

Abstract
Rapid isothermal nucleic acid amplification technologies can enable diagnosis of human pathogens and genetic variations in a simple, inexpensive, user-friendly format. The isothermal exponential amplification reaction (EXPAR) efficiently amplifies short oligonucleotides called triggers in less than 10 min by means of thermostable polymerase and nicking endonuclease activities. We recently demonstrated that this reaction can be coupled with upstream generation of trigger oligonucleotides from a genomic target sequence, and with downstream visual detection using DNA-functionalized gold nanospheres. The utility of EXPAR in clinical diagnostics is, however, limited by a nonspecific background amplification phenomenon, which is further investigated in this report. We found that nonspecific background amplification includes an early phase and a late phase. Observations related to late phase background amplification are in general agreement with literature reports of ab initio DNA synthesis. Early phase background amplification, which limits the sensitivity of EXPAR, differs however from previous reports of nonspecific DNA synthesis. It is observable in the presence of single-stranded oligonucleotides following the EXPAR template design rules and generates the trigger sequence expected for the EXPAR template present in the reaction. It appears to require interaction between the DNA polymerase and the single-stranded EXPAR template. Early phase background amplification can be suppressed or eliminated by physically separating the template and polymerase until the final reaction temperature has been reached, thereby enabling detection of attomolar starting trigger concentrations.
The detection and quantification of nucleic acid sequences for identification of pathogens or genetic markers is one of the fastest growing areas in clinical chemistry. Nucleic acid testing, which in most cases involves the polymerase chain reaction (PCR),1 is starting to move outside of the centralized laboratory environment toward the point of care (1–4). Simple, portable instrumentation can be implemented more readily if one avoids the need for thermocycling by using isothermal nucleic acid amplification methods, such as nucleic acid sequence-based amplification (NASBA) (5), transcription-mediated amplification (TMA) (6), strand displacement amplification (SDA) (7, 8), loop-mediated amplification reaction (LAMP) (9, 10), rolling circle amplification (RCA) (11), helicase-dependent amplification (HDA) (12, 13), and the exponential amplification reaction (EXPAR) (14–16). In contrast to most of these methods, which require amplification times from 30 min to several hours, EXPAR enables 106–109-fold amplification of short oligonucleotides in a manner of minutes (14), a distinct advantage in point of care applications.
EXPAR involves a combination of polymerase strand extension and single-strand nicking and occurs at 55 °C, a temperature which permits activity and stability of the thermophilic polymerase and nicking endonuclease required for amplification. The reaction amplifies a short oligonucleotide called trigger X serving as the analyte, which can be generated from a genomic target sequence. EXPAR is initiated when trigger X primes an amplification template consisting of 2 times the complementary sequence X′, separated by nine bases that enable generation of the nicking enzyme recognition and cleavage site (Figure 1). After trigger extension by the polymerase, the nicking enzyme nicks the top strand, generating another trigger oligonucleotide that either melts off or is displaced from the amplification template. The polymerase elongates the recessed 3′-hydroxyl created by the departing trigger, and another trigger is generated in the same manner. These triggers then prime other amplification templates, enabling true chain (exponential) reactions.
Figure 1.

Overview of the exponential amplification reaction (EXPAR), which rapidly and isothermally amplifies trigger X, a short oligonucleotide serving as the analyte. (a) Trigger X transiently binds to the complementary recognition sequence at the 3′-end of the amplification template. (b) The trigger sequence is extended by the DNA polymerase, forming the double-stranded nicking enzyme recognition site 5′-GAGTCNNNN-3′ on the top strand. (c) The top strand is cleaved through the nicking endonuclease Nt.BstNBI. (d) At the temperature of the reaction (55 °C), the newly formed trigger is released from the amplification template. The trigger-producing form of the template re-enters the linear amplification cycle, and new trigger oligonucleotides are generated through duplex extension, nicking, and release. (e) The newly formed trigger oligonucleotides activate additional template sequences, giving rise to exponential amplification of trigger X.
Trigger X can be generated from genomic target DNA through a reaction called Fingerprinting, which involves single-strand nicking at adjacent nicking enzyme recognition sites within the genomic DNA sequence, followed by polymerase extension and linear amplification similar to the linear amplification cycle described in Figure 1 (16). The Fingerprinting reaction occurs in the same Mastermix and under the same reaction conditions as EXPAR and generates a distinct set of trigger oligonucleotides specific for the target genomic DNA. To enable visual colorimetric detection of amplification products, we have coupled a two-stage EXPAR amplification reaction with DNA nanosphere aggregation (15). We further demonstrated proof of principle for coupling trigger generation via Fingerprinting with two-stage EXPAR amplification and visual colorimetric detection using DNA-functionalized gold nanospheres (16), using a recombinant plasmid with an insert containing a fingerprinting site derived from herpes simplex virus 1 (HSV 1) genomic DNA. The entire reaction can be performed in less than 10 min of total assay time using minimal instrumentation.
Despite its many positive attributes, the utility of EXPAR is currently hampered by a nonspecific amplification phenomenon observed in the absence of trigger, which causes the limit of detection to be comparatively high. We herein report an investigation of the cause of nonspecific DNA amplification observed in EXPAR, based on monitoring the reaction progression in real time via fluorescence and analyzing the reaction products using mass spectrometry, gel electrophoresis, and DNA sequencing. The results suggest that background amplification is caused by a novel unconventional DNA polymerase activity. This unconventional polymerase activity is particularly augmented in EXPAR due to the reaction design but is likely to occur to some degree in other nucleic acid amplification reactions as well, which places our observations into a broader biochemical context. In addition to describing this unconventional polymerase activity, we present a strategy for suppressing or eliminating nonspecific amplification, thereby increasing the sensitivity of EXPAR.
EXPERIMENTAL PROCEDURES
General Reagents
Nuclease-free water was purchased from Ambion (Austin, TX). Nt.BstNBI nicking enzyme, Bst large fragment polymerase, 10× Thermopol I buffer, 10× NE buffer 3, dNTPs, and 100 mM MgSO4 were purchased from New England Biolabs (Beverly, MA). SYBR Green II was purchased from Sigma (St. Louis, MO). Human genomic DNA was purchased from Promega (Madison, WI).
Oligonucleotides
Complete sequences for all template and trigger oligonucleotides used in this report are summarized in Table S1 (Supporting Information). Amplification templates were purchased from Integrated DNA Technologies, Inc. (Coralville, IA) and were in general capped at the 3′-end with an amino-C6 blocking group unless otherwise specified. The standard A and Ran2 templates were ordered capped and uncapped, as well as desalted, single-HPLC-purified, and dual-HPLC-purified. The randomer templates Ran1–Ran24 were ordered in single-HPLC-purified form. SNP templates SNP1–SNP10 were ordered in desalted form. To eliminate any possibility for cross contamination of amplification templates with triggers, the trigger oligonucleotides were purchased from MWG (High Point, NC) in desalted form.
Standard EXPAR Protocol
EXPAR amplification reactions were carried out in a reaction mixture containing each dNTP (250 μM), 1.2 mM EGTA, 0.5× NE buffer 3 [50 mM NaCl, 25 mM Tris-HCl (pH 7.9), 5 mM MgCl2, and 0.5 mM DTT], 1× Thermopol I [10 mM KCl, 10 mM (NH4)2SO4, 20 mM Tris-HCl (pH 8.8), 0.1% Triton-X-100, and 2 mM MgSO4], 0.4 unit/μL Nt.BstNBI nicking enzyme, and 0.08 unit/μL Bst polymerase. For real-time fluorescence-based monitoring of trigger amplification, SYBR Green II (10000× stock in DMSO) was added to the Mastermix to a final concentration of 10×. The Mastermix further contained 100 nM amplification template unless otherwise specified. Reactions were conducted in 50 μL volumes in eight-well PCR strips or a 96-well PCR microtiter plate. The desired amount of trigger oligonucleotide [10-fold dilution series with the final trigger concentration ranging between 10 pM and 100 aM (3 × 108 to 3 × 103 copies) or a subset thereof, plus a negative no trigger control (NTC), all prepared in 0.1× Thermopol I buffer] was added directly to each PCR well. EXPAR amplification was performed in a Bio-Rad (Hercules, CA) MJ Opticon I real-time thermocycler set to a constant temperature of 55 °C, and the fluorescence intensity was monitored in 10 s intervals using excitation at 488 nm over a period of 5 min to 2 h.
