

Abstract
The (salen) Co catalyst (4a) can be prepared as a mixture of cyclic oligomers in a short, chromatography-free synthesis from inexpensive, commercially available precursors. This catalyst displays remarkable enhancements in reactivity and enantioselectivity relative to monomeric and other multimeric (salen) Co catalysts in a wide variety of enantioselective epoxide ring-opening reactions. The application of catalyst 4a is illustrated in the kinetic resolution of terminal epoxides by nucleophilic ring-opening with water, phenols, and primary alcohols; the desymmetrization of meso epoxides by addition of water and carbamates; and the desymmetrization of oxetanes by intramolecular ring opening with alcohols and phenols. The favorable solubility properties of complex 4a under the catalytic conditions facilitated mechanistic studies, allowing elucidation of the basis for the beneficial effect of oligomerization. Finally, a catalyst selection guide is provided to delineate the specific advantages of oligomeric catalyst 4a relative to (salen) Co monomer 1 for each reaction class.
Keywords: kinetic resolution, salen, desymmetrization, epoxides, ring-opening
1. Introduction
Among the wide assortment of organic transformations catalyzed with high enantioselectivities by chiral (salen) metal complexes,1 epoxide ring-opening reactions have arguably proven most impactful from a synthetic standpoint in both academic and industrial contexts. In particular, the hydrolytic kinetic resolution (HKR) catalyzed by monomeric (salen) Co complex 1 provides a general approach to the preparation of both enantiopure terminal epoxides and highly enantioenriched 1,2-diols (Scheme 1).2,3,4,5 Monomeric complex 1 has also been applied with more limited success to the phenolytic kinetic resolution (PKR)6 and the carbamolytic kinetic resolution (CKR)7 of terminal epoxides to afford enantioenriched α-aryloxy alcohols and N-protected 1-amino-2-ols, respectively.8,9 Other classes of nucleophiles and epoxides have generally remained beyond the scope of this system.10,11 Moreover, although catalyst loadings required for HKR, PKR, and CKR reactions of most terminal epoxides are low (<5 mol% Co), additional improvements in catalytic reactivity are desirable in order to achieve highly efficient large-scale applications.12
Scheme 1.

The HKR of terminal epoxides with monomeric (salen)Co complex 1.
The recognition that the HKR13 and related14 epoxide ring-opening reactions proceed via cooperative bimetallic mechanisms, wherein both the epoxide electrophile and nucleophile are activated by separate (salen) Co complexes in the rate-limiting ring-opening event (Figure 1),15 has motivated the preparation and study of a wide variety of linked multi- (salen) metal complexes. The goal of these efforts has been to achieve higher catalytic activity by reducing the entropic cost of a second-order bimetallic pathway.16,17 In several cases, these studies succeeded in uncovering catalyst systems with enhanced reactivity relative to monomer 1 but that are also far more difficult to prepare and therefore less attractive from a practical standpoint. This is due in large part to the need to synthesize unsymmetrical salen ligand frameworks to allow dimerization as in Figure 2A.18 As an alternative to these catalysts that possess local C1 symmetry, multimeric (salen) Co complexes have been identified that are straightforward to prepare owing to the fact that they preserve the local C2 symmetry of each salen unit. As such, they can be synthesized simply by condensation of appropriate linked bis(salicylaldehydes), or by linkage of preformed C2-symmetric salen units (Figure 2B).
Figure 1.

Proposed rate-limiting transition structure in the hydrolytic kinetic resolution of terminal epoxides catalyzed by monomeric (salen)Co complex 1 (ref. 15).
Figure 2.

A) Linkage of metal salen complexes resulting in local C1 symmetry in the individual salen units. B) Strategies for the preparation of linked analogs of 1 with local C2 symmetry in the individual salen units.
Linked complexes 219 and 320 represented the first reported examples of these more readily accessible catalysts (Figure 3).21 These mixtures of cyclic, oligomeric linked (salen) Co units displayed not only dramatic reactivity improvements in the HKR and PKR of terminal epoxides relative to monomer 1, but also facilitated reactions that were impossible with monomeric catalyst such as the alcoholytic kinetic resolution (AKR) of terminal epoxides as well as the hydrolytic desymmetrization of cyclic meso epoxides. The basis for enhanced reactivity could be traced to cooperative reaction within the linked catalysts, as epoxide ring-opening reactions displayed a first-order kinetic dependence on catalyst concentration.19
Figure 3.

First- and second-generation cyclic oligomeric (salen)Co catalysts for epoxide ring-opening reactions.
Despite the very attractive properties of the cyclic oligomeric catalysts 2 and 3, their practical utility and the ability to study them systematically was limited by their poor solubility. Poor reproducibility both between runs employing the same catalyxst batch and between runs employing different catalyst batches was observed in several cases. For example, the conversion in the hydrolysis of cis-2-butene oxide model HKR reaction using different batches of catalyst 3b varied from 58–94% over 24 h. After systematic evaluation of various structural parameters, we discovered that the relatively minor perturbation of introducing an oxygen atom into the linker chain, as in 4 (Figure 4), resulted in catalysts with substantially improved physical properties, including solubility in common organic solvents such as CH2Cl2 and CH3CN.22 Catalyst 4a (X = OTf) was shown to be highly effective in a variety of HKR reactions, displaying both very high and reproducible activity and stereoselectivity with representative terminal epoxides. Tosylate derivative 4b was found to be less reactive, but most effective for highly reactive terminal epoxides such as styrene oxide.22,23 Catalyst 4a has found application in several advanced synthetic applications24 and new enantioselective ring-opening reactions.25
Figure 4.

Third-generation cyclic oligomeric (salen)Co catalysts for epoxide ring-opening reactions.
We provide here a full account of the scope of catalyst 4a in enantioselective epoxide ring-opening reactions, thereby establishing 4a as the most general and effective system for such reactions identified to date. We describe a practical, chromatography-free synthesis of catalyst 4a, and document the application of this catalyst in the kinetic resolution of terminal epoxides by nucleophilic ring-opening with water, phenols, and primary alcohols, as well as the desymmetrization of meso epoxides by addition of water and carbamates, and the desymmetrization of oxetanes by intramolecular ring opening with alcohols and phenols. The favorable solubility properties of complex 4a under the catalytic conditions were exploited in mechanistic studies, allowing elucidation of the basis for the beneficial effect of oligomerization. Finally, we provide a catalyst selection guide to delineate the specific advantages of oligomeric catalyst 4a relative to (salen) Co monomer 1 for each reaction class.
2. Results and Discussion
2.1 Catalyst Synthesis
A scalable, chromatography-free synthesis of catalyst 4a starting from commercially available tert-butylhydroquinone (6) and 2-cyanoethyl ether (8) has been developed and is outlined in Scheme 2. Selective protection of the less hindered hydroxyl group of 6 was followed by Sn(IV)-mediated directed formylation and subsequent deprotection to provide aldehyde 7 according to the previously established route.26 Acid-promoted hydrolysis of dinitrile 8 afforded diacid 9,27 which was coupled directly to salicylaldehyde 7 under carbodiimide-mediated conditions. Recrystallization from MeOH/H2O then afforded pure bis(aldehyde) 10 in 77–82% overall yield from 8. Following procedures analogous to those developed for the preparation of catalyst 3,20 bis(aldehyde) 10 was condensed with (1R,2R)-1,2-diaminocyclohexane generated in situ from its L-tartrate salt to yield the cyclic Schiff base ligand 5 as a mixture of oligomers (n=1,2) as determined by ESI MS and 1H-NMR analysis.28 This material was then subjected directly to one-pot Co(II) insertion and subsequent air oxidation in the presence of triflic acid to provide complex 4a in 100–102% yield based on the structure presented in Scheme 2. The catalyst thus obtained was thereby prepared in 60–66% yield from 6 on multigram scale. It was shown by 1H NMR to contain varying amounts (up to 9% by weight) of residual toluene, but applied without further purification to all the asymmetric ring-opening reactions described herein.29 Analyses of 4a by MS and NMR revealed that the mixture of oligomers consisted primarily of dimer (4an=1) and smaller amounts of trimer (4an=2).28 This material displayed excellent consistency across multiple batches, as illustrated in the PKR of 1,2-epoxyhexane with 2-bromophenol (Figure 5).
Scheme 2.

Optimized synthesis of catalyst 4a.a
aConditions: (a) t-BuCOCl, DMAP (0.15 equiv), imidazole, CH2Cl2, 0 → 4 °C, 97%; (b) SnCl4 (0.5 equiv), 2,6-lutidine, 0 °C, then (CH2O)n, toluene, 90 °C, 93%; (c) KOH, H2O/EtOH, 23 °C, 94%; (d) conc. HCl 50–55 °C; (e) EDC, DMAP (0.4 equiv), CH2Cl2/DMF, 0 °C → 23 °C, recrystallized from MeOH/H2O; (f) K2CO3, THF/H2O, reflux; (g) Co(OAc)2•4H2O, toluene/MeOH, N2, 23 °C, then TfOH, air, CH2Cl2, 23 °C.
Figure 5.

Batch consistency of catalyst 4a prepared under optimized synthetic conditions in the PKR of 1,2-epoxyhexane with 2-bromophenol. Conversions were determined by GC analysis relative to an internal standard. Enantiomeric excesses were determined by chiral HPLC analysis and are provided for the last data point of each curve.
2.2 Quantitative Comparison of Monomeric and Oligomeric Catalysts
The advantages of the 3rd generation oligomeric catalyst 4a relative to both the 2nd generation oligomeric (3b) and monomeric (1) catalysts are illustrated in model epoxide ring-opening reactions (Figure 6). The HKR of methyl glycidate (Figure 6A) could be carried out with very low catalyst loadings (0.03 mol%) under solvent-free conditions with 4a; in contrast, oligomer 3b was poorly soluble under these conditions, thus reducing its already lower inherent reactivity. Catalyst 1b was completely soluble under the same conditions, but displayed extremely low reactivity at such low concentrations.30,31 In the PKR of 1,2-epoxyhexane with 2-bromophenol, catalyst 4a induced both much faster rates and higher enantioselectivity than 3b in the formation of the ring-opened addition product (Figure 6B). The monomeric catalyst 1c that is optimal for PKR reaction was almost completely unreactive under the same conditions.32 A significant improvement in both reactivity and enantioselectivity of 4a relative to oligomer 3b and monomer 1a was also observed in the hydrolysis of cyclohexene oxide (Figure 6D).
Figure 6.

