

Abstract
Shigella flexneri, which replicates in the cytoplasm of intestinal epithelial cells, can use the Embden-Meyerhof-Parnas, Entner-Doudoroff, or pentose phosphate pathway for glycolytic carbon metabolism. To determine which of these pathways is used by intracellular S. flexneri, mutants were constructed and tested in a plaque assay for the ability to invade, replicate intracellularly, and spread to adjacent epithelial cells. Mutants blocked in the Embden-Meyerhof-Parnas pathway (pfkAB and pykAF mutants) invaded the cells but formed very small plaques. Loss of the Entner-Doudoroff pathway gene eda resulted in small plaques, but the double eda edd mutant formed normal-size plaques. This suggested that the plaque defect of the eda mutant was due to buildup of the toxic intermediate 2-keto-3-deoxy-6-phosphogluconic acid rather than a specific requirement for this pathway. Loss of the pentose phosphate pathway had no effect on plaque formation, indicating that it is not critical for intracellular S. flexneri. Supplementation of the epithelial cell culture medium with pyruvate allowed the glycolysis mutants to form larger plaques than those observed with unsupplemented medium, consistent with data from phenotypic microarrays (Biolog) indicating that pyruvate metabolism was not disrupted in these mutants. Interestingly, the wild-type S. flexneri also formed larger plaques in the presence of supplemental pyruvate or glucose, with pyruvate yielding the largest plaques. Analysis of the metabolites in the cultured cells showed increased intracellular levels of the added compound. Pyruvate increased the growth rate of S. flexneri in vitro, suggesting that it may be a preferred carbon source inside host cells.
INTRODUCTION
Shigella spp. are the causative agents of bacillary dysentery, an invasive disease of the colonic epithelium that is associated with as many as 1,000,000 deaths per year (1). Shigella species are closely related to Escherichia coli (2), but its virulence is facilitated by the presence of a large virulence plasmid which encodes a type III secretion system (T3SS) as well as secreted effector proteins. Pathogenesis of Shigella is best characterized in Shigella flexneri. Following ingestion, S. flexneri transits to the large intestine, and upon contact with colonic epithelial cells, it uses its T3SS to induce uptake of the bacterium into a vacuole (3). S. flexneri quickly lyses the vacuole membrane and replicates within the host cell cytoplasm. IcsA, which is located at a single pole on the surface of S. flexneri, polymerizes host cell actin, promoting movement of the bacteria into adjacent cells (4, 5). Subsequent damage to the epithelium and the resulting host inflammatory response are responsible for most of the symptoms of shigellosis (3, 6).
Studies of Shigella pathogenesis have benefitted from the availability of cell culture models that allow analysis of the interactions between S. flexneri and eukaryotic cells. Invasion of cultured epithelial cells can be visualized by microscopy, while intracellular replication and cell-to-cell spread are assessed by the formation of plaques in confluent cell monolayers (7). Analysis of S. flexneri mutants in invasion and plaque assays have allowed the identification of genes required for invasion, intracellular replication, and spread of bacteria (8–10), primarily those encoding the T3SS, secreted effector proteins, and IcsA. The expression of most of these genes requires the activator protein VirB and its upstream activator VirF. Expression of virF and virB is modulated in response to various environmental signals, including temperature, pH, osmolarity, and iron levels (11–18).
It is less well understood how Shigella adapts to the intracellular environment and which metabolic pathways it uses for growth and multiplication within the host cell. Extracellular bacteria microinjected into the eukaryotic cell cytoplasm fail to replicate (19), suggesting that intracellular cytosolic pathogens have adaptations for growth inside cells. This is supported by studies of the transcriptome and proteome of intracellular shigellae, which reveal specific responses to growth in the intracellular environment (20–24). For example, the increased expression of iron transport genes and high levels of iron transport proteins indicate that the eukaryotic cytosol is an iron-restricted environment.
The carbon sources available to bacteria growing in the eukaryotic cell cytosol and the pathways used to metabolize those carbon sources are not well understood. Changes in gene expression patterns suggest that the bacteria make adaptations to efficiently use the carbon sources available inside the cell (22, 23). One potential intracellular carbon source is glucose and related sugars, but the gene expression studies do not give a clear picture of the role of glucose metabolism in intracellular growth and plaque formation. Genes encoding proteins in the Embden-Meyerhof-Parnas (EMP) glycolytic pathway were downregulated in intracellular S. flexneri compared to in vitro-grown bacteria (20), as were the genes for the regulatory proteins CsrA, which promotes glycolysis while reducing gluconeogenesis and glycogen storage (25, 26), and Cra, which, conversely, promotes gluconeogenesis while reducing glycolysis (27). In spite of downregulation csrA and cra, S. flexneri strains carrying mutations in either of these genes failed to form normal plaques in Henle cell monolayers (28). This suggests that the properly regulated flow of carbon through glycolytic and gluconeogenesis pathways is required for virulence. Carbon flux analysis (29) of an intracellular pathogen closely related to Shigella, enteroinvasive E. coli, growing in the mammalian cell cytoplasm supports a model in which glucose and glucose phosphate are used by the bacteria, largely through glycolysis, but C3 compounds such as glycerol and pyruvate can be used by mutants blocked in glucose uptake.
Although the EMP pathway likely plays a major role in sugar catabolism by intracellular S. flexneri, these bacteria also have the genes for both the Entner-Doudoroff (ED) and the pentose phosphate (PP) pathways, shown in Fig. 1 (30, 31). However, the relative contribution of these pathways to carbon utilization by intracellular Shigella is unknown. In this work, we constructed strains blocked in each of these carbon catabolism pathways and tested their ability to form plaques in epithelial cell monolayers. Mutations blocking the EMP pathway prevented the formation of wild-type plaques, while the pentose phosphate pathway was not required for normal plaque phenotype. We also present evidence that the Entner-Doudoroff pathway is used, but is not essential, for intracellular replication. Supplementation of the cell culture medium with pyruvate increased the intracellular level of pyruvate and led to increased plaque size for both the wild type and all mutant strains tested.
FIG 1.
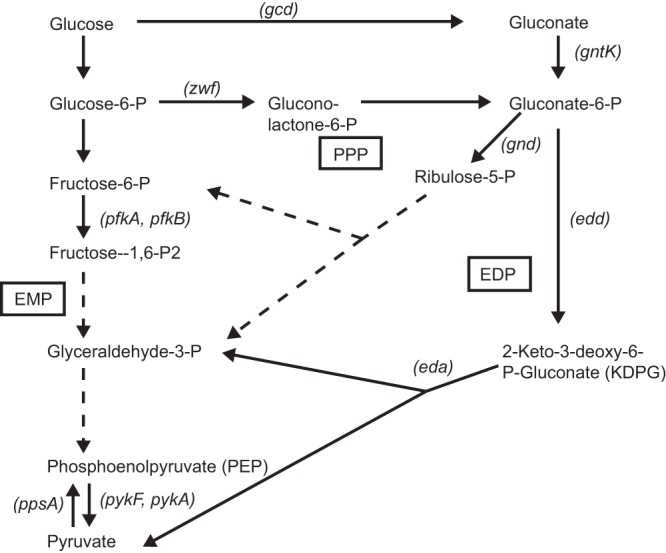
Major glycolytic pathways in S. flexneri. Pathways are indicated in boxes. Solid arrows indicate a single enzymatic reaction, while dashed arrows indicate multiple steps that are not shown. Genes discussed in this paper are indicated in parentheses beside the reaction. EMP, Embden-Meyerhof-Parnas pathway; PPP, pentose phosphate pathway; EDP, Entner-Doudoroff pathway.
MATERIALS AND METHODS
Media and growth conditions.
