

Abstract
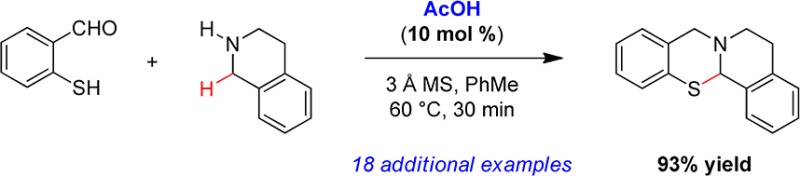
Secondary amines react with thiosalicylaldehydes in the presence of catalytic amounts of acetic acid to generate ring-fused N,S-acetals in redox-neutral fashion. A broad range of amines undergo α-sulfenylation, including challenging substrates such morpholine, thiomorpholine, and piperazines. Computational studies employing density functional theory indicate that acetic acid reduces the energy barriers of two separate steps, both of which involve proton transfer.
The N,S-acetal motif is common in nature and is present as a key functional group in pharmacologically active compounds (Figure 1).1N,S-Acetals have been investigated as sedatives (e.g., 1 and 2),1a antibacterials (e.g., 3),1d and cell growth inhibitors (e.g., 4).1c Penicillins such as amoxicillin (5) are widely used as antibacterial medicines.1i Traditional synthetic approaches to ring-fused N,S-acetals include the condensation of preformed imines with thiosalicylic acid, often requiring the addition of a coupling reagent (e.g., Scheme 1).1a,2,3 Here we report a new approach to N,S-acetals starting from thiosalicylaldehydes and secondary amines. The key feature of this process is a redox-neutral amine α-C–H bond functionalization with concurrent N-alkylation/α-sulfenylation.4,5
Figure 1.
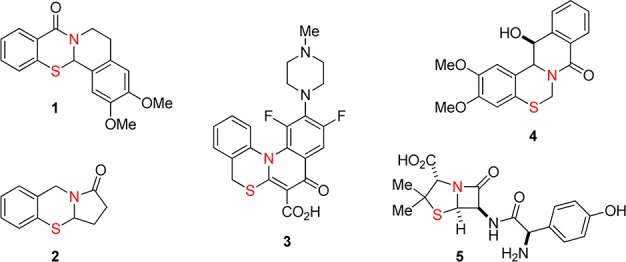
Examples of bioactive N,S-acetals.
Scheme 1. Selected Approaches to N,S-Acetals.

Previous work in our group has focused on the redox-neutral α-C–H bond functionalization of amines,6−8 including the α-amination of secondary amines with o-aminobenzaldehydes7 and α-oxygenation with salicylaldehydes.8 Through extensive experimental and computational studies, we have established the mechanisms of the α-amination and α-oxygenation reactions and revealed an important role for azomethine ylides as reactive intermediates.9,10 We recognized that an analogous α-sulfenylation of secondary amines with thiosalicylaldehydes would provide a practical entry to ring-fused N,S-acetals not easily accessible by other means. Based on the greater nucleophilicity of thiols compared to alcohols, we anticipated that α-sulfenylation might occur with a wider range of substrates.
The title reaction was evaluated using thiosalicylaldehyde (6-S) and 1,2,3,4-tetrahydroisoquinoline (THIQ) as the model substrates. Starting from conditions that were similar to those found ideal for the formation of the corresponding aminal and N,O-acetal analogues, a brief optimization survey was conducted (Table 1). Remarkably, the reaction of thiosalicylaldehyde (6-S) and THIQ was found to proceed in the absence of any additive at room temperature in ethanol solution to provide product 7a in 40% yield (entry 1). In toluene as the solvent, an increased yield of 51% was observed (entry 2). While higher temperatures served to improve the yield further (entries 3 and 4), the addition of acetic acid was found to have a more dramatic effect. With 10 mol % of acetic acid, 7a was obtained in 90% yield following a reaction time of just 2 h at room temperature (entry 5). Raising the reaction temperature to 60 °C in an otherwise identical experiment led to full conversion in only 30 min while allowing for the isolation of 7a in 93%, the highest yield observed (entry 6). As previously noted in the corresponding N,O-acetal formation,8 removal of water from the reaction mixture was crucial in order to achieve rapid conversion. A reaction conducted under otherwise optimal conditions but in the absence of molecular sieves led to the formation of 7a in only 46% yield after 1 h (entry 7). Interestingly, increasing the amount of acetic acid to one equivalent had a detrimental effect on conversion and product yield while leading to an increased formation of unidentified byproducts (entries 8 and 9). This observation is in contrast to what was seen for N,O-acetal formation where an increase in the amount of acid proved highly beneficial.8
Table 1. Evaluation of Reaction Conditions for α-Sulfenylation of 1,2,3,4-Tetrahydroisoquinoline with Thiosalicylaldehyde (6-S)a.
