

Abstract
Background
Precuneus choline acetytransferase activity reduction co-occurs with greater β-amyloid (Aβ) in Alzheimer's disease (AD). Whether this cholinergic deficit associates with alterations in nerve growth factor (NGF) signaling and its relation to Aβ plaque and neurofibrillary tangle (NFT) pathology during disease onset is unknown.
Methods
Precuneus NGF up and down stream signaling levels relative to Aβ and NFT pathology were evaluated using biochemistry and histochemistry in 62 subjects diagnosed premortem as non cognitively impaired (NCI, n=23), mild cognitively impaired (MCI, n=21) and mild to moderate Alzheimer's disease (AD, n=18)
Results
Immunoblots revealed increased levels of proNGF in AD but not MCI, whereas its cognate receptors were unchanged. There were no significant differences in protein level for the downstream survival kinase-signaling proteins Erk and phospho-Erk among groups. Apoptotic phospho-JNK, phospho-JNK/JNK ratio and Bcl-2 were significantly elevated in AD. Soluble Aβ1–42 and fibrillar Aβ measured by [3H] Pittsburgh compound-B (PiB) binding were significantly higher in AD compared to MCI and NCI. The density of plaques showed a trend to increase, but only 6-CN-PiB positive plaques reached significance in AD. AT8, TOC-1 and Tau C3 positive NFT density were unchanged, whereas only AT8 positive neuropil thread density was statistically higher in AD. A negative correlation was found between proNGF, phospho-JNK, Bcl-2 levels and phospho-JNK/JNK ratio and cognition, whereas proNGF correlated positively with 6-CN-PiB positive plaques during disease progression.
Conclusions
Data indicate that precuneus neurotrophin pathways are resilient to amyloid toxicity during the onset of AD.
Keywords: Alzheimer's disease, amyloid, mild cognitive impairment, neuropathology, neurotrophic factors, tau
Introduction
The precuneus, a component of the default mode network (DMN), is implicated in episodic memory retrieval (1) and displays high metabolic activity during conscious rest and selectively deactivates during non-self-directed cognitive tasks in the healthy brain (2, 3, 4), but is dysregulated in aging (5) and its ability to inactivate during cognitive tasks is compromised at the earliest stages of AD even before cognitive impairment (6, 7, 8). [3H]PiB (PiB) amyloid imaging reveals that the DMN is vulnerable to Aβ deposition in the earliest, pre-clinical stages of the disease (9, 10, 11). There is also an overlap between synaptic failure, functional disconnection and amyloid in the DMN in MCI (12), linking Aβ and connectivity disruptions within the DMN prior to clinical onset of dementia (13). The precuneus also displays a greater degree of atrophy in early compared to late onset AD (14, 15, 16) and a reduction in synapse number in AD but not MCI (17).
Precuneus cholinergic activity is reduced in AD but not MCI and co-occurs with increased amyloid burden (18). Aβ plays a key role in cholinergic dysfunction due to alterations in interactions between NGF/proNGF and its cognate high affinity TrkA and low affinity p75NTR receptors (19, 20, 21), which underlie cholinergic basal forebrain neuron survival. We have shown a shift in upstream and downstream NGF/proNGF signaling from cell survival to cell death within the lateral parietal cortex (22, 23) and hippocampus (24) during AD progression. Whether precuneus neurotrophic NGF signaling pathways are altered in relation to amyloid and NFT deposition during AD progression is not known. Therefore, expression levels of NGF/proNGF, TrkA and p75NTR as well as cell survival and pro-apoptotic downstream NGF activated pathways relative to Aβ and tau pathology were examined in precuneus tissue from people who died with a clinical diagnosis of NCI, MCI or AD. Neurochemical changes were correlated with cognitive and neuropathologic variables.
Subjects and Methods
Subjects
Study included 62 cases diagnosed as NCI (18F/5M), MCI (15F/6M) and AD (10F/8M) from the Rush Religious Order Study (RROS) (25, 26, 27) and University of Kentucky Alzheimer's Disease Center (UKADC) (28, 29) (see Table 1). Participants agreed to an annual premortem clinical evaluation and brain donation at death. Human Investigations Committees of Rush University and University of Kentucky approved the study.
Table 1. Clinical, demographic and neuropathological characteristics by clinical diagnosis category.
| NCI (N=23) |
MCI (N=21)a |
AD (N=18) |
Total (N=62) |
p-value | Pair-wise comparison | ||
|---|---|---|---|---|---|---|---|
| Age (years) at death: | Mean ± SD (Range) | 86.3 ± 3.9 (78-92) |
86.8 ± 5.1 (75-96) |
88.9 ± 7.1 (73-100) |
87.2 ± 5.4 (73-100) |
0.2b | -- |
| Number (%) of males: | 5(21.7%) | 6 (28.5%) |
8 (44.4%) |
18(29%) | 0.4c | -- | |
| Years of education: | Mean ± SD (Range) | 16.0 ± 3.1 (10-21) |
17.2 ± 3.1 (10-25) |
17.1 ± 3.4 (12-26) |
16.7 ± 3.2 (10-26) |
0.5b | -- |
| Number (%) with ApoE ε4 allele: | 1 (4.3%) | 10 (47.6%) | 5 (27.7%) | 16 (25.8%) | 0.001c | NCI < MCI | |
| MMSEd: | Mean ± SD (Range) | 28.3 ± 1.5 (26-30) |
27.0 ± 2.5 (22-30) |
18.0 ± 6.0 (10-28) |
24.9 ± 5.6 (10-30) |
<0.001b | (NCI, MCI) > AD |
| Global cognitive z-score (GCS)e: | Mean ± SD (Range) | 0.37 ± 0.17 (0.06, 0.59) |
0.17 ± 0.35 (-0.53,0.91) |
-0.62 ± 0.44 (-1.18,0.06) |
-0.03 ± 0.54 (-1.18,0.91) |
<0.001b | (NCI, MCI) > AD |
| Episodic memory z-scoree: | Mean ± SD (Range) | 0.73 ± 0.37 (0.08,1.37) |
0.36 ± 0.47 (-0.25, 1.68) |
-0.64 ± 0.76 (-1.99,0.65) |
0.12 ± 0.80 (-1.99,1.68) |
<0.001b | NCI > MCI > AD |
| Post-mortem interval (hours): | Mean ± SD (Range) | 4.3 ± 2.1 (1.0-9.0) |
4.8 ± 2.4 (2.0-10.6) |
4.5 ± 2.1 (1.5-8.6) |
4.5 ± 2.2 (1.0-10.6) |
0.9b | ∼ |
| Brain weight (grams): | Mean ± SD (Range) | 1174.1 ± 115.3 (940-1473) |
1163.2 ± 140.0 (890-1350) |
1132.7 ± 83.6 (975-1320) |
1158.4 ± 116.0 (890-1473) |
0.4b | ∼ |
| Distribution of Braak scores: | No AD | 3 | 0 | 0 | 3 | ||
| I/II | 6 | 8 | 0 | 15 | 0.008b | NCI < AD | |
| III/IV | 12 | 9 | 1 | 32 | |||
| V/VI | 2 | 4 | 6 | 12 | |||
| NIA Reagan diagnosis (likelihood of AD)f: | No AD | 6 | 0 | 0 | 6 | ||
| Low | 11 | 10 | 2 | 23 | <0.001b | NCI < MCI < AD | |
| Intermediate | 5 | 7 | 10 | 22 | |||
| High | 1 | 3 | 6 | 10 | |||
| CERAD diagnosisf: | No AD | 10 | 6 | 0 | 16 | ||
| Possible | 5 | 1 | 1 | 7 | <0.001b | NCI < AD | |
| Possible | 7 | 8 | 9 | 24 | |||
| Definite | 1 | 5 | 8 | 14 |
N=l 1 MCI were amnestic.
Kruskal-Wallis test, with Dunn's correction for multiple comparisons.
Fisher's exact test.
MMSE of 2 NCI cases were not available.
Cognitive z-scores were only available for the 12 NCI, 14 MCI, and 14 AD RROS cases with additional cognitive testing within 2 years before death. Among them, 1 AD case completed less than half of the testing battery and thus no GCS was computed for this case.
Reagan and CERAD diagnoses of 1 MCI case were not available
Clinical and Neuropathological Evaluations
Clinical criteria for diagnosis of AD, MCI and NCI have been reported (25, 27, 28, 29, 30). Five RROS and six UKADC cases were amnestic MCI (31). Mini Mental Score examination (MMSE) was performed within 2 years of death. Available for RROS cases were a global cognitive z-score (GCS) comprising 19 tests and an episodic memory z-score (see Supplement 1).
