

Abstract
Kallikrein 6 (Klk6) is a secreted serine protease preferentially expressed by oligodendroglia in CNS white matter. Elevated levels of Klk6 occur in actively demyelinating multiple sclerosis (MS) lesions and in cases of spinal cord injury (SCI), stroke and glioblastoma. Taken with recent evidence establishing Klk6 as a CNS-endogenous activator of protease-activated receptors (PARs), we hypothesized that Klk6 activates a subset of PARs to regulate oligodendrocyte physiology and potentially pathophysiology. Here, primary oligodendrocyte cultures derived from wild type or PAR1-deficient mice and the murine oligodendrocyte cell line, Oli-neu, were used to demonstrate that Klk6 mediates loss of oligodendrocyte processes and impedes morphological differentiation of oligodendrocyte progenitor cells (OPCs) in a PAR1-dependent fashion. Comparable gliopathy was also elicited by the canonical PAR1 agonist, thrombin, as well as PAR1-activating peptides (PAR1-APs). Klk6 also exacerbated ATP-mediated oligodendrogliopathy in vitro, pointing to a potential role in augmenting excitotoxicity. In addition, Klk6 suppressed the expression of proteolipid protein (PLP) RNA in cultured oligodendrocytes by a mechanism involving PAR1-mediated Erk1/2 signaling. Microinjection of PAR1 agonists, including Klk6 or PAR1-APs, into the dorsal column white matter of PAR+/+ but not PAR−/− mice promoted vacuolating myelopathy and a loss of immunoreactivity for myelin basic protein (MBP) and CC-1+ oligodendrocytes. These results demonstrate a functional role for Klk6-PAR1 signaling in oligodendroglial pathophysiology and suggest that PAR1 or PAR1-agonists may represent new targets to moderate demyelination and to promote myelin regeneration in cases of CNS white matter injury or disease.
Keywords: Thrombin, Protease Activated Receptor, Oligodendrocyte, Spinal Cord, Demyelination, Myelin
Introduction
Understanding the full complement of factors regulating oligodendrocyte dysfunction and demyelination is essential to the development of therapies to promote myelin regeneration. KLK6 is a secreted serine protease up regulated at sites of demyelination in MS and SCI and mediates oligodendrocyte process retraction at nanomolar concentrations (Blaber et al. 2004; Scarisbrick et al. 2002; Scarisbrick et al. 2006b). Despite the abundant expression of Klk6 by oligodendroglia (Scarisbrick et al. 2000; Scarisbrick et al. 1997) its functional role and mechanism of action in oligodendrocyte physiology and pathophysiology have not been established.
Tissue kallikreins comprise a family of 15 genes encoding for secreted serine proteases (biochemically ‘proteinase’, colloquially ‘protease’) with trypsin- or chymotrypsin-like activity (Clements et al. 2001; Yousef and Diamandis 2001). KLK6 is a more recently identified family member preferentially expressed in the CNS (Scarisbrick et al. 2006a; Scarisbrick et al. 2001; Scarisbrick et al. 1997). Klk6 readily hydrolyzes myelin proteins, including MBP and myelin oligodendrocyte glycoprotein (MOG), as well as components of the extracellular matrix, including laminin, fibronectin and aggrecan (Bernett et al. 2002; Blaber et al. 2002; Scarisbrick et al. 2006b) and is therefore positioned to play key roles in injury and disease affecting myelination.
Select serine proteases, including thrombin and trypsin, elicit hormone-like actions by activation of G-protein coupled receptors known as PARs (Nystedt et al. 1995; Vu et al. 1991). Recent studies indicate kallikreins also activate select PARs (Angelo et al. 2006; Oikonomopoulou et al. 2006a; Scarisbrick et al. 2011; Scarisbrick et al. 2012a; Vandell et al. 2008). PAR signaling is induced by proteolytic cleavage in the extracellular N-terminus revealing a tethered ligand that binds intramolecularly (Adams et al. 2011). Four PARs have been identified (PAR1-4) with PAR1, -2 and -4 activated by cleavage C-terminal to arginine (Ossovskaya 2004), the precise cleavage specificity of Klk6 (Bernett et al. 2002; Blaber et al. 2002). Klk6 activates PAR1 on neurons and both PAR1 and PAR2 on astrocytes to elicit Ca2+ and Erk1/2 signaling (Scarisbrick et al. 2012a; Vandell et al. 2008). PAR1 activation has been linked to astrocyte reactivity (Nicole et al. 2005; Scarisbrick et al. 2012a) and thrombin-induced neurotoxicity (Striggow et al. 2000). PAR1 is already known to be expressed by oligodendroglia (Wang et al. 2004b) and here we examine its potential role in oligodendrocyte pathophysiology.
In this study we specifically test the hypothesis that Klk6 regulates key aspects of oligodendrocyte biology and pathophysiology by activation of PARs. We demonstrate that Klk6-signaling at PAR1 plays a regulatory role in OPC process extension, in the maintenance of processes by mature oligodendrocytes and in the expression of both PLP and MBP RNA. Importantly, we demonstrate that other PAR1 agonists, namely thrombin and PAR1-AP mediate parallel oligodendrogliopathic effects. Moreover, we establish that Klk6- or PAR1-AP mediated PAR1 activation alone is sufficient to promote spinal cord myelinopathy, oligodendrogliopathy and MBP loss in vivo. Taken together, these data suggest that PAR1-agonists may serve as key regulators of oligodendrocyte differentiation, including process outgrowth and myelin gene expression and therefore that PAR1 or its agonists, such as Klk6, are important new targets for the development of therapies to treat or prevent demyelination in cases of CNS injury or disease.
Materials and Methods
Animal care and use
Eight- to ten-week-old C57BL6/J mice were obtained from Jackson Laboratories. Mice deficient in PAR1 (PAR1−/−, B6.129S4-F2rtm1Ajc/J) or PAR2 (PAR2−/−, B6.Cg-F2rl1(tm1Mslb)/J) were also obtained from Jackson and backcrossed to C57BL6/J for at least 20 generations, such that PAR1+/+ littermates served as controls in each experiment. All animal experiments were carried out with strict adherence to NIH Guidelines for animal care and safety and were approved by the Mayo Clinic Institutional Animal Care and Use Committee.
Oligodendrocyte cultures
Purified primary cortical oligodendrocyte progenitor cells (OPCs) and differentiated oligodendrocytes were isolated from mixed glial cultures derived from postnatal day 1 mice as previously described (McCarthy and de Vellis 1980). Glial cultures were grown in media containing DMEM, 2 mM Glutamax, 1 mM sodium pyruvate, 20 mM HEPES and 10% heat-inactivated fetal calf serum (Atlanta Biologicals, Lawrenceville, GA). OPCs were isolated from 10 days-in-vitro (DIV) mixed glial cultures using mechanical dissociation and purified by differential adhesion (Back et al. 1998). OPCs were seeded in defined Neurobasal A media containing 1% N2, 50 U/mL penicillin/streptomycin, 2 mM Glutamax, 1 mM sodium pyruvate, 0.45% glucose (Sigma Aldrich, USA) and 50 μM β-mercaptoethanol (Sigma Aldrich, USA). OPCs were seeded at 3×104/cm2 onto tissue culture treated plastic or 12 mm glass cover slips coated with poly-L-lysine (PLL, 10 μg/mL). At 24 h post-purification cultures contained 92–98% oligodendrocyte lineage cells as determined by sulfatide (O4) immunoreactivity (IR) and 4′,6-diamidino-2-phenylindole (DAPI). After 3 DIV, at least 80% of O4+ cells were also MBP+.
Oli-neu oligodendrocytes are derived from mouse primary oligodendrocyte cultures retrovirally transduced to constitutively express the t-neu oncogene and shown to express galatocerebroside (O1) and sulfatide (O4) (Jung et al. 1995). All morphology, signaling and myelin gene expression studies using Oli-neu oligodendrocytes were carried using cells cultured in media containing DMEM, 1% N2, 2 mM Glutamax, 1 mM sodium pyruvate, 20 mM HEPES buffer and 50 μM β-mercaptoethanol (Sigma Aldrich, USA). To evaluate PAR and Klk6 gene expression in Oli-neu (Table 2), cells were differentiated by treatment with 1 mM N6, 2′-O-dibutyryladenosine 3′, 5′-cyclic monophosphate disodium salt (dbcAMP) for 72 h prior to harvesting cells for RNA isolation. All cells were maintained at 37°C in 95% air and 5% CO2. Cell culture reagents were obtained from Life Technologies (Carlsbad, CA) unless otherwise indicated. All cell culture experiments were performed in triplicate and repeated at least twice.
Table 2. Quantitative PCR analysis demonstrates robust expression of Klk6, PAR1 and PAR2 by murine primary and Oli-neu oligodendrocytes.
The mean number of RNA copies encoding for Klk6, PAR1 and PAR2 RNA in 0.5 μg of total RNA isolated from immediately post-purification OPCs, mature oligodendrocytes (3DIV), undifferentiated (Oli-neu) or differentiated Oli-neu (Oli-neu + dbcAMP) oligodendrocytes is provided (± SEM). Amplification of GAPDH was carried out to verify equal loading.
| RNA Copy Number | ||||
|---|---|---|---|---|
|
| ||||
| Gene | Primary Oligodendrocyte Progenitor Cell | Primary Differentiated Oligodendrocyte | Oli-neu | Oli-neu + dbcAMP |
|
| ||||
| Klk6 | 6.0E+03 (± 2.2E+02) | 5.6E+03 (± 8.7E+01) | 6.4E+04 (± 2.2E+04) | 2.0E+04 (± 4.0E+03) |
| PAR1 | 1.1E+06 (± 5.7E+04) | 8.6E+05 (± 3.6E+04) | 1.5E+06 (± 2.2E+05) | 1.3E+06 (± 3.3E+05) |
| PAR2 | 2.5E+02 (± 2.3E+01) | 2.9E+02 (± 2.1E+01) | 3.1E+03 (± 1.6E+02) | 3.5E+03 (± 2.3E+01) |
| GAPDH | 6.0E+06 (± 1.3E+05) | 6.5E+06 (± 2.9E+05) | 2.4E+07 (± 6.7E+05) | 1.8E+07 (± 2.9E+05) |
Recombinant kallikreins and PAR agonists
Recombinant murine Klk6 and Klk1 were expressed using a baculovirus system and purified as described previously in detail (Blaber et al. 2002; Scarisbrick et al. 2011; Scarisbrick et al. 2012a). Concentrations of Klk6 used in these studies included 30 to 300 nM (1 to 10 μg/mL; 15,915 to 159,150 Units) and were based on previous work demonstrating concentrations of Klk6 that are sufficient to elicit Ca2+ signaling and activation of ERK in neural cells (Vandell et al. 2008). Klk1 was used at a comparable concentration (300nM (10 μg/mL; 173,000 Units). Levels of Klk6 are in the range of those reported to be present in human cerebrospinal fluid (40nM) (Zarghooni et al. 2002). An equivalent concentration of Human α-thrombin (270nM (10 μg/mL; 161,000 Units), Enzyme Research Laboratories, South Bend IN) was also examined. The specific activity of 1 ng of Klk6, Klk1 or thrombin was measured by analysis of the rate of hydrolysis against 100 μM t-Butyloxycarbonyl-Valine-Proline-Arginig-7-Amino-4-methylcoumarin (Boc-VP-AMC) fluorogenic peptide substrate (R&D Systems, Minneapolis, MN)(Michael et al. 2005; Oikonomopoulou et al. 2006b). PAR1-activating peptide (PAR1-AP) (TFLLR-amide, Peptides International, USA), that mimics the PAR1 tethered ligand, was used at 100 μM (100 μg/ml), a concentration known to elicit PAR1 signaling in CNS neurons and glia (Hollenberg et al. 1997; Maggio et al. 2008; Shirakawa et al. 2010; Vellani et al. 2010).
