

Abstract
There is a significant need for new antibiotics due to the rise in drug resistance. Drugs such as methicillin and vancomycin target bacterial cell wall biosynthesis, but methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant Enterococci (VRE) have now arisen and are of major concern. Inhibitors acting on new targets in cell wall biosynthesis are thus of particular interest since they might also restore sensitivity to existing drugs, and the cis-prenyl transferase undecaprenyl diphosphate synthase (UPPS), essential for lipid I, lipid II, and thus, peptidoglycan biosynthesis, is one such target. We used 12 UPPS crystal structures to validate virtual screening models and then assayed 100 virtual hits (from 450,000 compounds) against UPPS from S. aureus and Escherichia coli. The most promising inhibitors (IC50 ∼2 μM, Ki ∼300 nM) had activity against MRSA, Listeria monocytogenes, Bacillus anthracis, and a vancomycin-resistant Enterococcus sp. with MIC or IC50 values in the 0.25–4 μg/mL range. Moreover, one compound (1), a rhodanine with close structural similarity to the commercial diabetes drug epalrestat, exhibited good activity as well as a fractional inhibitory concentration index (FICI) of 0.1 with methicillin against the community-acquired MRSA USA300 strain, indicating strong synergism.
Introduction
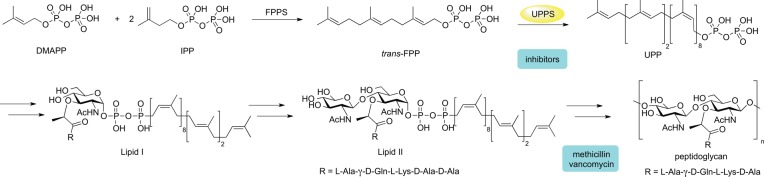
The need for new antibiotics has arisen due to the widespread resistance to current drugs.1 Despite this need, the antibiotic pipeline in the past few decades has been relatively dry in terms of new antibacterial classes when compared with progress against other diseases.2 One strategy to fight bacterial resistance is to inhibit enzymes that are not the targets of current antibiotics but, instead, act in the same pathways as existing drugs since this might enable the restoration of drug sensitivity via combination therapy. Undecaprenyl diphosphate synthase (UPPS) is one such target. The undecaprenyl diphosphate product (UPP) is essential for bacterial cell growth because of its role in the formation of bacterial cell wall peptidoglycan,1,3 Scheme 1, and it is not produced by humans.2,4
Scheme 1. Undecaprenyl Diphosphate Synthase Reaction and Relationship of UPP to Bacterial Cell Wall Biosynthesis.
SmithKline Beecham screened their compound collection against UPPS but reported no chemically tractable low micromolar hits.5 Novartis pursued tetramic and tetronic acids and dihydropyridin-2-ones, but noted issues associated with human serum albumin binding and a lack of in vivo activity.6,7 Previously, we reported several potent UPPS inhibitors together with X-ray crystallographic (or modeled) binding modes for a variety of chemical classes including lipophilic bisphosphonates,8 phthalic acids,9 diketo acids,10 anthranilic acids, benzoic acids,11,12 aryl phosphonates, bis-amines, and bis-amidines.12 The most promising of these compounds, a bis-amidine, was shown to have potent activity in biochemical assays, in cellular assays, and in a murine model of MRSA infection.12
Since UPPS must bind multiple substrates (IPP, FPP, or more elongated prenyl-PP intermediates) and many inhibitors are to some degree substrate mimics, it is common to observe numerous inhibitors simultaneously bound to UPPS, with up to 4 binding sites being occupied.8 However, it is unclear whether inhibitory activity is due to binding to one specific site or to multiple sites. It has been shown that some inhibitors occupy only site 4, an allosteric site distant from the catalytic center, while others bind to site 1, the substrate binding site,12 complicating docking studies and, regardless of the inhibitor-binding mode, the flexibility of UPPS creates challenges for virtual screening. Here, to help reduce these problems we employed the 12 crystallographic structures described in previous work8,12 to select those that provided maximal enrichment in retrospective virtual screening studies. We then made prospective predictions using these structures, leading to novel UPPS inhibitors, some with promising antibacterial activity.
Methods and Materials
Computational Aspects
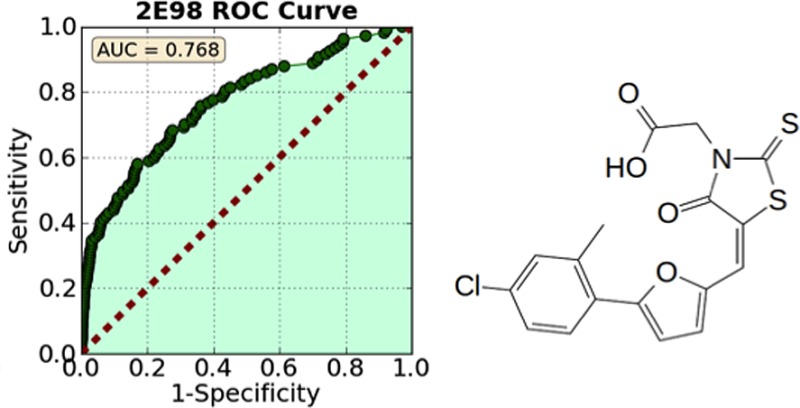
Following the methods described in previous work,12 we docked 112 known UPPS inhibitors having IC50 values <100 μM, together with 1000 decoys from the Schrödinger decoy collection (having an average molecular weight of 400 Da), to Escherichia coli UPPS (hereafter, EcUPPS). Docking was performed using the Glide13−15 program, and compounds were ranked by their Glide XP score. The proteins were prepared by stripping water and ligand molecules, capping, and neutralizing any unsolved loops, followed by preparation with the Schrödinger protein preparation wizard using standard parameters.16 After docking, compounds were ranked by their docking score, and then area under the curve (AUC) analyses were performed. Retrospective enrichment was quite good for 2/12 structures (PDB codes 2E98 and 4H3A), so we docked into these structures for the prospective studies (Figure 1). 2E98 is an EcUPPS X-ray structure containing four lipophilic bisphosphonates (BPH-629; IC50 ∼300 nM), which bind to sites 1–4, one inhibitor to each site.84H3A is an EcUPPS structure containing a diketo acid inhibitor (BPH-1330) which has a 2 μM IC50, and the inhibitor binds (in the solid state) only to site 4.10,12 These structures thus have significant differences: only site 4 is occupied in 4H3A, while in 2E98, all four sites are occupied and the protein is in a “wide-open” conformation (Figure 2).