DNase and RNase Treatment of Reaction Components
DNase treatment was performed using the Ambion DNAfree kit, which does not require heat denaturation of the DNase at temperatures incompatible with the polymerase and nicking enzyme used in EXPAR. DNase treatment consisted of incubating the enzymes and other Mastermix components with 1× DNase I buffer and rDNase I at 37 °C for 20 min, followed by addition of 0.1× DNase inactivation reagent and centrifugation at 10000g for 1.5 min. The supernatant was then transferred into a Mastermix containing the remainder of the EXPAR reagents, and the reaction progression at 55 °C was monitored using real-time fluorescence.
RNase treatment consisted of incubating the reaction enzymes (polymerase and/or nicking enzyme) with 0.25 or 0.025 unit/μL RNase A in 1× Thermopol buffer and 0.5× NE buffer 3 for 10 min at 37 °C. After incubation, the treated enzymes were added to a reaction mixture containing the remainder of the Mastermix components, and the reaction progression at 55 °C was monitored using real-time fluorescence.
EXPAR with a Preincubation Step
For the preincubation reactions, 25 μL of reaction mixtures containing a subset of the components of the general Mastermix was incubated at 55 °C for 5 min. In general, these preincubation mixtures did not contain EGTA and SYBR Green II. Other omitted components of the Mastermix are described in Results. After this preincubation period, the reaction tubes were put on ice, and 25 μL of a second reaction mixture was added. This second reaction mixture contained all components of the Mastermix that were present in the preincubation step at a concentration of 1×, and all components of the Mastermix that were absent in the preincubation step at a concentration of 2×, such that the composition of the final 50 μL reaction mixture equaled the composition of the Mastermix as described above. EXPAR amplification of this final reaction mixture was then performed at 55 °C and was monitored in real time, as described above.
DNA Analysis by Gel Electrophoresis
The products from the EXPAR amplification reactions were analyzed with the E-Gel system (Invitrogen, Carlsbad, CA) using 4% agarose gels. Gels were loaded with 10 μL of 0.1× Invitrogen “Blue Juice” sample loading buffer followed by addition of 10 μL of amplified EXPAR Mastermix or DNA ladder (10 or 100 bp ladder, Invitrogen), run for 30 min on the PowerBase, and imaged using a UVP transilluminator.
Mass Spectrometric Analysis of Oligonucleotides
For samples to be analyzed by mass spectrometry, EXPARs were performed in Mastermix containing no SYBR Green or EGTA, but otherwise identical in composition to the Mastermix described above. The Mastermix used further contained either 1 pM trigger, 100 nM template, and standard concentrations of both enzymes (positive), 100 nM template and standard concentrations of both enzymes with no trigger (negative), or 100 nM template with no trigger and no enzymes (blank). Reaction mixtures were heated to 55 °C in an Eppendorf thermomixer R for various amplification times, and the products were analyzed by LC–ESI MS, using a Micromass (Manchester, U.K.) LCT time-of-flight instrument connected to an Agilent (Palo Alto, CA) 1100 Series HPLC system. Buffer A was composed of 5 mM dimethylbutylamine acetate (DMBAA) in HPLC-grade water, and buffer B was composed of 5 mM DMBAA in a 50% (v/v) water/acetonitrile mixture. Samples were injected on a Waters Xterra MS column (C18 packing, 3.5 μm particle size, 125 Å pore size, 2.1 mm × 20 mm), which was heated to 30 °C, and separated using a gradient from 10 to 90% buffer B over 3 min, 90 to 20% buffer B over 30 s, and then 20 to 10% buffer B over 2 min. The first 60 s of this gradient served as a desalting step, and the eluent was diverted to waste. Mass spectra were acquired in electrospray negative ion mode, ranging from 800 to 2000 amu, with a scan time of 1 s. Spectra reported herein represent an integral of a certain region within the LC chromatogram, as indicated. Sequence Editor (Micromass) was used to determine the predicted charge states from the known sequences of different triggers. Spectral deconvolution was performed using MaxEnt1 (Micromass). With the current LC–MS protocols, the detection limit for short oligonucleotides is approximately 100 nM.
Sequencing of EXPAR Amplification Products
Template oligonucleotides SNP8 and Ran24 were used for the cloning and sequencing of background amplification products. EXPAR mixtures were incubated for 3 h as previously described using the standard EXPAR protocol with an increased level of EXPAR template (200 nM) and removing EGTA and SYBR (total volume of 100 μL). Enzymes, buffers, and excess nucleotides were removed using a QIAquick nucleotide removal kit (Qiagen) following the manufacturer’s protocol and resuspended in 50 μL of water.
To ensure that the DNA produced during EXPAR was double-stranded and blunt-ended for cloning, 5′-overhangs were filled using Klenow exo- and 3′-overhangs were removed using mung bean nuclease. The EXPAR mixture was processed initially using Klenow exo- as follows: 40 μL of purified EXPAR products was combined with NE buffer 2 [NEB (1× final concentration), 10 mM Tris-HCl, 50 mM NaCl, 10 mM MgCl2, and 1 mM dithiothreitol (pH 7.9) at 25 °C], 33 μM dNTPs, and 5 units/μL Klenow exo- (NEB) in a total volume of 60 μL and incubated at 37 °C for 1 h. After 3′-extension, the reaction mixture was processed using a QIAquick nucleotide removal kit following the manufacturer’s protocol and resuspended in 50 μL of water. The remaining 3′-overhangs were removed using the following protocol: 40 μL of purified EXPAR/Klenow reaction products was combined with mung bean nuclease buffer [NEB (1× final concentration), 50 mM sodium acetate, 30 mM NaCl, and 1 mM ZnSO4 (pH 5.0) at 25 °C] and 5 units/μL mung bean nuclease (NEB), in a total volume of 50 μL, and incubated for 30 min at 30 °C. The processed sample was analyzed on a 2% agarose gel, and the DNA was excised for cloning. The excised agarose sections were purified using the QIAquick gel extraction kit (Qiagen) following the manufacturer’s protocol.
The blunt-ended DNA was cloned into a SmaI-linerarized, 2× calf intestinal phosphatase, pUC19 plasmid using T4 DNA ligase: 25 fmol of linearized pUC19, 5 μL of blunted DNA, and 400 ceU/μL T4 DNA ligase (NEB) were combined in T4 DNA ligase buffer [NEB (1× final concentration), 50 mM Tris-HCl, 10 mM MgCl2, 1 mM ATP, and 10 mM dithiothreitol (pH 7.5) at 25 °C] in a total volume of 40 μL. The reaction was allowed to proceed at 16 °C for 12 h prior to the mixture being transformed into DH5α chemically competent cells (Invitrogen), plated on 25 μg/mL ampicillin plates, and incubated at 37 °C for 16 h. Single colonies were selected for colony PCR and inserts verified using M13 forward (−20) and M13 reverse (−27). Colonies with inserts were processed and sequenced using M13 forward (−20).
Manual Hot Start of EXPAR
Manual hot start was performed by separating the Mastermix into two separate solutions. The first contained the polymerase or other Mastermix components such as Mg2+, EXPAR template, or nicking enzyme, at a concentration of 2×, along with the reaction buffers at a concentration of 1×. The second contained the remainder of the Mastermix components at a concentration of 2×. Each portion was preheated to 55 °C in a standard thermocycler; then both solutions were combined, and the reaction progression was monitored using the Opticon real-time thermocycler.
RESULTS
Real-Time Fluorescence Monitoring of EXPAR
The progression of isothermal DNA amplification through EXPAR can be monitored in real time on the basis of fluorescence detection with the intercalating dye SYBR Green II. Although considered to be a single-strand RNA stain, SYBR Green II exhibits increased fluorescence intensity when bound to double-stranded DNA as opposed to single-stranded DNA.2 The real-time fluorescence intensity of EXPAR increases in a sigmoidal fashion as the amplification template is converted from single-stranded to partially or completely double-stranded DNA, eventually reaching a plateau once the majority of the single-stranded template is converted into active, trigger-producing forms (Figure 2A). The sigmoidal curves are therefore an indirect measure of trigger amplification, and we define the time at which exponential amplification occurs on the basis of the point of inflection (POI, the time corresponding to the maximum slope in the fluorescence curve). For EXPAR, the POI is similar in meaning to the cycle threshold in real-time PCR.
Figure 2.

EXPAR amplification monitored through real-time fluorescence, showing rapid amplification and correlation between the onset of amplification and the starting trigger concentration. (A) Amplification curves using template RAN2 for different starting trigger concentrations of (i) 10 pM, (ii) 1 pM, (iii) 100 fM, (iv) 10 fM, or (v) 1 fM or (vi) in the absence of trigger (no trigger control, or NTC). (B) Linear relationship between the inflection points of the amplification curves shown in panel A and the logarithm of the starting trigger concentration. Averages for the trigger dilution series and the no trigger control represent 10 replicates, taken from five experiments conducted in duplicate on five different days.