Expression of oligodendrocyte PARs and Klk6
Immunocytochemistry for PARs and Klk6
Oligodendrocyte cultures seeded on glass cover slips were immunostained with the following primary antibodies: rabbit anti-Klk6 (Blaber et al. 2004; Scarisbrick et al. 2012b), goat anti-PAR1 (C-18) or -PAR2 (C-17) (Santa Cruz Biotechnology Inc., Santa Cruz, CA) and mouse anti-sulfatide (O4) (gift from Dr. Ben Barres, Stanford University). In brief, immunostaining involved fixation of cultures with 2% paraformaldehyde (PFA) for 30 min prior to incubation with primary antibody, with the exception of O4 immunostaining, which was accomplished using live cells at 4°C using a HEPES-buffered DMEM solution containing the mouse hybridoma-derived O4 antibody, followed by fixation with 2% PFA. Cells were then incubated with affinity-purified, species-appropriate fluorochrome-conjugated secondary antibodies (1:100, Jackson Immunoresearch Laboratories, Westgrove, PA) and mounted using VECTASHIELD hardset with DAPI (Vector Laboratories, Burlingame, CA).
Real-time Quantitative PCR
Total RNA was isolated from cultured cells using RNA STAT-60 (Tel-Test Inc., Friendswood, TX). Klk6, PAR1 or PAR2 expression were determined in 0.5 μg of RNA with the iScript one-step RT-PCR kit with SYBR® Green and the iCycler iQ5 system (BioRad, Hercules, CA). Transcript copy number was determined using a standard curve prepared by parallel amplification of cDNA clones diluted to a known copy number as previously described (Christophi et al. 2004). Primers used for amplification of target gene transcripts are provided in Table1. Amplification of the housekeeping gene glyceraldehyde phosphate 3-dehydrogenase (GAPDH) was used to control for loading. The mean and standard error (SEM) of transcript copy number was determined and data expressed as RNA copy number on a logarithmic scale.
Table 1. Primers used for Quantitative PCR of murine protease activated receptors, Klk6 and myelin genes.
Glyceraldehyde phosphate 3-dehydrogenase (GAPDH); kallikrein-related peptidase 6 (Klk6); protease-activated receptor 1 (PAR1); protease-activated receptor 2 (PAR2); myelin basic protein (MBP); proteolipid protein (PLP).
| Gene | Entrez Accession number | Primer Sequence (F/R) |
|---|---|---|
|
| ||
| GAPDH | NM_008084.2 | ACCACCATGGAGAAGGC/GGCATGGACTGTGGTCATGA |
| KLK6 | NM_011177.2 | CCTACCCTGGCAAGATCAC/GGATCCATCTGATATGAGTGC |
| PAR1 | NM_010169.3 | CTTGCTGATCGTCGCCC/TTCACCGTAGCATCTGTCCT |
| PAR2 | NM_007974.4 | CCGGACCGAGAACCTTG/CGGAAGAAAGACAGTGGTCAG |
| MBP | NM_001025251 | CCAGTAGTCCATTTCTTCAAGAACAT/GCCGATTTATAGTCGGAAGCTC |
| PLP | NM_011123.2 | TCTTTGGCGACTACAAGACCAC/CACAAACTTGTCGGGATGTCCTA |
PAR agonist-induced changes in oligodendrocyte morphology
To determine the effects of recombinant Klk6 and other PAR agonists on cells of the oligodendrocyte lineage, 3 DIV PAR1+/+, PAR1−/− and PAR2−/− oligodendrocytes, PAR1+/+ OPCs immediately post-purification, or Oli-neu cells were grown on cover slips in the presence of recombinant protease (30–300 nM) for a period of 24 h. PAR-1 agonists studied included human α-thrombin (270 nM) and PAR1-AP (100 μM). For ATP toxicity assays, oligodendrocytes were incubated with ATP (50 μM, Sigma Aldrich, USA) in the presence of recombinant Klk6 or Klk1. Following each treatment, oligodendrocyte and OPC cultures were immunolabeled for O4 to visualize both the cell body and processes. Oli-neu processes were visualized by staining the actin cytoskeleton with Cy3-conjugated Phalloidin (Life Technologies, USA).
To assess the effects of Klk6 and other PAR1 agonists on oligodendrocyte morphology and process stability, 20X digital micrographs were overlaid with a 780 μm2 grid and ImageJ software 1.45r (National Institutes of Health, USA) used to record cell number and the number of processes crossing horizontal grid lines (Scarisbrick et al. 2002). Data are expressed as the mean number of O4+ or Phalloidin+ processes per DAPI+ cell (± SEM).
The extent of oligodendrocyte morphological differentiation was determined by scoring O4+/DAPI+ oligodendrocyte morphology as simple (only primary processes, no secondary branching), incomplete (one or more primary process without secondary branching), complete (all primary processes with secondary branching) or membrane (complete secondary branching with membrane sheets) (Adapted from Huang and colleagues (Huang et al. 2011). The mean number of cells in each morphology class was determined for each treatment group and expressed as percent of total O4+/DAPI+ cells.
Changes in OPC or oligodendrocyte morphology were quantified from five microscopic fields per cover slip with the mean and SEM calculated across three cover slips per experiment. Each experiment was repeated at least twice using independent cultures. Analysis of oligodendrocyte morphology was performed without knowledge of treatment groups.
Regulation of oligodendrocyte myelin gene expression by PAR1 agonists
Primary murine oligodendrocytes (3 DIV) or Oli-neu oligodendrocytes were seeded at 3.5×105 cells/well in 6-well tissue culture treated plates. Cells were then treated with recombinant Klk6 (30–300 nM), Klk1 (300 nM), thrombin (270 nM) or PAR1-AP (100 μM) for 24 h prior to RNA isolation. Expression levels of myelin-associated genes, PLP and MBP were then analyzed using real-time quantitative RT-PCR. Expression levels were obtained from triplicate treatment wells from at least two independent cultures. Expression levels of target genes were normalized to GAPDH and expressed as percent control.
Klk6-PAR Signaling Assays
To examine the ability of Klk6 to mediate PAR-dependent signaling in oligodendrocytes, Oli-neu oligodendrocytes were seeded at 3.5×105 cells/well in 6-well tissue culture treated vessels and stimulated with active recombinant Klk6. Cells were stimulated with Klk6 (150 nM) for 10 min then harvested for protein isolation. Cell lysates were analyzed for phosphorylated and non-phosphorylated Erk1/2 by Western blot. In experiments to determine the role of PARs in Klk6 signaling, Oli-neu were pre-incubated with the PAR-1 antagonist, SCH79797 dihydrochloride (50 nM, Tocris Biosciences, Minneapolis, MN USA) for 30 min prior to the addition of Klk6.
Western Blot
Oli-neu lysates were obtained using a lysis buffer containing 1% NP40, 0.5% deoxycholate, 10% glycerol and 20 mM Tris base and separated on SDS-polyacrylamide gels prior to transfer to nitrocellulose membranes. Membranes were blocked with 5% milk in TBS-T and incubated overnight at 4°C with primary antibodies including rabbit anti-(phospho-) extracellular signal-regulated kinases (Erk1/2) (1:1000, Cell Signaling Technology Inc., Danvers, MA), followed with a species-appropriate horseradish peroxidase-conjugated secondary antibody (1:20,000 GE Healthcare Unlimited, UK). Signal was detected using Chemiluminescence Supersignal Pico (Pierce, Rockford, IL). Western blots were repeated three times from independent cultures, scanned and quantified by densitometry using BioRad Quantity One 1-D Analysis Software (BioRad, Hercules, CA). Phosphorylated protein signal was normalized to the non-phosphorylated form of each protein. Equal loading was verified by re-probing blots for β-Actin (Novus Biologicals, Littleton, CO).
In vivo effects of excess Klkl6 or PAR1-AP in spinal cord white matter
All mice receiving a surgical procedure were administered pre-operative intraperitoneal (IP) injection of Buprenorphine (Buprenex, 0.03 mg/kg, Reckitt Benckise, Slough, UK) as an analgesic. Surgical plane anesthesia was induced in age-matched eight- to ten-week-old, male C57BL6/J or PAR1 deficient mice, using IP injection of Ketamine (Ketaset, 80 mg/kg, Fort Dodge Animal Health, Fort Dodge, IA) and Xylazine (Anased, 10 mg/kg, Llyod Laboratories, Shenendoah, IA). A thoracic (T11-T12) laminectomy was then performed under aseptic conditions. A 30–40 μm glass capillary microinjection needle backfilled with the experimental solution was inserted into a 10 μL gas-tight Hamilton Syringe (Hamilton Company, Reno, NV), and mounted to a stereotaxic injection system (Stoelting, Inc., Wood Dale, IL) with stereotaxic stage and digital manipulator (MyNeurolab, Rochmond, IL). Under micromanipulator control, the needle was inserted 350 μm into the dorsal column white matter and infusion of 2 μL was carried out over 5 min for each agonist including Klk6 (0.01 μg/μL (300 nM), PAR1-AP (0.01 μg/μL, (100 μM)) or vehicle (physiologic saline) alone (n = 3 for each treatment group). The needle was left in place for 3 min after the injection to prevent backflow, and the wound closed. All mice received 0.5 mL of sterile saline and Baytril (2.5 mg/kg, Bayer Healthcare, Shawnee Mission, KS) IP post-operatively. Buprenorphine (0.03 mg/kg) was given every 12 h for the first 48 h following surgery.