Figure 1.

AUC/ROC curves for 12 EcUPPS crystal structures. 4H3A and 2E98 were chosen for further study.
Figure 2.
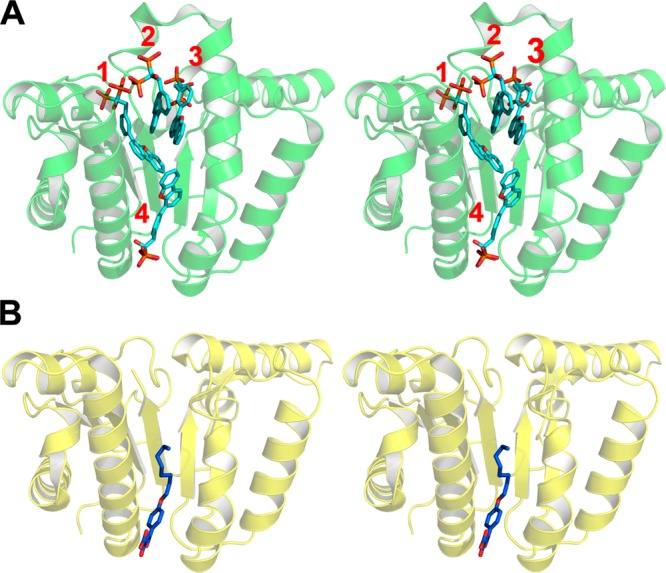
Stereo presentation of the X-ray structures chosen for further virtual screening from docking and ROC analysis. (A) 2E98 showing all four inhibitor binding sites. (B) 4H3A showing one inhibitor bound to site 4.
To find new inhibitors, we began with a library of ∼450,000 commercially available compounds, the ChemBridge Experimental Library. The library was filtered to exclude compounds that had undesired, toxic or reactive functional groups; known promiscuous binders; MW >460 Da or MW <250 Da; more than 4 chiral centers; polar surface area (PSA) >150 Å2 or PSA <50 Å2; number of rotatable bonds >10; or clogP >5 or clogP ≤2. Salts were also removed. Next, the selected compounds (∼100,000) were loaded into Schrödinger’s virtual screening workflow, where they were prepared with Ligprep and then docked using the filtering procedure for efficiency and only retaining the top 20% of compounds in the two rapid, initial docking modules HTVS (top 20% retained) and SP (top 20% retained). Finally, Glide XP was used to assign a final docking score to each molecule. AUC analyses on active and decoy data sets were previously performed using the Glide XP module, however, this was impractical for the large filtered ChemBridge library. Therefore, we relied on HTVS and SP modules to provide early filtering before employing the more time intensive XP protocol.
We then extracted the ∼400 top scoring compounds (docking score less than −7 kcal/mol). Binary Molprint2D fingerprints were generated using Canvas, and 40 clusters were generated using K-means clustering.17−19 Of these 40 clusters, the top scoring compounds from each cluster were visually inspected and a representative was chosen from each cluster, resulting in a final list of 100 compounds. These were purchased from ChemBridge (ChemBridge Corporation, San Diego, CA) and then assayed for UPPS inhibition activity. Three out of the 100 compounds were UPPS inhibitors. Similarity searches based on these active compounds were then performed using PubChem and SciFinder, and additional compounds were obtained and tested.
Enzyme and Cell Growth Inhibition Assays
Protein Expression and Purification
EcUPPS and SaUPPS were expressed and purified as described previously.8 Molecular weights and purities were verified by mass spectrometry and SDS–PAGE, respectively.
UPPS Inhibition Screening
The UPPS inhibition assays were carried out as described previously.8 Briefly, the condensation of FPP with IPP catalyzed by UPPS was monitored by using a continuous spectrophotometric assay20 in 96-well plates with 200 μL reaction mixtures containing 400 μM 2-amino-6-mercapto-7-methylpurine ribonucleoside (MESG), 350 μM IPP, 35 μM FPP, 20 mM Tris·HCl buffer (pH 7.5), 0.01% v/v Triton X-100, and 1 mM MgCl2. The IC50 values were obtained by fitting the inhibition data to a rectangular hyperbolic dose–response function using GraphPad PRISM 4.0 software (Graphpad Software, San Diego, CA). The IC50 values for the most active hits were verified using a radiometric assay21 with 2.5 μM FPP, 25 μM [3H]IPP, and 0.01% v/v Triton X-100.
Cell Strains
Bacillus subtilis subsp. subtilis (ATCC 6051), Escherichia coli (ATCC 29425), and Saccharomyces cerevisiae (ATCC 208352) were purchased from the American Type Culture Collection. Bacillus subtilis strain 168, Bacillus anthracis strain Sterne, Listeria monocytogenes strain 4b F2365, Staphylococcus aureus USA300 (methicillin-resistant), E. coli MC400, Pseudomonas putida, and Enterococcus faecalis U503 (vancomycin-resistant) were from our laboratory strain collection.22
E. coli ATCC 29425 Growth Inhibition Assay
IC50 values for E. coli growth inhibition were determined by using a microbroth dilution method. A 16 h culture of E. coli was diluted 50-fold into fresh Luria–Bertani (LB) broth and grown to an OD600 of ∼0.4. The culture was then diluted 500-fold into fresh LB medium, and 100 μL was inoculated into a 96-well flat bottom culture plate (Corning Inc., Corning, NY). The starting concentration of each compound was 0.3 mM, and this was 2-fold serially diluted. Plates were incubated for 3 h at 37 °C to midexponential phase. A 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell proliferation assay (ATCC) was then carried out to obtain bacterial viability dose–response curves. Ten microliters of MTT reagent was added into each well, followed by incubation for 2–4 h until a purple precipitate was visible. Then, 100 μL of detergent reagent was added and plates were further incubated in the dark at 23 °C for 2 h. The absorbance was recorded at 570 nm. A nonlinear regression analysis was then carried out using Origin 6.1. For each inhibitor, two independent experiments were performed and the IC50 values found were averaged.