Comparison of previous generations of (salen)Co(III) catalysts with oligomeric (salen)Co complex 4a. Conversions were determined by GC analysis relative to an internal standard or by 1H NMR analysis. Enantiomeric excesses were determined by chiral GC or HPLC analysis. Numerical conversion and ee values are provided for the last data point of each curve.
2.3 Applications of Oligomeric Catalyst 4a
2.3.1 HKR of Terminal Epoxides
The extraordinary effectiveness of (oligosalen)Co catalyst 4a in HKR reactions of terminal epoxides is illustrated in the representative examples in Table 1. All reactions were carried out under solvent-free conditions at ambient temperature. Extremely low catalyst loadings can be employed with several important classes of terminal epoxides including aliphatic epoxides (entry 1), epichlorohydrin (entry 2), glycidol derivatives (entry 3), glycidic esters (entry 4), vinyl epoxides (entry 5), and styrene derivatives (entry 6). The efficiency of this catalyst system is highlighted by considering the amounts of 4a required to effect HKR reactions: 1.5 mol of racemic propylene oxide was resolved within 24 h using only 3.6 mg (41 ppm by mass, 3 ppm on a molar basis) of 4a to yield 35 g (40%) of recovered epoxide in >99% ee (entry 1); similarly, only 18 mg of 4a are required to obtain one mole (93 g) of resolved epichlorohydrin. In addition, the HKR of epichlorohydrin can now be carried out at room temperature with 4a without any detectable signs of a secondary racemization pathway that is observed with monomeric catalyst 1.17b In certain cases, trace impurities present in the racemic epoxides may have a deleterious effect on catalyst efficiency. However, this problem may be circumvented by purification of the epoxide by standard protocols.33
Table 1.

| |||
|---|---|---|---|
|
| |||
| entry | R | Coc (mol%) | epoxide yieldd (%) |
| 1 e | Me | 0.0003 | 40 |
| 2 | CH2Cl | 0.001 | 44 |
| 3 | CH2OCH2CH=CH2 | 0.0025 | 43 |
| 4 | CO2Me | 0.015 | 44 |
| 5e,f | CH=CH2 | 0.025 | 35 |
| 6g | Ph | 0.04 | 40 |
Entry 1 was carried out on a 1.5 mol scale. Entries 2–7 were carried out on 80–130 mmol scale.
Enantiomeric excesses were determined by chiral GC or HPLC analysis.
Catalyst loading relative to racemic epoxide.
Isolated yield based on racemic epoxide (theoretical maximum = 50%).
Enantiomeric excess was determined by chiral HPLC analysis of the terminal addition product of 2-naphthalenethiol.
Employed 0.7 equiv H2O.
Employed catalyst 4b.
As noted above, the HKR of certain sensitive conjugated epoxides such as styrene oxide are best carried out with tosylate catalyst 4b (entry 6).23 Phenylacetaldehyde is present in variable amounts in commercially available racemic styrene oxide and is very difficult to remove completely. Because of the inhibitory effect of this impurity, the catalyst loading required to achieve >99% ee within 24 h was found to be somewhat variable (0.025–0.04 mol% Co).
The protocol for the HKR of terminal epoxides with catalyst 4a or 4b is operationally very simple. Water is added to a mixture of catalyst and epoxide,34 and the resulting reaction mixture is stirred at room temperature while the epoxide ee is monitored by chromatographic analysis of small aliquots. Once >99% ee has been attained, the epoxide is isolated by vacuum transfer and dried over an appropriate desiccant.35 In cases where the physical properties of the epoxide prevent resolution under solvent-free conditions (e.g. viscous or solid substrates), HKR reactions can be accomplished successfully by adding small amounts of CH3CN.36
2.3.2 Phenolic Kinetic Resolution (PKR) of Terminal Epoxides
The efficiency and scope of the PKR of terminal epoxides is expanded greatly relative to monomer 1 or oligomer 3 using catalyst 4a (Figure 6B). The ring-opening of epoxides with phenols bearing a broad range of electron-withdrawing and donating substituents at the ortho, meta, and para positions was accomplished in good yields and ≥97% ee at ambient temperatures with catalyst loadings under 0.1 mol% Co in nearly all cases (Table 2).37 Stericlly hindered ortho-substituted phenols generally required higher catalyst loadings than their meta- or para-substituted counterparts (e.g., entries 4 vs. 6), but the desired α-aryloxy alcohols could be obtained nonetheless in excellent yields and very high ee's. With sufficiently elevated catalyst loadings, even highly sterically hindered 2,6-disubstituted phenols could be engaged in the PKR of relatively unreactive epoxides such as methyl glycidate (entry 16). Epichlorohydrin is an excellent reaction partner in PKR reactions catalyzed by 4a (entries 10–13), providing practical access to a wide range of aryl glycidyl ethers. The PKR of styrene oxide with p-cresol (entry 15) provided a 25:1 ratio of desired terminally to undesired internally opened products.38 In contrast to the HKR of styrene oxide (vide supra),23 no advantage in regioselectivity was obtained by employing tosylate 4b in the PKR reaction.39
Table 2.
The PKR of terminal epoxides catalyzed by oligomeric (salen)Co complex 4a.a.

| ||||
|---|---|---|---|---|
|
| ||||
| entry | product | Cob (mol%) | yieldc (%) | eed (%) |
| 1 |

|
0.0075 | 92 | 99 |
| 2 |

|
0.05 | 97 | >99 |
| 3 |

|
0.075 | 96 | 99 |
| 4 |

|
0.5 | 89 | 98 |
| 5 |

|
0.05 | 95 | >99 |
| 6 |

|
0.0075 | 95 | 99 |
| 7 |

|
0.15 | 87 | 97 |
| 8 |

|
0.05 | 87 | 99 |
| 9 |

|
0.05 | 96 | >99 |
| 10 |

|
0.05 | >99 | 98 |
| 11 |

|
0.005 | 90 | >99 |
| 12 |

|
0.0075 | 92 | >99 |
| 13 |
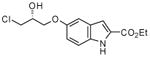
|
0.2 | 92 | 98 |
| 14 |

|
0.05 | 87 | 98 |
| 15 |

|
0.075 | 79 | 98 |
| 16 |

|
2.5 | 80 | 98 |
All entries were carried out on a 2.5 mmol scale relative to nucleophile.
Catalyst loadings calculated relative to the limiting reagent (phenol).
Isolated yield based on nucleophile.
Enantiomeric excesses were determined by chiral HPLC analysis.
As with the HKR, the experimental protocol for the PKR of terminal epoxides is straightforward. Reactions are carried out at ambient temperature with no special precautions to exclude air or moisture. Use of 2.2 equivalents of racemic epoxide relative to phenol is sufficient for hydrophobic epoxides such as 1,2-epoxyhexane, while more hygroscopic epoxides require the use of 2.5 equivalents to compensate for competitive HKR due to small amounts of adventitious water. Introduction of powdered 3Å molecular sieves led to significant decreases in reaction rate, and is therefore not recommended.
2.3.3 Alcoholic Kinetic Resolution (AKR) of Terminal Epoxides
Oligomeric complex 4a is an excellent catalyst for the kinetic resolution of terminal epoxides with primary alcohols, providing a variety of monoprotected 1,2-diols in good yields and ≥97% ee (Table 3).40 This methodology provides access to protected 1,2-diols, including benzyl, allyl, and 2-(trimethylsilyl)ethyl ethers (entries 1–3, 5–10), as well as methyl ethers (entry 4). Orthogonally protected chiral glycerol derivatives are prepared readily from racemic protected glycidols (entries 3–4), and enantioenriched glycidyl ethers can be accessed in two steps from epichlorohydrin after ring closure (entries 7–10).41 Activated epoxides again displayed excellent regioselectivity in ring opening, with styrene oxide affording a 24:1 ratio of terminally to internally opened products in reactions with allyl alcohol. Tosylate catalyst 4b afforded an even higher ratio of regioisomers (>100:1 terminal to internal opening) in this reaction, although the selectivity obtained with 4a was sufficient to allow isolation of pure product in 80% yield.
Table 3.
The AKR of terminal epoxides catalyzed by oligomeric (salen)Co complex 4a.a

| ||||
|---|---|---|---|---|
|
| ||||
| entry | product | Cob (mol%) | yieldc (%) | eed (%) |
| 1 |

|
0.1 | 92 | >99 |
| 2 |

|
0.2 | 91 | >99 |
| 3e,f |
|
0.1 | >99 | 98 |
| 4g |

|
0.01 | 92 | 97 |
| 5 |

|
0.1 | 94 | >99 |
| 6 |

|
0.5 | 80 | >99 |
| 7 |

|
0.1 | 98 | >99 |
| 8 |

|
0.02 | 94 | >99 |
| 9 |

|
0.1 | 94 | >99 |
| 10 |

|
0.5 | 91 | 98 |
All entries carried out on a 2.5 mmol scale relative to nucleophile.
Catalyst loadings and yields calculated relative to the limiting reagent (alcohol).
Enantiomeric excesses were determined by chiral HPLC analysis.
Enantiomeric excess was determined by chiral SFC analysis of the diol obtained from selective deprotection of the SEM ether.
R1=CH2OBn, R2=(TMS)CH2.
R1=CH2OBn, R2=H.
The basic experimental procedure for AKR of terminal epoxides is analogous to that employed in PKR reactions, except that AKR reactions are best carried out at reduced temperature. At ambient temperature, the active Co(III) catalyst is reduced by primary alcohols to the inactive Co(II) complex, presumably by one-electron transfer oxidation of the bound nucleophile. Lowering the temperature to 4 °C minimizes this catalyst deactivation pathway and allows efficient resolution to take place.
2.3.4 Hydrolytic Desymmetrization of Meso Epoxides
Hydrolysis of meso epoxides derived from cyclic alkenes (Table 4) represents an attractive approach to chiral diols that are not accessible via asymmetric alkene dihydroxylation. To our knowledge, 3 is the only non-enzymatic catalyst system that has been reported to date that promotes meso epoxide hydrolysis with high enantioselectivity.5,19,20,42,43 Oligomeric catalyst 4a confers significant improvements in both reactivity and enatioselectivity over monomeric catalyst 1 and oligomeric catalyst 3 in this reaction manifold (see Figure 6D, above). Meso epoxides derived from five- and six-membered ring cyclic alkenes undergo hydrolysis to provide the corresponding trans 1,2-diols in good yield and excellent ee (Table 4, entries 1–2, 6–10).44 Seven- and eight-membered endocyclic epoxides are also reactive, although with decreased enantioselectivity (entries 3–5). Cycloheptene oxide underwent ring-opening in 72% ee, but the diol could not be isolated cleanly from the crude reaction mixture (entry 3). Cyclooctene oxide was unreactive, presumably due to well known deactivating conformational effects (entry 4).45 In contrast, 1,5-cyclooctadiene mono epoxide underwent ring-opening in 80% ee (entry 5). Access to highly enantioenriched tetrahydrofuran and pyrrolidine derivatives from readily available precursors is illustrated in entries 6–7, while the stereoconvergent synthesis of 4-carboethoxy-1,2-cyclopentanol from diastereomeric epoxides is illustrated in entries 8–9. Similar enantioselectivity was observed in the hydrolysis of both the trans and cis epoxide isomers to yield the same enantiomer of diol in high ee.
Table 4.
The hydrolytic desymmetrization of meso epoxides catalyzed by oligomeric (salen)Co complex 4a.a

