

Abstract
Bacterial gene islands add to the genetic repertoire of opportunistic pathogens. Here, we perform comparative analyses of three Pseudomonas aeruginosa strains isolated sequentially over a 3-week period from a patient with ventilator-associated pneumonia (VAP) who received clindamycin and piperacillin-tazobactam as part of their treatment regime. While all three strains appeared to be clonal by standard pulsed-field gel electrophoresis, whole-genome sequencing revealed subtle alterations in the chromosomal organization of the last two strains; specifically, an inversion event within a novel 124-kb gene island (PAGI 12) composed of 137 open reading frames [ORFs]. Predicted ORFs in the island included metabolism and virulence genes. Overexpression of a gene island-borne putative β-lactamase gene was observed following piperacillin-tazobactam exposure and only in those strains that had undergone the inversion event, indicating altered gene regulation following genomic remodeling. Examination of a separate cohort of 76 patients with VAP for integration at this tRNAlys recombination site demonstrated that patients exhibiting evidence of integration at this site had significantly higher 28-day mortality. These findings provide evidence that P. aeruginosa can integrate, rapidly remodel, and express exogenous genes, which likely contributes to its fitness in a clinical setting.
INTRODUCTION
Pseudomonas aeruginosa is one of the leading causes of ventilator-associated pneumonia (VAP) and is associated with high mortality rates in intensive care unit patients (1, 2). This is due to several factors, including the organism's metabolic versatility, inherent as well as acquired resistance to antimicrobials, and a large repertoire of virulence factors. Although P. aeruginosa strains possess a relatively conserved core genome, insertions and deletions result in genome sizes ranging from 5.2 Mb to 7 Mb (3, 4), dramatically diversifying the genetic capacity of this species. One means of genome expansion is through the acquisition of exogenous DNA via intra- or interspecies horizontal transfer (4–6). This well-described phenomenon in P. aeruginosa appears to be a key factor in the macroevolution and genetic diversity of this species (3, 6).
Gene islands are segments of DNA integrated at discrete chromosomal recombination hot spots that are characteristically distinct from the core bacterial genome (typically due to GC content). These islands may be episomal in nature in that they can exist autonomously as self-replicating extrachromosomal elements, or they may integrate at site-specific recombination hot spots and replicate with the host bacterial chromosome. Interstrain transmission of a gene island from a pathogenic to a nonpathogenic strain, as well as autologous transmission from one chromosomal integration site to another, has been demonstrated experimentally (6). Gene islands confer the evolutionary advantage of simultaneously transferring several genes and/or operons encoding a variety of functions, with the majority described to date including genes that appear to enhance survival, virulence, and/or transmission of the organism (7–10).
As many as 11 distinct genomic islands have been identified in P. aeruginosa, several of which have been discovered in patient isolates (3, 6, 11–13), leading to the widely held premise that these exogenous genetic elements may increase bacterial fitness in the infected human host. P. aeruginosa islands described to date range in size from approximately 2 to 105 kb and carry a multitude of genes that display homology to structural and functional genes of P. aeruginosa as well as of other bacterial species, such as Cupriavidus (formerly Ralstonia) and Burkholderia species (13, 14). Genes within these islands typically encode proteins involved in metabolism, stress resistance mechanisms, and pathogenesis. For example, ExoU, a potent cytotoxin of the type III secretion system (TTSS), associated with acute infection, is believed to have been acquired via integration of a mobile genetic element primarily because of its proximity to a tRNAlys recombination site and the composition of surrounding genes (15). To date, several islands that encode this potent toxin have been described, all of which were discovered in patient isolates (15). The gene island phenomenon is not restricted to P. aeruginosa; several other human-pathogenic bacteria also harbor islands that contain genes encoding virulence factors of the type III or IV secretion systems (16), supporting the hypothesis that horizontal acquisition of ancillary genomic material contributes to pathogen fitness in the human host.