B. subtilis ATCC 6051 Growth Inhibition Assay
A 16 h culture of B. subtilis was diluted 50-fold into fresh Luria–Bertani (LB) broth and incubated to an OD600 of ∼0.4. The culture was then diluted 500-fold into fresh LB medium, and 100 μL was inoculated into a 96-well flat bottom culture plate (Corning Inc., Corning, NY). The starting concentration of each compound was 0.5 mM and was then serially diluted. Plates were incubated for 12–16 h at 37 °C. The absorbance was recorded at 570 nm. A nonlinear regression analysis was carried out on the data obtained using Origin 6.1. For each inhibitor, two independent experiments were performed and the IC50 values found were averaged.
S. cerevisiae Growth Inhibition Assay
The protocol was the same as with the B. subtilis assay protocol except that YPD medium was used and the 96-well plate was incubated for 36 h instead of 12–16 h.
Evaluation of 1 and 4 Inhibitory Activity and Synergy
B. subtilis strain 168, B. anthracis strain Sterne, E. coli MC4100, and P. putida were grown to stationary phase in 10 mL of LB broth at 37 °C. S. aureus USA300 (methicillin-resistant), E. faecalis U503 (vancomycin-resistant), and L. monocytogenes strain 4b F2365 were grown to stationary phase in 10 mL of brain–heart infusion (BHI) medium at 37 °C. The cultures were adjusted to an OD600 of 0.016 in the designated medium before being added to 96-well microplates. Successive 2-fold dilutions of compounds 1 and 4 were added to the cultures (0.25–64 μg mL–1). As a control, kanamycin (1–32 μg mL–1) was added to samples of E. coli, B. subtilis, B. anthracis, P. putida, and L. monocytogenes. Gentamycin was used as a control for S. aureus and E. faecalis with dilutions from 1 to 64 μg mL–1. As a negative control, an equal volume of DMSO lacking antibiotic was used. Plates were covered and incubated at 37 °C for 16 h with shaking. The minimum inhibitory concentration (MIC) that suppressed at least 99% of bacterial growth was established based on culture turbidity in the microbroth dilution assay. The assay was repeated in three replicates, and values were averaged, or a range was reported. Isobolograms were carried out as previously described.10,23
Inhibitor Characterization
The purities of the key compounds investigated, obtained from ChemBridge (1 and 4), were determined by high-performance liquid chromatography and structures verified by NMR spectroscopy and high resolution mass spectrometry (Supporting Information Figures S2–S9) and were consistent with the structures provided by the vendor. Purities were >95% by HPLC.
Results and Discussion
In previous work, we obtained moderate correlations between enzyme inhibition activity and docking scores within a congeneric series of UPPS inhibitors (lipophilic bisphosphonates)8,24 using docking methods, so we first examined whether we could obtain similar correlations between docking scores and experimentally determined IC50 values for the 112 known actives. There was no significant correlation between docking scores and pIC50 values (pIC50 = −logIC50), Figure S1 in the Supporting Information. The wide variety of potential binding modes (sites 1, 2, 3, and 48,12) and protein conformations would be expected to make it difficult to achieve a good correlation between a scoring function and the experimentally determined pIC50 values, in addition to the assumptions made in scoring functions that cause inaccuracy when compared to experimental affinities. Nevertheless, docking studies can provide enrichment of active compounds from large libraries, even though docking scores rarely correlate well with activity when structurally diverse compounds are involved. We thus next employed an area-under-the-curve (AUC) analysis, also known as the receiver-operating-characteristic (ROC), a method that has been shown to be useful in validating structure-based virtual screening protocols25 and is a standard method for evaluating such protocols.26
We therefore tested 12 EcUPPS X-ray structures for their ability to separate actives (IC50 <100 μM) from decoys (presumed inactive compounds in the decoy library). Several EcUPPS X-ray structures showed a good separation of active from decoy compounds, with AUC values of ∼0.8. These structures also demonstrated early enrichment, as evidenced by the steep initial slope of the curve. This means that the best scores were given primarily to active compounds and suggests that, in screening a large compound library, the best scoring compounds would be enriched in UPPS inhibitors. We thus picked the two X-ray structures (PDB codes 2E98 and 4H3A) that provided significant early enrichment and a high AUC in the validation studies, for predictive studies. Using these two X-ray structures, we screened the ChemBridge EXPRESS-pick compound library (after filtering) and determined ∼400 hits with GlideXP scores less than −7 kcal/mol (lower energy is better). Since many highly ranked compounds were chemically very similar, we clustered the top scoring compounds and selected representatives from each cluster to ensure chemical diversity among the compounds to be tested.
Discovery of Novel UPPS Inhibitor Cores
The screening of the ChemBridge EXPRESS-pick compound library using the validated docking protocol resulted in the discovery of three new UPPS inhibitor classes: the 4-oxo-2-thioxo-1,3-thiazolidines, also known as rhodanines (e.g., compound 1), dihydroxyphenyls (the resorcinol, compound 2), and pyrimidinetrione (a barbiturate analogue, compound 3). None of these have been previously reported to be UPPS inhibitors. All three compounds are predicted to bind in either site 1 or 3 of the 2E98 crystal structure (Figure 3), although X-ray crystallographic studies will be required to confirm this binding mode (and our attempts to obtain crystal structures of these systems have not been successful). In any case, the three new inhibitors discovered represent UPPS inhibitors with “drug-like” physicochemical properties, passing the common drug-like filters as described in the Methods and Materials section. The most potent of the 3 compounds was the 4-oxo-2-thioxo-1,3-thiazolidine 1 (IC50 ∼2.6 μM against S. aureus UPPS), which in an initial screen for bioactivity was also found to be active against B. subtilis, MIC (minimal inhibitory concentration) ∼3 μg/mL (Table 1). For this reason, we chose to next investigate analogues based on the 4-oxo-2-thioxo-1,3-thiazolidine core.