| entry | AcOH (equiv) | solvent | temp (°C) | time (h) | yield (%) |
|---|---|---|---|---|---|
| 1 | 0 | EtOH | rt | 9 | 40 |
| 2 | 0 | PhMe | rt | 18 | 51 |
| 3 | 0 | PhMe | 60 | 0.5 | 66 |
| 4 | 0 | PhMe | 120b | 0.17 | 60 |
| 5 | 0.1 | PhMe | rt | 2 | 90 |
| 6 | 0.1 | PhMe | 60 | 0.5 | 93 |
| 7c | 0.1 | PhMe | 60 | 1 | 46 |
| 8 | 1.0 | PhMe | rt | 36 | trace |
| 9 | 1.0 | PhMe | 60 | 1.5 | 18 |
Reactions were conducted on a 1 mmol scale. Yields correspond to isolated yields of chromatographically purified product.
Microwave irradiation in sealed vial.
Without molecular sieves.
The α-sulfenylation with thiosalicylaldehyde was evaluated with a broad range of secondary amines (Scheme 2). A number of cyclic amines such as pyrrolidine, piperidine, and azepane underwent the reaction with thiosalicylaldehyde at moderate temperatures to give products in generally good yields. Relatively electron-deficient amines such as morpholine and N-phenylpiperazine, substrates that are typically rather reluctant to undergo α-C–H bond functionalization, furnished the corresponding products at elevated temperatures (microwave irradiation at 120–150 °C). Initial attempts to synthesize these N,S-acetals at 60–90 °C required longer reaction times to reach complete consumption of the starting materials. In addition, it was found that for these substrates, oxidative dimerization of thiosalicylaldehyde to the corresponding disulfide was a competing process. This undesirable reaction pathway was reduced at elevated temperatures and further minimized by using a larger excess of the amine (3 equiv). Under these conditions, dibenzylamine, a representative open-chain substrate, generated the corresponding product 7i in good yield. Several other cyclic amines with benzylic α-C–H bonds, including the sterically demanding 1-phenyl-THIQ, underwent N,S-acetal formation under mild conditions. Finally, ring-substitution of thiosalicylaldehyde with either electron-donating or -withdrawing groups was well tolerated.
Scheme 2. Substrate Scope for the α-Sulfenylation.

Reactions were performed on a 1 mmol scale.
With 3 equiv of amine.
In order to explore the regioselectivity of the N,S-acetal formation for substrates with electronically similar α-C–H bonds, 6-S was allowed to react with 1-methyl pyrrolidine and 1-methylpiperidine (Scheme 3). Interestingly, in both cases the product distribution reflects a preference for functionalization of a secondary over an electronically favorable tertiary C–H bond. With the increased atomic radius size of sulfur relative to oxygen, steric constraints appear to have a far greater effect, outweighing electronic effects in these instances. This is in stark contrast to the corresponding aminal formation with 1-methylpyrrolidine and 1-methylpiperidine that exhibit a pronounced preference for tertiary C–H bond functionalization.7 Interestingly, the major products 7r and 7t were also obtained in higher diastereomeric ratios than their aminal counterparts.
Scheme 3. Regioselectivity of the α-Sulfenylation.

Reactions were performed on a 1 mmol scale.
To rationalize the enhanced reactivities in the N,S-acetal series compared to the corresponding N,O-acetals, we analyzed the model reaction between thiosalicylaldehyde (6–S) and THIQ by the same computational method described previously (M06-2X-D3/def2-TZVPP/IEFPCM(toluene)//TPSS-D2/6-31+G(d,p)/IEFPCM(toluene); see the Supporting Information for details of the computational method).8
For the uncatalyzed reactions without acetic acid, the calculated free energy profiles for the oxo- and thio pathways are summarized in Figure 2. The hemiaminals 8-O and 8-S as well as the transition states for the dehydration (TS1-O/S) are very similar for both systems. In contrast, a substantial difference was calculated for all other intermediates and transition states. While the sulfur-compound of 9, TS2, and 10 is 4–5 kcal mol–1 more stable than the oxygen analogue, differences of more than 10 kcal mol–1 were calculated for TS3, 11, and 7a. This stability difference can also be rationalized with the higher acidity of thiophenol compared to phenol in both DMSO (ΔpKa ≈ 8) and aqueous solution (ΔpKa ≈ 3).11 This difference in acidity might also be responsible for the fact that no thiosalicylaldehyde-mediated proton transfer (e.g., the thio-analogue of TS3-O-Sali)8 could be located.