We observe a linear correlation between the POI and the logarithm of the starting trigger concentration for a 10-fold trigger dilution series from 10 pM to 1 fM (Figure 2B). However, the no trigger control (NTC), which contains all components of the EXPAR Mastermix except for the trigger, also displays nonspecific background amplification, the timing of which depends on the template sequence and the Mastermix composition. Similar trigger dilution real-time fluorescence curves are observed for other template sequences (see Figure S1 of the Supporting Information). For suitable EXPAR templates and under the current reaction conditions, trigger concentrations of 10 fM (3 × 105 copies in a reaction volume of 50 μL) can be differentiated from background reproducibly using as a metric ≥10% difference between the POI for the trigger-containing (positive, P) and trigger-lacking (negative, N) samples [(N – P)/N in percent].
To investigate if nonspecific background amplification is caused by contamination with DNA or RNA sequences, we treated the enzymes and other Mastermix components with DNase or RNase A prior to use in the reaction. This treatment, however, did not reduce the level of nonspecific amplification. We further performed EXPAR in the presence of up to 60 ng of human genomic DNA, either in intact, high-molecular weight double-stranded (ds) form or as sheared DNA after cycles of heating to 95 °C, freezing at −80 °C, and sonication. Under the current conditions, the presence of intact DNA or sheared DNA (average size of 200 bases, size distribution of approximately 100–500 bases, according to gel electrophoresis) does not significantly accelerate background amplification and has little effect on EXPAR amplification in the presence of trigger.
Another hypothesis for nonspecific background amplification involves extension of the amplification template at the 3′-end by the polymerase, forming template dimers similar to “primer–dimer” type nonspecific background amplification observed in PCR. EXPAR in general is performed using a 3′-amine-capped amplification template, but the capping yield typically is only 99–99.5% (17). To investigate this hypothesis, we compared specific versus nonspecific amplification using 3′-capped versus uncapped templates of identical sequences, both in single-HPLC-purified form (Figure S2 of the Supporting Information). If template–dimer type extension was the predominant mechanism for nonspecific background amplification, use of an uncapped template would be expected to drastically accelerate background amplification. Experimentally observed acceleration of nonspecific background amplification using uncapped versus capped templates was however weak and not statistically significant. Similarly, template purity has only a minor effect on the progression of nonspecific background amplification, based on comparison of data from desalted, single-HPLC-purified, and dual-HPLC-purified templates of identical sequences.
Phases of Background Amplification
The nonspecific background amplification includes an early phase and a late phase (Figure 3). The early phase generally is observed within 5–10 min of isothermal amplification and interferes with the desired specific trigger generation and amplification. The late phase background amplification, if observed, appears after ≥20 min, depending on the template sequence and Mastermix composition, and results in a final fluorescence intensity much larger in magnitude than that following the early phase of background amplification. This late phase nonspecific background amplification does not, in most cases, directly interfere with specific amplification in the presence of trigger, which for suitable templates occurs in less than 10 min even for low copy numbers of trigger X.
Figure 3.

EXPAR in the absence of trigger monitored via real-time fluorescence over 2 h, showing early and late phase nonspecific amplification as a function of template sequence. (A) (i) A regular EXPAR template of structure X′rX′, containing a repeat of trigger complement X′ separated by the nicking enzyme recognition and four-base post cut sequence “r”, (ii and iii) altered templates X′rY′ and Y′rX′, respectively, in which the sequences at the 5′- and 3′-ends of the template are not identical but the nicking enzyme recognition site is intact, (iv) an altered template X′sX′, in which the sequences at the 5′- and 3′-ends of the template are identical but the nicking enzyme recognition and four-base post cut sequence “r” has been scrambled to yield sequence “s”, and (v) a no template control. (B) Amplification curves observed for template sequences (i) SNP8, (ii) StanA, (iii) Ran21, and (iv) Ran2.
We investigated how background amplification depends on the EXPAR template structure through a series of experiments using oligonucleotide templates containing at the 3′- and 5′-ends either the same or different trigger complement sequences, X′ and Y′. These two regions were joined by either the correct complement of the Nt.BstNBI recognition sequence followed by a four-nucleotide spacer (designated as “r”) or a scrambled version of this sequence “r”, designated as “s” (selected sequences given in Table 1; for full sequence information, see Table S1 of the Supporting Information). Reaction mixtures containing an amplification template of the structure X′rX′ or Y′rY′, which follows the EXPAR design rules (14), exhibit early as well as late phase background amplification (Figure 3A, curve i, for X′rX′; similar results obtained for Y′rY′). Reactions with templates containing two different trigger complement sequences (X′rY′ and Y′rX′, curves ii and iii of Figure 3A, respectively) do not exhibit early phase background amplification, and neither do template sequences containing the same trigger complement sequence joined by a scrambled nicking enzyme recognition site (X′sX′, curve iv of Figure 3A; similar results obtained for Y′sY′). Each reaction did, however, give rise to late phase background amplification. Without any templating DNA sequences, early phase nonspecific background amplification is again absent, but late phase background amplification is observed (Figure 3A, curve v).
Table 1.
Selected EXPAR Trigger and Template Sequencesa
| Name | Trigger | Template |
|---|---|---|
| Stan A | 5′-CAGTCGTAGG-3′ | 5′-CCTACGACTGAACAGACTCTCCTACGACTG-3′-NH2 |
| SNP 2 | 5′-CCGTCGTAGG-3′ | 5′-CCTACGACGGAACAGACTCTCCTACGACGG-3′-NH2 |
| SNP 8 | 5′-CAGTCGTTGG-3′ | 5′-CCAACGACTGAACAGACTCTCCAACGACTG-3′-NH2 |
| SNP 9 | 5′-CAGTCGTACG-3′ | 5′-CGTACGACTGAACAGACTCTCGTACGACTG-3′-NH2 |
| SNP 10 | 5′-CAGTCGTAGC-3′ | 5′-GCTACGACTGAACAGACTCTGCTACGACTG-3′-NH2 |
| Ran 2 | 5′-CTATTTCCCC-3′ | 5′-GGGGAAATAGGTGAGACTCTGGGGAAATAG-3′-NH2 |
| Ran 15b | 5′-TTATCACACG-3′ | 5′-CGTGTGATAAGAGCGACTCTCGTGTGATAA-3′-NH2 |
| Ran 17 | 5′-CCGCGCCCCG-3′ | 5′-CGGGGCGCGGTGCGGACTCTCGGGGCGCGG-3′-NH2 |
| Ran 21c | 5′-GGTTAAATCG-3′ | 5′-CGATTTAACCTGATGACTCTCGATTTAACC-3′-NH2 |
| Ran 23 | 5′-CAGACGATGT-3′ | 5′-ACATCGTCTGCGGCGACTCTACATCGTCTG-3′-NH2 |
| Ran 24 | 5′-GCTGCACGCT-3′ | 5′-AGCGTGCAGCGTGGGACTCTAGCGTGCAGC-3′-NH2 |
| X′ rY′ | N/A | 5′-CGTGTGATAAGAGCGACTCTCGATTTAACC-3′-NH2 |
| X′ sX′ | N/A | 5′-CGTGTGATAAATGACATATTCGTGTGATAA-3′-NH2 |
Template sequence design: underlined portions denote the trigger complement, bold portions the nicking enzyme recognition sequence, and italic portions the four-base spacer sequence after which the nicking enzyme cuts the top strand.
X′rX′ is equal to Ran 15.
Y′rY′ is equal to Ran 21.
It appears that rapid early phase nonspecific background amplification requires the possibility of exponentially amplifying trigger X via a suitable EXPAR template, such as X′rX′ or Y′rY′. In contrast, late phase nonspecific background amplification is observed in Mastermix that includes only polymerase, nicking enzyme, and dNTPs without any template or other oligonucleotides (Figure 3A, curve v), as well as in any reaction mixtures that contain either correct EXPAR templates or alternate template structures (Figure 3A, curves i–iv). However, for Mastermix with polymerase and dNTPs, but no nicking enzyme, template, or trigger oligonucleotides, no amplification is observed via real-time fluorescence monitoring in a 2 h time period (data not shown).
