

Abstract
Transforming growth factor (TGF)-β1 has been suggested to be involved in the recruitment of mesenchymal stem cells (MSCs) following arterial injury, but the role of downstream signaling and the contribution of the recruited MSCs are still unknown. The release of latent TGF-β1 from latent TGF-binding protein (LTBP) by matrix metallopeptidase-14 (MMP-14) proteolysis was demonstrated, which contributed to neointima formation, but the relationship between MMP-14 and activated TGF-β1 in the process of restenosis has yet to be explored. In this study, we observed the change in expression and distribution of TGF-β1/Smad signaling pathway proteins, MMP-14, and MSC markers in the process of neointima formation using a rat model for balloon-induced carotid artery injury. We found that the increase in downstream Smad signaling was consistent with the elevation of TGF-β1 levels and MSCs accumulated at the lumen side of neointima. Furthermore, the activation of MMP-14 in the injured artery was preceded by the increase in TGF-β1 levels. Herein, we conclude that MMP-14 induces an elevation in the levels of TGF-β1/Smad signaling proteins in injured arteries, and that MSCs are recruited by TGF-β1/Smad signaling and MMP-14, possibly differentiating into vascular smooth muscle cell (VSMC)-like cells and VSMC via modulation of TGF-β1/Smads signaling and MMP-14.
Keywords: TGF-β1/Smad signaling pathway, MMP-14, mesenchymal stem cells, balloon injury, restenosis
Introduction
Cardiovascular diseases are among the leading causes of death worldwide. Percutaneous transluminal coronary angioplasty (PTCA) treatments are frequently used but are often complicated by restenosis. The proliferation and differentiation of vascular smooth muscle cells (VSMCs) [1] and the dysregulation of extracellular matrix (ECM) [2] after intimal injury are contributing factors to restenosis. The recruitment of mesenchymal stem cells (MSCs) has recently been suggested to contribute to tissue repair where arterial injury is involved. Various cytokines, such as granulocyte colony-stimulating factor (G-CSF) and TGF-β1, have been proposed to be involved in the recruitment of MSCs, with TGF-β1 having an essential role [3]. However, the downstream signaling elements remain unknown.
Recent studies have suggested that TGF-β1 promotes the migration and recruitment of MSCs to lesions to participate in tissue repair and remodeling, such as bone remodeling [3]. Furthermore, transplantation of MSCs has been applied to demonstrate the contribution of MSCs to neointimal formation of vascular lesions in vivo [4]. Evidence has shown a high potential of MSCs to differentiate into cells with a muscular phenotype; meanwhile, several in vivo and in vitro studies have demonstrated the ability of MSCs to differentiate into VSMCs or VSMC-like cells [5], from which abundant ECM components are secreted. However, the specific underlying mechanism of recruitment and contribution of MSCs to the injured artery is not known.
Matrix metalloproteinases (MMPs), such as MMP-2 and MMP-9, cleave interstitial collagens and have recently been demonstrated to play a crucial role in intimal hyperplasia [4]. Sustained MMP activity is suggested to be associated with vascular pathologies such as hypertension restenosis and atherosclerosis [6]. Furthermore, MMPs were found to contribute to vascular remodeling by liberating cytokines from stromal matrices [7]. MMP-14 (MT1-MMP), a newly defined membranous MMP, was recently demonstrated to release latent TGF-β1 from latent TGF-binding protein (LTBP) by proteolysis [8] and to contribute to the migration and proliferation of arterial VSMCs [9,10]. MMP-14 was also revealed to regulate the migration, invasion, and proliferation of MSCs [11,12]. The correlations between MMP-14 and elevated levels of TGF-β1 and their potential roles on MSCs in the process of restenosis have not been studied in detail.
In this study, a rat model for left carotid arterial balloon injury was used to imitate the process of restenosis. The distribution and expression of TGF-β1/Smad signaling, MMP-14, and MSC markers were observed in the injured artery. The phenotype of neointima cells and newly accumulated ECM proteins were also detected. Based on our data, we concluded that TGF-β1/Smad signaling is stimulated by MMP-14, subsequently recruiting MSCs that then differentiate into VSMC-like cells and initiate proliferation.
Materials and methods
Materials
Forty-two male Sprague-Dawley rats (SD rats) weighing 350-400 g were purchased from the animal center of Xinjiang Medical University (Xinjiang, China). Percutaneous transluminal coronary angioplasty (PTCA) dilatation catheters (2.0 mm/15 mm) were purchased from Thermo Scientific (Waltham, MA, USA). Ophthalmic surgical instruments used in this study were purchased from 66vision (Suzhou, China). Chloral hydrate (0.1 g/mL) was used for anesthesia.