Seventy two h after microinjection of PAR1-agonists, mice were deeply anesthetized with Nembutal (50 mg/kg, IP, Lundbeck Inc. Deerfield, IL) and transcardially perfused with 4% PFA. 2 mm transverse segments of the spinal cord encompassing the microinjection epicenter as well as 2 mm rostral and caudal to the site of injection were embedded in paraffin and 6 μm sections cut and mounted serially on to Superfrost slides (Fisher Scientific, Pittsburgh, PA). Every sixth slide was stained with hematoxylin and eosin (H&E) to identify areas with tissue destruction, vacuolation and/or hemorrhage. For immunohistochemical staining, sections were deparaffinized and endogenous peroxidase quenched with 0.3% hydrogen peroxide in methanol. Oligodendrocytes were visualized using an antibody specific for CC-1/APC (ab16794, AbCam, Cambridge, MA)(Bhat et al. 1996), myelin was visualized with an antibody recognizing MBP (MAB386, Millipore, Bedford, MA) and standard avidin-biotin immunoperoxidase techniques as we have previously detailed (Scarisbrick et al. 2001; Scarisbrick et al. 2012b). Stained spinal cord sections were cover slipped with Permount (Fisher Scientific, Pittsburgh, PA) containing 2 μg/mL bisbenzamide to visualize nuclei (Sigma Aldrich, USA).
Measurements of white matter lesion area were based on signs of pathology (vacuolation, tissue destruction, hemorrhage) seen in H&E stained sections. Area measurements of dorsal column pathology were made from 20X digital images of H&E stained sections. The largest lesion area in each animal was used to determine mean maximal lesion area for each experimental condition and expressed in μm2. The integrity of MBP-immunoreactivity was assessed in sections for 2000 μm rostrocaudal surrounding the microinjection epicenter. The number of dorsal column CC-1+/DAPI+ cells was quantified in representative sections across 300 μm of spinal cord extending rostrocaudally to the epicenter in each case (Image J). The average area of dorsal column white matter of an intact mouse spinal cord was determined to be approximately 1.5 × 105 μm2. To put the number of CC-1+ oligodendrocytes into context therefore, the mean number CC-1+ cells per μm2 was evaluated as the mean number of per 1 × 105 μm2. Sections stained for CC-1 were also stained for GFAP (Sigma Aldrich, USA), allowing for the exclusion of CC-1+ astrocytes, however no examples of double labeled cells were observed.
Statistical analysis
Quantitative analysis of cell morphology, cell counts, qPCR RNA expression data, and histological measurements are presented as mean ± SEM. The Student’s t-test was used to determine the significance of differences between two treatment groups and the Mann-Whitney U test was used when data were not normally distributed. When carrying out comparisons between multiple groups, a One-Way Analysis of Variance (ANOVA) was used, followed by Student-Newman-Keuls (SNK) post-hoc analysis. When multiple comparison data was found to be not normally distributed, the Kruskal-Wallis ANOVA on Ranks was used with Dunn’s method for pairwise comparisons. Statistical significance was set at p < 0.05.
Results
Expression of Klk6, PAR1 and PAR2 in oligodendroglia and OPCs
Kallikrein 6 is known to be highly expressed by oligodendroglia of the rodent spinal cord, in vivo and in vitro (Scarisbrick et al. 2000; Scarisbrick et al. 1997). Here, Klk6 was shown to be densely expressed throughout the cytoplasm and processes of purified, O4+ murine primary oligodendrocytes (3 DIV) (Fig. 1). O4+ oligodendrocytes were also immunoreactive for PAR1 and PAR2. PAR1-immunorectivity was observed throughout the cell body and process network. PAR2-immunoreactivity was also dense in the cell body, but showed more limited staining in processes being most dense at nodule branch points (Fig. 1). A parallel pattern of Klk6, PAR1 and PAR2-immunoreactivity was observed in the Oli-neu oligodendrocyte cell line (data not shown).
Figure 1. Cellular localization of Klk6, PAR1 and PAR2 in primary oligodendrocytes.

Immunocytochemical labeling for Klk6, PAR1 and PAR2 in O4+ mouse oligodendrocyte cultures isolated from PAR1+/+ postnatal C57/BL6 mice (3 DIV). Abundant Klk6 immunoreactivity was observed in the cell soma and in some O4+ processes. PAR1 in the cell soma and throughout the oligodendrocyte process network. By contrast, PAR2 was localized primarily to process nodules. Scale bar = 25μm.
Quantitative real-time PCR was used to determine RNA expression levels of Klk6, PAR1, and PAR2 in primary cultures of OPCs and differentiated oligodendrocytes (3 DIV) (Table 2). Equivalent levels of Klk6 RNA were observed at both stages of oligodendrocyte differentiation. High levels of PAR1 RNA were detected in both OPC and oligodendrocyte cultures, though expression significantly declined with differentiation (Student’s t-test, p = 0.030). PAR2 RNA levels were nearly 4-log values lower than PAR1 and expressed by OPCs and oligodendrocytes at equivalent levels. Klk6, PAR1 and PAR2 RNA levels were also determined for the Oli-neu cell line, under resting and dbcAMP-differentiated conditions (Jung et al. 1995) (Table 2). Levels of Klk6 RNA were similar for resting and differentiated Oli-neu oligodendrocytes. PAR1 RNA expression was approximately 3-log values greater than PAR2 in Oli-neu oligodendrocytes, and each was expressed at equivalent levels in resting and differentiated culture conditions.
Klk6-mediated injury to mature oligodendroglial processes is a PAR1-dependent
In human cerebrospinal fluid (CSF), the concentration of KLK6 is approximately 40 nM, which is likely representative of that in the CNS (Zarghooni et al. 2002). However, Klk6 is elevated at sites of CNS injury due to expression by infiltrating immune cells and up regulation by resident CNS cells (Angelo et al. 2006; Blaber et al. 2002; Scarisbrick et al. 2002; Scarisbrick et al. 2012a; Scarisbrick et al. 2006b). We previously demonstrated that treatment of rat primary oligodendrocytes with 30 or 300 nM recombinant Klk6 for 24 h resulted in significant process retraction without oligodendrocyte degeneration (Scarisbrick et al. 2002). Here, we confirm this phenotype in PAR1+/+ murine oligodendrocyte cultures (3 DIV) (Fig. 2). Treatment of oligodendrocytes with either 30 (Fig. 2A, B; SNK, p = 9.0 × 10−4) or 300 nM Klk6 (SNK, p = 4.7 × 10−4) each produced significant and statistically equivalent oligodendrocyte process retraction compared to controls. To determine the involvement of PARs in Klk6-mediated oligodendrogliopathy, results were compared between primary PAR1+/+ oligodendrocyte cultures and those derived from PAR1−/− or PAR2−/− mice. PAR1−/− oligodendrocytes did not exhibit statistically significant process retraction in response to 30 (Fig. 2A, B) or 300 nM Klk6. By contrast, treatment of PAR2−/− oligodendrocytes with Klk6 resulted in process retraction comparable to that seen in PAR1+/+ cultures (Fig. 2B; 30 nM Klk6, SNK, p = 0.002; 300 nM Klk6, SNK, p = 1.2 × 10−4 vs. Control). No significant differences in the number O4+ PAR1+/+, PAR1−/− or PAR2−/− cells were observed in response to Klk6 treatment (Fig. 2C).
Figure 2. PAR1 is necessary for Klk6-induced oligodendrogliopathy in primary oligodendrocytes.

O4+ oligodendrocyte cultures from PAR1+/+, PAR1−/− or PAR2−/− mice were treated with active recombinant Klk6 for 24 h. A, B, Klk6 (30 nM) promoted significant process retraction in PAR1+/+ (SNK, p = 9.0 × 10−4) or PAR2−/− (SNK, p = 0.002) oligodendrocytes, but not in the absence of PAR1−/−. C, Klk6 treatment had no significant effect on oligodendrocyte number in cultures isolated from either PAR1+/+, PAR1−/− or PAR2−/− mice. Error bars indicate SEM. *p = < 0.005, **p = < 0.001, SNK. Scale bar = 100 μm.
To further evaluate the effects of excess Klk6 on oligodendroglial processes, primary oligodendrocyte cultures (3 DIV) were treated with recombinant Klk6 (150 nM) for 24 h and quantified with respect to morphological maturity, albeit simple, incomplete, complete or membrane morphologies (Huang et al. 2011) (Fig. 3A). Klk6 promoted a 2-fold increase in the number of simple (SNK, p = 0.007) and a greater than 4-fold increase in the number of incomplete (SNK, p = 4.7 × 10−4) oligodendrocyte morphologies. Additionally, Klk6 promoted a nearly 11-fold decrease in oligodendrocytes with complete morphology (Fig. 3C; SNK, p = 3.1 × 10−4) and the elimination of oligodendroglia with the most mature membrane morphology (SNK, p = 0.038). Klk6-treatment also resulted in 2-fold fewer processes per cell (Fig. 3E; SNK, p = 2.8 × 10−4), but there was no significant effect on oligodendrocyte number (Fig. 3F). By contrast, recombinant Klk1 (300 nM) did not significantly impact oligodendrocyte morphology (Fig. 3C), process (Fig. 3E) or cell number under the conditions of this study (Fig. 3F).
Figure 3. Klk6 reduces the maintenance of mature oligodendrocyte morphologies and exacerbates ATP-mediated toxicity.