Bacterial strains and plasmids used in this study are shown in Table 1. All strains were routinely maintained at −80°C in tryptic soy broth (TSB) supplemented with 20% (vol/vol) glycerol. Escherichia coli strains were grown in LB (1% tryptone, 0.5% yeast extract, 1% NaCl) or on LB agar plates. Shigella flexneri was grown on TSB agar containing 0.01% (wt/vol) Congo red (CR) to select for red colonies (CR+), indicative of a functional T3SS. When needed, antibiotics were added at final concentrations of 25 μg of ampicillin per ml, 50 μg of kanamycin per ml, or 20 μg of gentamicin per ml.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Characteristic(s) | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| DH5α | E. coli cloning strain; λpir | J. Kaper |
| JW0120 | gcd::Kan | Keio Collection |
| JW1666 | pykF::Kan | Keio Collection |
| JW1839 | eda::Kan | Keio Collection |
| JW1840 | edd::Kan | Keio Collection |
| JW1841 | zwf::Kan | Keio Collection |
| JW1843 | pykA::Kan | Keio Collection |
| JW3887 | pfkA::Kan | Keio Collection |
| JW5280 | pfkB::Kan | Keio Collection |
| JW3400 | gntK::Kan | Keio Collection |
| S. flexneri | ||
| 2457T | Wild-type serotype 2a | Walter Reed Army Institute of Research |
| EAWS132 | 2457T pfkA::Kan | This study |
| EAWS130 | 2457T pfkB::Kan | This study |
| EAWS118 | 2457T ΔpfkA | This study |
| EAWS122 | 2457T ΔpfkA pfkB::Kan | This study |
| EAWS105 | 2457T pykA::Kan | This study |
| EAWS107 | 2457T pykF::Kan | This study |
| EAWS120 | 2457T ΔpykF | This study |
| EAWS124 | 2457T ΔpykF pykA::Kan | This study |
| AGS200 | 2457T ppsA::Kan | 54 |
| EAWS134 | 2457T eda::Kan | This study |
| EAWS135 | 2457T edd::Kan | This study |
| EAWS144 | 2457T Δeda edd::Kan | This study |
| CFS103 | 2457T gntK::Kan | This study |
| CFS104 | 2457T gnd::Kan | This study |
| ARS100 | 2457T zwf::Kan | This study |
| ARS101 | 2457T gcd::Kan | This study |
| NHS102 | 2457T Δzwf | This study |
| NHS104 | 2457T Δzwf gcd::Kan | This study |
| Plasmids | ||
| pCP20 | Flp recombinase, Ampr, ts | 55 |
| pEda | eda cloned into SmaI site of pWKS30 | This study |
| pWKS30 | Low-copy-number cloning vector, Ampr | 56 |
| pPykF | pykF cloned into SmaI site of pWKS30 | This study |
| pWPfkA | pfkA cloned into pWSK29 | 28 |
To determine growth of the bacteria with glucose, pyruvate or gluconate as carbon sources, CR+ colonies were grown overnight at 30°C with aeration. Bacteria were harvested by centrifugation and resuspended in sterile saline. The optical density of the bacterial suspension was determined, and the bacteria were diluted to a final optical density at 650 nm (OD650) of 0.05 in M63 medium supplemented with 0.2 mg/ml of nicotinic acid and glucose (11.1 mM), pyruvate (22.2 mM), or a mixture of the two (5.55 mM glucose and 11.1 mM pyruvate). The concentrations were chosen to provide equal amounts of carbon. To test for growth in gluconate, M63 medium contained 0.2% gluconate. The cultures were grown at 37°C with aeration, and the optical density was evaluated at least hourly for 8 h total. The doubling time was calculated by exponential regression analysis as described by Roth (32).
Construction of bacterial mutant strains.
Most of the S. flexneri mutant strains were constructed using the in-frame E. coli mutants comprising the Keio Collection (33) via bacteriophage P1 transduction of the mutant allele into wild-type S. flexneri 2457T. CR+ strains were tested for the Kan insertion by PCR and confirmed by the change in size of the PCR product. Primers were gene specific for pykA (pykA_F and pykA_R), pykF (pykF-F-Alex and pykF-R-Alex), pfkB (pfkB_fwd and pfkB_rev), pfkA (pfkA_fwd and pfkA_rev), edd (edd_fwd and edd_rev), eda (eda_fwd3 and eda_rev3), gcd (gcd FWD and gcd REV), zwf (zwf_F3 and zwf_R3), and gntK (gntK For2 and gntK Rev). In the cases in which the gene was similar in size to the Kan cassette, internal Kan cassette primers kt and k2 (33) were used in conjunction with external gene-specific primers to confirm proper insertion of the Kan cassette.
Double pfkAB (EAWS122), pykAF (EAWS124), and zwf gcd (NHS104) mutants were constructed using a two-step procedure wherein the kanamycin resistance cassette that created the first mutation was excised using the plasmid pCP20 (30), forming EAWS118, EAWS120, and NHS102. These mutants had the same plaque phenotype as the parent strain (data not shown). The second mutation was then introduced by P1 transduction. PCR analysis was used to confirm excision of the first gene and proper Kan insertion into the second gene. The edd and eda genes are adjacent, and the method of Datsenko and Wanner (34) was used to create a single deletion that removed both genes. The primers used to create this deletion were edd_Keio_fwd and eda_Keio_rev (Table 2). The gnd mutant was also made by the method of Datsenko and Wanner (34), because differences in the flanking sequences between E. coli K-12 and S. flexneri prevented transfer of the mutation from E. coli by transduction. The primers for gnd were gnd Keio 3′ and gnd Keio 5′ (Table 2). All strains grew at similar rates in minimal medium M63 with sodium lactate as the sole carbon source, confirming that there was no overall defect in growth.
TABLE 2.
Primers used in this study
| Primer | Sequence (5′→3′) |
|---|---|
| pykA_F | ATCGCGGCGTTATTTCATTCG |
| pykA_R | ATCCGGCAACGTACTTACTC |
| pykF-F-Alex | CTCACACTGACAACTTCGGC |
| pykF-R-Alex | GCAGAGTAGAACCCAGGATTACC |
| pfkB_fwd | TTCGGTGCCAGACTGAAATC |
| pfkB_rev | AATGCTGGGGGAATGTTTTT |
| pfkA_fwd | AGCCAGACCCGCATTTTGTG |
| pfkA_rev | TTCATCGCCCGACTCTCTTATG |
| edd_fwd | TGAAACGCGTTGTGAATCAT |
| edd_rev | TCAGAGTTTTCTCTCGCCTGA |
| edd_Keio_fwd | CGCGGTGTGAATCATCCTGCTCTGACAACTCAATTTCAGGAGCCTTTATGATTCCGGGGATCCGTCGAC |
| eda_Keio_rev | GCCCGATCCTAGATCGGGCATTTTGACTTTTACAGCTTAGCGCCTTCTACTGTAGGCTGGAGCTGCTTC |
| eda_fwd3 | ACCTGCAAATCACCAAGCTG |
| eda_rev3 | TGCCGTTATTCTGCCACAAC |
| gcd FWD | AAAGCTCGAATTGTGATGACG |
| gcd REV | AAATATGCCCGGATCTCTCC |
| zwf_F3 | AAATTACAAGTATACCCTGGCTTAAGTA |
| zwf_R3 | ACAACGCGTTTCATTCAGAGGATTTAT |
| gntK For2 | ATGGAGCCACGACTTGCGAG |
| gntK Rev | ACTTCAAAGAACAGCGGTAGCG |
| gnd Keio 3′ | GCCTCAATTTTATTGTTGGTTAAATCAGATTAATCCAGCCATTCAGTATGTGTAGGCTGGAGCTGCTTCG |
| gnd Keio 5′ | TATAACGGTTAATAGACGGTTAATAATATAGAAGACAGGAGTAAACAATGATTCCGGGGATCCGTCGACC |
| kt | CGGCCACAGTCGATGAATCC |
| k2 | CGGTGCCCTGAATGAACTGC |
Plasmid construction.
pPykF and pEda were constructed by PCR amplification of the pykF and eda genes from wild-type S. flexneri strain 2457T DNA using the primer pairs pykF-F-Alex/pykF-R-Alex and eda_fwd/eda_rev, respectively (Table 2). The PCR product was cloned into the SmaI site of pWKS30. The correct sequence of the plasmid inserts was confirmed by DNA sequencing at the University of Texas DNA sequencing facility.
Cell culture assays.