| ||||
|---|---|---|---|---|
|
| ||||
| entry | product | Cob (mol%) | yieldc (%) | eed (%) |
| 1 |

|
1 | 85 | 96 |
| 2e |

|
1 | 77 | 96 |
| 3f,g |
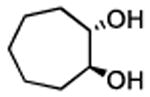
|
2.5 | >99% conv. | 72 |
| 4h |
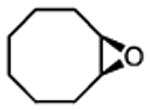
|
2.5 | <5% conv. | — |
| 5 |

|
2.5 | 87 | 80 |
| 6 |

|
2.5 | 76 | 99 |
| 7 |

|
2.5 | 91 | 99 |
| 8 |

|
1 | 98 | 99 |
| 9 |

|
1 | 92 | 98 |
| 10 |

|
1 | 89 | 97 |
Entries 1 and 2 carried out on a 101 and 99 mmol scale, respectively. Entries 3-10 were carried out on a 1 mmol scale.
Catalyst loadings and isolated product yields calculated relative to the limiting reagent (epoxide).
Enantiomeric excesses were determined by chiral GC analysis of the corresponding bis(trifluoroacetate) derivatives.
93% yield, 93% ee prior to recrystallization from hexanes/EtOAc. The crude product was suitable for most uses, containing only solvent and minor baseline impurities.
72 h reaction time.
Conversion determined by GC analysis of the reaction mixture.
48 h reaction time.
The hydrolytic desymmetrization reactions are conducted under ambient atmosphere at room temperature with epoxides used as received from commercial sources.46 Cyclopentene and cyclohexene oxide were hydrolyzed on a 101 and 99 mmol scale, respectively, to provide in each case 8.8 g (85% and 77% yield, respectively) of isolated product after purification. It should be noted that an exotherm was observed at larger scales (>1 mmol), resulting in slightly decreased enantioselectivity. Slow addition of epoxide to a solution of catalyst and water in CH3CN was therefore necessary in order to minimize this effect.
2.3.5 Carbamolytic Kinetic Resolution (CKR) of Terminal Epoxides and Carbamolytic Desymmetrization of Meso Epoxides
Bartoli and coworkers discovered that (salen)Co monomer 1 is a highly stereoselective catalyst for the kinetic resolution of terminal epoxides with carbamates, providing N-protected 1-amino-2-ols in good yields and excellent stereoselectivities (e.g., Scheme 3A).7 Oligomeric complex 4a displays similar stereoselectivity and significantly enhanced reactivity in the same transformation, allowing a greater than twentyfold reduction in catalyst loading (Scheme 3B).
Scheme 3.

The CKR of terminal epoxides catalyzed by monomeric (salen)Co complex 1 (ref. 7a) and oligomeric (salen)Co complex 4a.a-e
a The reaction with catalyst 1 was carried out on a 0.5 mmol scale relative to nucleophile; the reaction with catalyst 4a was carried out on a 2.5 mmol scale. bCatalyst loadings are relative to limiting reagent (BocNH2). cCo(II) is converted in situ to Co(III)(O2CC6H4NO2) in the presence of 4-NO2-C6H4CO2H and air. dIsolated yields are based on nucleophile. eEnantiomeric excesses were determined by chiral HPLC analysis of the O-benzyl derivative.
An even more significant advantage of the oligomeric catalyst relative to monomer 1 was seen in the ring opening of meso epoxides with phenyl carbamate.25b,47 Whereas monomer 1a (5 mol%) catalyzes addition to cyclohexene oxide to generate the corresponding cyclic carbamate in 33% yield and 21% ee for cyclohexene oxide at 5 mol% catalyst loading, oligomeric catalyst 4a (1 mol% Co) promotes the same transformation in high yield and 96% ee (Table 5). This methodology provides practical access to a variety of cyclic, protected trans-1,2-amino alcohols in high yield and enantioselectivity as demonstrated in the addition of phenyl carbamate to a variety of meso epoxides. Six-membered ring epoxides underwent clean addition with subsequent intramolecular cyclization to afford trans-4,5-disubstituted oxazolidinone products (entries 1–3). Phenyl carbamate addition to five-membered ring epoxides also proceeded with excellent enantioselectivity (entries 4–5), providing a mixture of the monomeric addition product together with carbamate-bridged oligomers. Here intramolecular cyclization appears to be impeded, presumably due to the unfavorable strain in trans-fused 5–5 ring systems.48 The free trans-1,2-amino alcohol product could subsequently be liberated in high overall yield through hydrolysis of the product mixture with base. This reaction is readily conducted under ambient atmosphere and has been used to prepare multigram quantities of trans-1,2-amino alcohol hydrochlorides.25b
Table 5.
The carbamolytic desymmetrization of endocyclic meso epoxides catalyzed by oligomeric (salen)Co complex 4a.a






All reactions were carried out on a 1 mmol scale.
Catalyst loadings and isolated yields calculated relative to the limiting reagent (phenyl carbamate).
Enantiomeric excesses were determined by chiral GC or HPLC analysis.
2.3.6 Intramolecular Alcoholytic and Phenolytic Desymmetrization of Oxetanes
In efforts to broaden the scope of electrophiles activated by (salen)Co(III) catalysts beyond epoxides, we demonstrated that the intramolecular alcoholytic and phenolytic desymmetrization of oxetanes could be catalyzed by oligomeric complex 4a to provide a variety of tetrahydrofuran, dihydrobenzofuran, and tetrahydropyran products in high enantioselectivity (Table 6).25a,49 Alkyl (entries 2, 4 and 6) and phenyl (entry 3) substitution at the 3-position of the oxetane was tolerated, affording products bearing quaternary stereocenters. Incorporation of a fluorine substituent yielded a tetrahydrofuran containing a fully substituted fluorine-bearing stereocenter (entry 5). Ring opening of phenolic substrates (entries 7–9) generated enantioenriched dihyrobenzofurans; however, higher catalyst loadings were required to attain high levels of enantioselectivity. Consistent with the cooperative bimetallic mechanism established for epoxide openings,13 use of oligomeric complex 4a lowered the required catalyst loading by 10–500-fold compared to (salen)Co monomer 1 (X=OTf) in all cases studied. Enantioselectivities were comparable with monomeric and oligomeric catalyst in oxetane ring-opening reactions, with the exception of the tetrahydropyran-forming reaction (entry 10) wherein the oligomeric catalyst 4a afforded much higher product ee.50
Table 6.
The intramolecular alcoholytic and phenolytic desymmetrization of oxetanes catalyzed by oligomeric (salen)Co complex 4a.a

| ||||
|---|---|---|---|---|
|
| ||||
| entry | product | Co (mol%) | yieldb (%) | eec (%) |
| 1d |

|
0.01 | 93 | 96 |
| 2 |

|
0.01 | 88 | 96 |
| 3 |

|
0.01 | 98 | 99 |
| 4 |

|
0.01 | 97 | 99 |
| 5d |

|
0.01 | 76 | 98 |
| 6 |

|
0.01 | 98 | 99 |
| 7 |

|
0.01 | 89 | 98 |
| 8 |

|
1 | 95 | 98 |
| 9 |

|
1 | 94 | 88 |
| 10e |

|
0.1 | 89 | 96 |
All entries were carried out on a 0.15–0.5 mmol scale.
Isolated yield.
Enantiomeric excesses were determined by chiral HPLC analysis of the corresponding benzoylated derivative unless noted otherwise.
Enantiomeric excess was determined by chiral GC analysis of the corresponding trifluoroacetylated derivative.
96 h reaction time.
2.4 Mechanistic Studies
2.4.1 Effects of Ring Size and Oligomeric Distribution
As noted, the (salen)Co complex 4a is prepared and used as a mixture of cyclic dimer and trimer. Extensive efforts to effect separation of these complexes were unsuccessful, so an independent synthesis of the two macrocycles, 4an=1 and 4an=2 was carried out in order to assess their relative contributions to the overall performance of the oligomeric catalyst. It proved possible to synthesize each macrocycle in pure form by condensation of the appropriate dialdehydes with diaminocyclohexane (Scheme 4).28
Scheme 4.

Synthesis of the individual components of oligomeric mixture 4a.
The pure dimeric and trimeric catalysts were analyzed in a series of model epoxide ring-opening reactions and compared to the mixture 4a that was prepared as described in Scheme 2. All reactions were conducted at the same loading of (salen)Co units to allow the most direct comparison between the mixture and its components. This resulted in the actual loading of catalyst molecules in runs with the cyclic trimer (4an=2) being two-thirds of that in runs with the cyclic dimer (4an=1). As illustrated in Figure 7, the cyclic trimer 4an=2 was more reactive than dimer 4an=1 in all cases. These differences were relatively small in the HKR and PKR reactions (Figure 7A and 7B), but quite significant in the more challenging reactions such as the AKR of terminal epoxides and the hydrolytic desymmetrization of meso epoxides (Figure 7C and 7D). As expected based on the characterization data discussed above that demonstrated the oligomer mixture 4a to be a mixture of mostly dimer and a small amount of trimer,28 4a displayed intermediate reactivity between 4an=1 and the more reactive 4an=2. It is particularly noteworthy, and perhaps somewhat unexpected, that catalyst stereoselectivity differed very little with ring size. Indeed, only in the hydrolytic desymmetrization of cyclohexene oxide could a measurable change in enantioselectivity be detected: 92% ee for cyclic dimer 4an=1 and 94% ee for cyclic trimer 4an=2. The mixture 4a exhibited intermediate behavior (93% ee), once again consistent with its composition being mostly dimer.
Figure 7.

Comparison of the cyclic dimer (4a n=1) and the cyclic trimer (4an=2) with the oligomeric mixture 4a in representative epoxide ring-opening reactions. Conversions were determined by GC analysis relative to an internal standard or by 1H NMR analysis. Enantiomeric excesses were determined by chiral GC analysis or HPLC analysis. Numerical conversion and ee values are provided for the last data point of each curve.
While the trimer 4an=2 displays measurable advantages in reactivity relative to the mixture 4a, it can only be accessed by a substantially more complicated synthesis (9 steps and 2 purifications by preparative HPLC for 4an=2 versus 4 steps and no chromatographic purifications for 4a). As such, there is no practical advantage to the pure trimeric catalyst, particularly since the stereoselectivities are so similar with the two catalyst compositions. While efforts to bias the synthesis of 4a toward the selective formation of cyclic trimer appear worthwhile, these have not yet been undertaken.
2.4.2. Structural Basis for the Enhanced Reactivity of 4a: Effect of Substituents, Tethering, and Cyclization
As illustrated in several contexts in section 2.2, oligomeric catalyst 4a displays consistently superior reactivity and often higher stereoselectivity relative to monomer 1 in a variety of epoxide ring-opening and related reactions. While 4a was designed to be essentially a covalently linked, multimeric version of 1, it in fact possesses three potentially important differences with the original monomeric catalyst: the replacement of t-butyl for carboxylate ligand substituents, the covalent linkage of the salen units, and the macrocyclic structure.
In order to assess how each of these differences is related to the enhanced catalytic properties of oligomeric complex 4a, we carried out a systematic study of closely related model systems 13-15 (Figure 9). Monomeric complex 13 was prepared and compared to 1a to evaluate the effect of the change in substituent. A significant effect was indeed observed in the HKR of 1,2-epoxyhexane (Figure 8), with catalyst 13 promoting complete resolution within 2h under conditions where monomer 1a required ca. 10h.
Figure 9.