Tissue
Frozen precuneus was homogenized (150 mg/mL) on ice in phosphate-buffered saline (PBS) and processed for [3H]PiB, Aβ ELISA and western blotting (see Supplement 1). Precuneus from the other hemisphere was dissected and fixed in 4% paraformaldehyde (pH 7.4) for 5 days, cryoprotected and cut frozen into 40 μm series and stored until processing (24, 30).
Antibodies
Antibody characterization and specificity are reported in Supplement 1 and Table S1 in Supplement 1.
Quantitative immunoblotting
Proteins were denatured in sodium dodecyl sulfate (SDS) loading buffer to a final concentration of 5 mg/mL. Proteins (50 μg/sample) were separated by 8%-16% or 7.5% SDS-PAGE and transferred to polyvinylidene fluoride membranes electrophoretically (24, 32) Protein signals were normalized to β-tubulin and samples analyzed in three independent experiments (see Supplement 1 for additional details).
In vitro [3H]PiB binding assay
[3H]PiB binding was performed in RROS cases (33). Unlabeled PiB was dissolved in dimethyl sulfoxide (DMSO) at 400μM to yield <1% DMSO. [3H]PiB (1nM; American Radiolabeled Chemicals, St. Louis, MO; specific activity 72.4 Ci/mM) was incubated with 100 μg of tissue in 1 mL PBS. Non-specific binding was defined as the number of counts remaining in the presence of 1 μM unlabeled PiB. Filters were counted in Cytoscint-ES, results corrected for non-specific, non-displaceable binding in the presence of 1 μM PiB and values expressed as pmol [3H]PiB bound per gram of wet tissue weight. Bound [3H]PiB correlates with total number of PiB binding sites (34) and mimics the low nanoM concentrations of [3H]PiB achieved in human positron emission tomography (PET) brain imaging.
Aβ enzyme linked immunoadsorbant assay (ELISA)
Aβ1–42 peptide concentration was quantified in diethylamine soluble Aβ fractions prepared by centrifuging precuneus homogenate (RROS samples) at 135,000 × g at 4° C for 1 hour and neutralizing the supernatant with 0.5M Tris-Cl. Aβ concentration was assayed using a fluorescent-based ELISA (Biosource, Camarillo, CA) with a capture antibody specific for the human Aβ NH2–terminus (amino acids 1–16), and detection antibodies specific for the 42 amino acid end of Aβ (18). Values were determined from standard curves using synthetic Aβ1–42 peptide (Biosource) and expressed as pmol/g wet brain tissue.
Histofluorescence and Immunohistochemistry
Precuneus sections from six randomly chosen RROS cases per clinical group were stained with X-34 and 6-CN-PiB to examine fibrillar plaques (33, 35). Sections were incubated in a 10 mM 6-CN-PiB solution (45 minutes), dipped in 0.1 M PBS, followed by a 1-min differentiation in 132.9 mM NaCl, 8.7 mM K2HPO4 and 1.5 mM KH2PO4 (pH 7.4) solution. Additional sections were processed for X-34 (0.04 g/L), which detects the full spectrum of amyloid pathology, or immunostained for 6E10 (APP/Aβ) as well as AT8 (early tau phosphorylation), TOC-1 (oligomeric tau) and Tau C3 (late tau truncation; see Table S1 in Supplement 1 and Supplement 1 for additional methods).
Plaque load
The two main morphological types of plaques included cored and diffuse (non-cored) plaques, which were quantified either separately or together in tissues sections processed for 6-CN-PiB or X-34. First the percent area was determined for all plaques, regardless of morphology, and this produced “total plaque load” for each of the two markers. Next, we quantified separately “diffuse plaque load” for each of the two markers; this was performed by manually deleting cored plaques when determining diffuse plaque load (see Supplement 1 for additional methods).
Aβ plaque, NFT and neuropil thread (NT) density
Density of Aβ plaques, NFTs and NTs were evaluated using 6E10, AT8, TOC-1 and Tau C3 antibodies. Plaques, NTs and NFTs were counted in one to two sections under a 10x objective in five random fields with a size of 1.13 mm2. Mean was calculated from counts of five different fields per section.
Statistical analysis
Data were compared across clinical groups using non-parametric tests, which are more robust to outliers and non-normality (i.e., Kruskal-Wallis test or Fisher's exact test, with Dunn's correction for multiple comparisons). Spearman rank correlation or Wilcoxon rank-sum test assessed variable associations. Plaque load and tau densities were log-transformed (adding 1 to original value and calculating the natural logarithm (ln)) to reduce outlier effects. Statistical significance was set at 0.05 (two-sided). Due to the large number of analyses performed, p-values between 0.01 and 0.05 were interpreted with caution (see Supplement 1 for additional details).
Results
Case demographics
Clinical diagnostic groups NCI (14 RROS and 9 UKADC), MCI (15 RROS and 6 UKADC) and mild to moderate AD (14 RROS and 4 UKADC) did not differ by age, gender, education, postmortem interval or brain weight (see Table 1). There were significantly more ApoE4 allele cases in the MCI (48%) than in NCI (4%) group. Cognitive function (MMSE, GCS, episodic memory z-score, and the 7 individual episodic memory tests) was significantly lower in AD compared to MCI and NCI, while differences between the latter two groups did not reach statistical significance (see Table 1). Neuropathology revealed that 94% of AD compared to ∼60% of NCI and MCI cases, were Braak stages III-VI. Using NIA-Reagan criteria, 26% of NCI, 50% of MCI, and 89% of AD cases were classified as intermediate to high likelihood of AD (Table 1). Consortium to Establish a Registry for Alzheimer's Disease (CERAD) diagnosis revealed that 35% of NCI, 65% of MCI, and 94% of AD cases were probable or definite AD. Among the 3 clinical groups we found significant differences in Braak scores, NIA-Reagan diagnosis, and CERAD diagnosis. While the differences between NCI and AD were statistically significant for these neuropathologic criteria, only the NIA-Reagan diagnosis showed significantly more advanced pathology in MCI compared to NCI, and significantly less pathology compared to AD.
ProNGF, TrkA, p75NTR and sortilin receptor levels
Precuneus proNGF levels were elevated 13% in MCI and a significant 30% in AD compared to NCI (Fig.1A). No differences in sortilin (Fig. 1B), TrkA (Fig. 1C and D) or p75NTR (Fig. 1C and E) levels were detected across groups (see Table 2). Sub-analysis revealed increased TrkA levels in amnestic compared to non-amnestic MCI subjects (p = 0.002). A second sub-analysis revealed no difference in proNGF levels between AD cases neuropathologically characterized as moderate Braak (III-IV) compared to severe Braak (V-VI) scores.
Figure 1.
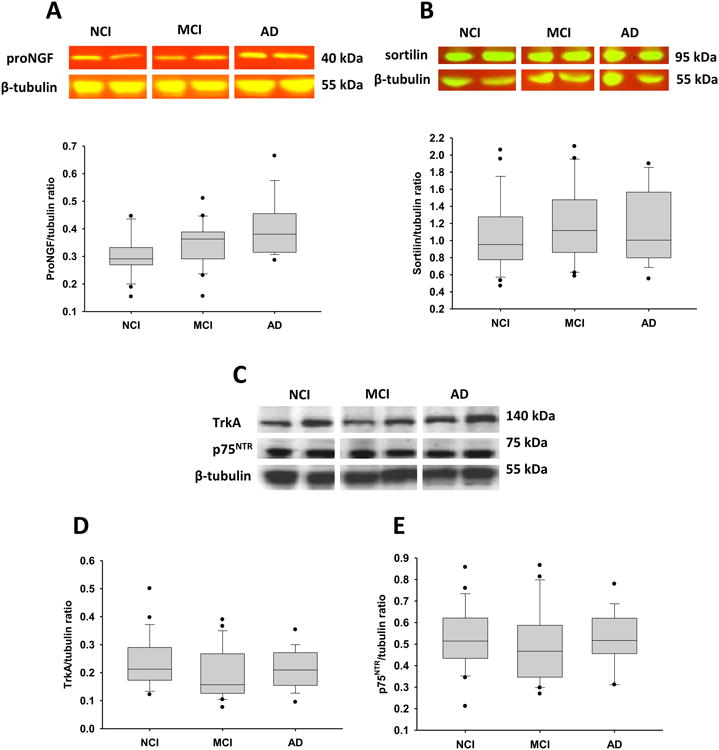
Representative immunoblots and box plots of precuneus levels of proNGF, TrkA, p75NTR and sortilin in cases clinically diagnosed as NCI, MCI and AD. β-tubulin probe was used to normalize the immunoreactive signal obtained by densitometry in the blots. Levels of proNGF in AD (A) were significantly higher compared to NCI (*p = 0.003), whereas the levels of sortilin (B), TrkA (C, D) and p75NTR (C, E) remained stable across the three clinical groups. Black dots in box plots indicate outliers.