Henle cells (intestinal 407; American Type Culture Collection) were cultured in minimal essential medium (MEM; Gibco) augmented with 10% Bacto tryptone phosphate broth (Difco, Becton, Dickinson and Company, Franklin Lakes, NJ), 10% fetal bovine serum (Gibco), 2 mM glutamine, and 1× nonessential amino acids (Gibco) (Henle medium). Henle cells were incubated at 37°C with 5% CO2. Invasion assays were performed using a modification of a previously described protocol (28). Briefly, CR+ colonies were inoculated into LB. After overnight growth at 30°C with shaking, the culture was diluted 1:100 into LB and grown at 37°C with shaking to mid-log phase (OD650 = 0.6 to 0.9). The bacteria were centrifuged and resuspended in sterile saline at a final concentration of 2 × 109 CFU/ml. One hundred microliters of this suspension was added per well to a subconfluent monolayer of Henle cells in 35-mm six-well polystyrene plates (Corning) and centrifuged for 10 min at 1,000 × g. The monolayers were incubated at 37°C with 5% CO2 for 30 min and then washed 4 times with phosphate-buffered saline (PBS-D; 1.98 g of KCl, 8 g of NaCl, 0.02 g of KH2PO4, and 1.4 g of K2HPO4 per liter; pH = 7.5) to remove extracellular bacteria. Fresh medium containing gentamicin, to prevent growth of extracellular bacteria, was added, and the infected monolayers were incubated for an additional 40 min at 37°C with 5% CO2. The monolayers were washed twice with PBS-D and stained with Wright-Giemsa stain (Camco, Ft. Lauderdale, FL). Internalized bacteria were visualized by microscopy. At least 300 Henle cells were examined per well, and cells containing three or more internal bacteria were scored positive for invasion. The invasion rate per assay was calculated based on the average of three wells, and each strain was assayed a minimum of three times. Dilution plate counts were made of the inoculum to ensure that similar numbers of bacteria were added.
Plaque assays were performed as previously described (24). Briefly, S. flexneri was grown as described for the invasion assay. The bacteria were centrifuged and resuspended in sterile saline to a final concentration of 105 CFU/ml, and 100 μl of this suspension was added per well to a confluent monolayer of Henle cells incubated in 35-mm six-well polystyrene plates (Corning). The plates were centrifuged for 10 min at 1,000 × g and incubated at 37°C with 5% CO2 for 1 h. Then the monolayers were washed four times with phosphate-buffered saline and incubated in Henle medium with gentamicin for 72 h at 37°C with 5% CO2, with a change of medium after 24 to 48 h. The monolayers were stained with Wright-Giemsa stain (Camco, Ft. Lauderdale, FL) to allow visualization of the plaques. Dilution plate counts were done on the inoculum to ensure that similar numbers of CR+ bacteria were added to each well.
For the competitive plaque assay, the wild-type and mutant strains were grown as described for the plaque assay and mixed in a 1:1 ratio, for a total bacterial count of 105 CFU/ml. Dilution plate counts were done on CR agar with or without kanamycin to determine the total number of bacteria and the number of mutant bacteria, respectively, in the inoculum. Bacterial infection and incubation were performed as described above for the plaque assay protocol, except that at 72 h, the infected Henle monolayers were washed twice with PBS-D and then lysed in 1 ml of deionized sterile water. The lysate was diluted and plated in parallel on CR agar with or without kanamycin. The competitive index was calculated by dividing the ratio of mutant bacteria recovered by the ratio of mutant bacteria in the inoculum.
To determine the effects of added glucose or pyruvate, the plaque assay was performed as described above, except that during the first 48 h of the assay, the Henle medium was left unsupplemented or was supplemented with either 44.4 mM glucose or pyruvate as indicated below.
Biolog phenotypic microarray.
The modified version of the Biolog (Hayward, CA) protocol was performed (35). In brief, bacteria were grown to late log phase at 37°C with aeration in LB and then centrifuged and resuspended in sterile saline. A master mix consisting of inoculating fluid, redox dye A, 5 × 107 CFU/ml of bacteria, and 2 μg of nicotinic acid per ml was prepared, and 100 μl of the mix was added to each well of plates PM1 and PM2A. Reduction of tetrazolium dye by respiring cells was measured by optical density. Data were collected every 15 min for 24 h.
Metabolomics analysis.
To assess changes in the relative concentration of intracellular carbon metabolites when cells were incubated in Henle medium with or without supplemental pyruvate or glucose, approximately 3 × 106 Henle cells were seeded onto tissue culture-treated plates (Cellstar; 100- by 20-mm TC dish) and incubated at 37°C with 5% CO2 in standard Henle medium. After overnight incubation, the medium was changed to fresh Henle medium or to Henle medium supplemented with 44.4 mM glucose or 44.4 mM pyruvate, and cells were incubated for 24 h at 37°C with 5% CO2. The medium was removed, and the cells were detached by trypsin treatment, resuspended in sterile PBS, and transferred to sterile 15-ml conical tubes. The Henle cells were pelleted, the PBS was removed and the cell pellet was flash-frozen in a dry ice-ethanol bath before storage at −80°C. The cells were lysed and the intracellular contents were analyzed by gas chromatography-mass spectrometry (GC-MS) at the University of Utah Metabolomics Core Facility. Internal standards were added for normalization of the data. The values of each metabolite were compared between samples from cells incubated in unsupplemented Henle medium with those incubated in Henle medium with supplemental glucose or pyruvate. The values of at least three biological samples were analyzed for each condition. A two-tailed Student t test was used to identify statistically significant differences.
RESULTS
The EMP pathway is essential for intracellular growth of S. flexneri.
To assess the role of carbon metabolism in S. flexneri virulence, isogenic strains with mutations blocking the Embden-Meyerhof-Parnas (EMP), pentose phosphate (PP), or Entner-Doudoroff (ED) glycolytic pathway (Fig. 1) were constructed. These were tested in a plaque assay to assess the ability of each mutant to invade epithelial cells, grow intracellularly, and spread to adjacent cells, characteristics of wild-type S. flexneri. To determine the role of the EMP pathway, we constructed mutants defective in the phosphofructokinase or pyruvate kinase enzymes, which catalyze reactions that occur near the beginning or the end of the pathway, respectively. Unlike other enzymes in the EMP pathway, these enzymes catalyze reactions that are irreversible and do not function in gluconeogenesis. Both the phosphofructokinase and pyruvate kinase activities are carried out by two isoenzymes, with one responsible for approximately 90% of the activity (PfkI and PykF) and the other performing a minor role (PfkII, PykA) (36–38). Analysis of strains with single mutations showed that loss of either of the major enzymes, PfkI (pfkA) or PykF (pykF), resulted in a small plaque phenotype, while those with mutations in genes encoding the minor enzymes, PfkII (pfkB) or PykA (pykA), formed plaques with wild-type morphology (Fig. 2). pfkAB and pykAF double mutants were also constructed, and each of these had a plaque phenotype that was more severe than that observed with the single mutants in the major enzyme gene, the pfkA and pykF mutants. The pykAF mutant formed plaques that were smaller than those formed by the pfkAB mutant (Fig. 2).
FIG 2.
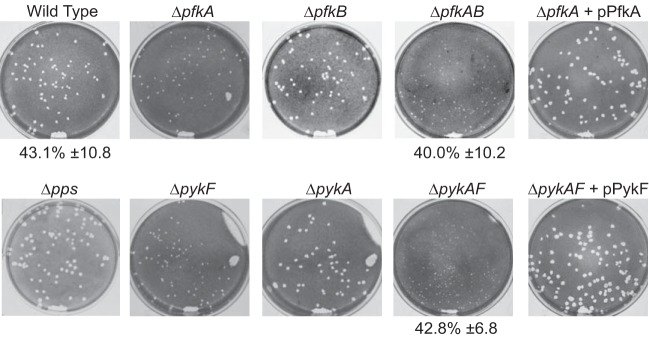
Effect of mutations in Embden-Meyerhof-Parnas pathway carbon metabolism genes on S. flexneri plaque formation. Mutants with defects in EMP pathway genes were tested for the ability to invade, replicate, and spread by assessing the plaque phenotype on a Henle cell monolayer. Where indicated, the mutant strain was carrying a plasmid encoding a wild-type gene. The invasiveness of wild-type and selected mutant strains was determined and is indicated below the picture as the percentage of Henle cells containing three or more intracellular bacteria (average ± standard deviation for ≥3 assays).