Mono-, di-, and trimeric acyclic analogs of oligomeric complex 4a.
Figure 8.

Comparison of monomer 13 to monomer catalyst 1a in the HKR of 1,2-epoxyhexane. Numerical conversion and ee values are provided for the last data point of each curve.
However, the substituent effect was relatively small compared to the benefit of linking two (salen)Co units covalently as in dimer 14 (Figure 10). At catalyst loadings where monomer 13 was virtually unreactive, linear dimer 14 proved to be highly active and stereoselective in representative epoxide ring-opening reactions (Figure 10A, B, and C). Similar to what was observed in comparisons of monomer 1b with oligomer 4a Figure 6D), monomer 13 catalyzed hydrolysis of cyclohexene oxide with poor enantioselectivity (19% ee), while dimer 14 was highly effective in the same reaction (94% ee, Figure 10D).
Figure 10.

Comparison of monomer 13 to linear dimer analog 14. Conversions were determined by GC analysis relative to an internal standard or by 1H NMR analysis. Enantiomeric excesses were determined by chiral GC or HPLC analysis. Numerical conversion and ee values are provided for the last data point of each curve. n.d. = not determined.
2.4.3 Effect of Cyclization
The effect of constraining the tethered salen units in a macrocycle was next analyzed by comparison of the cyclic oligomers 4an=1 and 4an=2 to linear dimer 14 and linear trimer 15. As illustrated in Figure 11, the differences in reactivity between the four catalysts vary considerably depending on the type of epoxide ring-opening reaction, but interesting patterns emerge. The acyclic catalysts 14 and 15 possess nearly identical reactivity in HKR and PKR reactions (Figures 11A and 11B), and the dimer is only slightly more reactive in the AKR and meso epoxide hydrolysis reactions (Figures 11C and 11D). Whereas macrocyclization has a negative impact on the reactivity of the dimeric catalysts (4an=1 versus 14), it results in considerable enhancement in reactivity of the trimeric catalysts (4an=2 versus 15). As a result, the cyclic trimer 4an=2 displays the highest level of reactivity of any of the catalysts in all four representative epoxide ring-opening reactions, with the greatest advantages seen in the AKR and meso epoxide hydrolysis reactions.
Figure 11.
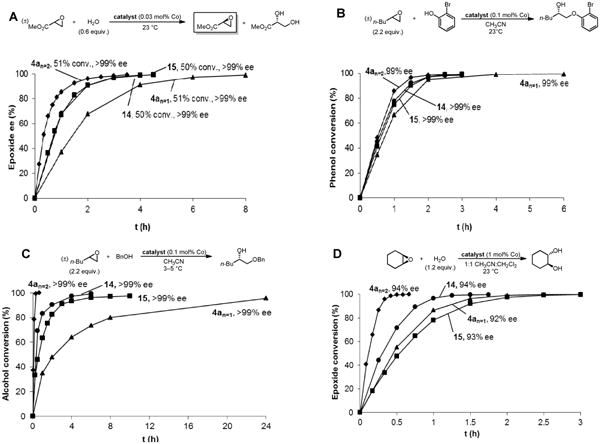
Comparison of the cyclic dimer 4a n=1 and the cyclic trimer 4an=2 with the linear dimer 14 and the linear trimer 15. Conversions were determined by GC analysis relative to an internal standard or by 1H NMR analysis. Enantiomeric excesses were determined by chiral GC analysis or HPLC analysis. Numerical conversion and ee values are provided for the last data point of each curve.
2.4.4 Summary of Mechanistic Findings
From the data obtained from this and earlier mechanistic studies, a clear picture emerges regarding the structural features responsible for the enhanced catalytic properties of tethered (salen)Co complexes:
1) The tether is crucial
Tethering the (salen)Co units through the etherdiester linker as in 4 is the predominant feature that leads to significant improvement in reactivity in all reactions examined (Figures 6 and 10). As established in earlier studies,19,20,22 the tether must be positioned and flexible enough to allow optimal relative positioning of the reacting (salen)Co units, balance opposing electronic requirements for nucleophile and electrophile activation, and provide adequate solubility of the catalyst for the reproducible preparation and practical application of the catalyst.
2) The cyclic trimer (4an=2) is the most reactive catalyst component
Catalyst 4a is composed of a mixture of dimeric and trimeric cyclic complexes, with the former being the dominant species present but the latter displaying generally higher reactivity. The macrocyclic nature of 4a has an unexpected effect on catalytic activity: the cyclic dimer 4an=1 is less reactive than its acyclic analog (14), whereas the cyclic trimer 4an=2 displays enhanced reactivity relative to its acyclic analog (15). However, the effects are quite complex and reaction-dependent, with small rate differences between the cyclic and acylic catalysts in reactions such as the phenolic ring-opening of epoxides (the PKR), but quite significant in reactions of primary alcohols with epoxides (the AKR).
3) Stereoselectivity is insensitive to oligomer size or cyclization
Perhaps the most unexpected finding is that very similar stereoselectivities are observed across a variety of epoxide ringopening reactions for either cyclic or acyclic dimeric and trimeric catalysts. This indicates that the stereoselectivity-inducing transition structure geometries in these reactions are largely insensitive to whether two or three catalyst units are present or to macrocyclization. This has the practical benefit that the mixture of oligomers in 4a affords consistently high stereoselectivity and is not sensitive to the precise ratio of its components.
Extensive studies on the mechanism of the HKR catalyzed by monomer catalyst 1 have led to a coherent stereochemical model wherein the two reacting (salen)Co complexes are canted relative to one another in the stereoselectivity-determining transition structure (Figure 12A).15 Given the high stereoselectivities observed with the cyclic, dimeric catalyst, the flexible tether linking the (salen)Co units in 4an=1 appears to allow access to a similar relative geometry (Figure 12C). The same canted geometry is not only accessible in the trimer (4an=2), but in fact may be preferred (Figure 12D). We propose that the trimer possesses not only a larger, more accessible active site, but also a higher proportion of low energy conformers with the appropriate geometric alignment of the two salen units, and these factors account for the higher reactivity this complex.
Figure 12.