Table 2. Summary of precuneus protein levels by clinical diagnosis category.
| NCI (N=23) |
MCI (N=21) |
AD (N=18) |
p-valuea | Pair-wise comparison | |
|---|---|---|---|---|---|
| proNGF | 0.30 ± 0.07b (0.15-0.44)c |
0.34 ± 0.08 (0.15-0.51) |
0.39 ± 0.09 (0.28-0.66) |
0.003 | NCI < AD |
| TrkA | 0.23±0.09; (0.12-0.50) |
0.19±0.09 (0.07-0.39) |
0.21±0.07 (0.09-0.35) |
0.2 | -- |
| p75NTR | 0.53 ± 0.14 (0.21-0.85) |
0.49 ± 0.17 (0.26-0.86) |
0.52 ± 0.12 (0.31-0.77) |
0.5 | -- |
| sortilin | 1.06 ± 0.40 (0.46-2.06) |
1.18 ± 0.43 (0.58-2.10) |
1.14 ± 0.43 (0.55-1.89) |
0.7 | -- |
| Erk | 0.51 ± 0.20 (0.13-0.91) |
0.46 ± 0.17 (0.28-1.03) |
0.48 ± 0.20 (0.20-0.93) |
0.6 | -- |
| p-Erk | 0.62 ± 0.22 (0.21-1.25) |
0.70 ± 0.39 (0.22-1.73) |
0.69 ± 0.34 (0.35-1.69) |
0.9 | -- |
| p-Erk/Erk ratio | 1.50 ± 1.02 (0.30-4.45) |
1.60 ± 0.91 (0.66-4.62) |
1.85 ± 1.59 (0.48-7.00) |
0.7 | -- |
| JNK | 0.43 ± 0.19 (0.18-0.82) |
0.37 ±0.07 (0.21-0.57) |
0.36 ± 0.13 (0.19-0.63) |
0.5 | -- |
| p-JNK | 0.13 ± 0.07 (0.04-0.38) |
0.16 ± 0.06 (0.06-0.25) |
0.24 ± 0.14 (0.06-0.56) |
0.015 | NCI < AD |
| p-JNK/JNK ratio | 0.34 ± 0.17 (0.15-0.90) |
0.45 ± 0.15 (0.19-0.74) |
0.67 ± 0.32 (0.19-1.29) |
<0.001 | NCI < (MCI, AD) |
| Bcl-2 | 0.41 ± 0.10 (0.15-0.58) |
0.46 ± 0.09 (0.30-0.68) |
0.52 ± 0.17 (0.29-1.02) |
0.043 | NCI < AD |
Kruskal-Wallis test, with Dunn's correction for multiple comparisons.
Mean ± standard deviation.
(Range).
Erk and phospho-Erk levels
Precuneus levels of total Erk (Fig. S1A), phospho-Erk (Fig. S1B) and the phospho-Erk to total Erk ratio were comparable across groups (Fig. S1C and Table 2). There was no difference between amnestic and non-amnestic MCI cases.
JNK, phospho-JNK and Bcl-2 levels
Although total JNK levels remained stable across clinical groups (Fig. 2A), phospho-JNK was increased 23% in MCI and significantly increased 85% in AD compared to NCI (Fig. 2B). The ratio of phospho-JNK to JNK was significantly increased in MCI (32%) and AD (97%) compared to NCI, while MCI and AD did not reach statistical significance (Fig. 2C). Bcl-2 levels showed a modest increase in MCI (12%) and AD (27%) compared to NCI, and only AD and NCI differed significantly (Fig. S2 and Table 2). No differences in these protein levels occurred between amnestic and non-amnestic MCI cases. A sub-analysis revealed no differences in phospho-JNK and the ratio of phospho-JNK to JNK or Bcl-2 levels between AD cases neuropathologically characterized as moderate Braak (III-IV) compared to severe Braak (V-VI) scores.
Figure 2.
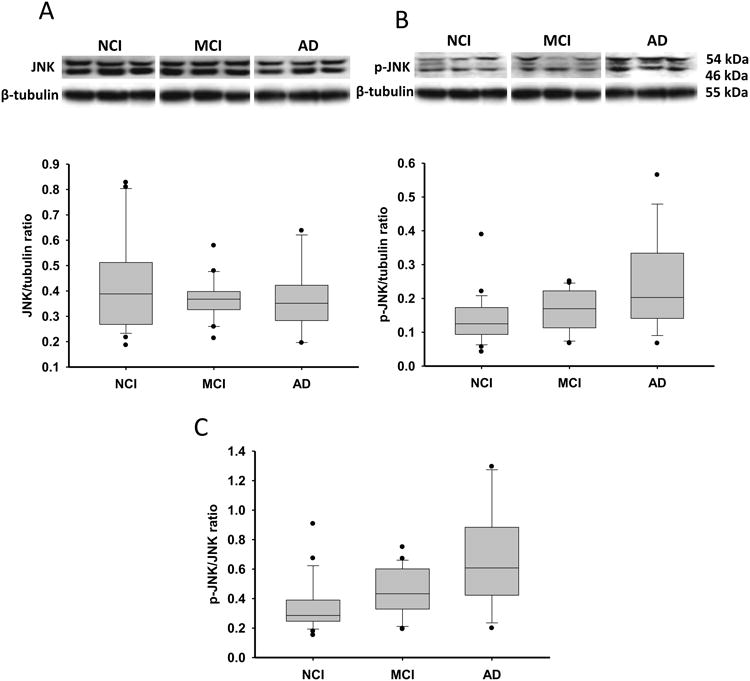
Representative immunoblots and box plots of precuneus levels of total JNK, phospho-JNK (p-JNK) in cases clinically diagnosed as NCI, MCI and AD. Box-plot of the phospho-JNK/JNK ratio is also presented. β-tubulin probe was used to normalize the immunoreactive signals obtained in the blots by densitometry. Levels of total JNK (A) were stable across clinical groups, while the level of phospho-JNK (B, *p = 0.015) and phospho-JNK/JNK ratio (C, *p < 0.001) were significantly increased in AD compared to NCI. Black dots in box-plots indicate outliers.
Aβ1–42 soluble and [3H]PiB binding levels
Both soluble Aβ1–42 levels and [3H]PiB binding levels were significantly higher in AD compared to NCI and MCI, but with no difference between the NCI and MCI groups (Table S2 in Supplement 1) as reported previously in a study using 19 of the current cases (18). No differences in these binding levels occurred between amnestic and non-amnestic MCI cases. A sub-analysis revealed no difference in Aβ1–42 soluble and [3H]PiB binding levels between AD cases neuropathologically characterized as moderate Braak (III-IV) compared to severe Braak (V-VI) scores.
Plaque pathology
Although qualitative examination of APP/Aβ (Fig. 3A-C), 6-CN-PiB (Fig. 5D-F) and X-34 (Fig. 3G-H) positive plaques suggest an increase in MCI and AD compared to NCI (Table S3 in Supplement 1), statistical analysis of total (compact/cored and diffuse) 6-CN-PiB and X-34 plaque load and APP/Aβ plaque density revealed no differences across groups (Table S3 in Supplement 1). Only the comparison for 6-CN-PiB compact/cored (neuritic) profiles reached statistical significance, with the AD plaque load significantly higher than NCI (Table S3 in Supplement 1). Compact/cored 6-CN-PiB-plaque load was significantly higher than diffuse 6-CN-PiB plaque loads in AD (p = 0.026), whereas no differences were observed in the other two groups.
Figure 3.
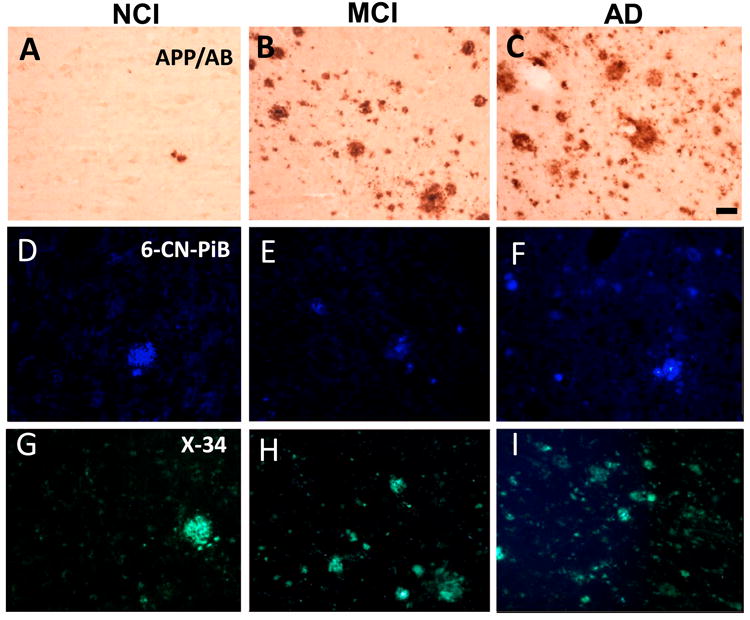
Photomicrographs of APP/Aβ (A-C), 6-CN-PiB (D-F) and X-34 (G-I) positive plaques in the precuneus in NCI, MCI and AD. Note the presence of many more APP/Aβ, 6-CN-PiB and X-34 plaques in AD (C, F and I) compared to NCI (A, D and G), while the difference with MCI (B, E and H) appeared intermediate between AD and NCI. Scale bar shown in C is same for A and B = 50 μm and in D-I = 40 μm.