Figure 3.

Three new classes of UPPS inhibitors discovered via virtual screening: (A) chemical structure and computed docking mode of compound 1, a rhodanine derivative; (B) chemical structure and docking mode of compound 2, a resorcinol derivative; (C) same for compound 3, a barbiturate.
Table 1. 4-Oxo-2-thioxo-1,3-thiazolidines Investigated in UPPS and Bacterial Cell Growth Inhibition Assaysa.
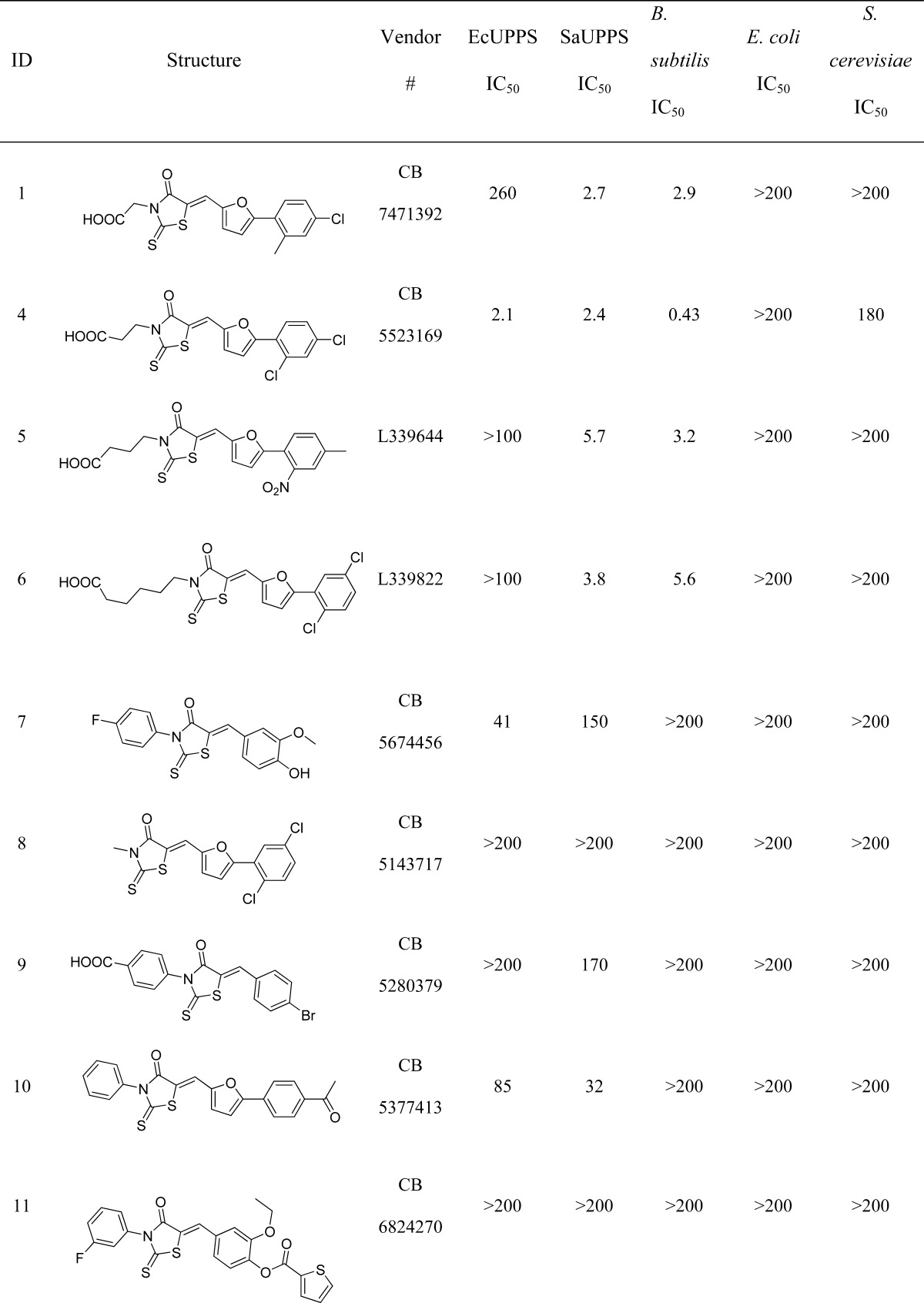
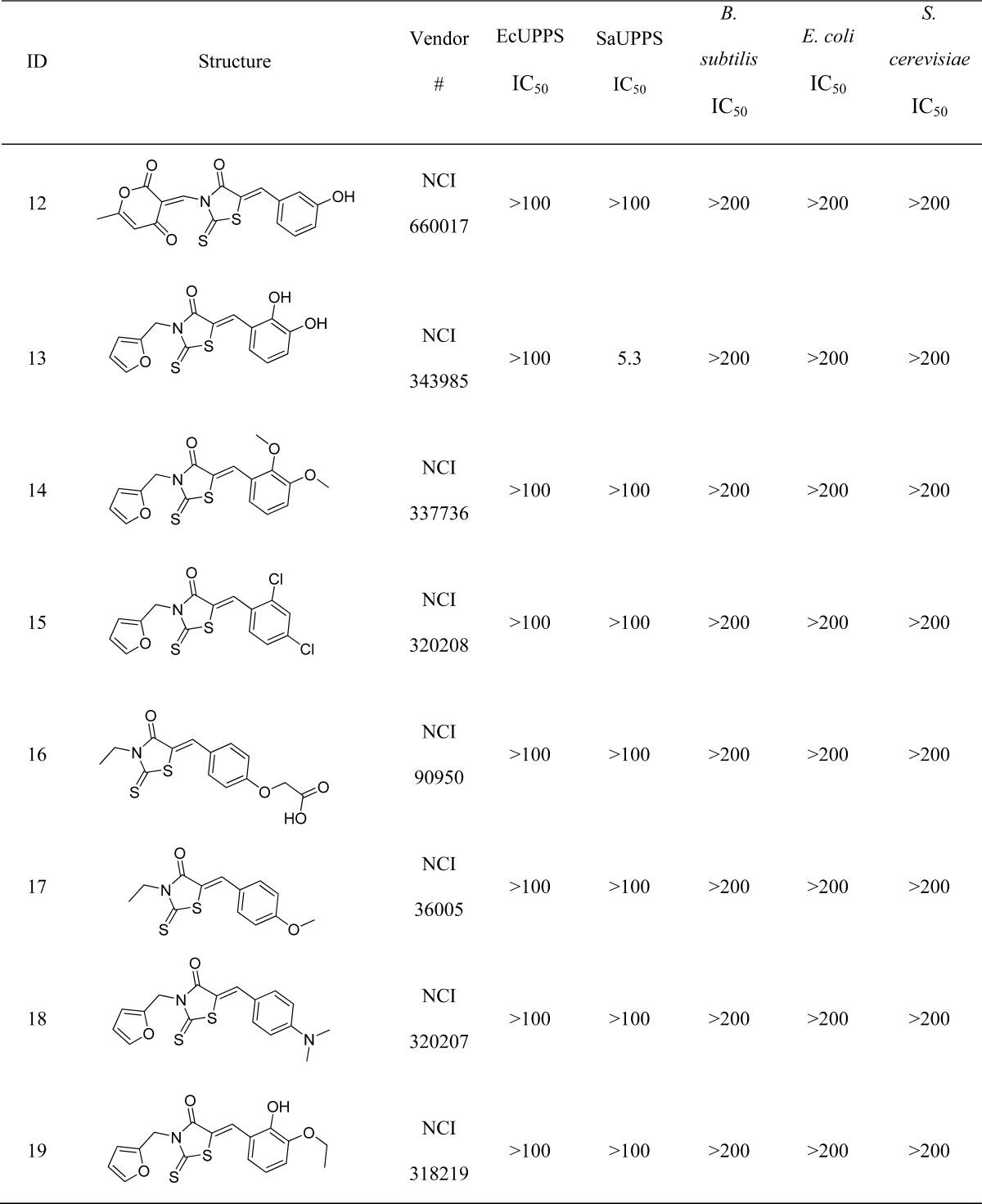
All concentrations are in μM.
Novel Core SAR
We next obtained 16 additional compounds from ChemBridge, from Sigma-Aldrich, and from the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute (4–19, Table 1), containing the 4-oxo-2-thioxo-1,3-thiazolidine core and tested them for activity against SaUPPS and EcUPPS, as well as a preliminary activity screen against B. subtilis, E. coli, and S. cerevisiae (the latter as a general cytotoxicity control, since it lacks UPPS). The alkyl carboxylic acid containing compounds with the 4-oxo-2-thioxo-1,3-thiazolidine core were active in assays against B. subtilis, and the most potent compound was 4 (an analogue of 1). 