A) Calculated, lowest energy transition structure for the HKR with monomeric catalyst, optimized at the B3LYP/6-31G(d) level of theory. The canted relationship of the (salen)Co units is clearly displayed (adapted from ref. 15). B) Schematic representation of the calculated structure in Figure 12A, with the thick lines symbolizing the salen frameworks. C) Schematic representation of the HKR transition structure with cyclic dimeric catalyst 4a n=1. D) Schematic representation of the HKR transition structure with cyclic trimeric catalyst 4an=2.
2.5 Catalyst Selection Guide
Even though the oligomeric catalyst 4a is readily prepared on any scale (Scheme 2) and is available commercially, the monomeric complex 1 is of course considerably simpler and more accessible. As such, the obvious practical question facing a chemist interested in performing a stereoselective epoxide ring-opening reaction is which catalyst to use to achieve the desired results. Since the relative advantages of the oligomeric catalyst 4a are reaction-dependent, we offer a comparative guide to aid in selecting the most appropriate catalyst for a given transformation (Table 7). The nucleophilic ring-opening reactions catalyzed by (salen)Co complexes can be divided into three categories.
Table 7.
Catalyst selection guide for epoxide ring-opening reactions mediated by monomeric (salen)Co complex 1 and/or oligomeric (salen)Co complex 4a.
| reaction class | electrophile | nucleophile | monomer 1 viable? | Co loading reduction with oligomer 4aa | Enhanced stereoselectivity or substrate scope with oligomer 4a?a |
|---|---|---|---|---|---|
| I | terminal epoxides | water | yes | 22–667-foldb | yes |
| carbamates | 22-fold c | n.d.d | |||
| oxetanes | intramolecular primary alcohols | 100-fold e | yes | ||
| intramolecular phenols | 10–500-fold e | no | |||
| II | terminal epoxides | phenols | substrate-dependent | 59–587-fold f | yes |
| III | terminal epoxides | primary alcohols | no | — | — |
| meso epoxides | water | ||||
| carbamates |
Based on reported values for reactions employing identical substrates under optimized conditions for each catalyst.
Based on ref. 2a, 2c, and 13a. Comparison for styrene oxide based on oligomer 4b.
Based on a single example from ref. 7a.
n.d. = not determined.
Based on ref. 25a.
Based on ref. 6.
Class I: Monomer 1 is viable but less efficient
The HKR of terminal epoxides2a,2c,13a and the intramolecular alcoholytic and phenolytic desymmetrization of oxetanes25,51 are catalyzed effectively by monomer complex 1. However, substantial reductions in catalyst loading are realized consistently with complex 4a, and improved stereoselectivity is observed in certain cases. Only a few examples of (salen)Co-catalyzed carbamate additions to terminal epoxides (CKR reactions) have been reported,7a but available data indicate that similarly high stereoselectivities can be obtained with monomeric or oligomeric catalysts. In the one case where a direct comparison was made, much higher catalytic efficiencies were achieved with oligomer 4a. For each of these Class I reactions, the ease of access to complex 1 must be weighed against the improved rates and turnover numbers achieved with complex 4a.
Class II: Reaction is substrate-dependent
The addition of phenols to terminal epoxides (PKR) is catalyzed with high stereoselectivity by monomer 1 as long as the phenol is relatively unhindered.6 Reactions of more hindered pheonols, particularly those bearing ortho substitutents, require the use of oligomer 4a. Even in cases where complex 1 is viable, complex 4a offers substantial improvements in reaction rate.
Class III: Oligomer 4 is uniquely effective
The addition of primary alcohols to terminal epoxides (AKR reactions) and the enantioselective opening of meso epoxides with either water11 or carbamates25b require the use of oligomer 4a, as monomer 1 is either unreactive or very poorly stereoselective in these reactions.
3. Conclusions and Outlook
The discovery that (salen)Co-catalyzed epoxide ring-opening reactions proceed by cooperative, bimetallic mechanisms laid the foundation for the development of simple, oligomeric catalysts possessing substantially improved reactivity, stereoselectivity, and substrate scope relative to the original monomeric catalysts. Oligomeric (salen)Co catalyst 4 is readily prepared in multigram quantities from inexpensive, commercially available materials and can be used under practical conditions at any scale. Analysis of the structural changes relative to monomeric complex 1 has revealed the key elements responsible for the enhanced catalytic properties of the oligomer and has provided new design principles for future generations of catalysts. In particular, the cyclic, trimeric component of catalyst 4a displays particularly high reactivity in certain reactions. Application of this insight to new catalyst designs and in other classes of bimetallic (salen)metal-catalyzed reactions appears highly worthwhile.
4. Experimental Section
Equipment
All reactions were performed open to the air unless otherwise noted. All reactions performed under N2 were conducted in round-bottomed flasks fitted with rubber septa. Air- and moisture-sensitive chemicals were transferred via syringe or stainless steel cannula. Flash chromatography was performed using silica gel 60 (230–400 mesh) purchased from EM Science. Preparatory TLC was performed on 20 × 20 cm silica gel 60 F254 plates (0.25 mm thickness) precoated with a fluorescent indicator purchased from EM Science.
Materials
Unless otherwise stated, all reagents were purchased from Acrôs, Sigma-Aldrich, Alfa Aesar, Lancaster, Pfaltz & Bauer, Strem, EM Science, or EMD Chemicals Inc. and used as received. Racemic methyl glycidate was received as a generous gift from Rhodia ChiRex. Solvent was distilled over CaH2 (CH2Cl2, CH3CN), Na (benzene, toluene), or Na/benzophenone ketone (THF) where indicated; otherwise, solvent was used directly from the bottle. Anhydrous DMF, MeOH, and EtOH were obtained from Sigma-Aldrich and packaged in a Sure/Seal™ bottle. Unless otherwise stated, catalyst loadings were not corrected for the presence of residual solvent. Measured values are rounded for convenience; calculations were performed prior to rounding.
Instrumentation
All solution state 1H NMR, 13C NMR, and 19F NMR spectra were recorded using an Inova-600 (600 MHz), an Inova-500 (500 MHz), or a Varian Mercury-400 (400 MHz) spectrometer. Chemical shifts for hydrogen are reported in parts per million downfield from tetramethylsilane and are referenced to the resonances of residual protium in the NMR solvent: pyridine-d5 (δ 8.71, 7.55, 7.19); CDCl3 (δ 7.26); D2O (δ 4.79); CD3OD (δ 4.78, 3.30); DMSO-d6 (δ 2.49); acetone-d6 (δ 2.04). H2O appears as a broad singlet between δ 4.5 and δ 5.5 in pyridine-d5. Chemical shifts for carbon are reported in parts per million downfield from tetramethylsilane and are referenced to the resonances of the NMR solvent, except D2O: pyridine-d5 (δ 149.9, 135.5, 123.5); CDCl3 (δ 77.0); CD3OD (δ 49.0); DMSO-d6 (δ 39.5); acetone-d6 (δ 206.0, 29.8). Chemical shifts for carbon in D2O are reported in parts per million downfield from the hydrogen resonances of the trimethylsilyl group of added of 3-(trimethy1si1yl)-1-propanesu1fonic acid, sodium salt (δ 0). IR spectra were recorded as KBr discs or as thin films on either NaCl or KBr plates on a Perkin-Elmer FTIR 1600 or a Galaxy Series FTIR 3000 spectrophotometer. Optical rotations were measured using a 2 mL cell with a 1 dm path length on a Jasco DIP 370 digital polarimeter. Mass spectrometric data was obtained at the Harvard University mass spectrometry facility or on a Waters Micromass® ZQ™ mass spectrometer. Preparative HPLC purification was performed on an Agilent 1100 Series instrument. Chiral GC analyses were performed on a Hewlett Packard 5890 Series II gas chromatograph Chiral HPLC analyses were performed on a Hewlett-Packard 1050 or a Shimadzu VP-series instrument. Chiral SFC analysis was performed on a Berger instrument.
4.1. Representative procedure for the HKR of terminalepoxides with catalyst (R,R)-4a: (R)-butadiene monoxide
A 50-mL round-bottomed flask equipped with a stir bar was charged with oligomeric cyclic (R,R)-(salen)Co(III) triflate 4a (26.0 mg, 0.033 mmol) and placed in a room temperature water bath. (±)-Butadiene monoxide (10.5 mL, 130 mmol) immediately followed by H2O (1.64 mL, 91 mmol) in one portion were added. The reaction flask was sealed with a septum secured by copper wire to prevent substrate evaporation. (CAUTION! An initial pressure buildup is observed due to the volatility of the epoxide under the exothermic reaction conditions. Care should be taken to use equipment adequate for elevated pressures.) After stirring 24 h at room temperature, resolved epoxide and excess H2O were vacuum transferred (0.45 mmHg, reaction pot: room temperature) to a −78 °C receiving flask. The epoxide was dried over MgSO4 and filtered through a sand plug to give 3.28 g of a clear liquid containing 1% by mass 3-butene-1,2-diol. The corrected yield was 35%. Observed >99% ee as determined by chiral HPLC analysis of the 2-naphthylsulfide derivative [obtained by ring opening with 0.8 equiv 2-naphthalenethiol and 0.8 equiv Et3N in MeOH (0.75 M) at 4 °C and subsequent purification of the terminal addition product by preparatory TLC (40% EtOAc/hexanes); Chiracel® OD, 5% i-PrOH/hexanes, 1 mL/min, 254 nm, tR (minor) = 18.4 min; tR (major) = 22.3 min]. [α]31D +6.19° (neat); lit.52 [α]22D +14.8° (c = 1.18, Et2O).