Rat carotid artery balloon injury model
Vascular injury was performed according to a protocol developed by David Anthony Tulis [13]. Intraperitoneal injection of chloral hydrate was used for anesthesia. A neck midline incision was made, and the internal and external carotid arteries of the left carotid artery were separated, which are closely adjacent to the trachea. The distal right common carotid artery and region of the bifurcation were exposed. A 2F Fogarty balloon was inflated with saline to 2.0 atm to distend the common carotid artery and was then pulled back to the external carotid artery. After three repetitions of this procedure, the artery intima was removed completely. The rats were randomly assigned into uninjured (control) group and 6 groups of left carotid artery balloon injury (6 rats per group). Arteries injured by balloon angioplasty were harvested at 0 h, 1 day, 3 days, 7 days, 14 days, and 21 days (experimental groups: control, 0 h, 1 d, 3 d, 7 d, 14 d, 21 d).
Western blot analysis
Common carotid artery segments from the control and injured groups were removed and disrupted. Radio-immunoprecipitation assay (RIPA) buffer with phenylmethanesulfonyl fluoride (100:1 dilution) was then added to the disrupted tissue, incubated on ice for 30 min. Supernatants were collected following centrifugation at 12,000 rpm for 20 min at 4°C to remove debris. Protein samples were mixed with loading buffer and heated to 100°C for 10 min, after which the samples were separated by 10% SDS-PAGE. Proteins were then transferred to polyvinyl difluoride membranes (Solarbio, Beijing, China). Membranes were blocked with 5% nonfat milk in TBST buffer for 1-2 h at room temperature. The blots were incubated with primary antibodies (Table 1) overnight at 4°C.
Table 1.
Antibodies used for western blotting analysis
| Antibody | Clone | Dilution | Source |
|---|---|---|---|
| TGF-β1 | Rabbit pAb | 1:200 | Santa Cruz |
| MMP-14 | Rabbit mAb | 1:1000 | Abcam |
| Nestin | Rabbit mAb | 1:400 | Abcam |
| p-Smad2/3 | Rabbit pAb | 1:100 | Santa Cruz |
| Smad2/3 | Rabbit pAb | 1:100 | Santa Cruz |
| Calponin | Mouse mAb | 1:3000 | Abcam |
| Vimentin | Mouse mAb | 1:1000 | Abcam |
| Collagen III | Rabbit pAb | 1:1000 | Abcam |
pAb, polyclonal antibody; mAb, monoclonal antibody.
Immunohistochemical staining
The EnVision two-step immunohistochemical (IHC) kit (Golden Bridge, Beijing, China) and DAB enhancer (DAKO, Carpinteria, CA) were used to detect specific target proteins. Briefly, paraffin embedded vascular sections were deparaffinized and rehydrated with ethanol and then quenched with 3% hydrogen peroxide for 10 min. Slides were soaked in 10.0 mM sodium citrate buffer and boiled in a pressure cooker for 8 min at 130°C for antigen retrieval. Slides were incubated with primary antibodies (Table 2) overnight at 4°C. Samples were then washed with PBS and subsequently incubated with secondary antibodies for 30 min. The sections were visualized by incubating with diaminobenzidine tetrahydrochloride and counterstained with hematoxylin. PBS was used in place of the primary antibody as negative control.
Table 2.
Antibodies used in the immunohistochemical study
| Antibody | Clone | Dilution | Source | Location |
|---|---|---|---|---|
| TGF-β1 | Rabbit pAb | 1:200 | Santa Cruz | Extracellular Matrix |
| TGF-β1-RI | Rabbit pAb | 1:200 | Santa Cruz | Cell membrane or cytoplasm |
| p-Smad2/3 | Rabbit pAb | 1:50 | Santa Cruz | Cell nucleus |
| MMP-14 | Rabbit mAb | 1:200 | Abcam | Cytoplasm |
| CD29 | Rabbit pAb | 1:600 | Abcam | Cell membrane |
| Nestin | Rabbit mAb | 1:400 | Abcam | Cell membrane |
| Calponin | Mouse mAb | 1:800 | DAKO | Cytoplasm |
| Vimentin | Rabbit mAb | 1:800 | DAKO | Cell membrane |
| Collagen III | Rabbit mAb | 1:600 | Abcam | Cytoplasm |
| Ki-67 | Rabbit pAb | 1:600 | DAKO | Cytoplasm |
pAb, polyclonal antibody; mAb, monoclonal antibody.
Immunofluorescence staining
Immunofluorescence staining was performed on the tissue sections. After blocking in 0.5% bull serum albumin for 30 min, sections were incubated with primary antibodies (anti-nestin and anti-CD29, Abcam, Cambridge, UK) followed by incubation with FITC-conjugated secondary antibodies (Zhongshan Golden Bridge Biotechnology Co., Beijing, China). Nuclei were counterstained with DAPI (Sigma) and observed by confocal microscopy (FLUOVIEW FV300, Olympus, Tokyo, Japan).