A, High power photomicrographs show four morphological phenotypes used to characterize the differentiation state of primary oligodendroglia under various treatment conditions (C, D). B, Low power photomicrographs show exacerbated loss of morphologic complexity following treatment ATP (50 μM) which was exacerbated by Klk6 (150 nM) (D to F) and to a lesser extent Klk1 (150 nM) (D). C, Treatment of 3 DIV oligodendrocyte cultures with Klk6 (150 nM) for 24 hr resulted in a significant increase in the immature oligodendrocyte phenotypes (simple, SNK, p = 0.007 or incomplete, SNK, p = 4.7 × 10−4), while the number of cells with a mature phenotype (complete, SNK, p = 3.1 × 10−4 or membrane, SNK, p = 0.038) was significantly reduced. Correspondingly, Klk6 treatment resulted in fewer O4+ processes per cell (E). Treatment with Klk1 alone did not significantly affect oligodendrocyte morphology (C) or process number (E). Treatment with ATP (50 μM) alone also decreased the number of oligodendroglia with complete morphologies (SNK, p = 0.002), increased the number with simple morphology (SNK, p = 0.028) and reduced the overall number of O4+ processes per cell (D, E, SNK, p = 2.4 × 10−4). D, Co-application of Klk6 and ATP further increased the loss of mature oligodendrocyte morphologies, increasing the percentage of cells with an immature phenotype, including simple (SNK, p = 0.039) and incomplete (SNK, p = 0.004), while eliminating cells with more mature complete (SNK, p = 2.0 × 10−4) and membrane morphologies (SNK, p = 0.001). The effects of Klk6 and ATP were also additive with regard to loss of O4+ process (E). Klk1 also exacerbated the effects of ATP, reducing the number of oligodendrocytes with a membrane morphology (SNK, p = 0.022). F, ATP promoted a significant loss of O4+ cells (SNK, p = 0.006), and this was exacerbated by the addition of Klk6 (SNK, p = 0.046), but not Klk1. * p < 0.05, ** p < 0.005, *** p ≤ 0.001, SNK. (Scale bar = 25 μm, A; 100 μm, B).
Elevated Klk6 exacerbates ATP-toxicity in oligodendrocytes
Aberrant ATP signaling causes oligodendrocyte excitotoxicity and high levels of ATP have been associated with pathophysiology in both SCI and MS (Matute et al. 2007; Wang et al. 2004a). Here, we investigated whether elevated levels of Klk6 augment ATP-induced excitotoxicity in oligodendroglia, using loss of morphological differentiation and cell number as measures of pathogenicity. Twenty-four h treatment of primary oligodendrocyte cultures (3 DIV) with ATP (50 μM) resulted in a significant increase in the percentage of cells with simple morphology (Fig. 3D; SNK, p = 0.028). Treatment with ATP alone also resulted in a significant decrease in cells with complete morphology (SNK, p = 0.002) and a small, but significant increase in cells with membrane morphology (SNK, p = 0.011). Klk6 (150 nM) amplified the gliopathic effects of ATP, further increasing the percentage of oligodendrocytes with ‘simple’ morphology relative to ATP alone (Fig. 3D; SNK, p = 0.039). The co-application of Klk6 and ATP, also resulted in a greater than 40% increase in the population of oligodendrocytes with incomplete morphologies compared to vehicle (SNK, p = 0.003) or ATP alone (SNK, p = 0.004). The addition of Klk6 and ATP also exacerbated the reduction in mature cells seen with ATP alone and resulted in the complete loss of oligodendroglia with the mature complete (Fig. 3D; SNK, p = 2.0 × 10−4, ATP vs. ATP + Klk6; SNK, p = 2.3 × 10−4, vehicle vs. ATP + Klk6) or membrane morphologies (SNK, p = 0.001, ATP vs. ATP + Klk6; SNK, p =0.033, vehicle vs. ATP + Klk6). Moreover, treatment with ATP resulted in a 2-fold reduction in the number of processes per oligodendrocyte (Fig. 3E; SNK, p = 2.3 × 10−4) and this increased to a 5-fold reduction with the co-application of Klk6 (Fig. 3E; SNK, p = 2.5 × 10−4). The co-application of Klk1 and ATP (Fig. 3D) did cause a significant decrease in the population of oligodendrocytes with membrane morphology (SNK, p = 0.022) but did not impact process loss per cell relative to treatment with ATP alone (Fig. 3E).
Treatment of murine oligodendrocytes (3 DIV) with ATP caused a significant decrease in oligodendrocyte number relative to control (Fig. 3F; SNK, p = 0.006). Treatment with Klk6 in addition to ATP caused a significant exacerbation of ATP-induced cell loss compared to ATP (Fig. 3F; SNK, p = 0.046) or to vehicle alone (SNK, p = 9.3 × 10−4). Klk1 did not increase ATP-mediated oligodendrocyte loss (Fig. 3F).
PAR1 agonists mediate process loss in Oli-neu oligodendrocytes
To determine the range of PAR1 agonists able to regulate oligodendrocyte process integrity, Oli-neu oligodendrocytes were treated with recombinant Klk6 (30, 150 and 300 nM), thrombin (270 nM) or PAR1-AP (100 μM) for 24 h (Fig. 4). A significant and dose-dependent (Fig. 4A, B; SNK, p =0.018, Klk6 30 vs. 150 nM; SNK, p =0.041, Klk6 30 vs. 300 nM) decrease in the number of processes per cell was observed in response to Klk6 (SNK, p = 1.5 × 10−4, Klk6 30–150 nM vs. control). A significant decrease in Oli-neu process number was also observed following treatment with thrombin (Fig. 4A, B; SNK, p = 1.1 × 10−4) or PAR1-AP (SNK, p = 2.7 × 10−4). A small but significant increase in Oli-neu cell number was also observed following treatment with the lowest concentration of Klk6 examined (Fig. 4C; 30 nM, SNK, p = 0.03). A similar increase in cell number was observed in the case of treatment with PAR1-AP (SNK, p = 0.003). Klk1 (300nM) had no effect on Oli-neu process stability (Fig. 4A, B) or cell number.
Figure 4. Klk6, thrombin and PAR1-AP promote process retraction in Oli-neu oligodendrocytes.
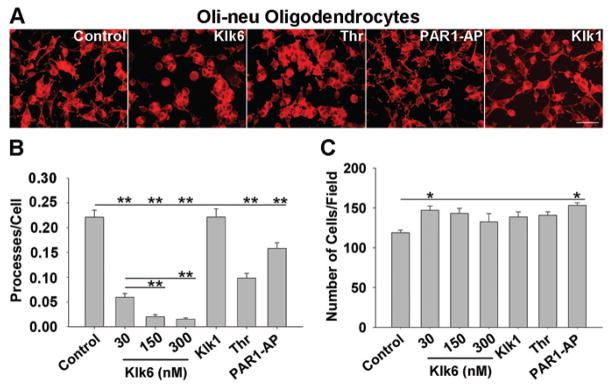
A, Photomicrographs show retraction of Oli-neu oligodendrocyte processes 24 h after treatment with Klk6, thrombin or PAR1-AP, but not Klk1. B, Histograms show counts of Cy3-Phalloidin stained Oli-neu processes per cell after 24 h treatment with Klk6 (30, 150, 300 nM), thrombin (270 nM), PAR1-AP (100 μM) or Klk1 (300 nM). Klk6 caused a dose-dependent retraction of Oli-neu processes, relative to control (SNK, p = 1.5 × 10−4). Thrombin (SNK, p = 1.1 × 10−4) and PAR1-AP (SNK, p = 2.7 × 10−4), but not Klk1, also promoted significant process retraction. C, Histogram shows counts of Oli-neu cells per field in each treatment condition, presented as mean number of Phalloidin+ processes/DAPI+ nuclei. 30 nM Klk6 and PAR1-AP treatment resulted in a small but significant increase in Oli-neu cell number (SNK, p = 0.03 and p = 0.003, respectively). Error bars indicate SEM. * p < 0.05; ** p < 0.001, SNK. Scale bar = 100 μm.
Klk6-blockade of OPC differentiation is PAR1-dependent
To determine whether elevated levels of Klk6 inhibit process outgrowth from OPCs, purified OPCs were treated with Klk6 (150 nM) just after plating for 24 h. Progenitor cells treated with Klk6 exhibited stunted morphological differentiation, having ~60% fewer processes per cell compared to controls (Fig. 5A, B; SNK, p = 2.3 × 10−4). Thrombin also inhibited OPC process extension (Fig. 5A, B; SNK, p = 2.0 × 10−4). OPCs treated with Klk1 developed processes comparable to controls (Fig. 5A, B). In the case of progenitors, Klk6 treatment also significantly reduced the number of OPCs (Fig. 5C; SNK, p = 0.032), while treatment with thrombin or Klk1 had no effect. To determine the role of PAR1 in mediating these effects, primary OPCs were differentiated in presence of a selective PAR1 inhibitor, SCH79797 (50 nM), for 3 h prior to the addition of Klk6. SCH79797 attenuated the ability of Klk6 to reduce OPC process outgrowth by ~34% (Fig. 5D; SNK, p = 0.006) while treatment with the SCH79797 alone had no significant effect relative to vehicle controls.
Figure 5. Klk6 impedes oligodendrocyte progenitor process outgrowth in a PAR1-dependent fashion.

A, Purified O4+ oligodendrocyte progenitor cell (OPC) cultures from PAR1+/+ mice were differentiated for 24 h in the presence of Klk6 (150 nM), thrombin (135 nM) or Klk1 (150 nM). B, Quantification of OPC process number per O4+ cell demonstrated a significant inhibition of OPC process outgrowth in the case of Klk6 and thrombin treatment, but not in response to Klk1 (SNK, p = 2.3 × 10−4, p = 2.0 × 10−4). Klk6 treated cells also developed significantly fewer processes than thrombin treated cultures (SNK, p = 2.3 × 10−4). C, Klk6 also promoted a significant decrease in cell number (SNK, p = 0.032). D, Klk6-mediated inhibition of process outgrowth was diminished in the presence of the PAR1 inhibitor, SCH79797 (50 nM) (SNK, p = 0.006, Klk6 vs. Klk6+SCH), albeit without returning process outgrowth to control levels (SNK, p = 2.4 × 10−4). Error bars indicate SEM. * P < 0.05; ** P < 0.001, SNK. Scale bar = 100 μm.