4.2. Representative procedure for the PKR of terminal epoxideswith catalyst (R,R)-4a: (2S)-1-phenoxy-hexan-2-ol
A 5-mL round-bottomed flask equipped with a stir bar was charged with phenol (235 mg, 2.5 mmol), (±)-1,2- epoxyhexane (0.67 mL, 5.6 mmol), and distilled CH3CN (0.27 mL). A stock solution of oligomeric cyclic (R,R)-(salen)Co(III) triflate 4a in distilled CH3CN (0.0125 M, 15 μL, 0.00019 mmol) was added, and the flask was sealed with a plastic cap. The reaction was then stirred for 16 h at room temperature, at which time pyridinium p-toluenesulfonate (1 mg, 0.004 mmol) was added to quench the catalyst and ensure complete oxidation to Co(III). The reaction was diluted with Et2O (3 mL) and applied to a pad of silica gel. The pad was eluted with Et2O (200 mL), and the filtrate washed with 1 N NaOH (3 × 25 mL) and brine, respectively. After drying over MgSO4, solvent was removed from the filtrate by rotary evaporation. The bulk of remaining epoxide was removed from the crude product under high vacuum. The product was purified by flash chromatography on silica gel (15% EtOAc/hexanes) to give 445 mg (92%) of a clear oil in 99% ee as determined by chiral HPLC analysis [Chiracel® OD, 5% EtOH/hexanes, 1 mL/min, 220 nm, tR (minor) = 6.9 min; tR (major)= 10.1 min]. 1H NMR (CD3OD) δ 7.22–7.27 (m, 2H), 6.88–6.94 (m, 3H), 3.82–3.92 (m, 3H), 1.58–1.67 (m, 1H), 1.45–1.58 (m, 2H), 1.31–1.45 (m, 3H), 0.94 (dd, J = 7.2, 7.2 Hz, 3H). 13C NMR (CD3OD) δ 160.5, 130.4, 121.7, 115.6, 73.2, 71.0, 34.3, 28.9, 23.8, 14.4. IR (thin film) ν 3407, 3063, 3042, 2957, 2932, 2872, 1599, 1497, 1458, 1379, 1335, 1300, 1244, 1173, 1136, 1078, 1040, 922, 883, 814, 754, 691, 613, 509 cm−1. [α]30D + 19.4° (c 2.03, CH2Cl2); lit.6 [α]25 D +18.7° (c 1.25, CH2Cl2). MS (ApCI) m/z calcd. for C12H17O 177.1, found 177.1 (100%) [M–OH]+; calcd. for C12H19O2 195.1, found 195.1 (26%) [M+H]+; calcd. for C12H22NO2 212.2, found 212.1 (9%) [M+NH4]+.
4.3. Representative procedure for the AKR of terminal epoxideswith catalyst (R,R)-4a: (2S)-1-benzyloxy-2-hexanol
A 5-mL round-bottomed flask equipped with a stir bar was charged with benzyl alcohol (0.26 mL, 2.5 mmol), (±)-1,2-epoxyhexane (0.67 mL, 5.6 mmol), and distilled CH3CN (0.066 mL). The flask was sealed with a plastic cap, and cooled to 4 °C. A room temperature stock solution of oligomeri P cyclic (R,R)-(salen)Co(III) triflate 4a in distilled CH3CN (0.0126 M, 200 μL, 0.0025 mmol) was added, and the flask was resealed with the plastic cap. The reaction was then stirred for 24 h at 4 °C, at which time pyridinium p-toluenesulfonate (2 mg, 0.008 mmol) was added to quench the catalyst and ensure complete oxidation to Co(III). The reaction was diluted with Et2O (3 mL) and applied to a pad of silica gel after warming to room temperature. The pad was eluted with Et2O (200 mL), and solvent was removed from the filtrate by rotary evaporation. The bulk of remaining epoxide was removed from the crude product under high vacuum. Purified by flash chromatography on silica gel (20% EtOAc/hexanes) to give 480 mg (92%) of a clear liquid in >99% ee as determined by chiral HPLC analysis [(R,R)-Whelk-01 (Pirkle), 2% i-PrOH/hexanes, l mL/min, 215 nm, tR (minor) = 10.6 min; tR (major) = 11.5 min]. 1H NMR (CD3OD) δ 7.29–7.37 (m, 4H), 7.26 (tt, J = 2.1, 6.8 Hz, 1H), 4.54 (d, J = 12.4 Hz, 1H), 4.52 (d, J = 12.5 Hz, 1H), 3.71 (dddd, J = 4.4, 6.5, 6.5, 7.4 Hz, 1H), 3.42 (dd, J = 4.2, 9.8 Hz, 1H), 3.37, (dd, J = 6.4, 9.8 Hz, 1H), 1.46–1.55 (m, 1H), 1.26–1.46 (m, 5H), 0.91 (dd, J = 7.1, 3H). 13C NMR δ 139.8, 129.3, 128.9, 128.6, 75.7, 74.3, 71.4, 34.5, 28.8, 23.8, 14.4. IR (thin film) ν 3442, 3088, 3064, 3030, 2955, 2931, 2860, 1496, 1466, 1454, 1378, 1364, 1331, 1311, 1270, 1204, 1101, 1028, 736, 698 cm−1 .[α]26 D +6.22° (c = 2.04, CH2Cl2); lit.19 [α]23 D +5.1° (c = 2.01, CH2Cl2). MS (ApCI) m/z calcd. for C6H13O2 117.1, found 117.0 (100%) [M–PhCH2]+; calcd. for C13H21O2 209.2, found 209.1 (24%) [M+H]+.
4.4. Representative procedure for the hydrolytic desymmetrization of endocyclic meso epoxide with catalyst (R,R)-4a: (1S)-trans-cyclopentane-l,2-diol
A 100-mL round-bottomed flask equipped with a stir bar was charged with oligomeric cyclic (R,R)-(salen)Co(III) triflate 4a (805 mg, 1.01 mmo1) and CH3CN (32 mL), respectively. To this solution was added H2O (2.18 mL, 121 mmo1). Cyc1opentene oxide (8.8 mL, 101 mmol) was then added over 1 h via syringe pump, and the reaction was stirred an additional 23 h at room temperature. At 24 h total time, pyridinium p-toluenesulfonate (760 mg, 3.02 mmol) was added to quench the catalyst and ensure complete oxidation to Co(III). The reaction mixture was diluted with EtOAc (400 mL) to precipitate out the bulk of the catalyst and then applied to a pad of silica gel. The pad was eluted with EtOAc (1 L), and solvent was removed from the filtrate by rotary evaporation. Residual solvent was then distilled away at 45 mm Hg over 15 min with a bath temperature of 50–55 °C. Short path distillation at 4 mm Hg into a 0 °C receiving flask then afforded 8.77 g (85%) of the product diol as a colorless low-melting solid in approximately 98% purity by 1H NMR analysis. The yield was uncorrected. Observed 96% ee as determined by chiral GC analysis of the bis(trifluoroacetate) derivative [obtained by treating product with neat TFAA, followed by evaporation of excess TFAA under a stream of N2; Chiraldex™ γ-TA, 65 °C, isothermal, tR (minor) = 4.4 min; tR (major) = 6.9 min]. 1H NMR (CD3OD) δ 3.89–3.93 (m, 2H), 1.90–1.99 (dtd, J = 5.5, 7.8, 13.0 Hz, 2H), 1.71 (td, J = 7.5, 15.2 Hz, 2H), 1.47–1.55 (m, 2H). 13C NMR (CD3OD) δ 79.7, 32.5, 21.4. IR (thin film) ν 3331, 2965, 1437, 1344, 1296, 1086, 1038, 974, 876 cm‒1. [α]29 D +24.7° (c 1.17, EtOH); lit.53 [α]20 D +24.54° (c 5.4, EtOH). MS (CI) m/z calcd. for C5H14NO2 120, found 120 (100%) [M+NH4]+; calcd. for C5H17N2O2 137, found 137 (9%) [M+NH4+NH3]+.
4.5. Representative procedure for the CKR of terminal epoxides with catalyst (S,S)-4a: ((R)-2-hydroxy-hexyl)-carbamic acid, tert-butyl ester
A 5-mL round-bottomed flask equipped with a stir bar was charged with oligomeric cyclic (S,S)-(salen)Co(III) triflate ent-4a (4.4 mg, contained 8% by mass toluene, 0.0051 mmol), tert-butyl carbamate (293 mg, 2.5 mmol, recrystallized from TBME/hexanes), distilled CH3CN (0.22 mL), and (±)-1,2-epoxyhexane (0.67 mL, 5.6 mmol, distilled from CaH2), respectively, and sealed with a plastic cap. The reaction was then stirred for 24 h at room temperature, at which time it was diluted with EtOAc (3 mL) and applied to a pad of silica gel. The pad was eluted with EtOAc (200 mL), and solvent was removed from the filtrate by rotary evaporation. The bulk of the remaining epoxide and residual carbamate were removed from the crude product under high vacuum. Purified by flash chromatography on silica gel (30% EtOAc/hexanes) to give 514 mg of a faintly yellow oil containing <1% by mass residual solvent by 1H NMR analysis. The corrected yield was 94%. Observe 98% ee as determined by chiral HPLC analysis of the O-benzyl derivative [obtained by etherification with 2.1 equiv NaH, 1.2 equiv benzyl bromide and 0.2 equiv tetrabutylammonium iodide in THF (0.18 M, distilled from Na/benzophenone ketone) at 0 °C to room temperature under an N2 atmosphere and subsequent purification by flash chromatography on silica gel (10% EtOAc/hexanes); Chiracel® AS, 2% i-PrOH/hexanes, 1 mL/min, 220 nm, tR (minor) = 4.9 min; tR (major) = 5.6 min]. 1H NMR (CD3OD) δ 3.52–3.60 (m, 1H), 3.11 (dd, J = 4.4, 13.7 Hz), 2.95 (dd, J = 6.8, 13.7 Hz, 1H), 1.40–1.50 (m, 2H), 1.43 (s, 9H), 1.27–1.40 (m, 4H), 0.92 (dd, J = 7.1 Hz, 3H). 13C NMR (CD3OD) δ 158.6, 80.1, 71.7, 47.5, 35.3, 28.8, 28.7, 23.8, 14.4. IR (thin film) ν 3367, 2960, 2933, 2873, 2863, 1692, 1520, 1457, 1392, 1367, 1275, 1252, 1173, 1090, 1042, 101, 92, 893, 880, 781 cm−1 . [α]26 D −11.7° (c = 1.24, CHCl3); lit.7a [α]rtD +14.2° (c = 0.9, CHCl3, S enantiomer). MS (ApCI) m/z calcd. for C7H16NO3 162.1, found 162.1 (100%) [M– t-Bu+2H]+; calcd. for C11H24NO3 found 218.1 (17%) [M+H]+.
Supplementary Material
Acknowledgments
This work was supported by the NIH (GM-43214), a predoctoral fellowship to D. E. W. from the Department of Defense, a postdoctoral fellowship to P. M. T. from the NIH, and a summer undergraduate research fellowship to Z. L. from Pfizer Inc. We thank Joe Ready, James Birrell, and Rebecca Loy for helpful discussions.
Dedicated to Professor Sarah Reisman on the occasion of her receipt of the Tetrahedron Young Investigator Prize
Footnotes
Supplementary Material: Experimental procedures and characterization data for the preparation of (salen)Co complexes 1a (X=OTf), 1c (X=(1S)-(+)-10-camphorsulfonate), 4a, 4an=1, 4an=2, 4b, and 13–15 as well as for previously unreported epoxide ring opening reactions (PDF). This material is available free of charge via the Internet.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Larrow JF, Jacobsen EN. Topics Organomet Chem. 2004;6:123–152. [Google Scholar]
- 2.(a) Tokunaga M, Larrow JF, Kakiuchi F, Jacobsen EN. Science. 1997;277:936–938. doi: 10.1126/science.277.5328.936. [DOI] [PubMed] [Google Scholar]; (b) Jacobsen EN. Acc Chem Res. 2000;33:421–431. doi: 10.1021/ar960061v. [DOI] [PubMed] [Google Scholar]; (c) Schaus SE, Brandes BD, Larrow JF, Tokunaga M, Hansen KB, Gould AE, Furrow ME, Jacobsen EN. J Am Chem Soc. 2002;124:1307–1315. doi: 10.1021/ja016737l. [DOI] [PubMed] [Google Scholar]