Figure 5.
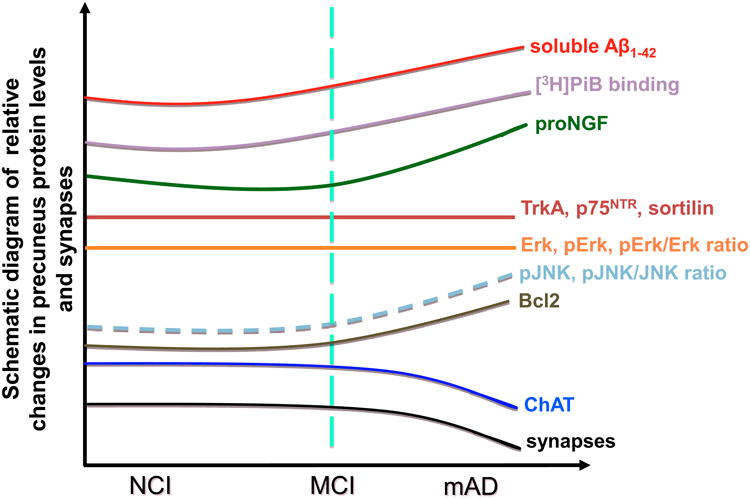
Schematic summary diagram showing the changes in neurotrophic upstream and downstream protein levels and synapses (see 17) in the precuneus during the progression of AD.
Tau pathology
Qualitative examination revealed very few AT8, TOC-1 and Tau C3 immunoreactive (ir) NFTs across clinical groups (Fig. 4 and Table 3). In fact, 14 (78%) of cases did not display Tau C3 positive NFTs. By contrast, AT8 and TOC-1-ir NTs were consistently more abundant in AD (Fig. 4C, F and Table 3). Median AT8-ir NT density was 7.2, 47.3, and 92.3 per mm2 in NCI, MCI and AD, respectively (Table 3). Post hoc analysis revealed that AT8-ir NT density was significantly higher in AD compared to NCI, but not with MCI. However, TOC-1 positive NT density did not differ statistically (Table 3). Although Tau C3 positive NT median density was lowest in MCI (0.3 per mm2) compared to NCI or AD (median 1.6 and 4.5 per mm2) (Table 3), statistical analysis did not reveal changes between clinical groups.
Figure 4.
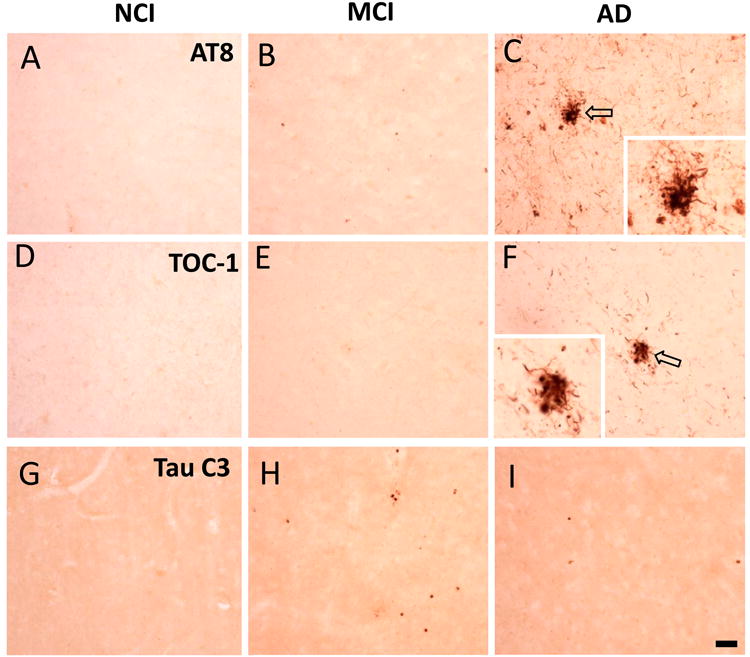
Photomicrographs showing phosphorylational AT8 (A-C), oligomeric TOC-1 (D-F) and truncated C3 (G-I) tau immunoreactivity in NCI, MCI and AD precuneus. Note the absence of AT8 (A-C), TOC-1 (D-F) or Tau C3 (G-I) positive NFTs across clinical groups, the abundance of AT8 (C) and TOC-1 (F) positive NTs in AD compared to NCI (A and D) or MCI (B and E) as well as a lack of Tau C3-ir NT in the three clinical subjects (G-I). Insets in C and F show clusters of dystrophic neurites indicated by unfilled arrows in their respective panels. Scale bars: 50 μm.
Table 3. Summary of NFT and NT density by clinical diagnosis.
| NCI (N=6) |
MCI (N=6) |
AD (N=6) |
p-value | Pair-wise comparison | ||
|---|---|---|---|---|---|---|
| AT8 NFT | Median (Range)* |
0 (0-0.88) |
0.46 (0-1.97) |
0.58 (0.34-2.00) |
0.091a | -- |
| TOC-1 NFT | Median (Range)* |
0 (0-0.96) |
0.51 (0-2.17) |
0.53 (0-1.95) |
0.6a | -- |
| Tau C3 NFT | # Absent | 6 | 4 | 4 | 0.5b | -- |
| # Present | 0 | 2 | 2 | |||
|
| ||||||
| AT8 NT | Median (Range)* |
1.92 (0-3.88) |
3.8 (1.34-5.96) |
4.54 (4.03-5.89) |
0.016a | NCI < AD |
| TOC-1 NT | Median (Range)* |
0.79 (0-4.19) |
3.64 (0-6.40) |
3.36 (1.10-4.89) |
0. 6a | -- |
| Tau C3 NT | Median (Range)* |
0.91 (0-1.44) |
0.24 (0-5.86) |
1.70 (0.88-4.73) |
0.2a | -- |
Based on log-transformed NFT and NT density values (number/mm2).
Kruskal-Wallis test, with Dunn's correction for multiple comparisons.
Fisher's exact test.
Biochemical, plaque and tau pathology associations
Increased proNGF levels were positively correlated with sortilin (r = 0.40, p = 0.0013) and negatively correlated with TrkA levels (r = -0.31, p = 0.013). There was a weak positive correlation between proNGF and phospho-Erk (r = 0.26, p = 0.043), but not between proNGF and the other proteins examined. ProNGF showed the strongest and most consistent correlation with plaque pathology (r = 0.30-0.54 for diffuse and neuritic 6-CN-PiB and X34 plaque loads). Phospho-JNK and the p-JNK/JNK ratio also correlated with plaque pathology (r = 0.26-0.36). Only the correlation between proNGF level and 6-CN-PiB neuritic plaque load reached significance (Fig. S3A, r = 0.54, p = 0.021).
The association between proNGF and NFTs and NTs was more pronounced. Precuneus proNGF levels were significantly correlated with the density of AT8 and TOC-1 positive NFTs and NTs (Fig. S3B-E, r = 0.54-0.60, p = 0.008-0.027) and to a lesser extent, the density of Tau C3 NTs (r = 0.47, p =0.049). No association was found between proNGF and the absence/presence of Tau C3-ir NTFs (Fisher's exact test, p = 0.2). We found that sortilin levels correlated with TOC-1 and Tau C3 NT density (r = 0.52-0.59, p = 0.016-0.031), though less with AT8 and TOC-1 NFT and AT8 NT densities (r = 0.37-0.46, p = 0.06-0.13). Phospho-JNK correlated with AT8 positive NFT and NT density (Fig. S3F, r = 0.57-0.60, p = 0.0086-0.013), but less with TOC-1 NFT and NT or Tau C3 positive NT density (r = 0.31-0.41, p = 0.10-0.21). Phospho-JNK/JNK ratio correlated with AT8 positive NFT and NT density (r = 0.48-0.56, p = 0.015-0.042); its correlation with TOC-1 and Tau C3 pathology was less evident (r < 0.30, p > 0.2).