The number of small plaques visible with both the pfkA and pykF mutants was similar to that observed with the wild type, suggesting that these mutants could invade the cell normally but had a defect in intracellular replication or spread. This was confirmed by measuring invasion rates of the pfkAB and pykAF mutants, both of which were similar to that of the wild-type parent (Fig. 2). This is in contrast to results with a pfkA mutant we isolated previously, which had reduced invasion (28). The reasons for the differences in invasion are not clear, but they likely indicate other differences in the two mutant strains. To confirm that the plaque defect was solely due to loss of the glycolytic enzymes, we complemented the pfkAB and pykAF double mutants with plasmids expressing pfkA and pykF, respectively, and observed restoration of the wild-type plaque phenotype (Fig. 2). In contrast, ppsA, which encodes the enzyme for the first committed step in gluconeogenesis, the conversion of pyruvate to phosphoenolpyruvate, was not required for plaque formation (Fig. 2).
The pentose phosphate pathway is not required for plaque formation.
The first enzyme in the pentose phosphate pathway, the glucose 6-phosphate dehydrogenase encoded by zwf, is rate limiting and tightly regulated (39–41). A zwf mutant was constructed and found to have a wild-type phenotype in the plaque assay (Fig. 3). To prevent a potential reroute from glucose to gluconate that would bypass the dehydrogenase (Fig. 1), a gcd mutant and a zwf gcd double mutant were constructed and tested in the plaque assay. Neither of these mutants showed a defect in plaque formation (Fig. 3). Carbon could also enter the pentose phosphate pathway via uptake of exogenous gluconate, which could be metabolized to gluconate-6 phosphate by gluconate kinase, encoded by gntK, and then to ribulose 5-phosphate by Gnd. Therefore, both gntK and gnd mutants were constructed to block the pentose phosphate pathway downstream of gluconate. Neither mutation prevented plaque formation (Fig. 3). These data indicate that the pentose phosphate pathway is not needed for plaque formation by S. flexneri. Further, we assayed S. flexneri for growth with gluconate as the sole carbon source. The wild-type strain grew poorly in minimal medium with gluconate (Fig. 4) and failed to grow on solid medium containing gluconate as the carbon source (data not shown). Thus, it is unlikely that the pentose phosphate pathway or gluconate metabolism plays a major role in growth of intracellular Shigella.
FIG 3.

Effect of mutations in pentose phosphate pathway or Entner-Doudoroff pathway genes on S. flexneri plaque formation. Mutants with defects in carbon metabolism genes were assayed for invasion and plaque formation as described for Fig. 2.
FIG 4.
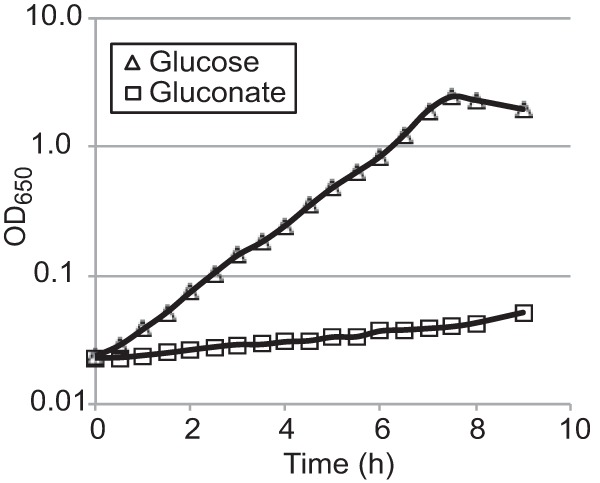
Growth of S. flexneri with glucose or gluconate as the sole carbon source. Wild-type S. flexneri 2457T was grown in minimal medium with glucose or gluconate (0.2%) as the sole carbon source. The OD650 was monitored every 30 min for 8 h.
The Entner-Doudoroff pathway is not essential but is utilized intracellularly.
The two genes encoding enzymes specific to the Entner-Doudoroff pathway, edd and eda, were mutated to assess their role in plaque formation. While the eda mutant formed very small plaques, the edd formed wild-type-size plaques. Both had rates of invasion similar to that of the wild type, suggesting that the defect in plaque formation occurred after invasion (Fig. 3). Complementation of eda on the low-copy-number plasmid pWKS30 restored the wild-type plaque size.
A possible explanation for the eda, but not the edd, mutant having a plaque defect is the accumulation of the bacteriostatic Entner-Doudoroff intermediate 2-keto-3-deoxy-6-phosphogluconic acid (KDPG) in the absence of Eda (42). To distinguish between the effect of loss of one or more products of Eda enzymatic activity and the potential buildup of its substrate KDPG, an eda edd double mutant was constructed. The eda edd mutant formed wild-type-size plaques, suggesting that the defect in the eda mutant is due to buildup of KDPG. This may indicate that there is sufficient carbon flow through this pathway during intracellular growth to build up toxic levels of KDPG but that other pathways for sugar utilization are sufficient for carbon metabolism in the double mutant.
Wild-type S. flexneri has a competitive advantage over pfkAB, pykAF, and eda mutants in cell culture.
To directly compare the relative fitness of the EMP and ED pathway mutants, we used a competition assay with the wild type in a Henle monolayer. Equal numbers of the mutant and its wild-type parent 2457T were mixed, inoculated onto a Henle cell monolayer, and allowed to form plaques. The monolayers were harvested at 72 h, and the ratio of mutant and wild-type bacteria recovered from the infected monolayers was determined and compared to the inoculum. A ratio of less than 1.0 for the mutant relative to the wild type indicates reduced fitness for growth in Henle cells. As shown in Table 3, the wild type had a competitive advantage over the pfkAB, pykAF, and eda mutants. The relative competitive index correlated with plaque size, and the pykAF mutant, which produced the smallest plaques, had the lowest competitive index.
TABLE 3.
pfk, pyk, and eda mutants have reduced fitness in the plaque assay
| Strain | Mutation(s) | Competitive indexa |
|---|---|---|
| EAWS122 | pfkAB | 0.379* |
| EAWS124 | pykAF | 0.043** |
| EAWS134 | eda | 0.268* |
The competitive index for intracellular growth of the mutants was determined by coinfection of Henle cells with equal numbers of the wild type and mutant and comparing the recovery of each strain after 72 h, as described in Materials and Methods. A competitive index of <1.0 indicates that the mutant has a disadvantage compared to the wild type. *, P ≤ 0.02, and **, P < 0.01, by t test.
Phenotype microarray reveals global defects in utilization of carbon sources.
The eukaryotic intracellular environment is expected to contain a number of potential carbon sources available for use by S. flexneri. To better characterize the carbon sources that can be used by wild-type S. flexneri and the carbon metabolism mutants, we used the Biolog Phenotypic MicroArray carbon source plates PM1 and PM2A to compare the pfkAB, pykAF, and eda mutants with the utilization pattern of wild-type 2457T (Fig. 5). The Biolog array measures cell respiration when the indicated compound is the sole carbon source. Both EMP pathway mutants failed to metabolize, or showed reduced metabolism of, a number of the carbon sources. The pfkAB mutant did not metabolize glucose, but the pykAF mutant, which is blocked at a later step in the EMP pathway than the pfkAB mutant, did use glucose, though at a reduced rate relative to the wild type. Both mutants displayed slower metabolism of glucose-6-phosphate and were defective in metabolism of fructose-6-phosphate, the second and third intermediates in the pathway (Fig. 1). E. coli pfkA mutants are defective in uptake of glucose (43), and it is likely that a similar defect in uptake is responsible for the poor growth of the S. flexneri pfkA mutant when glucose is the sole carbon source. The S. flexneri pykAF mutant may metabolize glucose in vitro through the Entner-Doudoroff pathway to bypass the requirement for pyruvate kinase (Fig. 1). Both the pykAF and the pfkAB mutants were able to metabolize pyruvate and grew well with this carbon source (Fig. 5).
FIG 5.