- 3.For reviews of applications of the HKR reaction in industrial and natural products synthesis, see: Larrow JF, Hemberger KE, Jasmin S, Kabir H, Morel P. Tetrahedron: Asymmetry. 2003;14:3589–3592. Schneider C. Synthesis. 2006:3919–3944.. Kumar P, Naidu V, Gupta P. Tetrahedron. 2007;63:2745–2785.. Furukawa Y, Suzuki T, Mikami M, Kitaori K, Yoshimoto H. J Synth Org Chem Japan. 2007;65:308–319.. Kumar P, Gupta P. Synlett. 2009:1367–1382.. Pellissier H. Adv Synth Catal. 2011;353:1613–1666..
- 4.For other examples of hydrolytic kinetic resolutions of terminal epoxides, see: Chengjun J. Kinetics and Catalysis. 2011;52:691–696.. Park J, Lang K, Abboud KA, Hong S. Chem Eur J. 2011;17:2236–2245. doi: 10.1002/chem.201002600.. Jiang CJ, Chen ZR. Kinetics and Catalysis. 2008;49:474–478.. Kawthekar RB, Kim GJ. Synth Commun. 2008;38:1236–1248.. Thakur SS, Chen SW, Li W, Shin CK, Koo YM, Kim GJ. Synth Commun. 2006;36:2371–2383.. Thakur SS, Li W, Shin CK, Kim GJ. Catal Lett. 2005;104:151–156.. Thakur SS, Li W, Kim SJ, Kim GJ. Tetrahedron Lett. 2005;46:2263–2266.. Shin CK, Kim SJ, Kim GJ. Tetrahedron Lett. 2004;45:7429–7433.. Cavazzini M, Quici S, Pozzi G. Tetrahedron. 2002;58:3943–3949.. Thakur SS, Li W, Kim SJ, Kim GJ. Stud Surf Sci Catal. 2006;159:205–208.. Thakur SS, Li WJ, Shin CK, Kim GJ. Chirality. 2006;18:37–43.. Thakur SS, Chen SW, Li W, Shin CK, Kin SJ, Koo YM, Kim GJ. J Organomet Chem. 2006;691:1862–1872.. Jain S, Venkatasubbaiah K, Jones CW, Davis RJ. J Mol Catal A Chemical. 2010;316:8–15.. Berkessel A, Ertürk E. Adv Synth Catal. 2006;348:2619–2625.. Dijk EW, Feringa BL, Roelfes G. Tetrahedron: Asymm. 2008;19:2374–2377..
- 5.For reviews on the enzymatic hydrolysis of epoxides, see: Schober M, Faber K. Trends in Biotechnology. 2013;31:468–478. doi: 10.1016/j.tibtech.2013.05.005.. Lin H, Liu JY, Wang HB, Ahmed AAQ, Wu ZL. J Mol Catal B Enzymatic. 2011;72:77–89.. Bala N, Chimni SS. Tetrahedron: Asymm. 2010;21:2879–2898.. Choi WJ. Appl Microbiol Biotechnol. 2009;84:239–247. doi: 10.1007/s00253-009-2110-9.. Smit MS, Labuschagne M. Curr Org Chem. 2006;10:1145–1161.. Choi WJ, Choi CY. Biotechnol Biopro Eng. 2005;10:167–179.. Smit MS. Trends in Biotechnology. 2004;22:123–129. doi: 10.1016/j.tibtech.2004.01.012.. de Vries EJ, Janssen DB. Curr Opin Biotechnol. 2003;14:414–420. doi: 10.1016/s0958-1669(03)00102-2.. Steinreiber A, Faber K. Curr Opin Biotechnol. 2001;12:552–558. doi: 10.1016/s0958-1669(01)00262-2.. Archelas A, Furstoss R. Curr Opin Chem Bio. 2001;5:112–119. doi: 10.1016/s1367-5931(00)00179-4.. Sinha SC, Keinan E, Reymond JL. J Am Chem Soc. 1993;115:4893–4894.
- 6.Ready JM, Jacobsen EN. J Am Chem Soc. 1999;121:6086–6087. [Google Scholar]
- 7.(a) Bartoli G, Bosco M, Carlone A, Locatelli M, Melchiorre P, Sambri L. Org Lett. 2004;6:3973–3975. doi: 10.1021/ol048322l. [DOI] [PubMed] [Google Scholar]; (b) Bartoli G, Bosco M, Carlone A, Locatelli M, Melchiorre P, Sambri L. Org Lett. 2005;7:1983–1985. doi: 10.1021/ol050675c. [DOI] [PubMed] [Google Scholar]
- 8.For other approaches to the phenolic kinetic resolution of terminal epoxides, see: Aral T, Karakaplan M, Hosgören H. Catal Lett. 2012;142:794–802.. Kawthekar RB, Lee YH, Kim GJ. J Porous Mater. 2009;16:367–378.. Kim YS, Guo XF, Kim GJ. Top Catal. 2009;52:197–204.. Lee KY, Lee YH, Shin CK, Kim GJ. Stud Surf Sci Catal. 2007;165:467–470.. Lee KY, Lee CY, Kim GJ. React Kinet Catal Lett. 2008;93:75–83.. Kawthekar RB, Ahn CH, Kim GJ. Catal Lett. 2007;115:62–69.. Solodenko W, Jas G, Kunz U, Kirschning A. Synthesis. 2007:583–589.. Karadeniz L, Koz G, Aydin K, Astley ST. Turk J Chem. 2010;34:711–718.
- 9.For additional examples of the aminolytic kinetric resolution of terminal and internal epoxides, see: Bao H, Wang Z, You T, Ding K. Chin J Chem. 2013;31:67–71.. Plancq B, Ollevier T. Chem Commun. 2012;48:3806–3808. doi: 10.1039/c2cc18032d.. Hu X, Gao B, Chu Y, Li W, Liu X, Lin L, Feng X. Chem Eur J. 2012;18:3473–3477. doi: 10.1002/chem.201103792.. Arai K, Lucarini S, Salter MM, Ohta K, Yamashita Y, Kobayashi S. J Am Chem Soc. 2007;129:8103–8111. doi: 10.1021/ja0708666.. Arai K, Salter MM, Yamashita Y, Kobayashi S. Angew Chem, Int Ed. 2007;46:955–957. doi: 10.1002/anie.200603787.. Kureshy RI, Kumar M, Agrawal S, Khan NH, Abdi SHR, Bajaj HC. Tetrahedron: Asymm. 2010;21:451–456.. Bartoli G, Bosco M, Carlone A, Locatelli M, Massaccesi M, Melchiorre P, Sambri L. Org Lett. 2004;6:2173–2176. doi: 10.1021/ol049372t.
- 10.Intramolecular additions of alcohols to terminal and meso epoxides could be effected with good enantioselectivity using monomer 1: Wu MH, Hansen KB, Jacobsen EN. Angew Chem, Int Ed. 1999;38:2012–2014. doi: 10.1002/(SICI)1521-3773(19990712)38:13/14<2012::AID-ANIE2012>3.0.CO;2-H.
- 11.The desymmetrization of meso epoxides with benzoic acid as nucleophile is catalyzed with moderate-to-high enantioselectivity using monomer 1: Jacobsen EN, Kakiuchi F, Konsler RG, Larrow JF, Tokunaga M. Tetrahedron Lett. 1997;38:773–776.
- 12.For a discussion of practical considerations in the HKR and other kinetic resolution processes, see: Keith JM, Larrow JF, Jacobsen EN. Adv Synth Catal. 2001;343:5–26.
- 13.(a) Nielsen LPC, Stevenson CP, Blackmond DG, Jacobsen EN. J Am Chem Soc. 2004;126:1360–1362. doi: 10.1021/ja038590z. [DOI] [PubMed] [Google Scholar]; (b) Nielsen LPC, Zuend SJ, Ford DD, Jacobsen EN. J Org Chem. 2012;77:2486–2495. doi: 10.1021/jo300181f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.The cooperative, bimetallic mechanism for epoxide ring-opening was first established in the (salen) Cr(III)-catalyzed asymmetric opening of meso epoxides with TMSN3: Hansen KB, Leighton JL, Jacobsen EN. J Am Chem Soc. 1996;118:10924–10925. Also see ref. 2b.
- 15.The transition structure representation in Figure 1 is based on detailed experimental and computational analyses reported in: Ford DD, Nielsen LPC, Zuend SJ, Musgrave CB, Jacobsen EN. J Am Chem Soc. 2013;135:15595–15608. doi: 10.1021/ja408027p..
- 16.For reviews, see ref. 1a and: Madhavan N, Jones CW, Weck M. Acc Chem Res. 2008;41:1153–1165. doi: 10.1021/ar800081y.. Haak RM, Wezenberg SJ, Kleij AW. Chem Commun. 2010:2713–2723. doi: 10.1039/c001392g.. Matsunaga S, Shibasaki M. Synthesis. 2013:421–437..
- 17.(a) Konsler RG, Karl J, Jacobsen EN. J Am Chem Soc. 1998;120:10780–10781. [Google Scholar]; (b) Peukert S, Jacobsen EN. Org Lett. 1999;1:1245–1248. doi: 10.1021/ol990920q. [DOI] [PubMed] [Google Scholar]; (c) Annis DA, Jacobsen EN. J Am Chem Soc. 1999;121:4147–4154. [Google Scholar]; (d) Breinbauer R, Jacobsen EN. Angew Chem, Int Ed. 2000;39:3604–3607. doi: 10.1002/1521-3773(20001016)39:20<3604::aid-anie3604>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]; (e) Belser T, Jacobsen EN. Adv Synth Catal. 2008;350:967–971. [Google Scholar]
- 18.For examples of tethered (salen)Co catalyst systems possessing C1 symmetric salen units, see ref. 13 and Kleij AW. Eur J Inorg Chem. 2009:193–205.. Kureshy RI, Khan NH, Abdi SHR, Patel ST, Jasra RV. J Mol Catal A: Chemical. 2002;179:73–77.. Song Y, Chen H, Hu X, Bai C, Zheng Z. Tetrahedron Lett. 2003;44:7081–7085.. Kureshy RI, Singh S, Khan NH, Abdi SHR, Ahmad I, Bhatt A, Jasra RV. Chirality. 2005;17:590–594. doi: 10.1002/chir.20196.. Holbach M, Weck M. J Org Chem. 2006;71:1825–1836. doi: 10.1021/jo051919+.. Zheng X, Jones CW, Weck M. Chem Eur J. 2006;12:576–583. doi: 10.1002/chem.200500786.. Rossbach BM, Leopold K, Weberskirch R. Angew Chem Int Ed. 2006;45:1309–1312. doi: 10.1002/anie.200503291.. Solodenko W, Jas G, Kunz U, Kirschning A. Synthesis. 2007:583–589.. Zheng X, Jones CW, Weck M. J Am Chem Soc. 2007;129:1105–1112. doi: 10.1021/ja0641406.. Kureshy RI, Prathap KJ, Singh S, Agrawal S, Khan NH, Abdi SHR, Jasra RV. Chirality. 2007;19:809–815. doi: 10.1002/chir.20472.. Gill CS, Venkatasubbaiah K, Phan NTS, Weck M, Jones CW. Chem Eur J. 2008;14:7306–7313. doi: 10.1002/chem.200800532.. Goyal P, Zheng X, Weck M. Adv Synth Catal. 2008;350:1816–1822.. Beigi M, Roller S, Haag R, Liese A. Eur J Org Chem. 2008:2135–2141.. Gill CS, Long W, Jones CW. Catal Lett. 2009;131:425–431.. Zhu X, Venkatasubbaiah K, Weck M, Jones CW. J Mol Catal A: Chemical. 2010;329:1–6.. Thomas RM, Widger PCB, Ahmed SM, Jeske RC, Hirahata W, Lobkovsky EB, Coates GW. J Am Chem Soc. 2010;132:16520–16525. doi: 10.1021/ja1058422.. Wezenberg SJ, Kleij AW. Adv Synth Catal. 2010;352:85–91.. Liu Y, Rawlston J, Swann AT, Takatani T, Sherrill CD, Ludovice PJ, Weck M. Chem Sci. 2011;2:429–438.. Kahn MGC, Weck M. Catal Sci Technol. 2012;2:386–389.. Kahn MGC, Stenlid JH, Weck M. Adv Synth Catal. 2012;354:3016–3024.. Zhu C, Yuan G, Chen X, Yang Z, Cui Y. J Am Chem Soc. 2012;134:8058–8061. doi: 10.1021/ja302340b.. Ma DY, Xiao ZY, Etxabe J, Warnmark K. ChemCatChem. 2012;4:1321–1329.. Matkiewicz K, Bukowska A, Bukowski W. J Mol Cat A: Chemical. 2013;368-369:43–52.. Venkatasubbaiah K, Feng Y, Arrowood T, Nickias P, Jones CW. ChemCatChem. 2013;5:201–209.. Ahmed SM, Poater A, Childers MI, Widger PCB, LaPointe AM, Lobkovsky EB, Coates GW, Cavallo L. J Am Chem Soc. 2013;135:18901–18911. doi: 10.1021/ja409521z..
- 19.Ready JM, Jacobsen EN. J Am Chem Soc. 2001;123:2687–2688. doi: 10.1021/ja005867b. [DOI] [PubMed] [Google Scholar]