Klk6-suppresses myelin gene expression in a PAR1-dependent fashion
To determine the impact of elevated Klk6 on other key aspects of oligodendrocyte biology, we evaluated the effects of treatment with Klk6 for 24 h on the expression of PLP and MBP and the involvement of PAR1 (Fig. 6). Treatment of PAR1+/+ but not PAR−/− oligodendrocyte cultures with recombinant Klk6 (300 nM) for 24 h resulted in a significant suppression of PLP (Fig. 6A; ± 36%, Student’s t-test, p = 5.9 × 10−4) and MBP RNA expression (Fig. 6B; ± 33%, Student’s t-test, p = 0.029). Treatment of either PAR1+/+ or PAR1−/− cells with Klk6 did not significantly alter GAPDH RNA expression (PAR1+/+-control, 1.5 × 106 ± 6.9 × 104 vs. PAR1+/+-Klk6, 1.4 × 106 ± 4.4 × 104 copies/0.5 μg RNA; PAR1−/−-control, 1.4 × 106 ± 1.3 × 104 vs. PAR1−/−-Klk6, 1.4 × 106 ± 6.5 × 103 copies/0.5 μg RNA).
Figure 6. Klk6 down regulates myelin gene expression in a PAR1-dependent manner in primary oligodendrocytes.
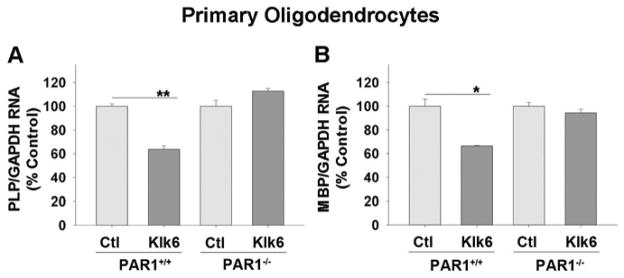
Histograms show quantitative PCR for A, proteolipid protein (PLP) and B, myelin basic protein (MBP) RNA expression in PAR1+/+ or PAR1−/− primary mouse oligodendrocytes (3 DIV) following 24 h treatment with Klk6 (300 nM) or vehicle alone. A significant Klk6-driven downregulation of PLP (Student’s t-test, p = 5.9 × 10−4) and MBP (Student’s t-test, p = 0.029) RNA expression was observed in PAR1+/+, but not PAR1−/− oligodendrocytes. Expression data were normalized to GAPDH and expressed as percent of control. Error bars indicate SEM. * P < 0.05, ** P < 0.001, Student’s t-test.
Parallel to the effects of Klk6 observed in primary oligodendrocytes, treatment of Oli-neu oligodendroglia with Klk6 for 24 h also significantly diminished PLP expression (Fig. 7A, B; SNK, p = 8.0 × 10−4), although no significant suppression of MBP RNA was observed at this time point. Notably, Klk6 caused significant and equivalent PLP RNA downregulation at 30, 150 and 300 nM (Fig. 7C; SNK, p = 0.002). PLP expression was also significantly decreased by treatment with either thrombin (Fig. 7A; 270 nM; SNK, p = 0.003, vs. Control) or PAR1-AP (100 μM; SNK, p = 7.4 × 10−4), but not recombinant Klk1 (300 nM) at the time point examined.
Figure 7. Klk6-PAR1 signals through Erk1/2 to suppress myelin gene expression.

Quantitative PCR analysis for A, proteolipid protein (PLP) or B, myelin basic protein (MBP) RNA expression in Oli-neu oligodendrocytes following 24 h treatment with Klk6 (300 nM), thrombin (270 nM), PAR1-AP (100 μM) or Klk1 (300 nM). A significant down regulation of PLP, but not MBP RNA expression was observed after treatment with Klk6 (SNK, p = 8.0 × 10−4), thrombin (SNK, p = 0.003) or PAR1-AP (SNK, p = 7.4 × 10−4). C, Down regulation of PLP was observed following 24 h treatment with as little as 30 nM recombinant Klk6 (SNK, p = 0.002). D, Klk6-induced downregulation of PLP mRNA was abolished in the presence of the MEK1/2 inhibitor, U0126 (10 μM) (SNK, p = 0.009, Klk6 vs. Klk6 + U0126). E, Western blot shows Erk1/2 phosphorylation in Oli-neu oligodendrocytes treated with Klk6 or Klk1 for 10 min, with or without the PAR1 inhibitor, SCH79797 dihydrochloride (50 nM). F, Histogram shows densitometric quantification of bands revealing a significant increase in Erk1/2 phosphorylation in response to treatment with Klk6 (SNK, p = 0.012), which was abolished in the presence of SCH79797. Band optical density measurements are expressed as percent of maximal response observed. β-actin levels were measured as a loading control. Error bars indicate SEM. * P < 0.05, ** P < 0.005, *** P ≤ 0.001, SNK.
Role of Erk1/2 in Klk6-regulation of myelin gene expression
Based on previous data demonstrating the Klk6-mediated MAPK signaling in neurons and astrocytes (Vandell et al. 2008), we examined whether Klk6 triggers similar signaling in Oli-neu oligodendrocytes and the possible role of this signaling in the regulation of myelin gene expression. Treatment of Oli-neu oligodendrocytes with Klk6 for 10 min elicited a nearly 4-fold increase in Erk1/2 phosphorylation (Fig. 7E, F; SNK, p = 0.012). The ability of Klk6 to induce MAPK signaling was blocked by the PAR1 inhibitor, SCH79797 (50 nM) (Fig. 7E, F). No significant changes in Erk1/2 signaling were observed following treatment with SCH79797 alone or Klk1. Linking Klk6-PAR1-mediated down regulation of PLP to Erk1/2 signaling, co-treatment of Oli-neu with Klk6 and a selective MEK1/2 inhibitor (U0126, 10 μM) for 24 h, significantly diminished the ability of Klk6 to suppress PLP RNA expression (Fig. 7D; SNK, p = 0.009, Klk6 vs. Klk6 + U0126), although suppression was not completely blocked (SNK, p = 0.034, Klk6 + U0126 vs. Control). No significant change in PLP RNA expression was observed following treatment with U0126 alone.
Klk6 promotes white matter pathology in a PAR1-dependent fashion
To determine whether elevated Klk6 or deregulated PAR1-agonism alone mediate white matter pathology in vivo, and the role of PAR1 in mediating these effects, recombinant Klk6 or PAR1-AP were microinjected unilaterally into the dorsal funiculus of PAR1+/+ or PAR1−/− murine spinal cord. Seventy-two h after microinjection of Klk6 (0.02 μg total) into PAR+/+ over 1200 μm of white matter surrounding the injection site presented with vacuolating myelinopathy, tissue destruction and hemorrhage in H&E stained sections, effects that were largely absent in PAR1−/− mice (Fig. 8A). Rostrocaudal white matter pathology mediated by Klk6 was 2-fold greater than that induced by saline alone (Fig. 8D; saline Control = 570.0 ± 39.3 μm vs. Klk6 = 1242.0 ± 95.3 μm; SNK, p = 6.9 × 10−4). Microinjection of PAR1-AP (0.02 μg) also induced significantly greater rostrocaudal white matter pathology relative to saline (Fig. 8A, D; PAR1-AP = 1026.0 ± 105.3 μm, SNK, p = 0.01) and these effects were also absent in PAR1−/− mice. In addition, Klk6 and PAR1-AP each caused significant and largely equivalent loss of MBP-immunoreactivity across multiple sections rostrocaudally (~45% loss relative to saline alone) (Fig. 8B, E; SNK, p = 4.1 × 10−4 and p = 0.001, respectively) in PAR+/+ but not in PAR−/− mice.
Figure 8. PAR1 plays a critical role in Klk6-driven myelinopathy in vivo.

A, B Photomicrographs illustrate the extent of white matter pathology observed 72 h after unilateral microinjection of 2 μL of physiologic saline, Klk6 (0.01 μg/μL) or PAR1-AP (0.1 μg/μL) into the dorsal column of PAR1+/+ or PAR1−/− mice (A, H&E; B, myelin basic protein (MBP) immunoreactivity). Dashed lines in H&E stained sections demarcate the site of maximal lesion in each case. D, Microinjection of Klk6 or PAR1-AP resulted in enhanced rostrocaudal white matter injury (Klk6, SNK, p = 6.9 × 10−4; PAR1-AP, SNK, p = 0.01), E, rostrocaudal MBP loss (Klk6, SNK, p = 4.1 × 10−4; PAR1-AP, SNK, p =0.001) and F, maximal lesion area (Klk6, SNK, p = 3.3 × 10−4; PAR1-AP, SNK, p = 3.2 × 10−4), relative to saline alone, in PAR+/+ but not in PAR−/− mice. G, Also, microinjection of either Klk6 or PAR1-AP resulted in a significant reduction in the number of CC-1+ oligodendrocytes counted per 1 × 105 μm2 (the approximate size of the dorsal column in a given section of spinal cord; Klk6, SNK, p = 0.001; PAR1-AP, SNK, p = 0.002), in PAR+/+ but not in PAR−/− mice. Error bars indicate SEM. * P < 0.05, ** P < 0.005, *** P ≤ 0.001, SNK. (Scale bar = 100 μm, A, B; 75 μm, C.)
To determine the effect of PAR1-agonists on white matter oligodendroglia, sections were stained for CC-1. Counts of CC-1+/DAPI+ cells in the dorsal columns in tissue sections encompassing the injection epicenter and for 300 μm rostrocaudally (Fig. 8C, G) indicated that Klk6 and PAR1-AP, each promoted a significant loss of CC-1+ cells throughout the dorsal column white matter (~15% reduction, SNK, p = 0.001 and p = 0.002, respectively) in PAR1+/+ but not in PAR1−/− mice. In PAR+/+ spinal cord injected with saline alone, approximately 92.4 ± 2.0 CC-1+ oligodendroglia were counted per 1.5 × 105, the approximate size of the dorsal column in a given tissue section. The number of CC-1+ oligodendroglia after saline microinjection in PAR−/− was nearly identical (93.4 ± 1.8). This number corresponds well with the number of CNPase+ oligodendroglia enumerated in prior studies of dorsal column white matter (Pitt et al. 2000).
Discussion
The present study demonstrates that Klk6 signals through PAR1 to regulate oligodendrocyte process stability and extension, myelin gene expression and cell survival in the presence of oligotoxic agents. Moreover, elevated levels of Klk6 and other PAR1 agonists were also shown to directly mediate white matter degeneration in a PAR1-dependent manner, in vivo. These findings therefore define PAR1-agonists such as Klk6 and thrombin as important mediators of white matter pathology and as potential new targets for the development of therapies to prevent oligodendrocyte injury and to promote myelin regeneration.