Late Phase Nonspecific Background Amplification
To further characterize late phase nonspecific background amplification in the presence of template sequences following the structure X′rX′, we analyzed the products formed for 35 different template sequences (selected sequences given in Table 1; for comprehensive sequence information, see Table S1 of the Supporting Information). The trigger–template sequences SNP1–SNP10 are based on our original EXPAR X′-X′ template Standard A (StanA) (15), introducing a single base change at each position of the 10 bp trigger complement region within the template. The template sequences Ran1– Ran 24 were constructed from trigger complement sequences and postrecognition site spacer sequences derived using a random sequence generator. In all cases, an additional T has been added in the trigger complement region located in the 3′-portion of the amplification template, adjacent to the nicking enzyme recognition site. This T serves to complement an extra A which may be added to the 3′-end of the amplified trigger by some polymerases due to terminal deoxynucleotidyl transferase activity (not observed for Bst polymerase).
There is considerable variability in the probability of observing significant late phase background amplification via real-time fluorescence, as well as in the timing of onset and in the real-time kinetic profile of this late phase amplification. In most cases, late phase background amplification is sigmoidal in shape, although in some cases biphasic (e.g., Figure 3B, curve ii) or even triphasic late phase amplification curves can be observed. Running the same template sequence multiple times under supposedly identical reaction conditions results in significantly different amplification profiles and onset times, even though some template sequences have a lower probability of exhibiting late phase background amplification than others.
Using agarose gel electrophoresis, we determined that different reactions generate products with considerable size variability after nonspecific EXPAR background amplification for 2 h (Figure 4A). Most reactions produce smears at molecular weights higher than the size of the EXPAR template (30mer). In some cases, distinct higher-molecular weight bands and occasionally even a ladder can be observed. Some sequences, such as Ran15, Ran21, and SNP9, predominantly produce distinct bands shorter than the EXPAR template, but again considerable variability is observed for product size distributions from late phase background amplification, as demonstrated by analysis of replicate background amplifications of the same template sequence under supposedly identical conditions (Figure 4B). If no late phase background amplification is observed via real-time fluorescence, then no product bands are observed on the gel.
Figure 4.

Agarose gel electrophoresis showing variability in products generated after isothermal amplification for 2 h at 55 °C in the presence of 100 nM template but no trigger. (A) Comparison of products formed using different template sequences: lane 1, 10 bp ladder; lanes 2 and 3, template Ran2; lanes 4 and 5, template Ran15; lanes 6 and 7, template Ran17; lanes 8 and 9, template Ran21; lanes 10 and 11, template Ran24; lane 12, 100 bp ladder. Replicates represent samples from two separate runs per template, performed on the same day. (B) Comparison of products formed in replicate runs using the same template sequences (Ran24): lane 1, 10 bp ladder; lanes 2–6, runs 1–5 on day 1; lanes 7–11, runs 1–5 on day 2; lane 12, 100 bp ladder.
We further analyzed amplification products generated after longer reaction times using negative ion mode LC–ESI MS. Mass spectra for reaction mixtures containing nicking enzyme, polymerase, and template but no trigger (Negative) were acquired following amplification for a total reaction time of 20 min or 2 h. In all cases, the amplified reaction mixtures contained a significant amount of trigger, observable as peak 1 in the LC trace (Figure 5), even though no trigger was originally present in the reaction mixture. This observation is discussed in more detail in the next section. After 20 min, reaction mixtures containing certain template sequences start to exhibit either a second peak (for example, Ran15 and Ran 23, Figure 5A,C) or a shoulder (Ran24) in the LC trace shortly after the trigger peak. A distinct peak 2 is observed most often for template sequences which produce a low-molecular weight gel electrophoresis band and which feature a linear increase in fluorescence intensity after the initial early phase amplification. Deconvolution of the mass spectrum corresponding to peak 2 reveals the mass of an oligonucleotide other than the trigger, with a predicted length of approximately 11 bases.
Figure 5.

LC–ESI MS analysis of products from nonspecific background amplification, with an emphasis on product peaks appearing after longer amplification times. Reaction mixtures contained polymerase, nicking enzyme, dNTPs, and template but no trigger. (A) LC trace (left) and mass spectrum (right) corresponding to peak 2 of a reaction mixture containing template Ran15 after amplification for 20 min. Peak 2 corresponds to an 11mer oligonucleotide (based on deconvolution) with characteristic masses distinct from the Ran15 trigger [m/z 1532 (2−) and 1021 (3−)]. This trigger is eluted under peak 1. (B and C) LC traces and mass spectra corresponding to peak 3 (~4.5 min retention time) of reaction mixtures containing templates Ran23 and Ran24, respectively, after amplification for 2 h. Peak 3 corresponds to longer oligonucleotide products, most likely poly d(AT) ladders.
After amplification for 2 h, all reactions exhibiting significant late phase background amplification via real-time fluorescence monitoring produce a third peak, observed as peak 3 at a retention time of 4 min on the LC traces. This includes reaction mixtures containing polymerase, nicking enzyme, and dNTPs, but no templating DNA sequences. Peak 3 usually (though not always) contains the same or similar ion traces regardless of the amplification template sequences. This third peak varies in magnitude and in many cases dominates the LC trace. The mass spectrometer used for these studies is able to accurately identify only masses for oligomers <30 bases in length. Longer sequences are likely to fragment inside the instrument.
We have cloned and sequenced the late phase EXPAR amplification products generated from reaction mixtures containing either the template sequence Ran24 or SNP8. Sequences for representative inserts are listed in Table 2. Late phase amplification products consist of mainly polyd( AT) sequences, with interspersed G in the position normally occupied by A, and with interspersed C in positions normally occupied by T. Incorporation of G instead of A opposite T is possible through the G-T wobble pair. Replication of this strand would lead to incorporation of a C in a position normally occupied by T. One notable exception is the repeated occurrence of the GAGAG(T/A)CTCTC sequence motif, which is almost palindromic, with the exception of the central T/A base. If the central base is a T, then the motif contains the sense strand of the 5′-GAGTC-3′ nicking enzyme recognition sequence. If the central base is an A, then the motif contains the antisense 5′-GACTC-3′ strand of the nicking enzyme recognition sequence.
Table 2.
Sequencing Results from Products of Late Phase Nonspecific Background Amplification
| template | sample | sequence |
|---|---|---|
| Ran24 | A | 5′-ATATATATGTATATATATGAGAGTCTCTCATATATATACATATATATATATATATATATATACATATATATATATATATATATGTATATATAT-3′ |
| B | 5′-ATATATACATATATATATATATATATATATATATATGAGAGTCTCTCATATATATATATATATATATATATA-3′ | |
| C | 5′-TATGTGTATATACATATATATATATATATA-3′ | |
| D | 5′-ATATATATACATATATATATGAGAGACTCTCATATATATATGTATATATATGAGAGTCTCTCGTATATATACATATATATAT-3′ | |
| SNP8 | A | 5′-ATATATATACATATATATATATATATATATATATATGTATATATAT-3′ |
| B | 5′-ATATATATATGTATATATATAT-3′ | |
| C | 5′-ATATATATACATATATATATATATATATATATATATATATATGTATATATAT-3′ | |
| D | 5′-TATATATATATGTATATATATGAGAGTCTCTCATATATATACATATATATATATATATATATATATGTATATATA-3′ | |
| E | 5′-TATACATATATATATATATACATATATACATATATATATATATATATATATGTATATAT-3′ |
These sequence motifs seem to correspond to product peaks observed in the mass spectrometric analysis of late phase amplification products. The 11mer GAGAGTCTCTC has a parent mass of approximately m/z 3000 and gives rise to a 2–charge state of m/z 1116, similar to the mass of m/z 1114.43 observed in the peak 2 trace (Figure 5A). A parent peak at approximately m/z 3000 is observed repeatedly when spectra corresponding to products eluted as peak 2 for different amplification templates are deconvoluted.
Deconvoluting the entire spectrum of Ran24, including peak 3, up to a mass of m/z 35000 revealed evenly spaced peaks at m/z 6166.7, 7708.9, and 9253.3 and two major peaks at m/z 15417.5 and 17603.5 (Figure S3 of the Supporting Information). These peaks correspond to different lengths of dAT sequences with sporadic substitutions of G for A and C for T. The mass accuracy of these peaks is within m/z 5, which is reasonable considering the inaccuracy of detecting large masses through deconvolution. In fact, a 57 bp poly-d(AT) sequence, with two C residues and one G substituted for T and A, respectively, has a mass of m/z 17604, which corresponds almost exactly to our deconvoluted peak at m/z 17603.5.