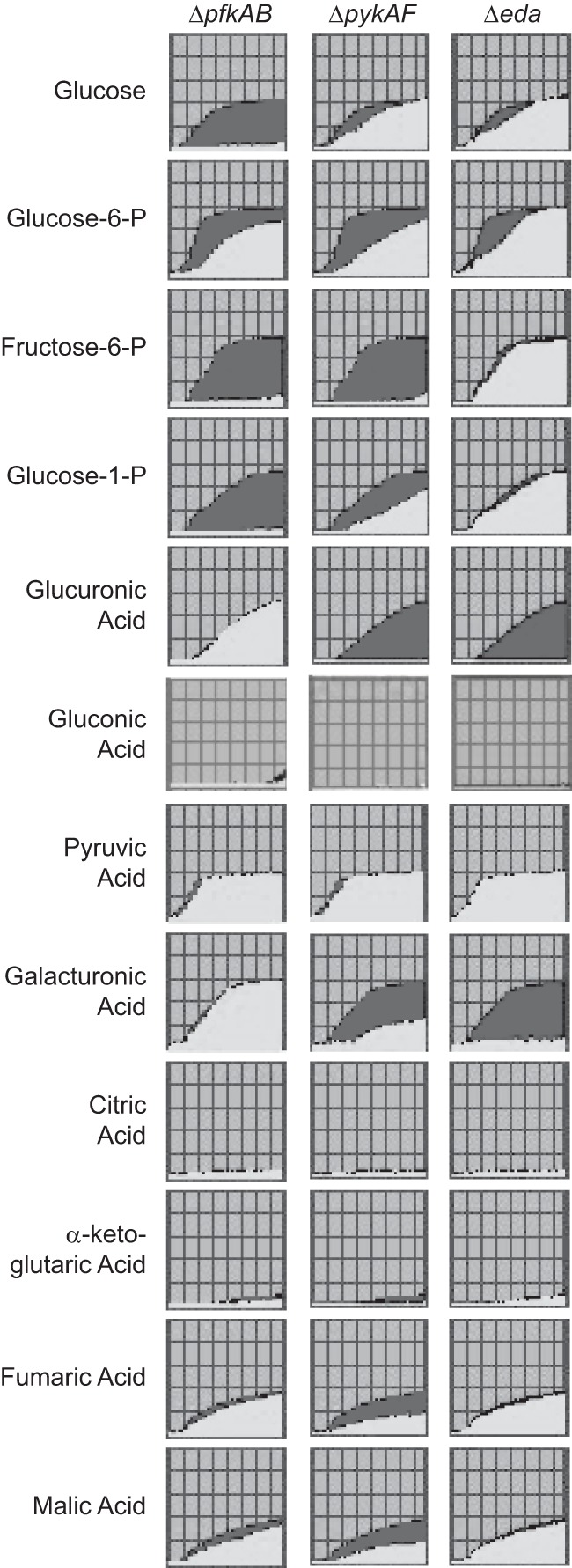
Use of carbon sources by wild-type and carbon metabolism mutants of S. flexneri. Phenotypic microarray carbon utilization plates PM1 and PM2A (Biolog, Hayward, CA) were used to measure use of the indicated carbon sources. Use of the compound resulted in a colorimetric change, and the optical density was measured every 15 min for 24 h. The data for the wild type were plotted in dark gray and overlaid with the data for the mutant in light gray. Carbon sources used by both the wild type and mutant strains are indicated by the plots filled with light gray, while sources used only by the wild-type strain are indicated by plots filled with dark gray. None of the carbon sources shown was preferentially used by a mutant strain compared with the wild type.
The eda mutant had a carbon utilization profile similar to the wild-type strain. However, the mutant failed to respire in the presence of d-glucuronic acid or d-galacturonic acid (Fig. 5), indicating a lack of metabolism of these compounds or inhibition by their metabolic product. In E. coli, these substrates are ultimately converted to 2-keto-3-deoxy-d-gluconic acid, which is phosphorylated by ATP to yield 2-keto-3-deoxy-6-phosphogluconic acid (KDPG), the substrate of Eda (44, 45). Accumulation of KDPG would be toxic to the bacteria.
Addition of exogenous pyruvate partially restores plaque formation by the pfkAB, pykAF, and eda mutants.
The pykAF mutant, which had the most severe plaque defect, is defective in the last step in the EMP pathway, the conversion of phosphoenolpyruvate to pyruvate. Therefore, we determined whether the addition of pyruvate to the cell culture medium could compensate for the reduction in pyruvate synthesis by the mutants during intracellular growth. As shown in Fig. 6, the presence of supplemental pyruvate, but not glucose, increased the size of the plaques formed by the pfkAB, pykAF, and eda mutants relative to the size of the plaques they formed in unsupplemented medium. This suggested that supplemental pyruvate increased the intracellular pyruvate pool and allowed the mutants to bypass the block in glycolysis. Interestingly, the addition of either supplemental glucose or pyruvate to the medium increased the size of the plaques formed by the wild-type bacteria, with pyruvate having the larger effect.
FIG 6.
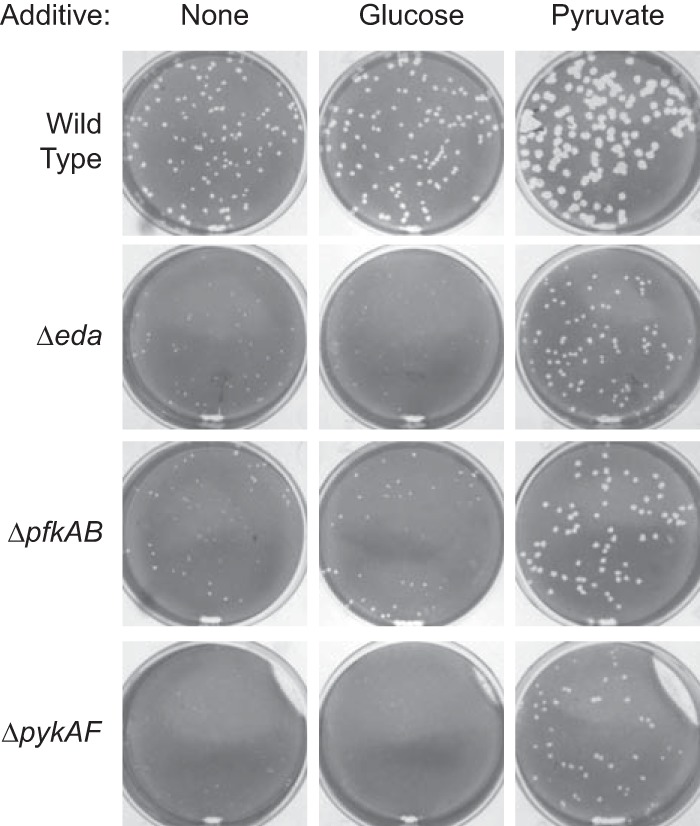
Effect of supplementation of the Henle cell medium with glucose and pyruvate on plaque size of the wild type and carbon metabolism mutants of S. flexneri. The indicated supplement was added to the Henle medium for the first 48 h. Incubation was continued in unsupplemented medium, and plaques were visualized at 72 h.
Exogenous pyruvate increases the level of intracellular pyruvate and of several glycolytic and TCA cycle intermediates.
Metabolomic analysis was used to determine the effects of glucose and pyruvate supplementation on potential carbon sources present within the Henle cell cytoplasm. The Henle cells were incubated in medium with or without glucose or pyruvate supplementation and washed to remove the extracellular medium prior to determination of the metabolome. The relative amounts of selected metabolites in the cells grown in the supplemented compared to unsupplemented medium are shown in Table 4, and the complete data from the metabolomic analysis is shown in the supplemental material. The glucose-treated cells had a much higher level of intracellular glucose than the untreated cells or pyruvate-supplemented cells. They also had elevated levels of several glycolysis and tricarboxylic acid (TCA) cycle intermediates but did not show an increase in intracellular pyruvate. The addition of pyruvate significantly increased intracellular pyruvate levels. Several of the glycolytic intermediates that were elevated, especially glucose-6-phosphate and fructose-6-phosphate, were metabolized by the wild type but not by the EMP pathway mutants in the phenotype arrays (Fig. 5), suggesting that the increases in these compounds do not explain the increased plaque size of the mutants. Several of the TCA cycle intermediates were higher in Henle cells grown in pyruvate-supplemented medium, but malic acid was the only one of these that was used with reasonable efficiency by any of the S. flexneri mutant strains tested in the phenotype analysis. Fumaric acid was used weakly, and neither 2-ketoglutaric acid nor citric acid was used by the bacteria (Fig. 5).
TABLE 4.