Although Aβ1–42 and [3H]PiB binding positively associated (r = 0.64, p < 0.0001) with each other, neither correlated with proNGF or other upstream or downstream proteins examined. [3H]PiB binding did exhibit a strong correlation with tau pathology, especially AT8 and TOC-1-ir NFT and NT densities (r = 0.52-0.60, p = 0.012-0.038). A similar pattern was observed between Aβ1–42 and AT8 and TOC-1 but correlations were weak (r = 0.34-0.42, p = 0.10-0.19). Total and diffuse X-34 and 6-CN-PiB plaque loads and APP/Aβ (6E10)-ir plaque density correlated with AT8 and TOC-1 pathology (r = 0.46-0.75, p = 0.0004-0.049). Correlation between 6-CN-PiB plaque loads and TOC-1 positive NT density was not significant (r = 0.45, p = 0.07).
Biochemical, clinical and neuropathological associations
Only levels of phospho-JNK and phospho-JNK/JNK ratio showed a negative association with cognitive function (GCS and episodic memory z-score, r = -0.41 and -0.33, p = 0.010 and 0.036, respectively; MMSE, r = -0.25, p = 0.060) (Fig. S4A and B). The association between phospho-JNK/JNK and episodic memory was due to a strong correlation (Fig. S4C, r = -0.45, p = 0.0036) between phospho-JNK/JNK and East Boston Memory (EBM) immediate recall test. EBM immediate recall showed a significant negative correlation with phospho-JNK and Bcl-2 (r = -0.36 and -0.35, p = 0.022 and 0.027, respectively) (Fig. S4D). Bcl-2 showed a negative correlation with MMSE (r = -0.30, p = 0.021). Only proNGF levels correlated with increased neuropathology (Braak scores (r = 0.32, p = 0.011); Reagan (r = 0.49, p < 0.0001); CERAD (r = 0.46, p = 0.0002).
Precuneus Aβ1–42 values correlated with reduced cognitive function (MMSE (r = -0.45, p = 0.004); GCS (r = -0.55, p < 0.001); episodic memory z-score (Fig. S4E, r = -0.62, p<0.001) and increased neuropathology (Braak scores (r = 0.38, p = 0.013); NIA-Reagan (r = 0.43, p= 0.0042); CERAD (r = 0.46, p = 0.0023). [3H]PiB binding values were negatively correlated with cognitive function (MMSE (r= -0.56, p < 0.0001); GCS (r = -0.53, p = 0.001); episodic memory z-score (Fig. S4F, r = -0.61, p < 0.0001) and positively with neuropathology (Braak scores (r = 0.62, p < 0.0001); Reagan (r = 0.69, p < 0.0001); CERAD (r = 0.61, p < 0.0001). Both Aβ1–42 and [3H]PiB binding correlated with episodic memory test scores (data not shown).
Discussion
The neurotrophin NGF is responsible for the differentiation, survival and maintenance of the cholinergic basal forebrain projection (CBF) neurons, which provide the major cholinergic innervation to the precuneus (36, 37). NGF is synthesized from its precursor molecule, proNGF, the major form found in human brain (38), which plays a key role in cellular apoptosis by binding with a higher affinity to p75NTR (39, 40, 41, 42, 43) than TrkA (44). We found that precuneus proNGF levels were increased in AD but not MCI, which contrasts to increased proNGF in lateral parietal cortex in both MCI and AD (23) but similar to frontal (38, 44, 45) and entorhinal cortex (46) and hippocampus (38, 44) in AD. These data suggest that alterations in proNGF are not uniform across brain regions during the onset of AD. The mechanisms underlying the differential changes in cortical proNGF remain unknown. One factor may be an alteration in NGF metabolic activity. Altered activity of components of the extracellular complex protease cascade (e.g., MMP-9 and plasmin), which is involved in the maturation and degradation of NGF (47) is upregulated in AD (48) resulting in an accumulation of proNGF. Perhaps these proteases are not dysregulated in the MCI precuneus allowing stable metabolic regulation. Another possibility may be related to the increase in amyloid load seen in our AD cases. In this regard, animal studies have shown that over-expression of APP/Aβ (49) and Aβ oligomers (50) increase hippocampal NGF/proNGF levels and activate MMP-9 (50). We found that proNGF levels were not associated with increased soluble Aβ1–42 or fibrillar Aβ [3H]PiB binding, but rather to compact/cored 6-CN-PiB positive plaque load, which was significantly higher in AD, suggesting that Aβ soluble forms are not needed for proNGF upregulation. Precuneus proNGF levels were positively associated with the AT8 and TOC-1 NFTs and NTs density (51) and Braak scores across clinical groups. We speculate that parenchymal changes induced by fibrillar Aβ deposits (52, 53, 54), such as dystrophic neurites, drive the local accumulation of precuneus proNGF and tau (55), due to disrupted transport mechanism(s) (51). Interestingly, the density of neuritic plaques correlate better with cognitive deficits than total plaque load highlighting the importance of these lesions in AD (56). Precuneus proNGF levels correlated negatively with Reagan and CERAD scores indicative of an association with disease pathology.
We found no changes in precuneus IrkA, p75NTR and sortilin levels across clinical groups. ProNGF binds with higher affinity to p75NTR, which is enhanced in the presence of sortilin to induce apoptosis (39, 40, 41, 42, 43, 46, 57). Homeostatic regulation of these receptors together with proNGF results in receptor internalization and retrograde transport of this signaling complex (58) to activate downstream pathways involved in cholinergic basal forebrain neuron survival and maintenance (38, 44). Under appropriate conditions proNGF alone activates neurotrophic activity (38, 44, 59) via TrkA binding (38, 44). Cortical TrkA expression levels are susceptible to regional and temporal differences during disease onset (32, 24). The anterior portion of the nucleus basalis provides the main source of cholinergic innervation to the precuneus and displays less pathology than posterior subfields (60). Gene array studies of single anterior cholinergic neurons revealed an intermediate down regulation of TrkA expression in MCI, and a significant reduction in mild to moderate AD, while p75NTR gene expression was unchanged (61). Since we have not found any change in the levels of p75NTR during disease progression, similar to other cortical regions (24, 32), it is unlikely that there would be a change in precuneus vulnerability to amyloid. Since both receptor levels were unchanged in the precuneus, it is possible that there is a compensatory upregulation of TrkA protein levels reminiscent of that found in the AD hippocampus (24). We reported reduced choline acetyltransferase (ChAT) activity in association with increased [3H]PiB binding and soluble Aβ1–42 only in the AD precuneus (18) which contrasts with ChAT activity upregulation in the superior frontal cortex and hippocampus in MCI (62, 63). Together these data reflect a selective cortical topographic vulnerability of CBF neuron projections sites to disease (60). Moreover, the resilience of neurotrophin signaling may mitigate the effects of the degenerative signaling within the precuneus.
Binding NGF/proNGF and TrkA triggers downstream cascades leading to intracellular events that promote neuronal survival. Erk is activated by TrkA phosphorylation and turns on nuclear effectors involved in gene transcription (64). Similar to the hippocampus (24), we found that precuneus levels of total Erk, phospho-Erk and phospho-Erk/Erk ratio were unchanged among the clinical groups examined. Conversely, the stress-activated kinase phospho-JNK, and the ratio of phospho-JNK to JNK were significantly increased in the AD precuneus, while total JNK levels remained stable across clinical groups similar to the AD hippocampus (24). We also found a significant upregulation in Bcl-2 levels in AD but not MCI. Bcl-2, a component of JNK signaling, is involved in the activation of apoptotic enzymes (65). We hypothesize that proNGF, which binds with high-affinity to p75NTR (46), could, at least in part, be responsible for the activation of JNK apoptotic pathways in AD. We detected a positive relationship between phospho-JNK and the density of AT8-ir NFTs and NTs during disease onset lending support to the observation that JNK activation mediates tau phosphorylation at Ser202/Thr205 (AT8) (66, 67), a pretangle marker (68). The correlation between phospho-JNK and tau suggests a role for the former in NFT and NT formation. Alternatively, we do not rule out that tau could induce JNK alterations. Our data revealed a relationship between Bcl-2, phospho-JNK and phospho-JNK/JNK ratio with the EBM test. Since the precuneus is involved in retrieval and formation of episodic memory, these data suggest that activation of JNK pro-apoptotic signaling plays a role in episodic memory disturbances in AD.