Early Phase Nonspecific Background Amplification
As mentioned previously, nonspecific background amplification appears to generate the trigger sequence for the EXPAR template present in the reaction, even though no trigger is included in the Mastermix. The trigger further is formed during early phase background amplification, as corroborated by mass spectrometric analysis of EXPAR products generated from the 35 different template sequence investigated in this study (Table 1 and Table S1 of the Supporting Information). For each template sequence, we analyzed amplified reactions of Mastermix containing 100 nM template, both enzymes, and 1 pM trigger (positive); 100 nM template and both enzymes with no trigger (negative); or 100 nM template with no enzymes and no trigger (blank). Each reaction was quenched shortly after the early phase nonspecific background amplification for the specific template sequence had reached its maximum plateau, as determined by real-time fluorescence. Mass analysis of the reaction products demonstrates that the trigger sequence specific for the template present in the Mastermix is produced as the predominant product for both the positive and negative reactions (Figure 6A,B). This was found to be true for all 35 template sequences analyzed. Blank reaction mixtures containing no enzymes show only background noise, indicating that the trigger oligonucleotides are generated through enzymatic amplification. Trigger is therefore generated by nonspecific background amplification regardless of its presence at the start of the reaction. For short amplification times, no other significant peaks besides the masses corresponding to the trigger sequence are observed. The sensitivity of the mass spectrometer is approximately 100 nM; thus, any sequences that may be present at lower concentrations cannot be detected.
Figure 6.

LC–ESI MS analysis of products from nonspecific background amplification, with an emphasis on product peaks appearing after short amplification times. LC traces (left) and corresponding mass spectra (right) for EXPARs using template sequence (A) StanA [trigger m/z 1573 (2−) and 1048 (3−)] and (B) Ran 2 [trigger m/z 1496 (2−) and 997 (3−)]. Positive reaction mixtures contained 1 pM trigger, enzymes at standard concentrations, and 100 nM template. Negative reaction mixtures contained no trigger but did contain enzymes at standard concentrations and 100 nM template. Blank reaction mixtures contained only 100 nM template.
The nonspecific amplification of trigger X is expected to involve polymerase, nicking enzyme, dNTPs, and template. To investigate which of these reagents contributes most significantly to nonspecific background amplification, we performed a series of experiments in which part of the Mastermix containing the reagents of interest was preincubated at 55 °C for 5 min. After this preincubation step, the remaining Mastermix components were added so that the final composition was equal to the composition of the standard EXPAR Mastermix, without the trigger. Reaction tubes were again heated to 55 °C, and the fluorescence intensity profile was monitored to obtain the POI times for background amplification. Significant differences were observed in the timing of background amplification during this second 55 °C reaction period, depending on which components of the Mastermix were present during the preincubation period (Figure 7). As a control, we used a sample containing template and dNTPs during the preincubation step, but no enzymes. The background amplification for this control sample is slightly accelerated compared to background amplification without preincubation.
Figure 7.

Acceleration of nonspecific amplification through preincubation of selected EXPAR Mastermix reagents at 55 °C for 5 min, compared to the regular reaction without preincubation (ix) or to preincubation of only dNTPs and template (viii). Following preincubation, the remaining Mastermix components were added, and point of inflection times for early phase background amplification were determined via real-time fluorescence monitoring during a second incubation at 55 °C. Results represent average POI times for three experiments conducted on three different days, each in duplicate. Preincubation reaction mixture iii contained polymerase, EXPAR template, dGTP, and dCTP (an asterisk denotes no A and T).
The compiled results of this study demonstrate that preincubating the polymerase together with the template most significantly accelerates nonspecific background amplification. The fastest nonspecific amplification occurs when polymerase, template, and dNTPs are preincubated together (Figure 7, i). Slightly slower but still significantly accelerated background amplification is observed when the polymerase is preincubated with the amplification template, either in the absence of dNTPs (Figure 7, ii) or with only dGTP and dCTP [no A and T (Figure 7, iii)]. The polymerase was also found to accelerate nonspecific background amplification if preincubated with dNTPs in the absence of template (Figure 7, iv), but the level of background acceleration is significantly reduced compared to that after preincubation of polymerase and template, with or without dNTPs. Interestingly, preincubating the polymerase and template in the presence of the nicking enzyme (Figure 7, v) leads to significantly less background acceleration compared to preincubation of polymerase and template without nicking enzyme (Figure 7, ii). No statistically significant difference was observed when preincubation of polymerase, and dNTPs, in the presence (Figure 7, vi) or absence (Figure 7, iv) of nicking enzyme was compared. Finally, preincubation of the nicking enzyme and template (Figure 7, vii) does not accelerate the background amplification compared to the control (Figure 7, viii). The p values for pairwise comparison of data shown in Figure 7 are listed in Table S2 of the Supporting Information.
These preincubation studies indicate that interactions between the polymerase and EXPAR template play a role in the early phase nonspecific background amplification. To further investigate this point, we pursued manual hot start EXPAR, in which two separate solutions containing different components of the Mastermix are heated separately to 55 °C in a thermal block, followed by rapid addition of one solution to the other, transfer of the final Mastermix into a real-time thermocycler preheated to 55 °C, and monitoring of the fluorescence intensity as a function of time. We have observed significant suppression or elimination of early phase nonspecific background amplification when the polymerase was separated from the template or preferably from the template and dNTPs during the initial warmup period (Figure 8). As in the preincubation experiments, we investigated the effect of separating other Mastermix components such as nicking enzyme and template from each other during manual hot start. These other permutations did not result in significant background suppression or elimination, consistent with our preincubation studies.
Figure 8.

Manual hot start of EXPAR. The separation of the polymerase from the EXPAR template until the reaction temperature is reached results in a significantly reduced level of background amplification. Reaction mixtures contained starting trigger concentrations of (i) 1 pM, (ii) 1 fM, or (iii) 10 aM or (iv) no trigger (NTC).
DISCUSSION
The nonspecific background amplification observed in EXPAR includes an early and late phase. Our data for late phase background amplification are consistent with literature reports of ab initio DNA synthesis by thermophilic polymerases in the absence of any templating or priming DNA strands (18–22), which is accelerated in the presence of restriction endonucleases (23) or nicking endonucleases (24). Original reports of such primer/template-independent DNA synthesis (25, 26) were questioned due to the lack of highly purified enzyme preparation (18, 20, 27), and the potential for contaminating DNA or RNA causing initiation of the reaction. These concerns were addressed in later studies by use of highly purified polymerases and rigorous control experiments used to rule out potential contamination (18, 20). On the basis of similar control studies performed in our laboratory, it is unlikely that nonspecific EXPAR background amplification is caused by DNA or RNA contamination, although contamination with very low copy numbers of short oligonucleotides cannot be ruled out with absolute certainty, especially if these sequences are tightly bound to the enzyme. Likewise, it is unlikely that nonspecific background amplification is linked to primer–dimer type extension of the EXPAR template, since nonspecific background amplification is not accelerated significantly when using a template uncapped at the 3′-end. The phenomenon is further not restricted to the Bst large fragment polymerase used herein. We have observed similar early and late phase background amplification with other polymerases that function under the reaction conditions and in the buffer used for EXPAR, such as for Vent (exo−) and 9°N.
Late phase EXPAR background amplification is far less linked to the specific EXPAR template present in the reaction mixture than the early phase of background amplification. While early phase background amplification requires the presence of a “proper” EXPAR template of sequence X′rX′, the late phase is observed in the presence of EXPAR templates or random DNA sequences, and most importantly also in the absence of any templating or priming DNA, thus termed ab initio DNA synthesis. Although some template sequences have a stronger tendency to give rise to late phase nonspecific background amplification, late phase amplification products of random sizes are produced in a stochastic manner, with considerable run-to-run variability. Late phase nonspecific background amplification in most cases produces smears of dsDNA both shorter and longer than the 30mer EXPAR amplification template, based on analysis via gel electrophoresis, with similar products or product clusters formed in a manner independent of the template sequence used, based on analysis via LC–ESI MS. Cloning and sequencing of these amplification products revealed mainly poly-d(AT) sequences with occasional T/G and A/C substitution, and with interspersed quasi-palindromic sequences containing the nicking enzyme recognition sites.
Ab initio synthesis of poly-d(AT) sequences by thermophilic polymerases has been reported (18, 19) and is thought to involve two different reactions. In a rate-limiting initiation step, a short precursor oligonucleotide which putative sequence d(AT)n is synthesized (n equals at least 5 or 6, resulting in an at least 10–12mer sequence), followed by rapid elongation of this sequence into long poly-d(AT) repeats. Elongation of d(AT)n and similar repetitive sequence motifs has been studied extensively and is thought to occur through strand slippage (28, 29), hairpin–coil transitions (30, 31), or end fraying and strand displacement polymerization (32). The GC content of sequences generated by ab initio DNA synthesis was further found to be correlated to reaction temperature, with predominantly AT-containing sequences observed at lower reaction temperatures (22), as is the case for EXPAR, which is performed at 55 °C, due to the instability of the nicking enzyme above 60 °C.