Statistical analysis
All experimental data were statistically analyzed using SPSS17.0 software (IBM Corp., Armonk, NY, USA). Quantitative data were presented as the means ± SEs. The differences in gray value were analyzed by Student’s t-test. P-values of less than 0.05 were considered statistically significant.
Results
Balloon injury induces thickening and cell accumulation in neointima
To explore the major components of hyperplasis neointima, we measured intima/media ratio and cell numbers of neointima. The I/M ratio increased significantly beginning at day 7, with the increase sustained through day 14 and reaching a maximum at day 21, after injury. The I/M ratios at 14 and 21 days (3.6 ± 0.1, 5.1 ± 0.2, respectively) were significantly higher (P < 0.01) than that at 7 days (0.9 ± 0.2). There was no neointima formation in control, 0 h, 1 d, and 3 d groups. Additionally, neointima cell numbers were calculated under high power field (HPF), and cell numbers at day 14 (150 ± 10/HPF, n=6) were significantly higher than the numbers at day 7 (53 ± 12/HPF, n=6) and day 21 (98 ± 15/HPF, n=6). The results revealed that neointima thickening was sustained from day 7 to day 21, and the number of cells in neointima peaked at 14 days after arterial injury, while at day 21, there were more intercellular subatances.
Levels of TGF-β1/Smad signaling proteins were elevated following arterial injury
To determine whether TGF-β1/Smad signaling was activated after arterial injury, western blot analysis was used to quantify the levels of the key pathway. TGF-β1 levels increased by day 7 after injury, reached a maximum at day 14, and then decreased at day 21 (Figure 1A and 1B). The TGF-β1 levels of the 14 d group were significantly higher (P < 0.01) than the levels in the 7 d and 21 d groups. Along with TGF-β1, the downstream Smads, including p-Smad2/3 and Smad2/3, were detected as well, to evaluate the activation of TGF-β1/Smad signaling and the results revealed protein levels that were consistent with TGF-β1, with Smad levels reaching a maximum at day 14 (Figure 2A). Smad7 was detected as the antagonist of TGF-β1/Smad signaling, with Smad7 expression significantly lower (P < 0.01) at 14 days than that at 7 days (Data not shown). These results demonstrated that the TGF-β1/Smad signaling was activated in neointima formation and peaked at 14 d after injury.
Figure 1.
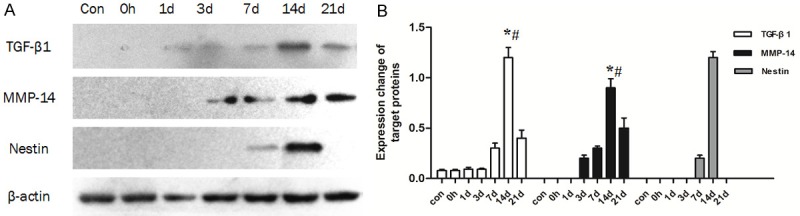
Expression of TGF-β1, MMP-14 and Nestin in arteries following balloon angioplasty as detected by western blot. A: Representative patterns of TGF-β1, MMP-14 and Nestin. B: Densitometry analysis of TGF-β1, MMP-14 and nestin (n=6 rats), *: compared with the 7 d group, P < 0.01; #: compared with the 21 d group, P < 0.01.
Figure 2.
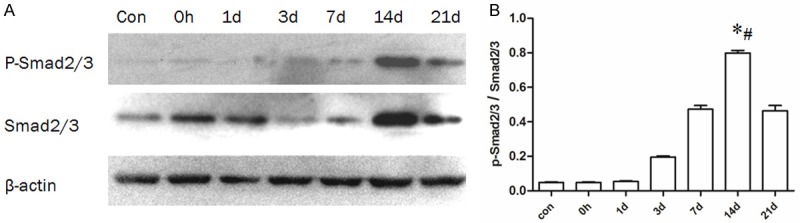
Expression of p-Smad2/3 and Smad2/3 in arteries following balloon angioplasty as detected by western blot. A: Representative patterns of p-Smad2/3 and Smad2/3. B: Quantification of the ratio of p-Smad2/3-to-Smad2/3 (n=6 rats), *: compared with the 7 d group, P < 0.01; #: compared with the 21 d group, P < 0.01.
IHC staining was used to examine if the activated TGF-β1/Smad signaling proteins were localized to the neointima. Neointima cells were strongly positive for TGF-β1, TGF-β1-RI(Data not shown), and p-Smad2/3 at 14 days after injury, and the staining intensity of those proteins in the 14 d group was much stronger than in the 7 d and 21 d groups, while no significant staining was observed in control, 0 h, 1 d, and 3 d groups (Figure 4A, 4B). These results confirm that TGF-β1/Smad signaling was localized to the neointima.
Figure 4.