Changes in levels of selected compounds in Henle cells grown with glucose or pyruvate
| Compound | Pathwaya | Ratio of amt of metabolite in cells grown in supplemented medium to amt in cells grown in unsupplemented mediumb |
|
|---|---|---|---|
| Glucose supplemented | Pyruvate supplemented | ||
| Glucose | Glycolysis | 18.9*** | 0.8 |
| Glucose-1-phosphate | Glycolysis | 1.7 | 1.3 |
| Glucose-6-phosphate | Glycolysis | 2.0 | 0.6 |
| Fructose-6-phosphate | EMP | 2.8 | 3.4* |
| 3-Phosphoglycerate | EMP | 2.9*** | 2.3* |
| 2-Phosphoglycerate | EMP | 4.1** | 2.8 |
| Phosphoenolpyruvate | EMP | 1.7* | 1.7 |
| Pyruvic acid | Glycolysis | 0.9 | 2.0* |
| Citric acid | TCA | 3.7*** | 12.9* |
| Aconitate | TCA | 3.6*** | 12.3** |
| Isocitric acid | TCA | 3.1*** | 5.9** |
| 2-Ketoglutaric acid | TCA | 1.8 | 6.1* |
| Succinic acid | TCA | 0.8 | 1.2 |
| Fumaric acid | TCA | 1.4 | 3.9** |
| Malic acid | TCA | 1.3*** | 2.1 |
| Gluconic acid | PP, ED precursor | 2.1* | 7.8* |
Compounds are substrates or intermediates in glycolysis, the TCA cycle, or specific glycolytic pathways: Embden-Meyerhof-Parnas (EMP), pentose phosphate (PP), or Entner-Doudoroff (ED).
Average ratios for ≥3 independent determinations. Differences that are statistically different using the Student two-tailed t test are shown by asterisks: *, P ≤ 0.05; **, P ≤ 0.01; and ***, P ≤ 0.001. The complete metabolome is shown in the supplemental material.
Pyruvate is a carbon source for both the wild type and glycolysis mutants.
The ability of these strains to use glucose or pyruvate as a carbon source was tested directly by measuring the growth of the strains in minimal medium with these carbon sources. As expected, the wild-type S. flexneri grew with glucose as the sole carbon source, while neither the pfkAB nor the pykAF mutants showed significant growth over the time course of the experiment (Fig. 7A). The eda mutant grew at an intermediate rate (Fig. 7A). In contrast, all strains grew similarly when pyruvate was the sole carbon source (Fig. 7B), suggesting that the elevated level of intracellular pyruvate in the pyruvate-supplemented cultures has the potential to stimulate the growth of the mutants. It is of interest that the plaque size of wild-type S. flexneri was increased by supplementation of the medium with either glucose or pyruvate. The increased plaque size with glucose supplementation may be due primarily to the large increase in the level of intracellular glucose (Table 4), which is known to be an excellent carbon source. To gain insight into the increased plaque size observed with pyruvate supplementation, we compared the growth of wild-type S. flexneri when either glucose, pyruvate, or an equal mixture of the two was the carbon source available in minimal medium. As seen in Fig. 7C, the bacteria grew faster with pyruvate than with glucose as the sole carbon source. The average doubling times for 2457T in minimal medium with glucose and with pyruvate were 2.3 h and 1.5 h, respectively (Fig. 7C). Both glucose and pyruvate should be available to the Henle cells, and when both carbon sources were present in the minimal medium, the bacteria grew more rapidly than with either carbon source alone (average doubling time of 1.1 h) (Fig. 7C).
FIG 7.
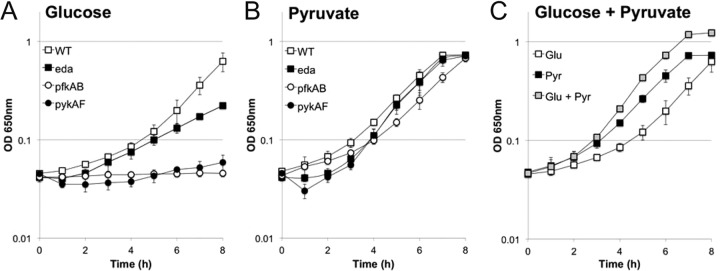
Growth of S. flexneri with pyruvate as the carbon source. The S. flexneri wild type or mutants (eda, pfkAB, and pykAF mutants) were grown in minimal medium with glucose (A), pyruvate (B), or a mixture of glucose and pyruvate (C) as the carbon source. Cultures were grown at 37°C with aeration for 8 h, and growth was monitored by measuring OD650 (average; bars indicate 1 standard deviation).
DISCUSSION
Catabolism of sugars plays an important role in colonization and pathogenesis of enteric pathogens. Most Enterobacteriaceae can use sugars via the Embden-Meyerhof-Parnas, Entner-Doudoroff, or pentose phosphate pathways (Fig. 1). Studies with Salmonella, various E. coli pathovars, and commensal E. coli have shown that at least one of these carbon pathways is required for initiation or maintenance of infection (29, 46–48). The intestine is rich in sugars due to degradation of intestinal mucins, releasing the sugars for potential use by bacteria. Thus, it is not surprising that intestinal pathogens or commensals would take advantage of this abundant carbon source when colonizing the intestine. In contrast, peptides and amino acids appear to be the primary carbon sources for uropathogenic E. coli during infection of the urinary tract (49), indicating that different strains are adapted to growth in different environments of the host.
Shigella spp. differ from most other human enteric bacteria in their ability to grow within the cytoplasm of human epithelial cells. Following invasion, the bacteria escape the vacuole and rapidly multiply within the cytoplasm. Although some of the pathways required for intracellular growth have been defined, the specific carbon metabolism pathways and carbon sources used by intracellular bacteria are poorly understood. We are beginning to gain a better understanding of the physiology and metabolism of bacteria growing within the host cell through an analysis of the bacterial genes and proteins expressed in this environment. In this study, we have focused on a genetic approach to analyzing the carbon metabolism pathways that play a role in the intracellular lifestyle.
A number of studies have indicated that sugar metabolism is important for Shigella intracellular replication, but the specific sugars used and the pathways for their catabolism in this environment are not fully characterized. There is reduced expression of genes encoding the glucose uptake proteins ptsG, manX, manY, and manZ during intracellular growth (22, 23). However, analysis of the proteome of intracellular S. flexneri showed that two of these proteins (ManZ and ManX), as well as other proteins involved in sugar transport (FruA and FruB), were present at high levels in intracellular bacteria (24). The relative abundance of these proteins suggests that the bacteria can import sugars during intracellular growth.
Shigella, like other Enterobacteriaceae, can metabolize sugars through the multiple pathways. Each of the sugar catabolism pathways has been shown to be important for certain enteric pathogens. Glycolysis through the EMP pathway is necessary for Salmonella virulence. A pfkAB double mutant was severely attenuated in macrophage replication, and it was recovered in lower numbers than the wild type from murine organs (50). Glycolysis was also important for initial colonization and maintenance of an enterohemorrhagic E. coli (EHEC) strain in the mouse intestine (47). There is no small-animal model for shigellosis that would allow testing the effect of mutations in glycolysis or other carbon pathways on intestinal colonization by S. flexneri, but analysis in cell culture shows that the EMP pathway is necessary for intracellular growth. In this study, we show that blocking either an early (Pfk) or late (Pyk) step in the EMP pathway resulted in loss of wild-type plaque formation. The importance of this glycolytic pathway for intracellular growth was supported by the observation that enzymes for the pathway were found at higher levels in intracellular S. flexneri than in bacteria grown in vitro (24), even though transcriptome analysis showed reduced expression of the genes (20). The EMP pathway may be important not only in using sugars as an energy source but also in the production of essential metabolic intermediates.
The pentose phosphate pathway contributes to virulence in the intracellular pathogen Salmonella enterica Typhimurium. A zwf mutant was attenuated in a mouse model, and the attenuation was due to increased susceptibility to reactive oxygen and nitrogen intermediates in the absence of glucose-6-phosphate dehydrogenase (51). The zwf mutant of S. flexneri was not affected in its ability to infect and spread in epithelial cells (Fig. 3) but has not been tested in macrophages. Taken together, these data suggest that the pentose pathway may generate NADPH for antioxidant defense but is not required for carbon assimilation during intracellular growth.