If ab initio DNA synthesis is performed in the presence of restriction endonucleases, the products are found to contain the restriction site of the endonuclease included in the reaction mixture (23). In this so-termed RePol reaction, nonspecific DNA amplification is exponentially accelerated due to successive cycles of elongation and restriction into smaller seed fragments that then become elongated again. Accelerated ab initio DNA synthesis has also been observed with Bst polymerase and the nicking endonuclease Nt.BspD6I (24). Nt.BspD6I is an isoschizomer of the nicking endonuclease Nt.BstNBI used herein. DNA sequences generated from ab initio DNA synthesis using Bst polymerase and Nt.BspD6I were reported to contain tandem repeats of the GAGTCA motif, which includes the nicking enzyme recognition GAGTC sequence extended by an additional A (24), but not palindromic or quasi-palindromic sequences as in our studies and most other reports of ab initio DNA synthesis. This discrepancy requires further investigation and may be related to differences in the enzymes used and in the general reaction conditions. Our late phase background reactions were further conducted in the presence of an EXPAR template, while no DNA is present in true ab initio DNA synthesis.
Our sequencing results indicate that products of late phase EXPAR background amplification consist primarily of (AT)n- GAGAG(T/A)CTCTC(AT)n. If the central base is a T, then the GAGAGTCTCTC motif contains the nicking enzyme recognition site (GAGTC) and four-base spacer TCTC so that the nicking enzyme would cut after the last base of this 11mer sequence (Figure 9A). Nicking at adjacent motifs oriented head to head or tail to tail [Table 2, Ran24 sample (D)] would lead to strand dissociation, while single-strand nicks can lead to elongation through hairpin–coil transitions or strand displacement amplification. The symmetry of this motif is striking, as is the fact that it extends beyond the five-base nicking enzyme recognition site. The reverse complement is the same sequence containing an A as a central base and therefore contains the degenerate nicking enzyme GAGAT recognition sequence, which differs from the enzyme’s cognate GAGCT sequence by a single base. If the nicking endonuclease were to exhibit relaxed sequence specificity, known as star activity (33–35) which can be observed for restriction endonucleases under nonideal buffer conditions, then this 11mer sequence motif could also be cut on the bottom strand (Figure 9B).
Figure 9.
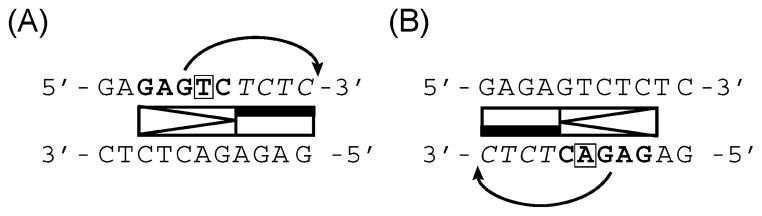
Schematic illustration of how the nicking enzyme recognizes and cuts the double-stranded quasi-palindromic 11mer GAGAG(T/A)CTCTC motif based on (A) regular activity involving the cognate Nt.BstNBI GAGTC recognition site on the top strand and (B) possible star activity involving the degenerate Nt.BstNBI recognition GAGAC site on the bottom strand. In each case, the nicking enzyme recognition site is shown in bold, and the four-base post recognition site spacers are shown in italic.
To test this hypothesis, we have hybridized oligonucleotides containing this 11mer motif and the trigger or trigger complement of StanA to each other and added the duplex to the EXPAR Mastermix containing template StanA but no trigger. The results indicate that the degenerate sequence is indeed recognized and cut by the nicking enzyme, albeit with slightly lower efficiency than the cognate sequence (Figure S4 of the Supporting Information). Other degenerate versions of only the five-base nicking enzyme recognition sequence not in the context of this 11mer motif, however, do not seem to be recognized and cut by the nicking enzyme (data not shown), indicating that the entire 11mer motif may have significance in enabling this reaction. Tandem repeats of this 11mer motif would be repeatedly cut by the nicking enzyme on the top and bottom strand, respectively, which could lead to amplification of the single-stranded GAGAGTCTCTC 11mer. An 11mer product is indeed formed during late phase background amplification, which can be observed in the LC–MS trace as peak 2, with base composition corresponding to GAGAGTCTCTC, especially in reactions that give rise to a low-molecular weight band via analysis by gel electrophoresis. Ran15 is one template prone to formation of such a low-molecular weight band during late phase background amplification. Not surprisingly, template Ran15 contains the GAGCGACTCTC sequence, which differs from the 11mer motif discussed above by only a single base, and therefore may promote formation of such amplification products. The GAGAG(T/A)CTCTC motif was, however, originally derived from late phase amplification products of templates Ran24 and SNP8, which are much less homologous to this 11mer sequence. Thus, we conclude that while certain EXPAR template sequences promote late phase background amplification, this process is overall relatively independent of the template. Ab initio DNA synthesis products generated using Bst and Nt.BspD6I were found to be branched (24), and formation of similar structures is possible in our case. This unconventional DNA synthesis further may involve alternative DNA structures (36, 37). The GAGAG(T/A)CTCTC motif contains a homopurine/homopyrimidine tandem repeat, which may be capable of forming a triple-helix H-DNA-like structure (36).
In contrast to previous reports of primer/template-independent DNA polymerization in the presence of thermophilic polymerases with or without restriction endonucleases, EXPAR also exhibits an early phase of nonspecific background amplification usually appearing in less than 10 min, which interferes with specific trigger generation and amplification. Early phase nonspecific background amplification is observed only in the presence of an EXPAR template of general structure X′rX′, where “r” stands for the nicking enzyme recognition site and four-base spacer. It generates the trigger sequence specific for the template present in the reaction mixture as the predominant product, as determined by MS analysis of reaction products formed from more than 30 different template sequences. Preincubating the polymerase with the template or with template and dNTPs significantly accelerates the first phase of background amplification. Less background acceleration is realized by preincubating the polymerase with dNTPs, but without the template. We therefore hypothesize that early phase nonspecific background amplification involves templated but unprimed DNA synthesis, wherein the polymerase binds to a single-stranded EXPAR template and synthesizes DNA complementary to this template in an unprimed fashion, or not primed in a conventional manner. The precise mechanism of the priming process is at present unclear. Generating even small numbers of short DNA sequences complementary to a template of the general structure X′rX′ can trigger EXPAR, resulting in exponential amplification of the trigger sequence X. An alternative explanation involves priming of the EXPAR template by short d(AT)n precursor oligonucleotide generated from ab initio poly-d(AT) synthesis. Early phase background amplification, however, also occurs efficiently for sequences with very high, almost exclusive GC content, such as Ran17, which would not be primed efficiently by a d(AT)n oligonucleotide. It is likely that such templated but unprimed DNA synthesis also takes place with low probability in the presence of other oligonucleotide sequences but is undetected due to the absence of exponential amplification through the unique EXPAR template structure and reaction design. It is possible that binding of the single-stranded EXPAR template to the polymerase is favored at low temperatures during the reaction setup, leading to a predisposition toward early phase nonspecific amplification. This hypothesis is supported by the observation that separating polymerase from template during the reaction setup through manual hot start causes reduction or elimination of early phase background amplification.
For the Bst polymerase used in our studies, it was reported that the rate of dATP and dTTP misincorporation was 5–50-fold greater than the rate of dCTP or dGTP misincorporation when reactions were carried out below the enzyme’s optimal reaction temperature of 65 °C, with a significant increase from 37 to 55 °C (38). This may explain the observed difference in the preincubation experiments with all dNTPs and with only dCTP and dGTP (Figure 7, i vs iii). We further observed that the nicking enzyme, when incubated with the polymerase and amplification template, slows the nonspecific amplification initiated by the polymerase and template. In other studies, we have observed that the nicking enzyme competitively inhibits EXPAR at high concentrations (data not shown). Restriction endonucleases are known to have a high affinity for their substrate (39), and the nicking enzyme is present in the reaction mixture at relatively high concentrations. It is therefore possible that binding of the nicking enzyme to the double-stranded recognition site inhibits or slows trigger amplification, either specific or nonspecific.
We have observed that further optimization of the EXPAR Mastermix composition leads to improved assay performance and significantly reduces the probability of observing late phase background amplification. Early phase background amplification is still observed in a reasonably consistent manner, but we have been able to delay its onset such that for suitable template sequences we are now able in more than 50% of our experiments to differentiate much lower trigger concentrations from background (10 aM, 180 copies of trigger X in 30 μL). The efficiency of early phase specific versus nonspecific amplification is further influenced by the particular trigger and template sequences present in the reaction mixture, a point currently under investigation.
