: New York, NY, 1901. [Google Scholar]
- a Heilbronner E.; Dunitz J. D.. Reflections on Symmetry in Chemistry...and Elsewhere; Verlag Helvetica Chimica Acta: Basel, Switzerland, 1993. [Google Scholar]; b Eliel E. L.; Wilen S. H.. Stereochemistry of Organic Compounds; John Wiley & Sons: New York, NY, 1994. [Google Scholar]; c Ramberg P. J.Chemical Structure, Spatial Arrangement: The Early History of Stereochemistry, 1874–1914; Ashgate Publishing Co.: Burlington, VT, 2003. [Google Scholar]
- Esposito L.; Vitagliano L.; Zagari A.; Mazzarella L. Protein Sci. 2000, 9, 2038–2042. [PMC free article] [PubMed] [Google Scholar]
- a Rondan N. G.; Paddon-Row M. N.; Caramella P.; Houk K. N. J. Am. Chem. Soc. 1981, 103, 2436–2438. [Google Scholar]; b Jeffrey G. A.; Houk K. N.; Paddon-Row M. N.; Rondan N. G.; Mitra J. J. Am. Chem. Soc. 1985, 107, 321–326. [Google Scholar]
- Eberhardt E. S.; Panasik N. Jr.; Raines R. T. J. Am. Chem. Soc. 1996, 118, 12261–12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bretscher L. E.; Jenkins C. L.; Taylor K. M.; DeRider M. L.; Raines R. T. J. Am. Chem. Soc. 2001, 123, 777–778. [DOI] [PubMed] [Google Scholar]; b DeRider M. L.; Wilkens S. J.; Waddell M. J.; Bretscher L. E.; Weinhold F.; Raines R. T.; Markley J. L. J. Am. Chem. Soc. 2002, 124, 2497–2505. [DOI] [PubMed] [Google Scholar]
- Lesarri A.; Cocinero E. J.; López J. C.; Alonso J. L. J. Am. Chem. Soc. 2005, 127, 2572–2579. [DOI] [PubMed] [Google Scholar]
- In contrast, a transannular C′i=Oi···H–N/O hydrogen bond from a 4S-configured NH or OH can enhance C′i=Oi as an acceptor of a frontal n→π* interaction. See: References (2l), (2r), andErdmann R. S.; Wennemers H. Angew. Chem., Int. Ed. 2011, 50, 6835–6838. [DOI] [PubMed] [Google Scholar]
- Panasik N. Jr.; Eberhardt E. S.; Edison A. S.; Powell D. R.; Raines R. T. Int. J. Pept. Protein Res. 1994, 44, 262–269. [DOI] [PubMed] [Google Scholar]
- Sonntag L.-S.; Schweizer S.; Ochsenfeld C.; Wennemers H. J. Am. Chem. Soc. 2006, 128, 14697–14703. [DOI] [PubMed] [Google Scholar]
- Hinderaker M. P.; Raines R. T. Protein Sci. 2003, 12, 1188–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a MacArthur M. W.; Thornton J. M. J. Mol. Biol. 1996, 264, 1180–1195. [DOI] [PubMed] [Google Scholar]; b Hu J.-S.; Bax A. J. Am. Chem. Soc. 1997, 119, 6360–6368. [Google Scholar]
- Lario P. I.; Vrielink A. J. Am. Chem. Soc. 2003, 125, 12787–12794. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.