There is a growing body of evidence that PARs are expressed at significant levels in the CNS and are likely to play important roles in pathogenesis (Boven et al. 2003; Hamill et al. 2009; Junge et al. 2004; Luo et al. 2007; Nicole et al. 2005; Suo et al. 2002; Vandell et al. 2008). Previously we showed that Klk6 is among the most abundant serine proteases in the adult CNS (Scarisbrick et al. 2006a; Scarisbrick et al. 2001; Scarisbrick et al. 1997), is up regulated at sites of CNS pathology (Christophi et al. 2004; Drucker et al. 2013; Scarisbrick et al. 2002; Scarisbrick et al. 2006b) and activates PAR1 on neurons, and both PAR1 and PAR2 on astrocytes to elicit Ca2+ and Erk1/2 signaling (Scarisbrick et al. 2012a; Vandell et al. 2008). Here we show that PAR1 is abundantly expressed by primary OPCs and oligodendroglia while PAR2 is expressed at substantially lower levels. Supporting the key role of PAR1 in oligodendrocyte pathophysiology, PAR1 but not PAR2 genetic deletion attenuated the oligotoxic effects of Klk6 toward oligodendroglia in culture. Taken with the significant levels of Klk6 known to be expressed by oligodendroglia in the rodent and human CNS (Scarisbrick et al. 2000; Scarisbrick et al. 2001; Scarisbrick et al. 1997) these studies suggest that Klk6 may operate in an autocrine or paracrine fashion to activate PAR1 signaling cascades that participate in oligodendrocyte differentiation, including regulation of process outgrowth, process stability and myelin gene expression.
Oligodendrocyte precursors undergo extensive differentiation involving polymerization of microtubules and microfilaments and extension of arborized processes that form the myelin sheaths (Wilson and Brophy 1989). We previously reported that Klk6 is elevated in actively demyelinating MS lesions, in the serum of progressive MS patients (Scarisbrick et al. 2002; Scarisbrick et al. 2008) and elicits a dying back of oligodendroglial processes in vitro (Scarisbrick et al. 2002). Here, we demonstrate that Klk6-mediated process retraction is dependent on PAR1, since Klk6-elicited process retraction was significantly reduced in cells derived from PAR1 genetically deficient mice. The ability of both thrombin and PAR1-AP to promote oligodendrocyte process retraction further supports the concept that PAR1 is a regulator of oligodendrocyte process stability and that in excess; PAR1-agonists can mediate oligodendrogliopathy. Thus, both PAR1 and its agonists may serve as targets for the development of new oligo-protective approaches. Importantly, while the present studies indicate that PAR2 does not play an essential role in Klk6-mediated oligodendrogliopathy in vitro, we cannot exclude other regulatory roles. At least low levels of PAR2 expression were observed throughout oligodendrocyte development and taken with the anti-apoptotic and proliferative functions established for PAR2 in astrocytes (Li et al. 2009; Wang et al. 2002), future studies will be needed to fully dissect the potential functional roles of PAR2 in oligodendrocyte and white matter biology.
Oligodendrocytes surviving demyelinating events generally do not contribute to remyelination (Keirstead and Blakemore 1997). OPCs, which hold the potential to differentiate and remyelinate axons are prevalent at sites of white matter injury (Chang et al. 2002; Kuhlmann et al. 2008; Tripathi and McTigue 2007; Wolswijk 1998) although in many cases remyelination stalls due in part to a failure of OPC maturation. While the mechanisms underpinning remyelination failure are not fully understood, explanations include a lack of available axons, a lack of soluble myelination promoting factors and the presence of factors that actively inhibit OPC differentiation (Franklin 2002; Kremer et al. 2011; Wolswijk 1998). We hypothesize that elevations in Klk6 and other PAR-activating proteases that can occur in the microenvironment of a wide range of CNS pathologies (Gingrich and Traynelis 2000), not only promote oligodendrogliopathy, but also actively inhibit OPC differentiation. Supporting this hypothesis, Klk6 was shown to impede OPC process outgrowth (Scarisbrick et al. 2002) and importantly, these growth-retarding effects were attenuated by a PAR1 small molecule inhibitor. The integral role of PAR1 in blocking the process of OPC differentiation is also suggested by data indicating that the canonical activator of this receptor, thrombin, also impedes OPC process outgrowth. Future studies will be needed to determine whether targeting PAR1 can be used to overcome what we hypothesize is a pathophysiological oligodendrocyte differentiation stop signal imparted by excess proteolytic signaling at PAR1.
Expression of myelin-associated genes are dynamically regulated with white matter insult (Wrathall et al. 1998) and are a hallmark of oligodendrocyte differentiation. Klk6 is already known to readily hydrolyze both MBP and MOG, suggestive of roles in myelin turnover (Blaber et al. 2004; Blaber et al. 2002; Scarisbrick et al. 2002). Here we demonstrate Klk6 also down regulates PLP and MBP expression at an RNA level and that this occurs in a PAR1-dependent fashion. Thrombin and PAR1-AP also diminished PLP RNA, suggesting a range of PAR1 agonists are capable of suppressing myelin gene expression. Supporting these findings, a previous study showed thrombin reduced the differentiation of subventricular zone-derived progenitors into MBP+ oligodendrocytes in vitro (Juliet et al. 2009). The robust effects mediated by PAR1-agonists in suppressing myelin gene expression support the hypothesis that proteolytic activation of PAR1 generates intracellular signals that mediate a shut down or slowing of oligodendroglial differentiation.
Multiple signaling pathways have been implicated in the regulation of myelination (Fancy et al. 2011) and here we provide evidence that Klk6-signals through PAR1 to regulate myelin gene expression using Erk1/2 as a signaling intermediate. We previously demonstrated Klk6 elicits Erk1/2 phosphorylation in both neurons and astrocytes (Vandell et al. 2008) and here show Klk6-induces a rapid increase in Erk1/2 activation in Oli-neu oligodendrocytes through activation of PAR1. Additionally, we show that Klk6-Erk activation is likely integral to its ability to suppress myelin gene expression since an inhibitor of Erk1/2 kinase, MEK1/2, attenuated the ability of Klk6 to suppress myelin gene expression. The generalizability of these findings in an oligodendrocyte cell line to primary oligodendroglia at progenitor and more mature stages of development will need to be determined in future studies.
Although elevated Klk6 suppressed myelin gene expression and promoted a dying back of oligodendrocyte processes, it did not result in loss of mature oligodendroglia (Scarisbrick et al. 2002). These data support a model in which elevations in Klk6 and other PAR1-agonists that occur with CNS injury or disease serve as a pathophysiologic luxury function “stop signal” that thereby may favor cell survival. Through the suppression of luxury metabolic activity involved in myelin biogenesis, such as the expression of PLP and MBP, as well as process outgrowth and maintenance, injured oligodendroglia may sustain functions vital for cell survival (Oldstone et al. 1982; Rodriguez 1985). Notably, suppression of luxury function has previously been proposed as an explanation for the reduction in PLP and MBP RNA in MS lesions and virally-induced demyelinating disease (Lavi and Constantinescu 2005).
While mature oligodendroglia are not vulnerable to cell loss when treated with Klk6 in vitro ( Scarisbrick et al. 2002), the same concentrations promoted significant degeneration of OPCs. Such stage-specific vulnerability to gliotoxic agents also occurs in the case of oxidative and excitotoxic oligodendrocyte death (Back et al. 1998; Fern and Moller 2000). In CNS traumatic and inflammatory conditions, elevations in Klk6 and other proteases occur in a milieu of excitotoxic agents already known to promote neurodegeneration and gliopathy (Matute 2011). Excitotoxic ATP signaling mediates white matter pathology in MS and SCI, causing oligodendrocyte death and demyelination (Matute et al. 2007; Wang et al. 2004a). Elevated Klk6 levels were shown to augment ATP-induced oligodendrogliopathy, including a greater loss of mature morphologies and oligodendrocyte numbers in vitro. While Klk1, which is best characterized for its kininogenase activity (Bhoola et al. 2001), was not oligotoxic on its own, in concert with ATP, it did promote a significant reduction in the number of oligodendrocytes with complex membranes in vitro. The potential involvement of bradykinin receptors or PAR(s) in Klk1-mediated effects will need to be examined in future studies. Injury-induced elevations in Klk6 and possibly other serine proteases may therefore not only suppress oligodendrocyte differentiation on their own, but may additionally amplify the effects of well recognized oligotoxic agents.
Though the precise effects of Klk6 in CNS white matter are not fully understood, previous and current in vitro and in vivo data suggest that elevated levels promote deleterious consequences (Blaber et al. 2004; Scarisbrick et al. 2002; Scarisbrick et al. 2012b). Herein, we modeled the specific effects of excess Klk6 in normal white matter by microinjecting recombinant protein into mouse dorsal column white matter. Klk6 microinjection into PAR+/+ mouse spinal cord elicited significant reductions in oligodendrocyte density, MBP loss and vacuolating myelinopathy that extended rostrocaudally. Importantly this pathology was mirrored by microinjection of PAR1-AP alone. Moreover, the pathogenic effects in each case were effectively blocked in mice in which the PAR1 gene is deleted. These results indicate that elevated levels of Klk6 alone are capable of directly mediating white matter pathology and that PAR1 is a critical component of the underlying pathogenic cascade. The pathogenic role of excess Klk6 in white matter pathology and its potential utility as a therapeutic target is supported by studies showing that Klk6-neutralizing antibodies attenuate MBP loss and white matter pathology in autoimmune and viral murine models of MS (Blaber et al. 2004; Scarisbrick et al. 2012b). Whether inhibitors of PAR1 can exert similar effects warrants future investigation.
Altogether these results demonstrate that the Klk6-PAR1 signaling axis impedes oligodendroglial differentiation thereby reducing key aspects of oligodendrocyte biology necessary for normal myelination, including the maintenance of axon wrapping processes and myelin gene expression. Moreover, PAR1-agonists exacerbate oligodendroglial excitotoxic injury in vitro and promote vacuolating myelinopathy and a loss of MBP and oligodendrocytes in vivo. These data suggest a model whereby therapeutic targeting of PAR1 may circumvent the oligotoxic properties of PAR1 agonists that can become deregulated at sites of CNS injury, including Klk6 and thrombin. Future studies will be needed to determine how PAR1 or its agonists may be therapeutically targeted to prevent demyelination and promote remyelination in cases of CNS injury or disease.