A significant observation made in this study is the possibility of reducing or eliminating early phase nonspecific background amplification by separating the template from the polymerase until the final reaction temperature has been reached. In PCR, nonspecific amplification is often caused by low-stringency priming events during the reaction setup, during which primers, target, and polymerase are preincubated below the optimal annealing temperature for the reaction. Thermophilic polymerases are known to possess residual activity at low reaction temperatures. A significant breakthrough in mitigating such nonspecific amplification in PCR was the implementation of hot start strategies, which prevent DNA polymerization from occurring until the final reaction temperature has been reached (40–43). The two most widely used strategies for hot start PCR, i.e., reversible inactivation of DNA polymerase through chemical modification and complexation with antipolymerase antibodies, are unsuitable for EXPAR and related reactions, partially due to the requirement of reactivating the polymerase through preincubation at 95 °C. To obtain proof of principle hot start EXPAR, we have manually separated the polymerase from the template until the reaction temperature has been reached. Manual hot start is, however, difficult to implement for EXPAR monitored in real time, because of the cumbersome addition and manual transfer step. We are therefore exploring other methods of performing hot start EXPAR in a more consistent and user-friendly manner, with promising preliminary results. Further development of hot start strategies for EXPAR, along with optimization of the Mastermix composition, refinement of template design rules, and the application of these studies to facilitate rapid visual colorimetric detection of HSV 1 from clinical isolates, is ongoing.
In conclusion, EXPAR has the potential to facilitate nucleic acid testing particularly in point of care settings due to its isothermal nature and rapid amplification kinetics. EXPAR is comparable to other isothermal amplification reactions in terms of complexity. For example, EXPAR, TMA, SDA, and HDA all require two enzymes, NASBA requires three enzymes, RCA often requires a preligation step thus introducing a second enzyme, and LAMP involves one enzyme but four primers with six recognition sites. A potential disadvantage of EXPAR is the requirement for generation of a trigger from genomic target DNA or RNA. Trigger generation through the Fingerprinting reaction occurs in the same Mastermix and under the same reaction conditions as EXPAR and, therefore, does not increase the complexity of the reaction. Fingerprinting, however, is limited to sequences within genomic DNA that contain adjacent nicking enzyme recognition sites. We are therefore developing alternate probe-based trigger generation methods applicable to arbitrary DNA as well as RNA sequences. One of the main advantages of EXPAR, the speed and efficiency of isothermal amplification, also contributes to one of its main limitations, the observed nonspecific background amplification discussed here. Most nucleic acid amplification reactions exhibit some form of nonspecific background amplification, and it is likely that the templated but unprimed DNA synthesis herein considered to be the cause of early phase background amplification occurs as a low-probability event in other nucleic acid amplification reactions as well. This phenomenon is, however, particularly prominent in EXPAR due to the feedback design of the reaction. Using hot start and other strategies, we are optimistic that this background amplification can be sufficiently suppressed or eliminated to ensure robust and reproducible detection of low copy numbers of trigger oligonucleotides without compromising speed or simplicity.
The herein presented results further contribute to a better understanding of unconventional DNA polymerase activities. Our observations related to late phase background amplification are in line with numerous literature reports of ab initio DNA synthesis in the absence of any templating and priming DNA. In addition, to the best of our knowledge, the early phase background amplification discussed herein is the first detailed description of templated but unprimed DNA synthesis. Although untemplated and unprimed ab initio DNA synthesis has been reported numerous times in the literature, and although the elongation step is fairly well understood, how the reaction is initiated is so far unknown. Likewise, it is currently unclear how the templated but unprimed DNA synthesis observed during early phase EXPAR is initiated. In ongoing efforts, we are using EXPAR as a model system to further investigate this phenomenon.
Supplementary Material
Footnotes
Funding for this work was provided by the National Institutes of Health through NIAID Grant 064804, by the National Science Foundation through Grants ECCS 0501629 and EEC 0552962, by the Keck Graduate Institute, and by the ARUP Institute for Clinical and Experimental Pathology.
Abbreviations: EXPAR, exponential amplification reaction; PCR, polymerase chain reaction; NASBA, nucleic acid sequence-based amplification; TMA, transcription-mediated amplification; SDA, strand displacement amplification; LAMP, loop-mediated amplification reaction; RCA, rolling circle amplification; HDA, helicase-dependent amplification; Bst, Bacillus stearothermophilus; POI, point of inflection; LC–ESI MS, liquid chromatography–electrospray ionization mass spectrometry.
In a control experiment, we determined that SYBR II exhibits a 6.42 times higher fluorescence intensity when bound to double-stranded DNA (two complementary 30mer oligonucleotides, 100 nM each) as opposed to single-stranded DNA (a single stranded 30mer oligonucleotide, 200 nM) in EXPAR buffer. In addition, the relative fluorescence intensities for the single-stranded and double-stranded samples are comparable to the intensities observed in real-time EXPAR experiments (see Figure 2A).
SUPPORTING INFORMATION AVAILABLE
Complete sequences for all template and trigger oligonucleotides used in this report, additional real-time fluorescence and mass spectrometry data, and statistical analysis of preincubation studies. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Holland CA, Kiechle FL. Point-of-care molecular diagnostic systems: Past, present and future. Curr Opin Microbiol. 2005;8:504–509. doi: 10.1016/j.mib.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 2.Easley CJ, Karlinsey JM, Bienvenue JM, Legendre LA, Roper MG, Feldman SH, Hughes MA, Hewlett EL, Merkel TJ, Ferrance JP, Landers JP. A fully integrated microfluidic genetic analysis system with sample-in-answer- out capability. Proc Natl Acad Sci USA. 2006;103:19272–19277. doi: 10.1073/pnas.0604663103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yeung SW, Lee TMH, Cai H, Hsing IM. A DNA biochip for on-the-spot multiplexed pathogen identification. Nucleic Acids Res. 2006;34:118–124. doi: 10.1093/nar/gkl702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu RH, Yang JN, Lenigk R, Bonanno J, Grodzinski P. Self-contained, fully integrated biochip for sample preparation, polymerase chain reaction amplification, and DNA microarray detection. Anal Chem. 2004;76:1824–1831. doi: 10.1021/ac0353029. [DOI] [PubMed] [Google Scholar]
- 5.Compton J. Nucleic-Acid Sequence-Based Amplification. Nature. 1991;350:91–92. doi: 10.1038/350091a0. [DOI] [PubMed] [Google Scholar]
- 6.Hill CS. Molecular diagnostic testing for infectious diseases using TMA technology. Expert Rev Mol Diagn. 2001;1:445–455. doi: 10.1586/14737159.1.4.445. [DOI] [PubMed] [Google Scholar]
- 7.McCartney RA, Walker J, Scoular A. Detection of Chlamydia trachomatis in genitourinary medicine clinic attendees: Comparison of strand displacement amplification and the ligase chain reaction. Br J Biomed Sci. 2001;58:235–238. [PubMed] [Google Scholar]
- 8.Walker GT, Linn CP, Nadeau JG. DNA detection by strand displacement amplification and fluorescence polarization with signal enhancement using a DNA binding protein. Nucleic Acids Res. 1996;24:348–353. doi: 10.1093/nar/24.2.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000;28:e63. doi: 10.1093/nar/28.12.e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagamine K, Watanabe K, Ohtsuka K, Hase T, Notomi T. Loop-mediated isothermal amplification reaction using a nondenatured template. Clin Chem. 2001;47:1742–1743. [PubMed] [Google Scholar]