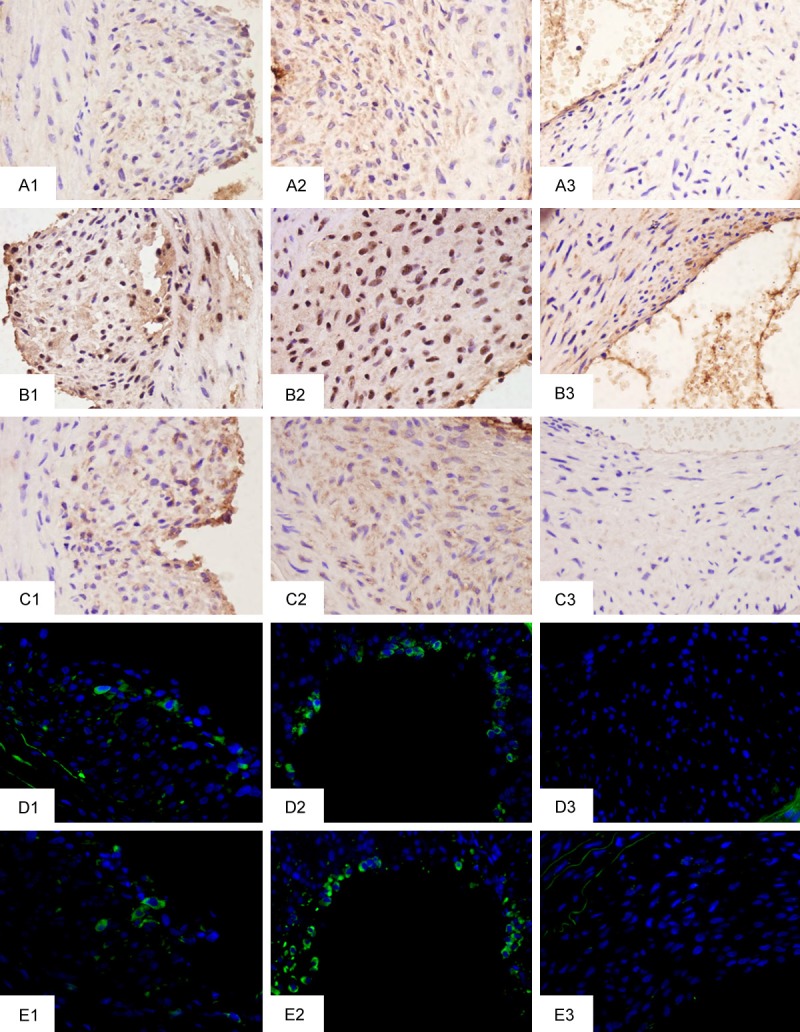
Representative IHC staining of target proteins in injured arteries (magnification 400×). 1, 2, 3 represent 7 d, 14 d, and 21 d groups, respectively. A: TGF-β1. B: p-Smad2/3. C: MMP-14. D: CD29. E: Nestin.
MMP-14 is activated after arterial injury
Next, we measured the levels of MMP-14 to explore the potential correlation with elevated levels of TGF-β1/Smad signaling. We found that MMP-14 levels began to rise at day 3 after injury (Figure 1A and 1B), which preceded that of TGF-β1/Smad signaling activation. MMP-14 levels reached a maximum at day 14, and decreased by day 21. The expression level at 14 days was significantly higher than at day 3, day 7, and day 21 (P < 0.01). There was no detectable expression observed in the control, 0 h, and 1 d groups.
To examine the distribution of MMP-14 within the developing carotid neointima, IHC staining was used. Positive staining of MMP-14 was observed throughout the thickness of the neointima, and the staining intensity at 14 days was much stronger than that in the 7 d and 21 d groups, and no significant staining was apparent in the control, 0 h, 1 d, and 3 d groups (Figure 4C).
MSCs accumulated in neointima after arterial injury
In order to determine whether MSCs accumulated in the injured artery, Nestin was detected as an MSC marker. Nestin exhibited significant expression in the 7 d and 14 d groups only (Figure 1A), and the expression at 14 days was significantly higher (P < 0. 01) than that at 7 days (Figure 1B). To locate the recruited MSCs, immunofluorescence was used to detect the expression of CD29 and Nestin. Results revealed that Nestin- and CD29-positive cells were located at the lumen side of the neointima, with a greater number of positive cells observed at day 14 compared to day 7 samples, while no positive cells were detected in the other groups (Figure 4D and 4E).