Figure 2.
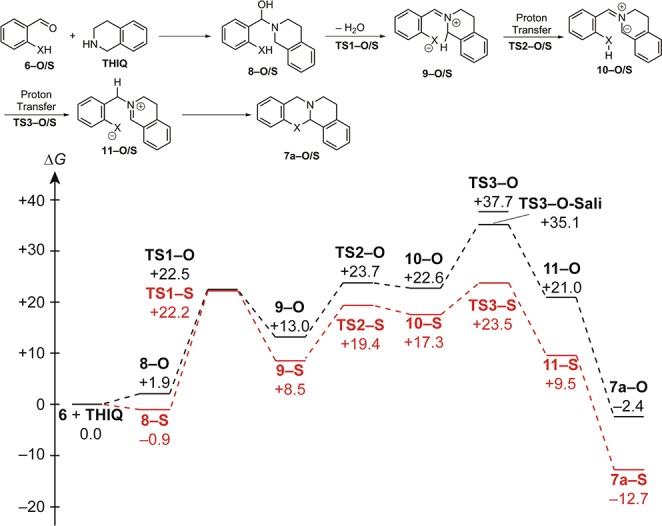
Free energy profile [in kcal·mol–1, M06-2X-D3/def2-TZVPP/IEFPCM//TPSS-D2/6-31+G(d,p)/IEFPCM] for uncatalyzed transformations of 6-O (black) and 6-S (red) and THIQ in toluene.
Next, we analyzed whether acetic acid has the same catalytic effect for the synthesis of N,S-acetals as previously described for the corresponding N,O-acetals.8 Figure 3 summarizes the calculated transition states for the uncatalyzed and acetic-acid-catalyzed N,S-acetal formation. Similar to the formation of N,O-acetals, acetic acid stabilizes the transition states TS1-S (ΔΔG⧧ = −1.5 kcal mol–1) and TS3–S (ΔΔG⧧ = −2.6 kcal mol–1). As previously reported for the formation of N,O-acetals,8 transition state TS2–S for the endergonic transformation of 9-S to 10-S is actually destabilized by acetic acid (ΔΔG⧧ = +10.9 kcal mol–1). Again, a small barrier (with respect to 10-S) and the entropic penalty (−TΔS) render TS2-S-HOAc less favorable than TS2-S and are responsible for the preference of the intramolecular proton transfer over the intermolecular process for this step.
Figure 3.

Calculated transition-state structures [M06-2X-D3/def2-TZVPP/IEFPCM//TPSS-D2/6-31+G(d,p)/IEFPCM], relative free energies (in kcal·mol–1), and selected bond lengths (in Å) for the uncatalyzed and acetic-acid-catalyzed transformation of 6-S and THIQ.
Because of the higher acidities of thiols, the rate-determining step (TS3-S) is lowered to a much smaller extent than in the N,O-acetal series (2.6 vs 13.1 kcal mol–1). These computational findings are also reflected in the experimental data of Table 1, as acetic acid is not necessarily required as a catalyst for the formation of N,S-acetals but is ultimately needed in the N,O-acetal series.
In summary, a relatively mild and effective approach to ring-fused N,S-acetals has been developed. Further exploration of related processes is under active investigation.
Acknowledgments
Financial support from the NIH–NIGMS (Grant R01GM101389-01 to D.S.) is gratefully acknowledged. We thank Alena Yu. Platonova (Rutgers University) for conducting preliminary experimental studies. This work used resources from the UCLA Institute for Digital Research and Education (IDRE) and the Cologne High Efficiency Operating Platform for Sciences (CHEOPS). We are grateful to the Alexander von Humboldt foundation (Feodor Lynen fellowship to M.B.), the Fonds der Chemischen Industrie (Liebig scholarship to M.B.), and the National Science Foundation (Grant No. CHE-1059084 to K.N.H.) for support of this research.