The presence of gluconate in murine cecal material (52) suggests that the Entner-Doudoroff pathway may be induced in bacteria that colonize the intestinal tract. S. flexneri does not use exogenous gluconate as a carbon source, but carbon could enter this pathway from glucose or glucose-phosphate. In E. coli, the Entner-Doudoroff pathway is necessary for colonization initiation and maintenance in a mouse model (45). The Entner-Doudoroff pathway also contributes to colonization and production of disease by Vibrio cholerae, as an edd mutant strain failed to colonize in a mouse model or to induce fluid accumulation in ligated rabbit ileal loops (53). Analysis of the S. flexneri edd eda double mutant indicated that the pathway is not essential for intracellular growth, since it produced wild-type plaques in cultured cells (Fig. 3). However, the eda mutant had a small plaque phenotype, indicative of accumulation of the intermediate KDPG, resulting in a bacteriostatic effect. This suggests that the pathway is used intracellularly and that sufficient carbon flows through the pathway to generate toxic levels of KDPG in the eda mutant in the intracellular environment.
Although sugars appear to represent a major source of carbon for intracellular bacteria, the eukaryotic cytosol has multiple potential carbon sources, and it is likely that Shigella uses a variety of compounds for growth inside the host cell. The proteome analysis of intracellular shigellae showed that enzymes for mixed acid fermentation were present at high levels in the intracellular bacteria, and mutational analysis confirmed that this pathway was essential for normal intracellular growth (24). The pyruvate metabolized by this pathway could be a product of glycolysis, but exogenous pyruvate can also be used as a carbon source (Fig. 7), indicating that it could also be obtained from the host cell. The addition of pyruvate to the cell culture medium supported plaque formation by mutants blocked in the EMD or ED pathway, and analysis of the metabolites within the Henle cell showed that the addition of exogenous pyruvate significantly increased the pool of pyruvate within the cell. Pyruvate supplementation also increased the size of plaques produced by wild-type S. flexneri, suggesting that this is a good carbon source for the intracellular bacteria. This is consistent with our previous studies showing that genes for mixed-acid fermentation of pyruvate are necessary for wild-type plaque formation by S. flexneri (24). The increased amount of pyruvate may lead to a larger number of bacteria in the cell by increasing the carrying capacity. This could result in more rapid spread of the bacteria to adjacent cells, creating larger plaques. In vitro analysis showed that S. flexneri grows faster when pyruvate is the sole carbon source than with glucose alone, but the highest growth rate was achieved when the bacteria were grown on a mixture of pyruvate and glucose. This likely reflects the preferred carbon sources for the intracellular bacteria, which could use the intracellular sugars along with pyruvate to grow rapidly within the intestinal epithelial cells. Our results indicate that Shigella has the ability to use varied and multiple carbon sources when growing intracellularly. This allows it to adapt to the changing environment within the cell cytoplasm and maximize its ability to extract nutrients and energy sources from the host cell.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grant R01 AI16935 from the National Institutes of Health.
We thank Alexandra Mey for helpful discussions and critical reading of the manuscript.
Footnotes
Published ahead of print 14 April 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01575-13.
REFERENCES
- 1.Kotloff KL, Winickoff JP, Ivanoff B, Clemens JD, Swerdlow DL, Sansonetti PJ, Adak GK, Levine MM. 1999. Global burden of Shigella infections: implications for vaccine development and implementation of control strategies. Bull. World Health Organ. 77:651–666 [PMC free article] [PubMed] [Google Scholar]
- 2.Wei J, Goldberg MB, Burland V, Venkatesan MM, Deng W, Fournier G, Mayhew GF, Plunkett G, Rose DJ, Darling A, Mau B, Perna NT, Payne SM, Runyen-Janecky LJ, Zhou S, Schwartz DC, Blattner FR. 2003. Complete genome sequence and comparative genomics of Shigella flexneri serotype 2a strain 2457T. Infect. Immun. 71:2775–2786. 10.1128/IAI.71.5.2775-2786.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sasakawa C. 2010. A new paradigm of bacteria-gut interplay brought through the study of Shigella. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 86:229–243. 10.2183/pjab.86.229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goldberg MB, Bârzu O, Parsot C, Sansonetti PJ. 1993. Unipolar localization and ATPase activity of IcsA, a Shigella flexneri protein involved in intracellular movement. J. Bacteriol. 175:2189–2196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goldberg MB, Theriot JA. 1995. Shigella flexneri surface protein IcsA is sufficient to direct actin-based motility. Proc. Natl. Acad. Sci. U. S. A. 92:6572–6576. 10.1073/pnas.92.14.6572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carayol N, Tran Van Nhieu G. 2013. Tips and tricks about Shigella invasion of epithelial cells. Curr. Opin. Microbiol. 16:32–37. 10.1016/j.mib.2012.11.010 [DOI] [PubMed] [Google Scholar]
- 7.Oaks EV, Wingfield ME, Formal SB. 1985. Plaque formation by virulent Shigella flexneri. Infect. Immun. 48:124–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.High N, Mounier J, Prevost MC, Sansonetti PJ. 1992. IpaB of Shigella flexneri causes entry into epithelial cells and escape from the phagocytic vacuole. EMBO J. 11:1991–1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ménard R, Sansonetti PJ, Parsot C. 1993. Nonpolar mutagenesis of the ipa genes defines IpaB, IpaC, and IpaD as effectors of Shigella flexneri entry into epithelial cells. J. Bacteriol. 175:5899–5906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sansonetti PJ, Ryter A, Clerc P, Maurelli AT, Mounier J. 1986. Multiplication of Shigella flexneri within HeLa cells: lysis of the phagocytic vacuole and plasmid-mediated contact hemolysis. Infect. Immun. 51:461–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prosseda G, Fradiani PA, Di Lorenzo M, Falconi M, Micheli G, Casalino M, Nicoletti M, Colonna B. 1998. A role for H-NS in the regulation of the virF gene of Shigella and enteroinvasive Escherichia coli. Res. Microbiol. 149:15–25. 10.1016/S0923-2508(97)83619-4 [DOI] [PubMed] [Google Scholar]
- 12.Falconi M, Prosseda G, Giangrossi M, Beghetto E, Colonna B. 2001. Involvement of FIS in the H-NS-mediated regulation of virF gene of Shigella and enteroinvasive Escherichia coli. Mol. Microbiol. 42:439–452. 10.1046/j.1365-2958.2001.02646.x [DOI] [PubMed] [Google Scholar]
- 13.Prosseda G, Falconi M, Giangrossi M, Gualerzi CO, Micheli G, Colonna B. 2004. The virF promoter in Shigella: more than just a curved DNA stretch. Mol. Microbiol. 51:523–537. 10.1046/j.1365-2958.2003.03848.x [DOI] [PubMed] [Google Scholar]
- 14.Durand JM, Dagberg B, Uhlin BE, Bjork GR. 2000. Transfer RNA modification, temperature and DNA superhelicity have a common target in the regulatory network of the virulence of Shigella flexneri: the expression of the virF gene. Mol. Microbiol. 35:924–935. 10.1046/j.1365-2958.2000.01767.x [DOI] [PubMed] [Google Scholar]
- 15.Nakayama S, Watanabe H. 1995. Involvement of cpxA, a sensor of a two-component regulatory system, in the pH-dependent regulation of expression of Shigella sonnei virF gene. J. Bacteriol. 177:5062–5069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Porter ME, Dorman CJ. 1997. Differential regulation of the plasmid-encoded genes in the Shigella flexneri virulence regulon. Mol. Gen. Genet. 256:93–103. 10.1007/s004380050550 [DOI] [PubMed] [Google Scholar]