Complex phenotype of neointima cells were observed and extracellular matrix protein was secreted from neointima
To initially define the cell type associated with neointima and dissect the effects of MSCs on neointima formation, the expression of vimentin and calponin were examined. Both vimentin and calponin were expressed after injury (Figure 3A), and expression levels were significantly higher in the 14 d group than in the 7 d and 21 d groups (Figure 3B). The expression pattern of vimentin and calponin were similar to TGF-β1/Smad signaling. Furthermore, the immunolocalization of vimentin and calponin showed positive staining in the majority of neointima cells (Figure 5D and 5E). To examine the relationship between the stimulation of TGF-β1/Smad signaling and MMP-14 activation and the production of a new extracellular matrix material in neointima, the expression of collagen III was examined in the injured artery. Collagen III expression increased dramatically in 7 d, 14 d, and 21 d groups, reaching a maximum at 21 days, but showed a noticeable decrease at day 14 relative to day 7 (Figures 3A, 3B and 5G-I). Moreover, Ki-67 was detected to display the proliferation of neointima cells, and the expression peaked at 14 days as well (Figure 5A-C). CD31 was detected to observe whether new endothelial cells were generated, however, no positive cells were observed at day 7, day 14, or day 21 (Figure 5F).
Figure 3.
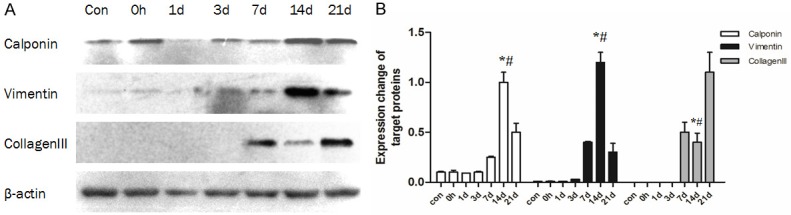
Expression of calponin, vimentin, and collagen III in arteries following balloon angioplasty as detected by western blot. A: Representative patterns of calponin, vimentin, and collagen III. B: Densitometry analysis of calponin, vimentin and collagen III (n=6 rats), *: compared with the 7 d group, P < 0.01; #: compared with the 21 d group, P < 0.01.
Figure 5.
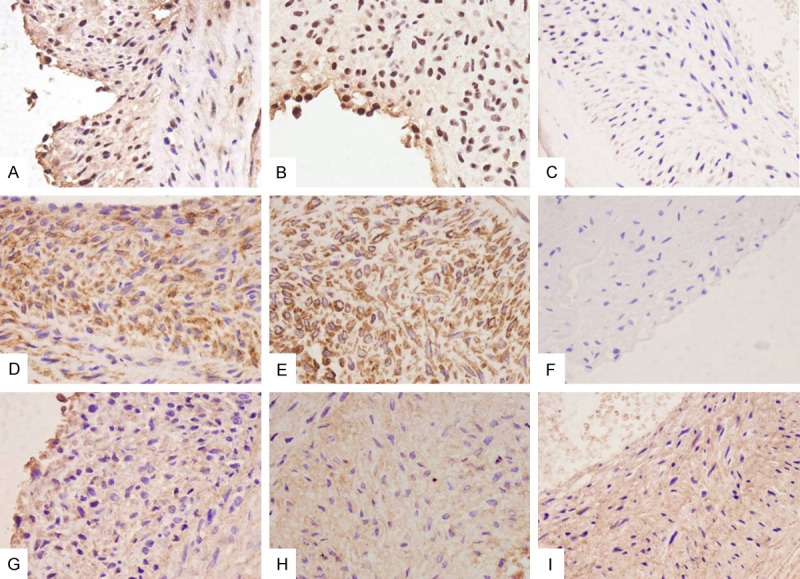
Representative IHC staining of target proteins related with cell type and ECM (magnification 400×). A-C: Staining of Ki-67 in 7 d, 14 d, and 21 d groups. D: Calponin in 14 d group. E: Vimentin in 14 d group. F: CD31 in 21 d group. G-I: Staining of collagen III in neointima in 7 d, 14 d, and 21 d groups.
Discussion
In our present study, western blot analysis and IHC staining were used to examine the protein expression patterns of TGF-β1/Smad signaling components, MSC markers, and MMP-14 at different time points following arterial injury. In addition, the characteristics of neointima cells were explored as well. Based on our present data, we inferred that the influence of TGF-β1 upon neointima formation is achieved via Smad signaling, leading to the recruitment of MSCs to the arterial lesion, and thereby modulating the formation of neointima. This is the first demonstration of the underlying relationship between MMP-14 and TGF-β1/Smad signaling and the potential role of MMP-14 in the progression of neointima hyperplasia.