Supporting Information Available
Experimental procedures, characterization data, details of computational methods, Cartesian coordinates, and energies of all reported structures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Lombardino J. G.; McLamore W. M.; Lanbach G. D.. Derivatives of thiabenzopyrrocoline, thiabenzopyridocoline and thiazepine. US2985649A, 1961.; b Sammes P. G. Chem. Rev. 1976, 76, 113. [Google Scholar]; c Fodor L.; Szabó J.; Bernáth G.; Sohár P.; Maclean D. B.; Smith R. W.; Ninomiya I.; Naito T. J. Heterocycl. Chem. 1989, 26, 333. [Google Scholar]; d Cecchetti V.; Filipponi E.; Fravolini A.; Tabarrini O.; Xin T. Bioorg. Med. Chem. 1997, 5, 1339. [DOI] [PubMed] [Google Scholar]; e Deziel R.; Malenfant E. Bioorg. Med. Chem. Lett. 1998, 8, 1437. [DOI] [PubMed] [Google Scholar]; f Hilton S. T.; Motherwell W. B.; Potier P.; Pradet C.; Selwood D. L. Bioorg. Med. Chem. Lett. 2005, 15, 2239. [DOI] [PubMed] [Google Scholar]; g Song Z.-C.; Ma G.-Y.; Lv P. C.; Li H. Q.; Xiao Z. P.; Zhu H. L. Eur. J. Med. Chem. 2009, 44, 3903. [DOI] [PubMed] [Google Scholar]; h Gomez- Monterrey I.; Bertamino A.; Porta A.; Carotenuto A.; Musella S.; Aquino C.; Granata I.; Sala M.; Brancaccio D.; Picone D.; Ercole C.; Stiuso P.; Campiglia P.; Grieco P.; Ianelli P.; Maresca B.; Novellino E. J. Med. Chem. 2010, 53, 8319. [DOI] [PubMed] [Google Scholar]; i Kaur S. P.; Rao R.; Nanda S. Int. J. Pharm. Pharm. Sci. 2011, 3, 30. [Google Scholar]
- Selected examples for the synthesis of ring-fused N,S-acetals:; a Marsden R.; MacLean D. B.; Fodor L. Can. J. Chem. 1984, 62, 2682. [Google Scholar]; b Okada E.; Masuda R.; Hojo M.; Tone H.; Tomifuji T. Heterocycles 1994, 37, 157. [Google Scholar]; c Hucher N.; Decroix B.; Daïch A. J. Org. Chem. 2001, 66, 4695. [DOI] [PubMed] [Google Scholar]; d Hucher N.; Pesquet A.; Netchitaïlo P.; Daïch A. Eur. J. Org. Chem. 2005, 2758. [Google Scholar]; e Unsworth W. P.; Kitsiou C.; Taylor R. J. K. Org. Lett. 2013, 15, 258. [DOI] [PubMed] [Google Scholar]
- Other selected examples of N,S-acetal formation:; a Rothe M.; Steinberger R. Tetrahedron Lett. 1970, 11, 649. [Google Scholar]; b Rothe M.; Steinberger R. Tetrahedron Lett. 1970, 11, 2467. [Google Scholar]; c Mahata P. K.; Venkatesh C.; Syam Kumar U. K.; Ila H.; Junjappa H. J. Org. Chem. 2003, 68, 3966. [DOI] [PubMed] [Google Scholar]; d Ingle G. K.; Mormino M. G.; Wojtas L.; Antilla J. C. Org. Lett. 2011, 13, 4822. [DOI] [PubMed] [Google Scholar]; e Stojanović M.; Marković R.; Kleinpeter E.; Baranac-Stojanović M. Org. Biomol. Chem. 2012, 10, 575. [DOI] [PubMed] [Google Scholar]; f Li X.; Qin Z.; Yang T.; Zhang H.; Wei S.; Li C.; Chen H.; Meng M. Bioorg. Med. Chem. Lett. 2012, 22, 2712. [DOI] [PubMed] [Google Scholar]