4 was roughly equipotent against SaUPPS and EcUPPS with an IC50 of ∼2 μM. Additionally, 4 was active against B. subtilis with an MIC ∼0.43 μg/mL and was very weakly active (∼200 μg/mL) against S. cerevisiae, indicating that 4 was not generally cytotoxic. Since 1 and 4 showed significant activity in enzymatic and bacterial growth assays, we subsequently tested them against several pathogens. Both 1 and 4 gave MIC values in the high ng/mL to low μg/mL range against B. anthracis Sterne, MRSA, VRE, and L. monocytogenes, Table 2. This promising antibacterial activity suggested the potential utility of these UPPS inhibitors in synergizing with other cell wall agents but where significant resistance has emerged, such as with methicillin (MRSA) and vancomycin (VRE).
Table 2. MIC Values for Two 4-Oxo-2-thioxo-1,3-thiazolidine Analogues, Compounds 1 and 4, Tested in Diverse Bacterial Cell Growth Inhibition Assaysa.
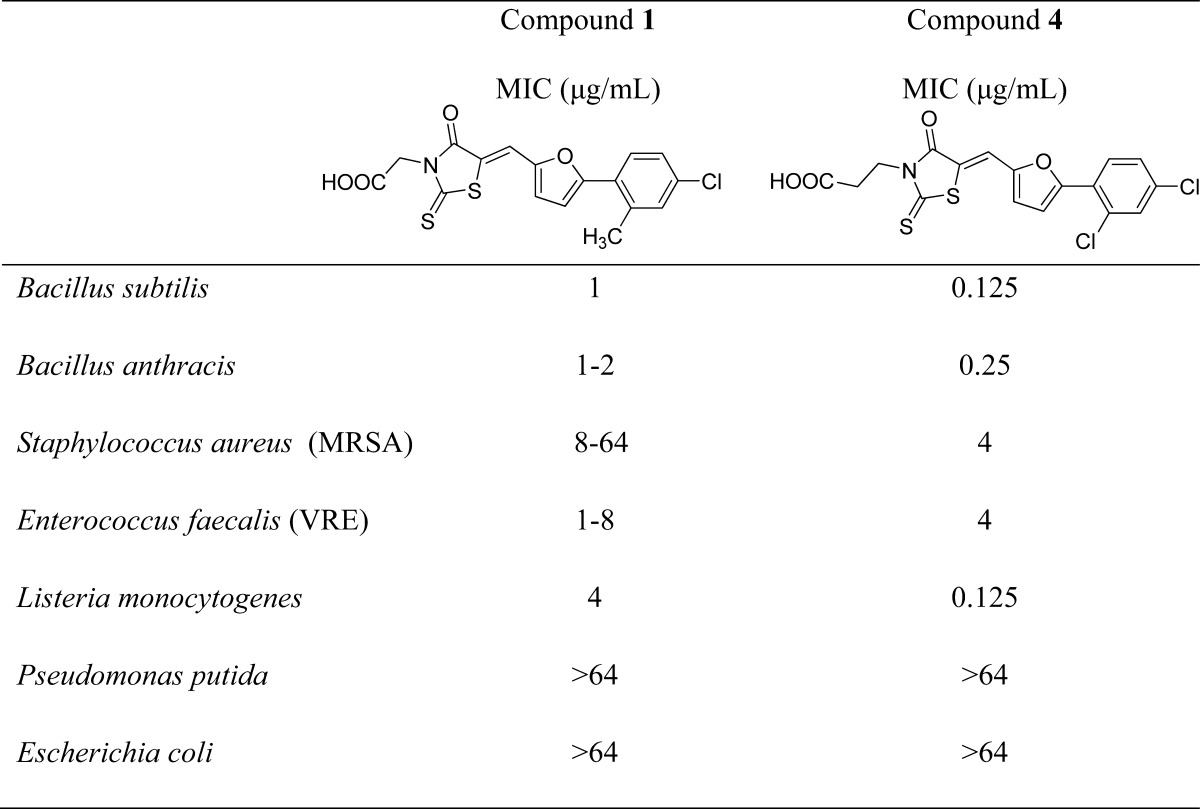
The compounds were tested against a panel of both Gram-positive (top five) and Gram-negative (lower two) bacteria.