Acknowledgments
These studies were supported by R01NS052741, The Christopher and Dana Reeve Paralysis Foundation, RG3367 and a Collaborative MS Research Award CA1060A11-02 from the National Multiple Sclerosis Society (IAS). The technical assistance of Jianmin Wu Ph.D., Christi Galardi and Swarna Submaranian is also gratefully acknowledged.
Literature Cited
- Adams MN, Ramachandran R, Yau MK, Suen JY, Fairlie DP, Hollenberg MD, Hooper JD. Structure, function and pathophysiology of protease activated receptors. Pharmacol Ther. 2011;130(3):248–82. doi: 10.1016/j.pharmthera.2011.01.003. [DOI] [PubMed] [Google Scholar]
- Angelo PF, Lima AR, Alves FM, Blaber SI, Scarisbrick IA, Blaber M, Juliano L, Juliano MA. Substrate specificity of human kallikrein 6: salt and glycosaminoglycan activation effects. The Journal of biological chemistry. 2006;281(6):3116–26. doi: 10.1074/jbc.M510096200. [DOI] [PubMed] [Google Scholar]
- Back SA, Gan X, Li Y, Rosenberg PA, Volpe JJ. Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J Neurosci. 1998;18(16):6241–53. doi: 10.1523/JNEUROSCI.18-16-06241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernett MJ, Blaber SI, Scarisbrick IA, Dhanarajan P, Thompson SM, Blaber M. Crystal structure and biochemical characterization of human kallikrein 6 reveals that a trypsin-like kallikrein is expressed in the central nervous system. J Biol Chem. 2002;277(27):24562–70. doi: 10.1074/jbc.M202392200. [DOI] [PubMed] [Google Scholar]
- Bhat RV, Axt KJ, Fosnaugh JS, Smith KJ, Johnson KA, Hill DE, Kinzler KW, Baraban JM. Expression of the APC tumor suppressor protein in oligodendroglia. Glia. 1996;17(2):169–74. doi: 10.1002/(SICI)1098-1136(199606)17:2<169::AID-GLIA8>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Bhoola K, Ramsaroop R, Plendl J, Cassim B, Dlamini Z, Naicker S. Kallikrein and kinin receptor expression in inflammation and cancer. Biol Chem. 2001;382(1):77–89. doi: 10.1515/BC.2001.013. [DOI] [PubMed] [Google Scholar]
- Blaber SI, Ciric B, Christophi GP, Bernett MJ, Blaber M, Rodriguez M, Scarisbrick IA. Targeting kallikrein 6 proteolysis attenuates CNS inflammatory disease. FASEB J. 2004;18(7):920–2. doi: 10.1096/fj.03-1212fje. [DOI] [PubMed] [Google Scholar]
- Blaber SI, Scarisbrick IA, Bernett MJ, Dhanarajan P, Seavy MA, Jin Y, Schwartz MA, Rodriguez M, Blaber M. Enzymatic properties of rat myelencephalon-specific protease. Biochemistry. 2002;41(4):1165–73. doi: 10.1021/bi015781a. [DOI] [PubMed] [Google Scholar]
- Boven LA, Vergnolle N, Henry SD, Silva C, Imai Y, Holden J, Warren K, Hollenberg MD, Power C. Up-regulation of proteinase-activated receptor 1 expression in astrocytes during HIV encephalitis. J Immunol. 2003;170(5):2638–46. doi: 10.4049/jimmunol.170.5.2638. [DOI] [PubMed] [Google Scholar]
- Chang A, Tourtellotte WW, Rudick R, Trapp BD. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N Engl J Med. 2002;346(3):165–73. doi: 10.1056/NEJMoa010994. [DOI] [PubMed] [Google Scholar]
- Christophi GP, Isackson PJ, Blaber S, Blaber M, Rodriguez M, Scarisbrick IA. Distinct promoters regulate tissue-specific and differential expression of kallikrein 6 in CNS demyelinating disease. J Neurochem. 2004;91(6):1439–49. doi: 10.1111/j.1471-4159.2004.02826.x. [DOI] [PubMed] [Google Scholar]
- Clements J, Hooper J, Dong Y, Harvey T. The expanded human kallikrein (KLK) gene family: genomic organisation, tissue-specific expression and potential functions. Biol Chem. 2001;382(1):5–14. doi: 10.1515/BC.2001.002. [DOI] [PubMed] [Google Scholar]
- Drucker KL, Paulsen AR, Giannini C, Decker PA, Blaber SI, Blaber M, Uhm JH, O’Neill BP, Jenkins RB, Scarisbrick IA. Clinical significance and novel mechanism of action of kallikrein 6 in glioblastoma. Neuro-oncology. 2013;15(3):305–18. doi: 10.1093/neuonc/nos313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancy SP, Chan JR, Baranzini SE, Franklin RJ, Rowitch DH. Myelin regeneration: a recapitulation of development? Annu Rev Neurosci. 2011;34:21–43. doi: 10.1146/annurev-neuro-061010-113629. [DOI] [PubMed] [Google Scholar]
- Fern R, Moller T. Rapid ischemic cell death in immature oligodendrocytes: a fatal glutamate release feedback loop. J Neurosci. 2000;20(1):34–42. doi: 10.1523/JNEUROSCI.20-01-00034.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin R. Why does remyelination fail in multiple sclerosis? Nature Reviews Neuroscience. 2002;3(9):705–714. doi: 10.1038/nrn917. [DOI] [PubMed] [Google Scholar]
- Gingrich MB, Traynelis SF. Serine proteases and brain damage - is there a link? Trends Neurosci. 2000;23(9):399–407. doi: 10.1016/s0166-2236(00)01617-9. [DOI] [PubMed] [Google Scholar]
- Hamill CE, Mannaioni G, Lyuboslavsky P, Sastre AA, Traynelis SF. Protease-activated receptor 1-dependent neuronal damage involves NMDA receptor function. Exp Neurol. 2009;217(1):136–46. doi: 10.1016/j.expneurol.2009.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenberg MD, Saifeddine M, al-Ani B, Kawabata A. Proteinase-activated receptors: structural requirements for activity, receptor cross-reactivity, and receptor selectivity of receptor-activating peptides. Can J Physiol Pharmacol. 1997;75(7):832–41. [PubMed] [Google Scholar]
- Huang JK, Jarjour AA, Nait Oumesmar B, Kerninon C, Williams A, Krezel W, Kagechika H, Bauer J, Zhao C, Evercooren AB, et al. Retinoid X receptor gamma signaling accelerates CNS remyelination. Nat Neurosci. 2011;14(1):45–53. doi: 10.1038/nn.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliet PA, Frost EE, Balasubramaniam J, Del Bigio MR. Toxic effect of blood components on perinatal rat subventricular zone cells and oligodendrocyte precursor cell proliferation, differentiation and migration in culture. Journal of Neurochemistry. 2009;109(5):1285–99. doi: 10.1111/j.1471-4159.2009.06060.x. [DOI] [PubMed] [Google Scholar]
- Jung M, Kramer E, Grzenkowski M, Tang K, Blakemore W, Aguzzi A, Khazaie K, Chlichlia K, von Blankenfeld G, Kettenmann H. Lines of murine oligodendroglial precursor cells immortalized by an activated neu tyrosine kinase show distinct degrees of interaction with axons in vitro and in vivo. The European journal of neuroscience. 1995;7(6):1245–65. doi: 10.1111/j.1460-9568.1995.tb01115.x. [DOI] [PubMed] [Google Scholar]
- Junge CE, Lee CJ, Hubbard KB, Zhang Z, Olson JJ, Hepler JR, Brat DJ, Traynelis SF. Protease-activated receptor-1 in human brain: localization and functional expression in astrocytes. Exp Neurol. 2004;188(1):94–103. doi: 10.1016/j.expneurol.2004.02.018. [DOI] [PubMed] [Google Scholar]
- Keirstead HS, Blakemore WF. Identification of post-mitotic oligodendrocytes incapable of remyelination within the demyelinated adult spinal cord. J Neuropathol Exp Neurol. 1997;56(11):1191–201. doi: 10.1097/00005072-199711000-00003. [DOI] [PubMed] [Google Scholar]
- Kremer D, Aktas O, Hartung HP, Kury P. The complex world of oligodendroglial differentiation inhibitors. Ann Neurol. 2011;69(4):602–18. doi: 10.1002/ana.22415. [DOI] [PubMed] [Google Scholar]
- Kuhlmann T, Miron V, Cui Q, Wegner C, Antel J, Bruck W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain. 2008;131(Pt 7):1749–58. doi: 10.1093/brain/awn096. [DOI] [PubMed] [Google Scholar]
- Lavi E, Constantinescu CS. Experimental models of multiple sclerosis. New York, NY: Springer; 2005. p. xiii.p. 901. [Google Scholar]
- Li R, Rohatgi T, Hanck T, Reiser G. Alpha A-crystallin and alpha B-crystallin, newly identified interaction proteins of protease-activated receptor-2, rescue astrocytes from C2-ceramide- and staurosporine-induced cell death. Journal of Neurochemistry. 2009;110(5):1433–44. doi: 10.1111/j.1471-4159.2009.06226.x. [DOI] [PubMed] [Google Scholar]
- Luo W, Wang Y, Reiser G. Protease-activated receptors in the brain: receptor expression, activation, and functions in neurodegeneration and neuroprotection. Brain research reviews. 2007;56(2):331–45. doi: 10.1016/j.brainresrev.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Maggio N, Shavit E, Chapman J, Segal M. Thrombin induces long-term potentiation of reactivity to afferent stimulation and facilitates epileptic seizures in rat hippocampal slices: toward understanding the functional consequences of cerebrovascular insults. J Neurosci. 2008;28(3):732–6. doi: 10.1523/JNEUROSCI.3665-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matute C. Glutamate and ATP signalling in white matter pathology. J Anat. 2011;219(1):53–64. doi: 10.1111/j.1469-7580.2010.01339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matute C, Torre I, Perez-Cerda F, Perez-Samartin A, Alberdi E, Etxebarria E, Arranz AM, Ravid R, Rodriguez-Antiguedad A, Sanchez-Gomez M, et al. P2X(7) receptor blockade prevents ATP excitotoxicity in oligodendrocytes and ameliorates experimental autoimmune encephalomyelitis. J Neurosci. 2007;27(35):9525–33. doi: 10.1523/JNEUROSCI.0579-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85(3):890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael IP, Sotiropoulou G, Pampalakis G, Magklara A, Ghosh M, Wasney G, Diamandis EP. Biochemical and enzymatic characterization of human kallikrein 5 (hK5), a novel serine protease potentially involved in cancer progression. J Biol Chem. 2005;280(15):14628–35. doi: 10.1074/jbc.M408132200. [DOI] [PubMed] [Google Scholar]