- 11.Demidov VV. Rolling-circle amplification in DNA diagnostics: The power of simplicity. Expert Rev Mol Diagn. 2002;2:542–548. doi: 10.1586/14737159.2.6.542. [DOI] [PubMed] [Google Scholar]
- 12.An LX, Tang W, Ranalli TA, Kim HJ, Wytiaz J, Kong HM. Characterization of a thermostable UvrD helicase and its participation in helicase-dependent amplification. J Biol Chem. 2005;280:28952–28958. doi: 10.1074/jbc.M503096200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vincent M, Xu Y, Kong HM. Helicase-dependent isothermal DNA amplification. EMBO Rep. 2004;5:795–800. doi: 10.1038/sj.embor.7400200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Ness J, Van Ness LK, Galas DJ. Isothermal reactions for the amplification of oligonucleotides. Proc Natl Acad Sci USA. 2003;100:4504–4509. doi: 10.1073/pnas.0730811100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tan E, Wong J, Nguyen D, Zhang Y, Erwin B, VanNess LK, Baker SM, Galas DJ, Niemz A. Isothermal DNA Amplification Coupled with DNA Nanosphere-Based Colorimetric Detection. Anal Chem. 2005;77:7984–7992. doi: 10.1021/ac051364i. [DOI] [PubMed] [Google Scholar]
- 16.Tan E, Erwin B, Dames S, Voelkerding KV, Niemz A. Isothermal DNA Amplification with Gold Nanosphere- Based Visual Colorimetric Readout for Herpes Simplex Virus Detection. Clin Chem. 2007;53:2017–2020. doi: 10.1373/clinchem.2007.091116. [DOI] [PubMed] [Google Scholar]
- 17.Dames S, Margraf RL, Pattison DC, Wittwer CT, Voelkerding KV. Characterization of Aberrant Melting Peaks in Unlabeled Probe Assays. J Mol Diagn. 2007;9:290–296. doi: 10.2353/jmoldx.2007.060139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanaki K, Odawara T, Muramatsu T, Kuchino Y, Masuda M, Yamamoto K, Nozaki C, Mizuno K, Yoshikura H. Primer/template-independent synthesis of poly d(A-T) by Taq polymerase. Biochem Biophys Res Commun. 1997;238:113–118. doi: 10.1006/bbrc.1997.7197. [DOI] [PubMed] [Google Scholar]
- 19.Hanaki K, Odawara T, Nakajima N, Shimizu YK, Nozaki C, Mizuno K, Muramatsu T, Kuchino Y, Yoshikura H. Two different reactions involved in the primer/template-independent polymerization of dATP and dTTP by Taq DNA polymerase. Biochem Biophys Res Commun. 1998;244:210–219. doi: 10.1006/bbrc.1998.8237. [DOI] [PubMed] [Google Scholar]
- 20.Ogata N, Miura T. Genetic information ‘created’ by archaebacterial DNA polymerase. Biochem J. 1997;324:667–671. doi: 10.1042/bj3240667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ogata N, Miura T. Creation of genetic information by DNA polymerase of the thermophilic bacterium Thermus thermophilus. Nucleic Acids Res. 1998;26:4657–4661. doi: 10.1093/nar/26.20.4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ogata N, Miura T. Creation of genetic information by DNA polymerase of the archaeon Thermococcus litoralis: Influences of temperature and ionic strength. Nucleic Acids Res. 1998;26:4652–4656. doi: 10.1093/nar/26.20.4652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liang XG, Jensen K, Frank-Kamenetskii MD. Very efficient template/primer-independent DNA synthesis by thermophilic DNA polymerase in the presence of a thermophilic restriction endonuclease. Biochemistry. 2004;43:13459–13466. doi: 10.1021/bi0489614. [DOI] [PubMed] [Google Scholar]
- 24.Zyrina NV, Zheleznaya LA, Dvoretsky EV, Vasiliev VD, Chernov A, Matvienko NI. N.BspD6I DNA nickase strongly stimulates template-independent synthesis of non-palindromic repetitive DNA by Bst DNA polymerase. Biol Chem. 2007;388:367–372. doi: 10.1515/BC.2007.043. [DOI] [PubMed] [Google Scholar]
- 25.Schachman HK, Adler J, Radding CM, Lehman IR, Kornberg A. Enzymatic Synthesis of Deoxyribonucleic Acid: VII Synthesis of a Polymer of Deoxyadenylate and Deoxythymidylate. J Biol Chem. 1960;235:3242–3249. [PubMed] [Google Scholar]
- 26.Okazaki T, Kornberg A. Enzymatic Synthesis of Deoxyribonucleic Acid: XV. Purification and Properties of a Polymerase from Bacillus subtilis. J Biol Chem. 1964;239:259–268. [PubMed] [Google Scholar]
- 27.Nazarenko IA, Bobko LE, Romashchenko AG, Khripin YL, Salganik RI. Study on the Unprimed Poly (dA-dT) Synthesis Catalyzed by Preparations of Escherichia coli DNA Polymerase I. Nucleic Acids Res. 1979;6:2545–2560. doi: 10.1093/nar/6.7.2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elson E. Kinetics of Synthesis of Polymeric dAT on Oligomeric Templates. Biopolymers. 1968;6:269–283. doi: 10.1002/bip.1968.360060302. [DOI] [PubMed] [Google Scholar]
- 29.Schlotterer C, Tautz D. Slippage Synthesis of Simple Sequence DNA. Nucleic Acids Res. 1992;20:211–215. doi: 10.1093/nar/20.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ogata N, Miura T. Elongation of tandem repetitive DNA by the DNA polymerase of the hyperthermophilic archaeon Thermococcus litoralis at a hairpin-coil transitional state: A model of amplification of a primordial simple DNA sequence. Biochemistry. 2000;39:13993–14001. doi: 10.1021/bi0013243. [DOI] [PubMed] [Google Scholar]
- 31.Ogata N, Morino H. Elongation of repetitive DNA by DNA polymerase from a hyperthermophilic bacterium Thermos thermophilus. Nucleic Acids Res. 2000;28:3999–4004. doi: 10.1093/nar/28.20.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tuntiwechapikul W, Salazar M. Mechanism of in vitro expansion of long DNA repeats: Effect of temperature, repeat length, repeat sequence, and DNA polymerases. Biochemistry. 2002;41:854–860. doi: 10.1021/bi0110950. [DOI] [PubMed] [Google Scholar]
- 33.Robinson CR, Sligar SG. Changes in solvation during DNA binding and cleavage are critical to altered specificity of the EcoRI endonuclease. Proc Natl Acad Sci USA. 1998;95:2186–2191. doi: 10.1073/pnas.95.5.2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nasri M, Thomas D. Alteration of the Specificity of PvuII Restriction Endonuclease. Nucleic Acids Res. 1987;15:7677–7687. doi: 10.1093/nar/15.19.7677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Polisky B, Greene P, Garfin DE, Mccarthy BJ, Goodman HM, Boyer HW. Specificity of Substrate Recognition by EcoRI Restriction Endonuclease. Proc Natl Acad Sci USA. 1975;72:3310–3314. doi: 10.1073/pnas.72.9.3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mirkin SM, Frank-Kamenetskii MD. H-Dna and Related Structures. Annu Rev Biophys Biomol Struct. 1994;23:541–576. doi: 10.1146/annurev.bb.23.060194.002545. [DOI] [PubMed] [Google Scholar]
- 37.Sinden RR, Potaman VN, Oussatcheva EA, Pearson CE, Lyubchenko YL, Shlyakhtenko LS. Triplet repeat DNA structures and human genetic disease: Dynamic mutations from dynamic DNA. J Biosci. 2002;27:53–65. doi: 10.1007/BF02703683. [DOI] [PubMed] [Google Scholar]
- 38.Stenesh J, McGowan GR. DNA polymerase from mesophilic and thermophilic bacteria: III. Lack of fidelity in the replication of synthetic polydeoxyribonucleotides by DNA polymerase from Bacillus licheniformis and Bacillus stearothermophilus. Biochim Biophys Acta. 1977;475:32–41. doi: 10.1016/0005-2787(77)90336-7. [DOI] [PubMed] [Google Scholar]
- 39.Hinsch B, Kula MR. Reaction-Kinetics of Some Important Site-Specific Endonucleases. Nucleic Acids Res. 1981;9:3159–3174. doi: 10.1093/nar/9.13.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chou Q, Russell M, Birch DE, Raymond J, Bloch W. Prevention of Pre-PCR Mis-Priming and Primer Dimerization Improves Low-Copy-Number Amplifications. Nucleic Acids Res. 1992;20:1717–1723. doi: 10.1093/nar/20.7.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hebert B, Bergeron J, Potworowski EF, Tijssen P. Increased PCR Sensitivity by Using Paraffin Wax As A Reaction Mix Overlay. Mol Cell Probes. 1993;7:249–252. doi: 10.1006/mcpr.1993.1036. [DOI] [PubMed] [Google Scholar]
- 42.Moretti T, Koons B, Budowle B. Enhancement of PCR amplification yield and specificity using AmpliTaq Gold (TM) DNA polymerase. BioTechniques. 1998;25:716–722. [PubMed] [Google Scholar]
- 43.Sharkey DJ, Scalice ER, Christy KG, Atwood SM, Daiss JL. Antibodies As Thermolabile Switches: High- Temperature Triggering for the Polymerase Chain-Reaction. Bio/Technology. 1994;12:506–509. doi: 10.1038/nbt0594-506. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.