- 20.Ready JM, Jacobsen EN. Angew Chem, Int Ed. 2002;41:1374–1377. doi: 10.1002/1521-3773(20020415)41:8<1374::aid-anie1374>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 21.For other examples of tethered (salen)Co catalyst systems possessing C2 symmetric salen units, see: Song Y, Yao X, Chen H, Bai C, Hu X, Zheng Z. Tetrahedron Lett. 2002;43:6625–6627.. Kwon MA, Kim GJ. Catal Today. 2003;87:145–151.. Gianneschi NC, Bertin PA, Nguyen ST, Mirkin CA, Zakharov LN, Rheingold AL. J Am Chem Soc. 2003;125:10508–10509. doi: 10.1021/ja035621h.. Shin CK, Kim SJ, Kim GJ. Tetrahedron Lett. 2004;45:7429–7433.. Gianneschi NC, Cho SH, Nguyen ST, Mirkin CA. Angew Chem Int Ed. 2004;43:5503–5507. doi: 10.1002/anie.200460932.. Welbes LL, Scarrow RC, Borovik AS. Chem Commun. 2004:2544–2545. doi: 10.1039/b408553a.. Kureshy RI, Singh S, Khan NH, Abdi SHR, Agrawal S, Jasra RV. Tetrahedron: Asymm. 2006;17:1638–1643.. Kawthekar RB, Lee YH, Kim GJ. J Porous Mater. 2009;16:367–378.. Haak RM, Belmonte MM, Escudero-Adan EC, Benet-Buchholz J, Kleij AW. Dalton Trans. 2010;39:593–602. doi: 10.1039/b911856j.. Zulauf A, Mellah M, Schulz E. Chem Eur J. 2010;16:11108–11114. doi: 10.1002/chem.201001012.. Kureshy RI, Prathap KJ, Kumar M, Bera PK, Khan NH, Abdi SHR, Bajaj HC. Tetrahedron. 2011;67:8300–8307.. Sadhukhan A, Khan NH, Roy T, Kureshy RI, Abdi SHR, Bajaj HC. Chem Eur J. 2012;18:5256–5260. doi: 10.1002/chem.201103574.. Hong X, Mellah M, Bordier F, Guillot R, Schulz E. ChemCatChem. 2012;4:1115–1121.. Kumar M, Kureshy RI, Shah AK, Das A, Khan NH, Abdi SHR, Bajaj HC. J Org Chem. 2013;78:9076–9084. doi: 10.1021/jo4012656.. Hong X, Billon L, Mellah M, Schulz E. Catal Sci Technol. 2013;3:723–729..
- 22.White DE, Jacobsen EN. Tetrahedron: Asymm. 2003;14:3633–3638. [Google Scholar]
- 23.The improved performance of catalyst 4b in the HKR of styrene oxide can be ascribed to its lower Lewis acidity relative to catalyst 4a. The more Lewis acidic catalyst promotes competitive and less enantiomer-selective opening of the epoxide at the internal position.
- 24.Selected examples of the application of 4a in the synthesis of complex targets: Peloruside A, McGowan MA, Stevenson CP, Schiffler MA, Jacobsen EN. Angew Chem, Int Ed. 2010;49:6147–6150. doi: 10.1002/anie.201002177.. Snider BB, Zhou J. Org Lett. 2006;8:1283–1286. doi: 10.1021/ol052948+. Sch 642305:. Moenomycin A, Adachi M, Zhang Y, Leimkuhler C, Sun B, LaTour JV, Kahne DE. J Am Chem Soc. 2006;128:14012–14013. doi: 10.1021/ja065905c.. Covell DJ, Vermeulen NA, Labenz NA, White MC. Angew Chem Int Ed. 2006;45:8217–8220. doi: 10.1002/anie.200603321. Hexose derivatives:. Chae J, Buchwald SL. J Org Chem. 2004;69:3336–3339. doi: 10.1021/jo035819k. Indolodioxane U86192A:. Rajapaksa NS, McGowan MA, Rienzo M, Jacobsen EN. Org Lett. 2013;15:706–709. doi: 10.1021/ol400046n. Reserpine:.
- 25.(a) Loy RN, Jacobsen EN. J Am Chem Soc. 2009;131:2786–2787. doi: 10.1021/ja809176m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Birrell JA, Jacobsen EN. Org Lett. 2013;15:2895–2897. doi: 10.1021/ol401013s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.An unoptimized version of the first two steps of this procedure has been reported: Vachal P, Jacobsen EN. Org Lett. 2000;2:867–870. doi: 10.1021/ol005636+..
- 27.Samat A, Bibout MEM, Elguéro J. Heterocycles. 1982;19:469–472. [Google Scholar]
- 28.Details of the synthetic procedures and characterization studies are provided in the Supporting Information.
- 29.In all of the synthetic applications described herein, catalyst loadings are calculated without correcting for residual solvent, and therefore represent upper limits.
- 30.While it was possible in certain cases to employ solvent-free conditions in the PKR and AKR of terminal epoxides and the hydrolytic desymmetrization of meso epoxides when using 4a, generally superior results were obtained using CH3CN as solvent.
- 31.Measurement of conversions for runs employing complexes 1 and 3b show both catalysts to be highly selective under the reaction conditions.
- 32.The (1S)-(+)-10-camphorsulfonate counterion provides superior results relative to tosylate in the PKR catalyzed by monomeric complex 1 (ref.6).
- 33.Substrates in entries 1–2 and 4–6 were used as received from commercial suppliers. Allyl glycidyl ether (entry 3) was purified by distillation from CaH2.
- 34.In order to avoid catalyst deactivation, it is important to not let catalyst and epoxide sit together for extended periods prior to the addition of nucleophile. The mechanistic basis for this deactivation has been elucidated (ref. 13b) and involves formation of less reactive (but still highly stereoselective) (salen)Co–OH complexes.
- 35.In the case of methyl glycidate, considerable foaming hampered distillation of the product mixture on the laboratory scale. However, removal of the diol product by aqueous extraction prior to distillation circumvented the foaming problem, and the epoxide was thus isolated in good yield: Stevenson CP, Nielsen LPC, Jacobsen EN. Org Synth. 2006;83:162–169..
- 36.Other solvents are also useful in this regard. For example, tert-butyl ethylene oxide was resolved to >99% ee at 56% conversion in 24 h employing 4a with CH2Cl2 as solvent.
- 37.Catalyst loadings were optimized to correspond to the lowest loadings possible to achieve >95% conversion of nucleophile within 24 h.
- 38.With monomer 1b•2,6-lutidine, the resolution of styrene oxide with phenol provided a 3:2 ratio of the undesired internal addition product to the desired α-aryloxy alcohol: Ready JM. PhD Thesis. Harvard University; Cambridge, MA: 2001. .
- 39.The PKR of styrene with p-cresol catalyzed by 4b afforded a 21:1 ratio of terminal to internal opening products.
- 40.Secondary alcohols were found to be unreactive under these conditions.
- 41.For example, see: Dishong DM, Diamond CJ, Cinoman MI, Gokel GW. J Am Chem Soc. 1983;105:586–593..
- 42.For examples of phenolic desymmetrization of meso epoxides, see: Iida T, Yamamoto N, Matsunaga S, Woo HG, Shibasaki M. Angew Chem, Int Ed. 1998;37:2223–2226. doi: 10.1002/(SICI)1521-3773(19980904)37:16<2223::AID-ANIE2223>3.0.CO;2-Z.. Vogl EM, Matsunaga S, Kanai M, Iida T, Shibasaki M. Tetrahedron Lett. 1998;39:7917–7920.. Matsunaga S, Das J, Roels J, Vogl EM, Yamamoto N, Iida T, Yamaguchi K, Shibasaki M. J Am Chem Soc. 2000;122:2252–2260.. Matsunaga S, Ohshima T, Shibasaki M. Adv Synth Catal. 2002;344:3–15.. For examples of the alcoholic desymmetrization of meso epoxides, see: Schneider C, Skreekanth AR, Mai E. Angew Chem, Int Ed. 2004;43:5691–5694. doi: 10.1002/anie.200460786.. Tschöp A, Marx A, Sreekanth AR, Schneider C. Eur J Org Chem. 2007:2318–2327.. For the catalytic asymmetric co-polymerization of carbon dioxide and meso epoxides to form polycarbonates, see: Nozaki K, Nakano K, Hiyama T. J Am Chem Soc. 1999;121:11008–11009.. Cheng M, Darling NA, Lobkovsky EB, Coates GW. Chem Commun. 2000:2007–2008..
- 43.For examples of enzymatic hydrolytic desymmetrization of meso epoxides, see: Zhao L, Han B, Huang Z, Miller M, Huang H, Malashock DS, Zhu Z, Milan A, Robertson DE, Weiner DP, Burk MJ. J Am Chem Soc. 2004;126:11156–11157. doi: 10.1021/ja0466210., and references therein. Weijers CAGM. Tetrahedron: Asymm. 1997;8:639–647.. Hodgson DM, Gibbs AR, Lee GP. Tetrahedron. 1996;52:14361–14384., and references therein.
- 44.Meso epoxides derived from acylic alkenes are relatively poor substrates for hydrolysis catalyzed by 4a, with lower rates and enantioselectivities obtained in all cases examined. Primary alcohols and phenols were found to be unreactive toward cyclohexene oxide in the presence of 4a.
- 45.Servis KL, Noe EA. J Am Chem Soc. 1973;95:171–174. , and references therein. [Google Scholar]
- 46.The commercially available epoxides employed in entries 1–2 and 4–6 were used as received. For the preparation and purification of the epoxides employed in entries 3 and 7–10, see the supporting information.
- 47.For examples of the carbamolytic desymmetrization of meso epoxides, see: Sekine A, Ohshima T, Shibasaki M. Tetrahedron. 2002;58:75–82.. Hou XL, Wu J, Dai LX, Xia LJ, Tang MH. Tetrahedron: Asymm. 1998;9:1747–1752.. Fu XL, Wu SH. Synth Commun. 1997;27:1677–1683.. Regati S, He Y, Thimmaiah M, Li P, Xiang S, Chen B, Zhao JCG. Chem Commun. 2013;49:9836–9838. doi: 10.1039/c3cc45988h.. Azoulay S, Manabe K, Kobayashi S. Org Lett. 2005;7:4593–4595. doi: 10.1021/ol051546z.. Mai E, Schneider C. Synlett. 2007:2136–2138.. Carrée F, Gil R, Collin J. Org Lett. 2005;7:1023–1026. doi: 10.1021/ol0475360..
- 48.(a) Chang S, McNally D, Shary-Tehrany S, Hickey MJ, Boyd RH. J Am Chem Soc. 1970;92:3109–3118. [Google Scholar]; (b) Allinger NL, Tribble MT, Miller MA, Wertz DH. J Am Chem Soc. 1971;93:1637–1648. [Google Scholar]
- 49.For examples of the desymmetrization of oxetanes with other nucleophiles, see: Wang Z, Chen Z, Sun J. Angew Chem, Int Ed. 2013;52:6685–6688. doi: 10.1002/anie.201300188.. Chen Z, Wang Z, Sun J. Chem Eur J. 2013;19:8426–8430. doi: 10.1002/chem.201301065.. Mizuno M, Kanai M, Iida A, Tomioka K. Tetrahedron: Asymm. 1996;7:2483–2484.. Mizuono M, Kanai M, Iida A, Tomioka K. Tetrahedron. 1997;53:10699–10708..
- 50.For entry 10, (salen)Co monomer 1a (10 mol%) afforded product in 7% ee at 38% conversion after 96 h. In tetrahydrofuran- and dihydrobenzofuran-forming reactions (entries 1–9), comparable ee's ±5% were obtained using monomeric or oligomeric catalsyt.
- 51.The sole exception to this statement observed thus far is in the formation of tetrahydropyrans as described in section 2.3.7 and ref. 25a.
- 52.Chenault KM, Kim MJ, Akiyama A, Miyazawa T, Simon ES, Whitesides GW. J Org Chem. 1987;52:2608–2611. [Google Scholar]
- 53.Cunningham AF, Jr, Kündig EP. J Org Chem. 1988;53:1823–1825. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.