- Nicole O, Goldshmidt A, Hamill CE, Sorensen SD, Sastre A, Lyuboslavsky P, Hepler JR, McKeon RJ, Traynelis SF. Activation of protease-activated receptor-1 triggers astrogliosis after brain injury. J Neurosci. 2005;25(17):4319–29. doi: 10.1523/JNEUROSCI.5200-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nystedt S, Emilsson K, Larsson AK, Strombeck B, Sundelin J. Molecular cloning and functional expression of the gene encoding the human proteinase-activated receptor 2. Eur J Biochem. 1995;232(1):84–9. doi: 10.1111/j.1432-1033.1995.tb20784.x. [DOI] [PubMed] [Google Scholar]
- Oikonomopoulou K, Hansen KK, Saifeddine M, Tea I, Blaber M, Blaber SI, Scarisbrick I, Andrade-Gordon P, Cottrell GS, Bunnett NW, et al. Proteinase-activated receptors, targets for kallikrein signaling. The Journal of biological chemistry. 2006a;281(43):32095–112. doi: 10.1074/jbc.M513138200. [DOI] [PubMed] [Google Scholar]
- Oikonomopoulou K, Hansen KK, Saifeddine M, Vergnolle N, Tea I, Diamandis EP, Hollenberg MD. Proteinase-mediated cell signalling: targeting proteinase-activated receptors (PARs) by kallikreins and more. Biological Chemistry. 2006b;387(6):677–85. doi: 10.1515/BC.2006.086. [DOI] [PubMed] [Google Scholar]
- Oldstone MB, Sinha YN, Blount P, Tishon A, Rodriguez M, von Wedel R, Lampert PW. Virus-induced alterations in homeostasis: alteration in differentiated functions of infected cells in vivo. Science. 1982;218(4577):1125–7. doi: 10.1126/science.7146898. [DOI] [PubMed] [Google Scholar]
- Ossovskaya V. Protease-Activated Receptors: Contribution to Physiology and Disease. Physiological reviews. 2004;84(2):579–621. doi: 10.1152/physrev.00028.2003. [DOI] [PubMed] [Google Scholar]
- Pitt D, Werner P, Raine CS. Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med. 2000;6(1):67–70. doi: 10.1038/71555. [DOI] [PubMed] [Google Scholar]
- Rodriguez M. Virus-induced demyelination in mice: “dying back” of oligodendrocytes. Mayo Clin Proc. 1985;60(7):433–8. doi: 10.1016/s0025-6196(12)60865-9. [DOI] [PubMed] [Google Scholar]
- Scarisbrick IA, Asakura K, Blaber SI, Blaber M, Isackson PJ, Bieto T, Rodriguez M, Windebank AJ. Preferential expression of myelencephalon-specific protease by oligodendrocytes of the adult rat spinal cord white matter. Glia. 2000;30(3):219–30. doi: 10.1002/(sici)1098-1136(200005)30:3<219::aid-glia2>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Scarisbrick IA, Blaber SI, Lucchinetti CF, Genain CP, Blaber M, Rodriguez M. Activity of a newly identified serine protease in CNS demyelination. Brain. 2002;125(Pt 6):1283–96. doi: 10.1093/brain/awf142. [DOI] [PubMed] [Google Scholar]
- Scarisbrick IA, Blaber SI, Tingling JT, Rodriguez M, Blaber M, Christophi GP. Potential scope of action of tissue kallikreins in CNS immune-mediated disease. J Neuroimmunol. 2006a;178(1–2):167–76. doi: 10.1016/j.jneuroim.2006.05.022. [DOI] [PubMed] [Google Scholar]
- Scarisbrick IA, Epstein B, Cloud BA, Yoon H, Wu J, Renner DN, Blaber SI, Blaber M, Vandell AG, Bryson AL. Functional role of kallikrein 6 in regulating immune cell survival. PLoS One. 2011;6(3):e18376. doi: 10.1371/journal.pone.0018376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarisbrick IA, Isackson PJ, Ciric B, Windebank AJ, Rodriguez M. MSP, a trypsin-like serine protease, is abundantly expressed in the human nervous system. The Journal of comparative neurology. 2001;431(3):347–61. [PubMed] [Google Scholar]
- Scarisbrick IA, Linbo R, Vandell AG, Keegan M, Blaber SI, Blaber M, Sneve D, Lucchinetti CF, Rodriguez M, Diamandis EP. Kallikreins are associated with secondary progressive multiple sclerosis and promote neurodegeneration. Biol Chem. 2008;389(6):739–45. doi: 10.1515/BC.2008.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarisbrick IA, Radulovic M, Burda JE, Larson N, Blaber SI, Giannini C, Blaber M, Vandell AG. Kallikrein 6 is a novel molecular trigger of reactive astrogliosis. Biol Chem. 2012a;393(5):355–67. doi: 10.1515/hsz-2011-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarisbrick IA, Sabharwal P, Cruz H, Larsen N, Vandell AG, Blaber SI, Ameenuddin S, Papke LM, Fehlings MG, Reeves RK, et al. Dynamic role of kallikrein 6 in traumatic spinal cord injury. Eur J Neurosci. 2006b;24(5):1457–69. doi: 10.1111/j.1460-9568.2006.05021.x. [DOI] [PubMed] [Google Scholar]
- Scarisbrick IA, Towner MD, Isackson PJ. Nervous system-specific expression of a novel serine protease: regulation in the adult rat spinal cord by excitotoxic injury. J Neurosci. 1997;17(21):8156–68. doi: 10.1523/JNEUROSCI.17-21-08156.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarisbrick IA, Yoon H, Panos M, Larson N, Blaber SI, Blaber M, Rodriguez M. Kallikrein 6 Regulates Early CNS Demyelination in a Viral Model of Multiple Sclerosis. Brain Pathol. 2012b doi: 10.1111/j.1750-3639.2012.00577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirakawa H, Sakimoto S, Nakao K, Sugishita A, Konno M, Iida S, Kusano A, Hashimoto E, Nakagawa T, Kaneko S. Transient receptor potential canonical 3 (TRPC3) mediates thrombin-induced astrocyte activation and upregulates its own expression in cortical astrocytes. J Neurosci. 2010;30(39):13116–29. doi: 10.1523/JNEUROSCI.1890-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striggow F, Riek M, Breder J, Henrich-Noack P, Reymann KG, Reiser G. The protease thrombin is an endogenous mediator of hippocampal neuroprotection against ischemia at low concentrations but causes degeneration at high concentrations. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(5):2264–9. doi: 10.1073/pnas.040552897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suo Z, Wu M, Ameenuddin S, Anderson HE, Zoloty JE, Citron BA, Andrade-Gordon P, Festoff BW. Participation of protease-activated receptor-1 in thrombin-induced microglial activation. J Neurochem. 2002;80(4):655–66. doi: 10.1046/j.0022-3042.2001.00745.x. [DOI] [PubMed] [Google Scholar]
- Tripathi R, McTigue DM. Prominent oligodendrocyte genesis along the border of spinal contusion lesions. Glia. 2007;55(7):698–711. doi: 10.1002/glia.20491. [DOI] [PubMed] [Google Scholar]
- Vandell AG, Larson N, Laxmikanthan G, Panos M, Blaber SI, Blaber M, Scarisbrick IA. Protease-activated receptor dependent and independent signaling by kallikreins 1 and 6 in CNS neuron and astroglial cell lines. J Neurochem. 2008;107(3):855–70. doi: 10.1111/j.1471-4159.2008.05658.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vellani V, Kinsey AM, Prandini M, Hechtfischer SC, Reeh P, Magherini PC, Giacomoni C, McNaughton PA. Protease activated receptors 1 and 4 sensitize TRPV1 in nociceptive neurones. Mol Pain. 2010;6:61. doi: 10.1186/1744-8069-6-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu TK, Hung DT, Wheaton VI, Coughlin SR. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991;64(6):1057–68. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- Wang H, Ubl JJ, Reiser G. Four subtypes of protease-activated receptors, co-expressed in rat astrocytes, evoke different physiological signaling. Glia. 2002;37(1):53–63. doi: 10.1002/glia.10012. [DOI] [PubMed] [Google Scholar]
- Wang X, Arcuino G, Takano T, Lin J, Peng WG, Wan P, Li P, Xu Q, Liu QS, Goldman SA, et al. P2X7 receptor inhibition improves recovery after spinal cord injury. Nat Med. 2004a;10(8):821–7. doi: 10.1038/nm1082. [DOI] [PubMed] [Google Scholar]
- Wang Y, Richter-Landsberg C, Reiser G. Expression of protease-activated receptors (PARs) in OLN-93 oligodendroglial cells and mechanism of PAR-1-induced calcium signaling. Neuroscience. 2004b;126(1):69–82. doi: 10.1016/j.neuroscience.2004.03.024. [DOI] [PubMed] [Google Scholar]
- Wilson R, Brophy PJ. Role for the oligodendrocyte cytoskeleton in myelination. J Neurosci Res. 1989;22(4):439–48. doi: 10.1002/jnr.490220409. [DOI] [PubMed] [Google Scholar]
- Wolswijk G. Chronic stage multiple sclerosis lesions contain a relatively quiescent population of oligodendrocyte precursor cells. J Neurosci. 1998;18(2):601–9. doi: 10.1523/JNEUROSCI.18-02-00601.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrathall JR, Li W, Hudson LD. Myelin gene expression after experimental contusive spinal cord injury. J Neurosci. 1998;18(21):8780–93. doi: 10.1523/JNEUROSCI.18-21-08780.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousef GM, Diamandis EP. The new human tissue kallikrein gene family: structure, function, and association to disease. Endocrine Reviews. 2001;22(2):184–204. doi: 10.1210/edrv.22.2.0424. [DOI] [PubMed] [Google Scholar]
- Zarghooni M, Soosaipillai A, Grass L, Scorilas A, Mirazimi N, Diamandis EP. Decreased concentration of human kallikrein 6 in brain extracts of Alzheimer’s disease patients. Clin Biochem. 2002;35(3):225–31. doi: 10.1016/s0009-9120(02)00292-8. [DOI] [PubMed] [Google Scholar]