We used western blot analysis and IHC staining to explore whether the activation of TGF-β1 was time-dependent. Protein expression of TGF-β1 was detectable at 7 days, reaching maximum levels at 14 days and decreasing by 21 days. To measure the activation of Smad signaling, we measured the expression of p-Smad2/3 and Smad2/3 (Figure 2A and 2B) and found their expression profiles similar to that of TGF-β1. These results indicate that TGF-β1, as well as downstream signaling through Smads, were activated in a time-dependent manner in intima-denuded artery. Previously, Majeski et al. [14] reported that mRNA expression of TGF-β1 was increased at 6 h after carotid balloon injury and reached a maximum at 24 h. The increased TGF-β1 mRNA levels were sustained over the next 14 days, during which time a mass of neointimal thickening was formed. And our study demonstrated that activation of TGF-β1/Smad signaling increased and sustained for more than 21 d, and located in neointima, in which neointimal thickening performed a dramatic increasing as well. The result strongly indicated that the biological effect of TGF-β1 in neointima formation after arterial injury may be achieved by Smad signaling.
TGF-β1 has been shown to be involved in restenosis, recruiting MSCs to the injured tissue, affecting lesion repair [3]. CD29 [15] and nestin [16] have been shown to be markers for MSCs. In this study, CD29- or nestin-positive cells were observed in the lumen side of neointima at days 7 and 14 following arterial injury via immunofluorescence, and the number of those cells in the 14 d group was significantly higher than those in the 7 d group. No CD29- or nestin-positive cells were observed in the other groups. Meanwhile, when the expression of CD29 was examined by western blot, the highest levels were observed at 14 days after injury. In consideration of the expression profile of TGF-β1 and p-Smad2/3, which expressed maximally at day 14, we hypothesized that the accumulation of MSCs in the lumen side of neointima was likely regulated by TGF-β1 via Smad signaling. However, the specific role of MSCs in neointima formation is still unknown. To preliminarily explore the contribution of MSCs, we detected other neointima components by using calponin, vimentin, and collagen III. We found that both the smooth muscle cell (SMC) marker calponin and the mesenchymal marker vimentin were expressed in neointima cells, with maximum expression at day 14 while the neointima cell number and Ki-67 index got the maximum as well, similar to the expression pattern of TGF-β1 and Smad. The expression of collagen III, which acts as an ECM component, also increased by day 7, coinciding with the increase in signaling through the TGF-β1/Smad pathway. We inferred from those results that TGF-β1/Smad signaling recruited MSCs to the injured intima and modulated the differentiation and proliferation of MSCs into VSMC-like cells in part and from which ECMs were secreted. Transplantation of MSCs was once applied to demonstrate the contribution of MSCs to neointimal formation due to vascular lesion in vivo [4,16]. Furthermore, various studies suggest that TGF-β1 promotes the migration and recruitment of MSCs to the lesions to participate in tissue repair and remodeling. Evidence shows that a higher potential of MSCs differentiate into cells with a muscular phenotype than hematopoietic stem cells [17]. Although several studies, both in vivo and in vitro, have demonstrated the ability of MSCs differentiation into VSMCs or VSMC-like cells, the underlying mechanism of the cellular or molecular events is still unclear. It has been observed that MSCs co-cultured with VSMCs could acquire a VSMC-like phenotype when stimulated with TGF-β [18]. It has already been demonstrated that TGF-β1 can stimulate the synthesis of type I and III collagen in VSMCs through the Smad3 signaling pathway, resulting in the deposition of ECM in the neointima [19], leading to restenosis. A myofibroblast (MF)-a VSMC-like cell-possessing both an SMC and MSC phenotype, can produce large amounts of ECM components such as fibronectins, collagens, and MMPs. The implantation of MSCs was demonstrated to promote myofibroblast congregation via the induction of TGF-β1/Smad2-signaling in myocardial infarction [20]. It has also been shown that excessive TGF-β1 activation induces myofibroblast differentiation from various precursor cell types, stimulating the secretion of collagens [21]. Collectively, these data suggest that the sustained activation of the TGF-β1/Smad pathway locally may recruit MSCs to injured arteries and induce the differentiation of MSCs to both VSMCs and MFs, leading to the enhanced accumulation of neointima cells and the deposition of ECM after balloon injury.
While examining collagen III expression in hyperplasia neointima, protein expression increased significantly at day 7, day 14, and day 21 compared with other groups. We unexpectedly observed a decrease in collagen III expression at day 14. Based on this observation, we hypothesized that some unknown factor(s) reversed ECM deposition. We first suspected that MMPs, regulators of ECM, were involved in this process. MMP-14, which has been newly found and rarely studied in restenosis, was demonstrated to release latent TGF-β1 from the ECM by proteolytic processing of LTBPs. In our study, MMP-14 was detected by western blot analysis and IHC staining and expression was observed beginning at 3 days after injury, with a sustained increase that reached a maximum at 14 days. This led to a decrease in collagen III while TGF-β1/Smad signaling peaked. It has been suggested that MMP-14 not only cleaves the ECM but also activates signaling pathways such as TGF-β, which is associated with cell migration in skin and cornea wound healing [22]. MMP-14 was demonstrated to promote fibrosis and collagen remodeling, leading to pancreatic fibrosis through TGF-β1/Smad2 signaling [23]. Moreover, the bioactivity of TGF-β1 was regulated by MMP-14 and the balance of the modulated vascular stability [24]. One study revealed the directional role of MMP-14 in neointima formation via the stimulation of the differentiation of VSMC to an invasive and proliferative cell phenotype [25]. Lately, studies demonstrated that MMP-14 not only regulates hMSC collagenolytic activity, but also trafficking and invasion in vitro. All these studies focused on the MT1-MMP-TGF axis, and our results also indicated that MSCs may be not only activated by TGF-β1/Smad signaling but also contribute to the recruitment of MSCs via the proliferation of neointima cells and remodeling of ECM directly or through alternative pathways. Based on the findings from the present study, a more comprehensive examination of specific MMP14-TGFβ1 proteolytic interactions would be warranted.