We found that fibrillar amyloid detected by [3H]PiB binding and soluble Aβ1–42 levels were significantly higher in AD compared to NCI, whereas no differences were found between MCI and AD or MCI and NCI. In vivo [11C]PiB PET amyloid imaging has revealed precuneus Aβ deposition prior to clinical AD (9, 11). Histological examination revealed that 6-CN-PiB positive compact/cored plaque load was significantly higher in AD compared to NCI, whereas no differences in X-34 or 6-CN-PiB diffuse or total plaque load, or number of APP/Aβ (6E10)-ir plaques were found across clinical groups. An association between precuneus [11C]PiB binding retention and 6E10-ir plaque number occurs in non-demented subjects (69). Our 6E10-ir plaque counts in MCI do not support high in vivo PiB binding levels prior to clinical AD (9, 11). This discrepancy could be explained by differences in cohorts, techniques (in vivo versus in vitro), or that [11C]PiB binds to other non-fibrillar Aβ related proteins. Our data support previous observations that values of [3H]PiB binding and soluble Aβ1–42 in the precuneus correlate negatively with MMSE, CERAD, episodic memory, indicating an interaction between amyloid and cognition late in AD (18). A fundamental question related to PiB binding is whether visualized amyloid accumulation is linked etiologically to cognitive impairment or secondarily to some other pathologic process.
Here the density of neurons expressing AT8, oligomeric TOC-1 and truncated tau C3 were unchanged across clinical groups, whereas the AT8-ir NTs density was significantly higher in AD. The significant increase in the density of NTs may indicate that tau phosphorylation is initiated first in neurites prior to somal expression (68) suggesting that precuneus NFTs are in an early developmental stage. Density of precuneus tau pathology is associated with diffuse rather than compact/cored plaques, 6E10-ir plaques and [3H]PiB binding, but not with soluble Aβ1–42 across groups. Although we found a parallel increase and interaction between plaque pathology and tau changes, differences among the clinical groups were not significant until the onset of clinical AD, similar to earlier studies (70, 71). Figures 5 and 6 summarize our findings and suggest that Aβ was insufficient to drive proNGF signaling dysfunction in MCI, perhaps reflecting a slow progression of AD pathology or resilience of the precuneus to Aβ toxicity (17, 72).
Figure 6.
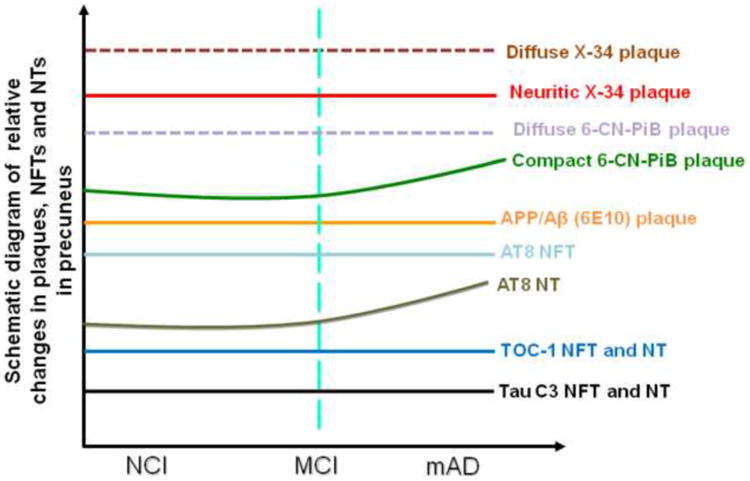
Summary diagram of the pathobiology found within the precuneus during AD progression.
Supplementary Material
Acknowledgments
The authors thank the nuns, priests, and brothers from across the country that participated in the Religious Orders Study and the staff of the Rush Alzheimer's Disease Center. The authors also thank patients and research participants at the University of Kentucky Alzheimer's Disease Center. This study was supported by NIA grants PO1AG14449, PO1AG9466, P30AG10161, RO1AG025204 and RO1AG043375.
Footnotes
Financial Disclosures: Dr. Ikonomovic has consulted for GE Healthcare, which holds the commercial licensing and distribution rights for PiB PET imaging. Drs. Mufson, Perez, Bin He, Nadeem, Scheff, and Abrahamson as well as Ms. Wuu reported no biomedical financial interests or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wagner AD, Shannon BJ, Kahn I, Buckner RL. Parietal lobe contributions to episodic memory retrieval. Trends Cogn Sci. 2005;9:445–453. doi: 10.1016/j.tics.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 2.Buckner RL, Andrews-Hanna JR, Schacter DL. The brain's default network: anatomy, function, and relevance to disease. Ann N Y Acad Sci. 2008;1124:1–38. doi: 10.1196/annals.1440.011. [DOI] [PubMed] [Google Scholar]
- 3.Raichle ME, MacLeod AM, Snyder AZ, Powers WJ, Gusnard DA, Shulman GL. A default mode of brain function. Proc Natl Acad Sci U S A. 2001;98:676–682. doi: 10.1073/pnas.98.2.676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sperling RA, Dickerson BC, Pihlajamaki M, Vannini P, LaViolette PS, Vitolo OV, et al. Functional alterations in memory networks in early Alzheimer's disease. Neuromolecular Med. 2010;12:27–43. doi: 10.1007/s12017-009-8109-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andrews-Hanna JR, Snyder AZ, Vincent JL, Lustinq C, Head D, Raichle ME, et al. Disruption of large-scale brain systems in advanced aging. Neuron. 2007;56:924–935. doi: 10.1016/j.neuron.2007.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herholz K, Salmon E, Perani D, Baron JC, Holthoff V, Frölich L, et al. Discrimination between Alzheimer dementia and controls by automated analysis of multicenter FDG PET. Neuroimage. 2002;17:302–316. doi: 10.1006/nimg.2002.1208. [DOI] [PubMed] [Google Scholar]
- 7.Matsuda H. The role of neuroimaging in mild cognitive impairment. Neuropathology. 2007;27:570–577. doi: 10.1111/j.1440-1789.2007.00794.x. [DOI] [PubMed] [Google Scholar]
- 8.Rombouts SA, Barkhof F, Goekoop R, Stam CJ, Scheltens P. Altered resting state networks in mild cognitive impairment and mild Alzheimer's disease: an fMRI study. Hum Brain Mapp. 2005;26:231–239. doi: 10.1002/hbm.20160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mintun MA, Larossa GN, Sheline YI, Dence CS, Lee SY, Mach RH, et al. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology. 2006;67:446–452. doi: 10.1212/01.wnl.0000228230.26044.a4. [DOI] [PubMed] [Google Scholar]
- 10.Sheline YI, Raichle ME, Snyder AZ, Morris JC, Head D, Wang S, et al. Amyloid plaques disrupt resting state default mode network connectivity in cognitively normal elderly. Biol Psychiatry. 2010;676:584–587. doi: 10.1016/j.biopsych.2009.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sperling RA, Laviolette PS, O'Keefe K, O'Brien J, Rentz DM, Pihlajamaki M, et al. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron. 2009;63:178–188. doi: 10.1016/j.neuron.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drzezga A, Becker JA, Van Dijk KR, Sreenivasan A, Talukdar T, Sullivan C, et al. Neuronal dysfunction and disconnection of cortical hubs in non-demented subjects with elevated amyloid burden. Brain. 2011;134:1635–1646. doi: 10.1093/brain/awr066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yan H, Zhang Y, Chen H, Wang Y, Liu Y. Altered effective connectivity of the default mode network in resting-state amnestic type mild cognitive impairment. J Int Neuropsychol Soc. 2013;19:400–409. doi: 10.1017/S1355617712001580. [DOI] [PubMed] [Google Scholar]
- 14.Frisoni GB, Testa C, Sabattoli F, Beltramello A, Soininen H, Laakso MP. Structural correlates of early and late onset Alzheimer's disease: voxel based morphometric study. J Neurol Neurosurg Psychiatry. 2005;76:112–114. doi: 10.1136/jnnp.2003.029876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishii K, Kawachi T, Sasaki H, Kono AK, Fukuda T, Kojima Y, et al. Voxel-based morphometric comparison between early- and late-onset mild Alzheimer's disease and assessment of diagnostic performance of z score images. AJNR Am J Neuroradiol. 2005;26:333–340. [PMC free article] [PubMed] [Google Scholar]
- 16.Karas G, Scheltens P, Rombouts S, van Schijndel R, Klein M, Jones B, et al. Precuneus atrophy in early-onset Alzheimer's disease: a morphometric structural MRI study. Neuroradiology. 2007;49:967–976. doi: 10.1007/s00234-007-0269-2. [DOI] [PubMed] [Google Scholar]