- Reviews on redox-economy:; a Burns N. Z.; Baran P. S.; Hoffmann R. W. Angew. Chem., Int. Ed. 2009, 48, 2854. [DOI] [PubMed] [Google Scholar]; b Newhouse T.; Baran P. S.; Hoffmann R. W. Chem. Soc. Rev. 2009, 38, 3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selected reviews on amine α-functionalization, including redox-neutral approaches:; a Murahashi S.-I. Angew. Chem., Int. Ed. Engl. 1995, 34, 2443. [Google Scholar]; b Matyus P.; Elias O.; Tapolcsanyi P.; Polonka-Balint A.; Halasz-Dajka B. Synthesis 2006, 2625. [Google Scholar]; c Campos K. R. Chem. Soc. Rev. 2007, 36, 1069. [DOI] [PubMed] [Google Scholar]; d Li C.-J. Acc. Chem. Res. 2009, 42, 335. [DOI] [PubMed] [Google Scholar]; e Jazzar R.; Hitce J.; Renaudat A.; Sofack-Kreutzer J.; Baudoin O. Chem.—Eur. J. 2010, 16, 2654. [DOI] [PubMed] [Google Scholar]; f Jones K. M.; Klussmann M. Synlett 2012, 23, 159. [Google Scholar]; g Pan S. C. Beilstein J. Org. Chem. 2012, 8, 1374. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Mitchell E. A.; Peschiulli A.; Lefevre N.; Meerpoel L.; Maes B. U. W. Chem.—Eur. J. 2012, 18, 10092. [DOI] [PubMed] [Google Scholar]; i Platonova A. Y.; Glukhareva T. V.; Zimovets O. A.; Morzherin Y. Y. Chem. Heterocycl. Compd. 2013, 49, 357. [Google Scholar]; j Prier C. K.; Rankic D. A.; MacMillan D. W. C. Chem. Rev. 2013, 113, 5322. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Peng B.; Maulide N. Chem.—Eur. J. 2013, 19, 13274. [DOI] [PubMed] [Google Scholar]; l Qin Y.; Lv J.; Luo S. Tetrahedron Lett. 2014, 55, 551. [Google Scholar]; m Wang L.; Xiao J. Adv. Synth. Catal. 2014, 356, 1137. [Google Scholar]; n Girard S. A.; Knauber T.; Li C.-J. Angew. Chem., Int. Ed. 2014, 53, 74. [DOI] [PubMed] [Google Scholar]; o Vo C.-V. T.; Bode J. W. J. Org. Chem. 2014, 79, 2809. [DOI] [PubMed] [Google Scholar]; p Haibach M. C.; Seidel D. Angew. Chem., Int. Ed. 2014, 53, 5010. [DOI] [PubMed] [Google Scholar]
- a Zhang C.; Murarka S.; Seidel D. J. Org. Chem. 2009, 74, 419. [DOI] [PubMed] [Google Scholar]; b Murarka S.; Zhang C.; Konieczynska M. D.; Seidel D. Org. Lett. 2009, 11, 129. [DOI] [PubMed] [Google Scholar]; c Murarka S.; Deb I.; Zhang C.; Seidel D. J. Am. Chem. Soc. 2009, 131, 13226. [DOI] [PubMed] [Google Scholar]; d Deb I.; Seidel D. Tetrahedron Lett. 2010, 51, 2945. [Google Scholar]; e Zhang C.; Seidel D. J. Am. Chem. Soc. 2010, 132, 1798. [DOI] [PubMed] [Google Scholar]; f Zhang C.; Das D.; Seidel D. Chem. Sci. 2011, 2, 233. [Google Scholar]; g Haibach M. C.; Deb I.; De C. K.; Seidel D. J. Am. Chem. Soc. 2011, 133, 2100. [DOI] [PubMed] [Google Scholar]; h Deb I.; Das D.; Seidel D. Org. Lett. 2011, 13, 812. [DOI] [PubMed] [Google Scholar]; i Deb I.; Coiro D. J.; Seidel D. Chem. Commun. 2011, 47, 6473. [DOI] [PubMed] [Google Scholar]; j Das D.; Richers M. T.; Ma L.; Seidel D. Org. Lett. 2011, 13, 6584. [DOI] [PubMed] [Google Scholar]; k Ma L.; Chen W.; Seidel D. J. Am. Chem. Soc. 2012, 134, 15305. [DOI] [PubMed] [Google Scholar]; l Das D.; Sun A. X.; Seidel D. Angew. Chem., Int. Ed. 2013, 52, 3765. [DOI] [PMC free article] [PubMed] [Google Scholar]; m Das D.; Seidel D. Org. Lett. 2013, 15, 4358. [DOI] [PMC free article] [PubMed] [Google Scholar]; n Chen W.; Wilde R. G.; Seidel D. Org. Lett. 2014, 16, 730. [DOI] [PMC free article] [PubMed] [Google Scholar]; o Seidel D. Org. Chem. Front. 