Taken together, our study findings indicate that TGF-β1/Smad signaling was activated post intima denudation, and the activation correlated with the recruitment and differentiation of MSCs, proliferation of neointima cells, and deposition of ECM. We provide a new insight on the mechanisms of neointima formation and the role of TGF-β1/Smad signaling, likely induced by MMP-14, which may also recruit MSCs and induce VSMC proliferation directly or through other pathways. Additional studies are necessary to further explore the specific relationships between TGF-β1, MMP14, and MSCs in arterial injury.
Acknowledgements
National Natural Science Foundation of China (No. 81160018); The Corps Doctor Foundation (No. 2014BB018); Outstanding Youth Science and Technology Talent Cultivation Plan of Shihezi University (2013ZRKXJQ05); One Thousand Youth Talents Plan.
Disclosure of conflict of interest
None.
References
- 1.Hiroyuki H, Giulio G, Marie-Luce BP. Arterial smooth muscle cell heterogeneity implications for atherosclerosis and restenosis development. Arterioscler Thromb Vasc Biol. 2003;23:1510–1520. doi: 10.1161/01.ATV.0000090130.85752.ED. [DOI] [PubMed] [Google Scholar]
- 2.ten Dijke P, Arthur HM. Extracellular control of TGFbeta signalling in vascular development and disease. Nat Rev Mol Cell Biol. 2007;8:857–869. doi: 10.1038/nrm2262. [DOI] [PubMed] [Google Scholar]
- 3.Wan M, Li C, Zhen G, Jiao K, He W, Jia X, Wang W, Shi C, Xing Q, Chen YF, Beur SJD, Yu B, Cao X. Injury-activated TGFβcontrols mobilization of MSCs for tissue remodeling. Stem Cells. 2012;10:1066–1087. doi: 10.1002/stem.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Camussi G, Li M, Li S, Yu L, Wu J, She T, Gan Y, Hu Z, Liao W, Xia H. Bone Mesenchymal Stem Cells Contributed to the Neointimal Formation after Arterial Injury. PLoS One. 2013;8:e82743. doi: 10.1371/journal.pone.0082743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abedin M. Mesenchymal Stem Cells and the Artery Wall. Circulation Res. 2004;95:671–676. doi: 10.1161/01.RES.0000143421.27684.12. [DOI] [PubMed] [Google Scholar]
- 6.Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodeling. Nat Rev Mol Cell Biol. 2007;8:221–233. doi: 10.1038/nrm2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang M, Zhao D, Spinetti G, Zhang J, Jiang LQ, Pintus G, Monticone R, Lakatta EG. Matrix metalloproteinase 2 activation of transforming growth factor-beta1 (TGF-beta1) and TGF-beta1-type II receptor signaling within the aged arterial wall. Arterioscler Thromb Vasc Biol. 2006;26:1503–1509. doi: 10.1161/01.ATV.0000225777.58488.f2. [DOI] [PubMed] [Google Scholar]
- 8.Tatti O, Vehvilainen P, Lehti K, Keskioja J. MT1-MMP releases latent TGF-β1 from endothelial cell extracellular matrix via proteolytic processing of LTBP-1. Exp Cell Res. 2008;314:2501–2514. doi: 10.1016/j.yexcr.2008.05.018. [DOI] [PubMed] [Google Scholar]
- 9.Mountain DJ, Kirkpatrick SS, Freeman MB, Stevens SL, Goldman MH, Grandas OH. Role of MT1-MMP in estrgen-mediated cellular processes of intimal hyperplasia. J Surg Res. 2012;173:224–231. doi: 10.1016/j.jss.2011.05.037. [DOI] [PubMed] [Google Scholar]
- 10.Ding Q, Chai H, Mahmood N, Tsao J, Mochly-Rosen D, Zhou W. Matrix metalloproteinases modulated by protein kinase Cε mediate resistin-induced migration of human coronary artery smooth muscle cells. J Vasc Surg. 2011;53:1044–1051. doi: 10.1016/j.jvs.2010.10.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Campeau PM, Rafei M, Boivin MN, Sun Y, Grabowski GA, Galipeau J. MT1MMP14 controls human MSCs trafficking and differentiantion. Blood. 2010;115:221–229. [Google Scholar]