- 17.Scheff SW, Price DA, Schmitt FA, Roberts KN, Ikonomovic MD, Mufson EJ. Synapse stability in the precuneus early in the progression of Alzheimer's disease. J Alzheimers Dis. 2013;35:599–609. doi: 10.3233/JAD-122353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ikonomovic MD, Klunk WE, Abrahamson EE, Wuu J, Mathis CA, Scheff SW, et al. Precuneus amyloid burden is associated with reduced cholinergic activity in Alzheimer disease. Neurology. 2011;77:39–47. doi: 10.1212/WNL.0b013e3182231419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bulbarelli A, Lonati E, Cazzaniga E, Re F, Sesana S, Barisani D, et al. TrkA pathway activation induced by amyloid-beta (Abeta) Mol Cell Neurosci. 2009;40:365–373. doi: 10.1016/j.mcn.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 20.Fombonne J, Rabizadeh S, Banwait S, Mehlen P, Bredesen DE. Selective vulnerability in Alzheimer's disease: amyloid precursor protein and p75(NTR) interaction. Ann Neurol. 2009;65:294–303. doi: 10.1002/ana.21578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yaar M, Zhai S, Pilch PF, Doyle SM, Eisenhauer PB, Fine RE, et al. Binding of beta-amyloid to the p75 neurotrophin receptor induces apoptosis. A possible mechanism for Alzheimer's disease. J Clin Invest. 1997;100:2333–2340. doi: 10.1172/JCI119772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mufson EJ, Binder L, Counts SE, DeKosky ST, de Toledo-Morrell L, Ginsberg SD, et al. Mild cognitive impairment: pathology and mechanisms. Acta Neuropathologica. 2012;123:13–30. doi: 10.1007/s00401-011-0884-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peng S, Wuu J, Mufson EJ, Fahnestock M. Increased proNGF levels in subjects with mild cognitive impairment and mild Alzheimer disease. J Neuropathol Exp Neurol. 2004;63:641–649. doi: 10.1093/jnen/63.6.641. [DOI] [PubMed] [Google Scholar]
- 24.Mufson EJ, He B, Nadeem M, Perez SE, Counts SE, Leurgans S, et al. Hippocampal proNGF signaling pathways and β-amyloid levels in mild cognitive impairment and Alzheimer disease. J Neuropathol Exp Neurol. 2012;71:1018–1029. doi: 10.1097/NEN.0b013e318272caab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bennett DA, Schneider JA, Bienias JL, Evans DA, Wilson RS. Mild cognitive impairment is related to Alzheimer disease pathology and cerebral infarctions. Neurology. 2005;64:834–841. doi: 10.1212/01.WNL.0000152982.47274.9E. [DOI] [PubMed] [Google Scholar]
- 26.Bennett DA, Schneider JA, Arvanitakis Z, Kelly JF, Aggarwal NT, Shah RC, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology. 2006;66:1837–1844. doi: 10.1212/01.wnl.0000219668.47116.e6. [DOI] [PubMed] [Google Scholar]
- 27.Mufson EJ, Chen EY, Cochran EJ, Beckett LA, Bennett DA, Kordower JH. Entorhinal cortex beta-amyloid load in individuals with mild cognitive impairment. Exp Neurol. 1999;158:469–490. doi: 10.1006/exnr.1999.7086. [DOI] [PubMed] [Google Scholar]
- 28.Davis DG, Schmitt FA, Wekstein DR, Markesbery WR. Alzheimer neuropathologic alterations in aged cognitively normal subjects. J Neuropathol Exp Neurol. 1999;58:376–388. doi: 10.1097/00005072-199904000-00008. [DOI] [PubMed] [Google Scholar]
- 29.Schmitt FA, Nelson PT, Abner E, Scheff S, Jicha GA, Smith C, et al. University of Kentucky Sanders-Brown healthy brain aging volunteers: donor characteristics, procedures and neuropathology. Curr Alzheimer Res. 2012;9:724–733. doi: 10.2174/156720512801322591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perez SE, Getova DP, He B, Counts SE, Geula C, Desire L, et al. Rac1b increases with progressive tau pathology within cholinergic nucleus basalis neurons in Alzheimer's disease. Am J Pathol. 2012;180:526–540. doi: 10.1016/j.ajpath.2011.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmitt FA, Davis DG, Wekstein DR, Smith CD, Ashford JW, Markesbery WR. “Preclinical” AD revisited: neuropathology of cognitively normal older adults. Neurology. 2000;55:370–376. doi: 10.1212/wnl.55.3.370. [DOI] [PubMed] [Google Scholar]
- 32.Counts SE, Nadeem M, Wuu J, Ginsberg SD, Saragovi HU, Mufson EJ. Reduction of cortical TrkA but not p75(NTR) protein in early-stage Alzheimer's disease. Ann Neurol. 2004;56:520–531. doi: 10.1002/ana.20233. [DOI] [PubMed] [Google Scholar]
- 33.Ikonomovic MD, Klunk WE, Abrahamson EE, Mathis CA, Price JC, Tsopelas ND, et al. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer's disease. Brain. 2008;131:1630–1645. doi: 10.1093/brain/awn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klunk WE, Lopresti BJ, Ikonomovic MD, Lefterov IM, Koldamova RP, Abrahamson EE, et al. Binding of the positron emission tomography tracer Pittsburgh compound-B reflects the amount of amyloid-beta in Alzheimer's disease brain but not in transgenic mouse brain. Neurosci. 2005;25:10598–10606. doi: 10.1523/JNEUROSCI.2990-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ikonomovic MD, Abrahamson EE, Isanski BA, Debnath ML, Mathis CA, Dekosky ST, et al. X-34 labeling of abnormal protein aggregates during the progression of Alzheimer's disease. Methods Enzymol. 2006;412:123–144. doi: 10.1016/S0076-6879(06)12009-1. [DOI] [PubMed] [Google Scholar]
- 36.Lad SP, Neet KE, Mufson EJ. Nerve growth factor: structure, function and therapeutic implications for Alzheimer's disease. Curr Drug Targets CNS Neurol Disord. 2005;2:315–334. doi: 10.2174/1568007033482724. [DOI] [PubMed] [Google Scholar]
- 37.Mesulam MM, Mufson EJ, Levey AI, Wainer BH. Cholinergic innervation of cortex by the basal forebrain: cytochemistry and cortical connections of the septal area, diagonal band nucleus, nucleus basalis (substantia innominata), and hypothalamus in the rhesus monkey. J Comp Neurol. 1983;214:170–197. doi: 10.1002/cne.902140206. [DOI] [PubMed] [Google Scholar]
- 38.Fahnestock M, Michalski B, Xu B, Coughlin MD. The precursor pro-nerve growth factor is the predominant form of nerve growth factor in brain and is increased in Alzheimer's disease. Mol Cell Neurosci. 2001;18:210–220. doi: 10.1006/mcne.2001.1016. [DOI] [PubMed] [Google Scholar]
- 39.Al-Shawi R, Hafner A, Chun S, Raza S, Crutcher K, Thrasivoulou C, et al. ProNGF, sortilin, and age-related neurodegeneration. Ann N Y Acad Sci. 2007;1119:208–215. doi: 10.1196/annals.1404.024. [DOI] [PubMed] [Google Scholar]
- 40.Lee R, Kermani P, Teng KK, Hempstead BL. Regulation of cell survival by secreted proneurotrophins. Science. 2001;294:1945–1948. doi: 10.1126/science.1065057. [DOI] [PubMed] [Google Scholar]
- 41.Masoudi R, Ioannou MS, Coughlin MD, Pagadala P, Neet KE, Clewes O, et al. Biological activity of nerve growth factor precursor is dependent upon relative levels of its receptors. J Biol Chem. 2009;284:18424–18433. doi: 10.1074/jbc.M109.007104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nykjaer A, Lee R, Teng KK, Jansen P, Madsen P, Nielsen MS, et al. Sortilin is essential for proNGF-induced neuronal cell death. Nature. 2004;427:843–848. doi: 10.1038/nature02319. [DOI] [PubMed] [Google Scholar]
- 43.Pedraza CE, Podlesniy P, Vidal N, Arévalo JC, Lee R, Hempstead B, et al. Pro-NGF isolated from the human brain affected by Alzheimer's disease induces neuronal apoptosis mediated by p75NTR. Am J Pathol. 2005;166:533–543. doi: 10.1016/S0002-9440(10)62275-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fahnestock M, Yu G, Michalski B, Mathew S, Colquhoun A, Ross GM, et al. The nerve growth factor precursor proNGF exhibits neurotrophic activity but is less active than mature nerve growth factor. J Neurochem. 2004;89:581–592. doi: 10.1111/j.1471-4159.2004.02360.x. [DOI] [PubMed] [Google Scholar]