2014, 1, 426. [DOI] [PMC free article] [PubMed] [Google Scholar]; p Chen W.; Kang Y.; Wilde R. G.; Seidel D. Angew. Chem., Int. Ed. 2014, 53, 5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zhang C.; De C. K.; Mal R.; Seidel D. J. Am. Chem. Soc. 2008, 130, 416. [DOI] [PubMed] [Google Scholar]; b Zhang C.; De C. K.; Seidel D. Org. Synth. 2012, 89, 274. [Google Scholar]; c Dieckmann A.; Richers M. T.; Platonova A. Y.; Zhang C.; Seidel D.; Houk K. N. J. Org. Chem. 2013, 78, 4132. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Richers M. T.; Deb I.; Platonova A. Y.; Zhang C.; Seidel D. Synthesis 2013, 45, 1730. [PMC free article] [PubMed] [Google Scholar]; e Richers M. T.; Zhao C. F.; Seidel D. Beilstein J. Org. Chem. 2013, 9, 1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richers M. T.; Breugst M.; Platonova A. Y.; Ullrich A.; Dieckmann A.; Houk K. N.; Seidel D. J. Am. Chem. Soc. 2014, 136, 6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recent examples of related redox-neutral amine C–H functionalizations that likely involve azomethine ylides as reactive intermediates:; a Oda M.; Fukuchi Y.; Ito S.; Thanh N. C.; Kuroda S. Tetrahedron Lett. 2007, 48, 9159. [Google Scholar]; b Zheng L.; Yang F.; Dang Q.; Bai X. Org. Lett. 2008, 10, 889. [DOI] [PubMed] [Google Scholar]; c Pahadi N. K.; Paley M.; Jana R.; Waetzig S. R.; Tunge J. A. J. Am. Chem. Soc. 2009, 131, 16626. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Mao H.; Xu R.; Wan J.; Jiang Z.; Sun C.; Pan Y. Chem.—Eur. J. 2010, 16, 13352. [DOI] [PubMed] [Google Scholar]; e Xue X. S.; Yu A.; Cai Y.; Cheng J. P. Org. Lett. 2011, 13, 6054. [DOI] [PubMed] [Google Scholar]; f Zheng Q.-H.; Meng W.; Jiang G.-J.; Yu Z.-X. Org. Lett. 2013, 15, 5928. [DOI] [PubMed] [Google Scholar]; g Lin W.; Cao T.; Fan W.; Han Y.; Kuang J.; Luo H.; Miao B.; Tang X.; Yu Q.; Yuan W.; Zhang J.; Zhu C.; Ma S. Angew. Chem., Int. Ed. 2014, 53, 277. [DOI] [PubMed] [Google Scholar]; h Haldar S.; Mahato S.; Jana C. K. Asian J. Org. Chem. 2014, 3, 44. [Google Scholar]; i Rahman M.; Bagdi A. K.; Mishra S.; Hajra A. Chem. Commun. 2014, 50, 2951. [DOI] [PubMed] [Google Scholar]; j Li J.; Wang H.; Sun J.; Yang Y.; Liu L. Org. Biomol. Chem. 2014, 12, 2523. [DOI] [PubMed] [Google Scholar]
- Selected reviews on azomethine ylides:; a Padwa A.; Pearson W. H.. Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products; Wiley: Chichester, 2002; Vol. 59. [Google Scholar]; b Najera C.; Sansano J. M. Curr. Org. Chem. 2003, 7, 1105. [Google Scholar]; c Coldham I.; Hufton R. Chem. Rev. 2005, 105, 2765. [DOI] [PubMed] [Google Scholar]; d Pandey G.; Banerjee P.; Gadre S. R. Chem. Rev. 2006, 106, 4484. [DOI] [PubMed] [Google Scholar]; e Nyerges M.; Toth J.; Groundwater P. W. Synlett 2008, 1269. [Google Scholar]; f Adrio J.; Carretero J. C. Chem. Commun. 2011, 47, 6784. [DOI] [PubMed] [Google Scholar]
- a Bordwell F. G.; Hughes D. L. J. Org. Chem. 1982, 47, 3224. [Google Scholar]; b Bordwell F. G.; McCallum R. J.; Olmstead W. N. J. Org. Chem. 1984, 49, 1424. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.