- 17.Broach WH, Egan N, Wing HJ, Payne SM, Murphy ER. 2012. VirF-independent regulation of Shigella virB transcription is mediated by the small RNA RyhB. PLoS One 7:e38592. 10.1371/journal.pone.0038592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murphy ER, Payne SM. 2007. RyhB, an iron-responsive small RNA molecule, regulates Shigella dysenteriae virulence. Infect. Immun. 75:3470–3477. 10.1128/IAI.00112-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goetz M, Bubert A, Wang G, Chico-Calero I, Vazquez-Boland JA, Beck M, Slaghuis J, Szalay AA, Goebel W. 2001. Microinjection and growth of bacteria in the cytosol of mammalian host cells. Proc. Natl. Acad. Sci. U. S. A. 98:12221–12226. 10.1073/pnas.211106398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Headley VL, Payne SM. 1990. Differential protein expression by Shigella flexneri in intracellular and extracellular environments. Proc. Natl. Acad. Sci. U. S. A. 87:4179–4183. 10.1073/pnas.87.11.4179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bartoleschi C, Pardini MC, Scaringi C, Martino MC, Pazzani C, Bernardini ML. 2002. Selection of Shigella flexneri candidate virulence genes specifically induced in bacteria resident in host cell cytoplasm. Cell. Microbiol. 4:613–626. 10.1046/j.1462-5822.2002.00216.x [DOI] [PubMed] [Google Scholar]
- 22.Lucchini S, Liu H, Jin Q, Hinton JC, Yu J. 2005. Transcriptional adaptation of Shigella flexneri during infection of macrophages and epithelial cells: insights into the strategies of a cytosolic bacterial pathogen. Infect. Immun. 73:88–102. 10.1128/IAI.73.1.88-102.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Runyen-Janecky LJ, Payne SM. 2002. Identification of chromosomal Shigella flexneri genes induced by the eukaryotic intracellular environment. Infect. Immun. 70:4379–4388. 10.1128/IAI.70.8.4379-4388.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pieper R, Fisher CR, Suh M-J, Huang S-T, Parmar P, Payne SM. 2013. Analysis of the proteome of intracellular Shigella flexneri reveals pathways important for intracellular growth. Infect. Immun. 81:4635–4648. 10.1128/IAI.00975-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Romeo T. 1998. Global regulation by the small RNA-binding protein CsrA and the non-coding RNA molecule CsrB. Mol. Microbiol. 29:1321–1330. 10.1046/j.1365-2958.1998.01021.x [DOI] [PubMed] [Google Scholar]
- 26.Sabnis NA, Yang H, Romeo T. 1995. Pleiotropic regulation of central carbohydrate metabolism in Escherichia coli via the gene csrA. J. Biol. Chem. 270:29096–29104. 10.1074/jbc.270.49.29096 [DOI] [PubMed] [Google Scholar]
- 27.Saier MH, Ramseier TM. 1996. The catabolite repressor/activator (Cra) protein of enteric bacteria. J. Bacteriol. 178:3411–3417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gore AL, Payne SM. 2010. CsrA and Cra influence Shigella flexneri pathogenesis. Infect. Immun. 78:4674–4682. 10.1128/IAI.00589-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Götz A, Eylert E, Eisenreich W, Goebel W. 2010. Carbon metabolism of enterobacterial human pathogens growing in epithelial colorectal adenocarcinoma (Caco-2) cells. PLoS One 5:e10586. 10.1371/journal.pone.0010586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muñoz-Elías EJ, McKinney JD. 2006. Carbon metabolism of intracellular bacteria. Cell. Microbiol. 8:10–22. 10.1111/j.1462-5822.2005.00648.x [DOI] [PubMed] [Google Scholar]
- 31.Fraenkel DG. 1996. Glycolysis, p 189–198 In Neidhardt FC, Curtiss R, III, Ingraham JL, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE. (ed), Escherichia coli and Salmonella: cellular and molecular biology. ASM Press, Washington, DC [Google Scholar]
- 32.Roth V. 2006. Doubling time online calculator. http://www.doubling-time.com/compute.php [Google Scholar]
- 33.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:2006.0008. 10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645. 10.1073/pnas.120163297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bochner BR. 2009. Global phenotypic characterization of bacteria. FEMS Microbiol. Rev. 33:191–205. 10.1111/j.1574-6976.2008.00149.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ponce E, Flores N, Martinez A, Valle F, Bolívar F. 1995. Cloning of the two pyruvate kinase isoenzyme structural genes from Escherichia coli: the relative roles of these enzymes in pyruvate biosynthesis. J. Bacteriol. 177:5719–5722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vinopal RT, Fraenkel DG. 1975. pfkB and pfkC loci of Escherichia coli. J. Bacteriol. 122:1153–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Daldal F. 1983. Molecular cloning of the gene for phosphofructokinase-2 of Escherichia coli and the nature of a mutation, pfkB1, causing a high level of the enzyme. J. Mol. Biol. 168:285–305. 10.1016/S0022-2836(83)80019-9 [DOI] [PubMed] [Google Scholar]
- 39.Fawcett WP, Wolf RE., Jr 1995. Genetic definition of the Escherichia coli zwf “soxbox,” the DNA binding site for SoxS-mediated induction of glucose 6-phosphate dehydrogenase in response to superoxide. J. Bacteriol. 177:1742–1750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jair KW, Martin RG, Rosner JL, Fujita N, Ishihama A, Wolf RE., Jr 1995. Purification and regulatory properties of MarA protein, a transcriptional activator of Escherichia coli multiple antibiotic and superoxide resistance promoters. J. Bacteriol. 177:7100–7104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rowley DL, Wolf RE., Jr 1991. Molecular characterization of the Escherichia coli K-12 zwf gene encoding glucose 6-phosphate dehydrogenase. J. Bacteriol. 173:968–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fuhrman LK, Wanken A, Nickerson KW, Conway T. 1998. Rapid accumulation of intracellular 2-keto-3-deoxy-6-phosphogluconate in an Entner-Doudoroff aldolase mutant results in bacteriostasis. FEMS Microbiol. Lett. 159:261–266. 10.1111/j.1574-6968.1998.tb12870.x [DOI] [PubMed] [Google Scholar]
- 43.Roehl RA, Vinopal RT. 1976. Lack of glucose phosphotransferase function in phosphofructokinase mutants of Escherichia coli. J. Bacteriol. 126:852–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ashwell G. 1962. Enzymes of glucuronic and galacturonic acid metabolism in bacteria. Methods Enzymol. 5:190–208 [Google Scholar]
- 45.Richard P, Hilditch S. 2009. D-galacturonic acid catabolism in microorganisms and its biotechnological relevance. Appl. Microbiol. Biotechnol. 82:597–604. 10.1007/s00253-009-1870-6 [DOI] [PubMed] [Google Scholar]
- 46.Chang DE, Smalley DJ, Tucker DL, Leatham MP, Norris WE, Stevenson SJ, Anderson AB, Grissom JE, Laux DC, Cohen PS, Conway T. 2004. Carbon nutrition of Escherichia coli in the mouse intestine. Proc. Natl. Acad. Sci. U. S. A. 101:7427–7432. 10.1073/pnas.0307888101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miranda RL, Conway T, Leatham MP, Chang DE, Norris WE, Allen JH, Stevenson SJ, Laux DC, Cohen PS. 2004. Glycolytic and gluconeogenic growth of Escherichia coli O157:H7 (EDL933) and E. coli K-12 (MG1655) in the mouse intestine. Infect. Immun. 72:1666–1676. 10.1128/IAI.72.3.1666-1676.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sweeney NJ, Laux DC, Cohen PS. 1996. Escherichia coli F-18 and E. coli K-12 eda mutants do not colonize the streptomycin-treated mouse large intestine. Infect. Immun. 64:3504–3511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alteri CJ, Smith SN, Mobley HL. 2009. Fitness of Escherichia coli during urinary tract infection requires gluconeogenesis and the TCA cycle. PLoS Pathog. 5:e1000448. 10.1371/journal.ppat.1000448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bowden SD, Rowley G, Hinton JC, Thompson A. 2009. Glucose and glycolysis are required for the successful infection of macrophages and mice by Salmonella enterica serovar Typhimurium. Infect. Immun. 77:3117–3126. 10.1128/IAI.00093-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lundberg BE, Wolf RE, Jr, Dinauer MC, Xu Y, Fang FC. 1999. Glucose 6-phosphate dehydrogenase is required for Salmonella typhimurium virulence and resistance to reactive oxygen and nitrogen intermediates. Infect. Immun. 67:436–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Peekhaus N, Conway T. 1998. What's for dinner?: Entner-Doudoroff metabolism in Escherichia coli. J. Bacteriol. 180:3495–3502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Patra T, Koley H, Ramamurthy T, Ghose AC, Nandy RK. 2012. The Entner-Doudoroff pathway is obligatory for gluconate utilization and contributes to the pathogenicity of Vibrio cholerae. J. Bacteriol. 194:3377–3385. 10.1128/JB.06379-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gore AL. 2010. Carbon metabolism influences Shigella flexneri pathogenesis. Ph.D. dissertation University of Texas. [Google Scholar]
- 55.Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14. 10.1016/0378-1119(95)00193-A [DOI] [PubMed] [Google Scholar]
- 56.Wang RF, Kushner SR. 1991. Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 100:195–199. 10.1016/0378-1119(91)90366-J [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.