Synergistic Interactions
To investigate the possibility of synergistic interactions with known cell wall biosynthesis inhibitors, we determined the fractional inhibitory concentration index (FICI) values for three systems: MRSA, using 1 + methicillin; VRE, using 1 + vancomycin; and B. anthracis, using 1 + ampicillin. The FICI is defined as
where FIC(A) and FIC(B) are the fractional inhibitory concentrations of drugs A and B, MIC(A) and MIC(B) are the MIC values of drugs A and B acting alone, and MIC(AB) and MIC(BA) are the MIC values of the most effective combination of drug A or B in the presence of drug B or A. Using this method, FICI values of <0.5 represent synergism, >0.5 and <1.0 represent additivity, >1 and <2 represent an indifferent effect, and >2 represents drug antagonism. Isobolograms are shown in Figure 4. As can be seen in Figure 4B, the FICI for 1 + methicillin in MRSA is 0.11, which indicates strong synergism during late stage growth. However, with both VRE (1 + vancomycin) and B. anthracis (1 + ampicillin) the FICI values are in the 1–2 range, which indicates an indifferent effect.
Figure 4.
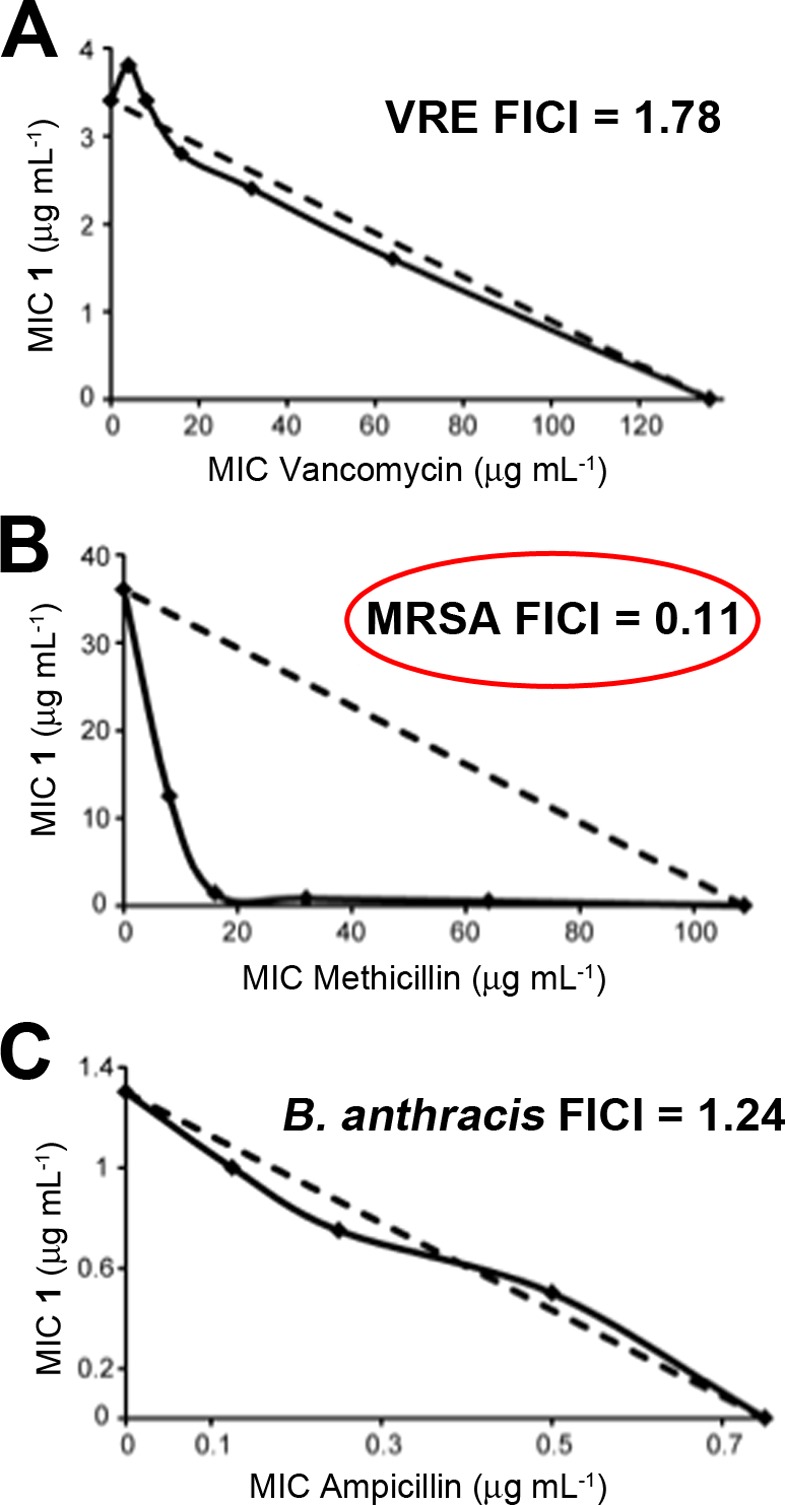
In vitro synergy assays. Isobolograms for growth inhibition of VRE, MRSA, and B. anthracis strain Sterne. (A) 1 and vancomycin inhibition of E. faecalis U503 (vancomycin-resistant, VRE). FICI = 1.78. (B) 1 and methicillin inhibition of S. aureus (USA300). FICI = 0.11. (C) 1 and ampicillin inhibition of B. anthracis strain Sterne. FICI =1.24.