- 12.Sun X, Gao X, Zhou L, Sun L, Lu C. PDGFBB induced MT1-MMP expression regulates proliferation and invasion of MSCs in 3D clloagen via MEK ERK12 and PI3K AKT signaling. Cell Signal. 2013;25:1279–1287. doi: 10.1016/j.cellsig.2013.01.029. [DOI] [PubMed] [Google Scholar]
- 13.Tulis DA. Rat Carotid Artery Balloon Injury Model. Methods Mol Med. 2007;139:1–30. doi: 10.1007/978-1-59745-571-8_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Majesky MW, Lindner V, Twardzik DR, Schwartz SM, Reidy MA. Production of transforming growth factorβ1 during repair of arterial injury. J Clin Invest. 1991;88:904–910. doi: 10.1172/JCI115393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shoji M, Koba S, Kobayashi Y. Roles of Bone-Marrow-Derived Cells and Inflammatory Cytokines in Neointimal Hyperplasia after Vascular Injury. Biomed Res Int. 2014;2014:1–8. doi: 10.1155/2014/945127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Méndez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, MacArthur BD, Lira SA, Scadden DT, Ma’ayan A, Enikolopov GN, Frenette PS. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466:829–834. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawada H, Fujita J, Kinjo K, Matsuzaki Y, Tsuma M, Miyatake H, Muguruma Y, Tsuboi K, Itabashi Y, Ikeda Y, Ogawa S, Okano H, Hotta T, Ando K, Fukuda K. Nonhematopoietic mesenchymal stem cells can be mobilized and differentiate into cardiomyocytes after myocardial infarction. Blood. 2004;104:3581–3587. doi: 10.1182/blood-2004-04-1488. [DOI] [PubMed] [Google Scholar]
- 18.Vallabhaneni KC, Tkachuk S, Kiyan Y, Shushakova N, Haller H, Dumler I, Eden G. Urokinase receptor mediates mobilization, migration, and differentiation of mesenchymal stem cells. Cardiovasc Res. 2011;90:113–121. doi: 10.1093/cvr/cvq362. [DOI] [PubMed] [Google Scholar]
- 19.Lu P, Wang S, Cai W, Sheng C. Role of TGF-β1/Smad3 Signaling Pathway in Secretion of Type I and III Collagen by Vascular Smooth Muscle Cells of Rats Undergoing Balloon Injury. J Biomed Biotechnol. 2012;2012:1–8. doi: 10.1155/2012/965953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Du Y, Yao R, Pu S, Zhao X, Liu G, Zhao L, Chen Q, Li L. Mesenchymal stem cells implantation increases the myofibroblasts congregating in infarct region in a rat model of myocardial infarction. Zhonghua Xin Xue Guan Bing Za Zhi. 2012;40:1045–1050. [PubMed] [Google Scholar]
- 21.Forte A, Della Corte A, De Feo M, Cerasuolo F, Cipollaro M. Role of myofibroblasts in vascular remodelling: focus on restenosis and aneurysm. Cardiovasc Res. 2010;88:395–405. doi: 10.1093/cvr/cvq224. [DOI] [PubMed] [Google Scholar]
- 22.Joo CK, Seomun Y. Matrix metalloproteinase (MMP) and TGFβ1-stimulated cell migration in skin and cornea wound healing. Cell Adh Migr. 2008;2:252–253. doi: 10.4161/cam.2.4.6772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krantz SB, Shields MA, Dangi-Garimella S, Cheon EC, Barron MR, Hwang RF, Rao MS, Grippo PJ, Bentrem DJ, Munshi HG. MT1-MMP Cooperates with KrasG12D to Promote Pancreatic Fibrosis through Increased TGF-β Signaling. Mol Cancer Res. 2011;9:1294–1304. doi: 10.1158/1541-7786.MCR-11-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sounni NE, Dehne K, van Kempen L, Egeblad M, Affara NI, Cuevas I, Wiesen J, Junankar S, Korets L, Lee J, Shen J, Morrison CJ, Overall CM, Krane SM, Werb Z, Boudreau N, Coussens LM. Stromal regulation of vessel stability by MMP14 and TGFbeta. Dis Model Mech. 2010;3:317–332. doi: 10.1242/dmm.003863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lehti K, Rose NF, Valavaara S, Weiss SJ, Keski-Oja J. MT1-MMP promotes vascular smooth muscle dedifferentiation through LRP1 processing. J Cell Sci. 2008;122:126–135. doi: 10.1242/jcs.035279. [DOI] [PubMed] [Google Scholar]