- 45.Podlesniy P, Kichev A, Pedraza C, Saurat J, Encinas M, Perez B, et al. Pro-NGF from Alzheimer's disease and normal human brain displays distinctive abilities to induce processing and nuclear translocation of intracellular domain of p75NTR and apoptosis. Am J Pathol. 2006;169:119–131. doi: 10.2353/ajpath.2006.050787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Al-Shawi R, Hafner A, Olsen J, Chun S, Raza S, Thrasivoulou C, et al. Neurotoxic and neurotrophic roles of proNGF and the receptor sortilin in the adult and ageing nervous system. Eur J Neurosci. 2008;27:2103–2114. doi: 10.1111/j.1460-9568.2008.06152.x. [DOI] [PubMed] [Google Scholar]
- 47.Bruno MA, Cuello AC. Activity-dependent release of precursor nerve growth factor, conversion to mature nerve growth factor, and its degradation by a protease cascade. Proc Natl Acad Sci U S A. 2006;103:6735–6740. doi: 10.1073/pnas.0510645103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bruno MA, Mufson EJ, Wuu J, Cuello AC. Increased matrix metalloproteinase 9 activity in mild cognitive impairment. J Neuropathol Exp Neurol. 2009;68:1309–1318. doi: 10.1097/NEN.0b013e3181c22569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Salehi A, Delcroix JD, Belichenko PV, Zhan K, Wu C, Valletta JS, et al. Increased App expression in a mouse model of Down's syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron. 2006;51:29–42. doi: 10.1016/j.neuron.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 50.Bruno MA, Leon WC, Fragoso G, Mushynski WE, Almazan G, Cuello AC. Amyloid beta-induced nerve growth factor dysmetabolism in Alzheimer disease. J Neuropathol Exp Neurol. 2009;68:857–869. doi: 10.1097/NEN.0b013e3181aed9e6. [DOI] [PubMed] [Google Scholar]
- 51.Ward SM, Himmelstein DS, Lancia JK, Binder LI. Tau oligomers and tau toxicity in neurodegenerative disease. Biochem Soc Trans. 2012;40:667–671. doi: 10.1042/BST20120134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Knowles JK, Rajadas J, Nguyen TV, Yang T, LeMieux MC, Vander Griend L, et al. The p75 neurotrophin receptor promotes amyloid-beta(1-42)-induced neuritic dystrophy in vitro and in vivo. J Neurosci. 2009;29:10627–10637. doi: 10.1523/JNEUROSCI.0620-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lombardo JA, Stern EA, McLellan ME, Kajdasz ST, Hickey GA, Bacskai BJ, et al. Amyloid-beta antibody treatment leads to rapid normalization of plaque-induced neuritic alterations. J Neurosci. 2003;23:10879–10883. doi: 10.1523/JNEUROSCI.23-34-10879.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vehmas AK, Kawas CH, Stewart WF, Troncoso JC. Immune reactive cells in senile plaques and cognitive decline in Alzheimer's disease. Neurobiol Aging. 2003;24:321–331. doi: 10.1016/s0197-4580(02)00090-8. [DOI] [PubMed] [Google Scholar]
- 55.Götz J, Schild A, Hoerndli F, Pennanen L. Amyloid-induced neurofibrillary tangle formation in Alzheimer's disease: insight from transgenic mouse and tissue-culture models. Int J Dev Neurosci. 2004;22:453–465. doi: 10.1016/j.ijdevneu.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 56.Nagy Z, Esiri MM, Jobst KA, Morris JH, King EM, McDonald B, et al. Relative roles of plaques and tangles in the dementia of Alzheimer's disease: correlations using three sets of neuropathological criteria. Dementia. 1995;6:21–31. doi: 10.1159/000106918. [DOI] [PubMed] [Google Scholar]
- 57.Skeldal S, Matusica D, Nykjaer A, Coulson EJ. Proteolytic processing of the p75 neurotrophin receptor: A prerequisite for signalling?: Neuronal life, growth and death signalling are crucially regulated by intra-membrane proteolysis and trafficking of p75(NTR) Bioassays. 2011;33:614–625. doi: 10.1002/bies.201100036. [DOI] [PubMed] [Google Scholar]
- 58.Mufson EJ, Kroin JS, Sendera TJ, Sobreviela T. Distribution and retrograde transport of trophic factors in the central nervous system: functional implications for the treatment of neurodegenerative diseases. Prog Neurobiol. 1999;574:451–484. doi: 10.1016/s0301-0082(98)00059-8. [DOI] [PubMed] [Google Scholar]
- 59.Lakshmanan J, Beattie GM, Hayek A, Burns C, Fisher DA. Biological actions of 53 kDa nerve growth factor as studied by a blot and culture technique. Neurosci Lett. 1989;99:263–267. doi: 10.1016/0304-3940(89)90457-6. [DOI] [PubMed] [Google Scholar]
- 60.Mufson EJ, Ginsberg SD, Ikonomovic MD, DeKosky ST. Human cholinergic basal forebrain: chemoanatomy and neurologic dysfunction. J Chem Neuroanat. 2003;26:233–242. doi: 10.1016/s0891-0618(03)00068-1. [DOI] [PubMed] [Google Scholar]
- 61.Ginsberg SD, Che S, Wuu J, Counts SE, Mufson EJ. Down regulation of trk but not p75NTR gene expression in single cholinergic basal forebrain neurons mark the progression of Alzheimer's disease. J Neurochem. 2006;97:475–487. doi: 10.1111/j.1471-4159.2006.03764.x. [DOI] [PubMed] [Google Scholar]
- 62.DeKosky ST, Ikonomovic MD, Styren SD, Beckett L, Wisniewski S, Bennett DA, et al. Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann Neurol. 2002;51:145–155. doi: 10.1002/ana.10069. [DOI] [PubMed] [Google Scholar]
- 63.Ikonomovic MD, Mufson EJ, Wuu J, Cochran EJ, Bennett DA, DeKosky ST. Cholinergic plasticity in hippocampus of individuals with mild cognitive impairment: correlation with Alzheimer's neuropathology. J Alzheimers Dis. 2003;5:39–48. doi: 10.3233/jad-2003-5106. [DOI] [PubMed] [Google Scholar]
- 64.Niewiadomska G, Mietelska-Porowska A, Mazurkiewicz M. The cholinergic system, nerve growth factor and the cytoskeleton. Behav Brain Res. 2011;221:515–526. doi: 10.1016/j.bbr.2010.02.024. [DOI] [PubMed] [Google Scholar]
- 65.Bhakar AL, Howell JL, Paul CE, Salehi AH, Becker EB, Said F, et al. Apoptosis induced by p75NTR overexpression requires Jun kinase-dependent phosphorylation of Bad. J Neurosci. 2003;23:11373–11381. doi: 10.1523/JNEUROSCI.23-36-11373.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Goedert M, Hasegawa M, Jakes R, Lawler S, Cuenda A, Cohen P. Phosphorylation of microtubule-associated protein tau by stress-activated protein kinases. FEBS Lett. 1997;409:57–62. doi: 10.1016/s0014-5793(97)00483-3. [DOI] [PubMed] [Google Scholar]
- 67.Reynolds CH, Nebreda AR, Gibb GM, Utton MA, Anderton BH. Reactivating kinase/p38 phosphorylates tau protein in vitro. J Neurochem. 1997;69:191–198. doi: 10.1046/j.1471-4159.1997.69010191.x. [DOI] [PubMed] [Google Scholar]
- 68.Vana L, Kanaan NM, Ugwu IC, Wuu J, Mufson EJ, Binder LI. Progression of tau pathology in cholinergic Basal forebrain neurons in mild cognitive impairment and Alzheimer's disease. Am J Pathol. 2011;179:2533–2550. doi: 10.1016/j.ajpath.2011.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Driscoll I, Troncoso JC, Rudow G, Sojkova J, Pletnikova O, Zhou Y, et al. Correspondence between in vivo (11)C-PiB-PET amyloid imaging and postmortem, region-matched assessment of plaques. Acta Neuropathol. 2012;124:823–831. doi: 10.1007/s00401-012-1025-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 71.Nelson PT, Abner EL, Scheff SW, Schmitt FA, Kryscio RJ, Jicha GA, et al. Alzheimer's-type neuropathology in the precuneus is not increased relative to other areas of neocortex across a range of cognitive impairment. Neurosci Lett. 2009;450:336–339. doi: 10.1016/j.neulet.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Arnold SE, Louneva N, Cao K, Wang LS, Han LY, Wolk DA, et al. Cellular, synaptic, and biochemical features of resilient cognition in Alzheimer's disease. Neurobiol Aging. 2013;34:157–168. doi: 10.1016/j.neurobiolaging.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.