What is particularly interesting about the most active species
investigated here (1) is that it has a structure that
is very similar to that found in the drug epalrestat,  an aldolase reductase inhibitor27 that
is used to treat diabetic neuropathy, and is approved for clinical
use in Japan, China, and India. This is encouraging because rhodanines
as a class are known to often have activity in widely different assays,
and indeed computer programs such as PAINS28 categorize, e.g., 1–4 (as well
as epalrestat) as possible “pan assay interference compounds”.
This can mean that the compounds cause false positives in assays,
or that they may be multitarget inhibitors. In some cases multitargeting
may be undesirable; however, in the context of anti-infective development,
multitargeting is expected to increase efficacy as well as decrease
the possibility of resistance development,29 both very desirable features.
an aldolase reductase inhibitor27 that
is used to treat diabetic neuropathy, and is approved for clinical
use in Japan, China, and India. This is encouraging because rhodanines
as a class are known to often have activity in widely different assays,
and indeed computer programs such as PAINS28 categorize, e.g., 1–4 (as well
as epalrestat) as possible “pan assay interference compounds”.
This can mean that the compounds cause false positives in assays,
or that they may be multitarget inhibitors. In some cases multitargeting
may be undesirable; however, in the context of anti-infective development,
multitargeting is expected to increase efficacy as well as decrease
the possibility of resistance development,29 both very desirable features.
Conclusions
The results described herein are of interest for several reasons. First, we carried out an in silico screen of ∼100 known UPPS inhibitors and 1000 decoys using 12 reported UPPS X-ray structures. The two X-ray structures providing the best enrichment in an AUC-ROC analysis were then used to screen a subset of ∼100,000 compounds selected for drug-like activity from an initial ChemBridge library of ∼450,000 compounds. We then tested the ∼100 in silico hits in vitro against SaUPPS and EcUPPS, leading to several μM UPPS inhibitors (as deduced from both PPi release and radioactive assays). The most potent lead was 1, which is structurally quite similar to epalrestat, in clinical use to treat diabetic neuropathy. 1 (and its analogue 4) inhibited the growth of Gram positives; they did not inhibit the growth of Gram negatives (important with E. coli in the context of maintaining commensal microflora), and they had no activity against S. cerevisiae. Activity against B. anthracis, a vancomycin-resistant Enterococcus spp., and Listeria monocytogenes was good, in the 0.125–4 μg/mL range, and there was very strong synergy (FICI = 0.11) with methicillin and 1 in a MRSA strain of S. aureus, suggesting that 1 could be a promising lead (in combination therapies) for treating staph infections.
Acknowledgments
This work was supported by the United States Public Health Service (National Institutes of Health Grants GM065307, CA158191, GM08326, GM31749, and HD071600); the National Science Foundation (Grant MCB-1020765); a Packard Fellowship for Science and Engineering (D.A.M.), the National Biomedical Computation Resource, the UCSD Center for Theoretical Biological Physics, the Howard Hughes Medical Institute, and the NSF Supercomputer Centers. F.F. acknowledges financial support of the Beatriu de Pinós program from AGAUR for a postdoctoral grant (2010 BP_A 00339). We thank the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute, for providing chemicals, and Professors James Wells and Paul Hergenrother for providing bacteria.
Glossary
Abbreviations Used
- MRSA
methicillin-resistant Staphylococcus aureus
- VRE
vancomycin-resistant Enterococci
- UPPS
undecaprenyl diphosphate synthase
- MIC
minimal inhibitory concentration
- FICI
fractional inhibitory concentration index
- UPP
undecaprenyl diphosphate
- IPP
isopentyl diphosphate
- FPP
farnesyl diphosphate
- BPH
bisphosphonate
- ROC
receiver-operating-characteristic
- AUC
area under the curve
- LB
Luria–Bertani
- PAINS
pan assay interference compounds
Supporting Information Available
Correlation of pIC50 with docking scores analysis, as well as 1H NMR, HPLC analysis, mass spectra, and high-resolution mass spectra of compounds 1 and 4. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Fischbach M. A.; Walsh C. T. Antibiotics for emerging pathogens. Science 2009, 325, 1089–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler M. S.; Cooper M. A. Antibiotics in the clinical pipeline in 2011. J. Antibiot. 2011, 64, 413–425. [DOI] [PubMed] [Google Scholar]
- van Heijenoort J. Lipid intermediates in the biosynthesis of bacterial peptidoglycan. Microbiol. Mol. Biol. Rev. 2007, 71, 620–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apfel C. M.; Takacs B.; Fountoulakis M.; Stieger M.; Keck W. Use of genomics to identify bacterial undecaprenyl pyrophosphate synthetase: cloning, expression, and characterization of the essential uppS gene. J. Bacteriol. 1999, 181, 483–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne D. J.; Gwynn M. N.; Holmes D. J.; Pompliano D. L. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discovery 2007, 6, 29–40. [DOI] [PubMed] [Google Scholar]
- Peukert S.; Sun Y.; Zhang R.; Hurley B.; Sabio M.; Shen X.; Gray C.; Dzink-Fox J.; Tao J.; Cebula R.; Wattanasin S. Design and structure-activity relationships of potent and selective inhibitors of undecaprenyl pyrophosphate synthase (UPPS): tetramic, tetronic acids and dihydropyridin-2-ones. Bioorg. Med. Chem. Lett. 2008, 18, 1840–1844. [DOI] [PubMed] [Google Scholar]
- Lee L. V.; Granda B.; Dean K.; Tao J.; Liu E.; Zhang R.; Peukert S.; Wattanasin S.; Xie X.; Ryder N. S.; Tommasi R.; Deng G. Biophysical investigation of the mode of inhibition of tetramic acids, the allosteric inhibitors of undecaprenyl pyrophosphate synthase. Biochemistry 2010, 49, 5366–5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo R. T.; Cao R.; Liang P. H.; Ko T. P.; Chang T. H.; Hudock M. P.; Jeng W. Y.; Chen C. K.; Zhang Y.; Song Y.; Kuo C. J.; Yin F.; Oldfield E.; Wang A. H. Bisphosphonates target multiple sites in both cis- and trans-prenyltransferases. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 10022–10027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant J. D.; Cao R.; Gorfe A. A.; Zhu W.; Li J.; Sankovsky A.; Oldfield E.; McCammon J. A. Non-bisphosphonate inhibitors of isoprenoid biosynthesis identified via computer-aided drug design. Chem. Biol. Drug Des. 2011, 78, 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Lin F.-Y.; Li K.; Zhu W.; Liu Y.-L.; Cao R.; Pang R.; Lee E.; Axelson J.; Hensler M.; Wang K.; Molohon K. J.; Wang Y.; Mitchell D. A.; Nizet V.; Oldfield E. HIV-1 integrase inhibitor-inspired antibacterials targeting isoprenoid biosynthesis. ACS Med. Chem. Lett. 2012, 3, 402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindert S.; Zhu W.; Liu Y.-L.; Pang R.; Oldfield E.; McCammon J. A. Farnesyl diphosphate synthase inhibitors from in silico screening. Chem. Biol. Drug Des. 2013, 81, 742–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W.; Zhang Y.; Sinko W.; Hensler M. E.; Olson J.; Molohon K. J.; Lindert S.; Cao R.; Li K.; Wang K.; Wang Y.; Liu Y.-L.; Sankovsky A.; de Oliveira C. A. F.; Mitchell D. A.; Nizet V.; McCammon J. A.; Oldfield E. Antibacterial drug leads targeting isoprenoid biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 123–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halgren T. A.; Murphy R. B.; Friesner R. A.; Beard H. S.; Frye L. L.; Pollard W. T.; Banks J. L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Murphy R. B.; Repasky M. P.; Frye L. L.; Greenwood J. R.; Halgren T. A.; Sanschagrin P. C.; Mainz D. T. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [DOI] [PubMed] [Google Scholar]
- Schrödinger L. L. C.Suite 2011: Glide, version 5.7; Schrödinger LLC: New York, 2011. [Google Scholar]
- Sastry G. M.; Adzhigirey M.; Day T.; Annabhimoju R.; Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [DOI] [PubMed] [Google Scholar]
- Suite 2012: Canvas version 1.5; 2012. [Google Scholar]
- Duan J.; Dixon S. L.; Lowrie J. F.; Sherman W. Analysis and comparison of 2D fingerprints: insights into database screening performance using eight fingerprint methods. J. Mol. Graphics Modell. 2010, 29, 157–170. [DOI] [PubMed] [Google Scholar]
- Sastry M.; Lowrie J. F.; Dixon S. L.; Sherman W. Large-scale systematic analysis of 2D fingerprint methods and parameters to improve virtual screening enrichments. J. Chem. Inf. Model. 2010, 50, 771–784. [DOI] [PubMed] [Google Scholar]
- Webb M. R. A continuous spectrophotometric assay for inorganic phosphate and for measuring phosphate release kinetics in biological systems. Proc. Natl. Acad. Sci. U.S.A. 1992, 89, 4884–4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Huang J.; Jiang X.; Seefeld M.; McQueney M.; Macarron R. The effect of triton concentration on the activity of undecaprenyl pyrophosphate synthase inhibitors. J. Biomol. Screening 2003, 8, 712–715. [DOI] [PubMed] [Google Scholar]
- Molohon K. J.; Melby J. O.; Lee J.; Evans B. S.; Dunbar K. L.; Bumpus S. B.; Kelleher N. L.; Mitchell D. A. Structure determination and interception of biosynthetic intermediates for the plantazolicin class of highly discriminating antibiotics. ACS Chem. Biol. 2011, 6, 1307–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leon A.; Liu L.; Yang Y.; Hudock M. P.; Hall P.; Yin F.; Studer D.; Puan K.-J.; Morita C. T.; Oldfield E. Isoprenoid biosynthesis as a drug target: bisphosphonate inhibition of Escherichia coli K12 growth and synergistic effects of fosmidomycin. J. Med. Chem. 2006, 49, 7331–7341. [DOI] [PubMed] [Google Scholar]
- Sinko W.; de Oliveira C.; Williams S.; Van Wynsberghe A.; Durrant J. D.; Cao R.; Oldfield E.; Andrew McCammon J. Applying molecular dynamics simulations to identify rarely sampled ligand bound conformational states of undecaprenyl pyrophosphate synthase, an antibacterial target. Chem. Biol. Drug Des. 2011, 77, 412–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triballeau N.; Acher F.; Brabet I.; Pin J.-P.; Bertrand H.-O. Virtual screening workflow development guided by the “receiver operating characteristic” curve approach. Application to high-throughput docking on metabotropic glutamate receptor subtype 4. J. Med. Chem. 2005, 48, 2534–2547. [DOI] [PubMed] [Google Scholar]
- Christofferson A. J.; Huang N. How to benchmark methods for structure-based virtual screening of large compound libraries. Methods Mol. Biol. 2012, 819, 187–195. [DOI] [PubMed] [Google Scholar]
- Ramirez M. A.; Borja N. L. Epalrestat: an aldose reductase inhibitor for the treatment of diabetic neuropathy. Pharmacotherapy 2008, 28, 646–645. [DOI] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
- Li K.; Schurig-Briccio L. A.; Feng X.; Upadhyay A.; Pujari V.; Lechartier B.; Fontes F. L.; Yang H.; Rao G.; Zhu W.; Gulati A.; No J. H.; Cintra G.; Bogue S.; Liu Y. L.; Molohon K. J.; Orlean P.; Mitchell D. A.; Freitas-Junior L. H.; Ren F.; Sun H.; Jiang T.; Li Y.; Guo R. T.; Cole S. T.; Gennis R. B.; Crick D. C.; Oldfield E. Multi-target drug discovery for tuberculosis and other infectious diseases. J. Med. Chem. 2014, 57, 3126–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.