In this study, we examined sequential P. aeruginosa strains isolated from endotracheal aspirates (ETAs) of a patient who developed VAP. In a previously published study, we performed bacterial community analysis by clone library 16S rRNA sequencing of these same endotracheal aspirates. Despite antimicrobial administration, P. aeruginosa numbers did not decline; the number of CFU increased from 1.1 × 107 (± 9 × 106) to 3.8 × 107 (± 2 × 106) (P < 0.03) CFU ml−1 during this period, which was concomitant with a dramatic increase in the relative abundance of P. aeruginosa in the airway community from 4% in the first ETA to 43% in the last ETA collected over a 3-week period (17). These microbiological trends were coincident with the clinical development of P. aeruginosa VAP in this patient. We therefore embarked on a series of experiments to better understand how, despite antimicrobial exposure, P. aeruginosa persisted and proliferated in the airways of this patient, focusing particularly on genomic analyses of a series of sequential strains isolated over this 3-week period.
MATERIALS AND METHODS
P. aeruginosa isolation from endotracheal aspirates.
P. aeruginosa strains were cultured on Pseudomonas isolation agar (Difco, KS) and isolated from ETAs collected sequentially from a single patient with VAP (who consented as part of a VAP study, which was approved by the University of California—San Francisco [UCSF] Institutional Review Board) over a 3-week period (Fig. 1A). Multiple glycerol stocks of each strain were generated to ensure that experiments were always performed with a fresh stock (to avoid the effects of multiple freeze-thaw cycles). Genomic DNA was extracted from overnight cultures grown at 37°C in Luria-Bertani [LB] broth using the Wizard genomic DNA purification kit (Promega, WI) according to the manufacturer's instructions.
FIG 1.

(A) Collection of P. aeruginosa isolates during hospitalization and antibiotic treatment of patient. No abx, no antibiotics. (B) RAPD PCR profiling demonstrates that strains collected at each time point are identical. (C) Pulsed-field gel electrophoresis of SpeI-digested P. aeruginosa chromosomal DNA performed under standard conditions on each representative isolate collected at each time point demonstrates that they are clonal.
RAPD analysis and sequencing of band of interest.
Random amplification of polymorphic DNA (RAPD) PCR using the RAPD 272 primer was performed as described previously (18). Banding patterns were visualized on a 1% agarose gel using ethidium bromide and UV illumination.
The RAPD PCR amplified band of interest was excised from a 1% agarose gel and purified using the Qiagen gel extraction kit (Qiagen, CA). The purified band was cloned using the Topo-TA PCR4 cloning kit (Invitrogen, CA). Forward and reverse sequences were generated using M13 primers at the UC Berkeley DNA Sequencing Center (Berkeley, CA). A BLAST search (http://blast.ncbi.nlm.nih.gov/Blast.cgi) was performed to identify sequences homologous to the query sequence.
PFGE and Southern hybridization analysis.
P. aeruginosa strains from frozen glycerol stocks were grown overnight at 37°C in LB with shaking. Agarose plugs containing 5 × 108 cells ml−1 of overnight culture were prepared according to the Bio-Rad CHEF bacterial genomic DNA plug kit (Bio-Rad Laboratories, CA) with subsequent overnight digestion using 40 units of SpeI at 37°C. Pulsed-field gel electrophoresis (PFGE) was carried out using a Lambda ladder size standard (Bio-Rad Laboratories, CA) and a CHEF-DR III apparatus (Bio-Rad Laboratories, CA). Gels were run under the following conditions: initial switch time, 1.0 s; final switch time, 25 s; total run time, 24 h; included angle, 120°; electric field, 6 V/cm; 0.5× Tris-borate-EDTA (TBE) buffer at 14°C, on a 1% pulsed-field certified agarose gel (Bio-Rad Laboratories, CA).
For high-resolution PFGE, agarose plugs were prepared as described above. Using a Midrange II PFG marker (New England BioLabs, MA), gels were run under the following conditions: initial switch time, 7.2 s; final switch time, 7.5 s; total run time, 36 h; included angle, 120°; electric field, 6 V/cm; 0.5× TBE buffer at 14°C on a 1% pulsed-field certified agarose gel. Southern hybridization of these high-resolution gels was performed using a digoxigenin (DIG)-labeled (Roche, IN) 855-bp probe. The probe was generated using the amplified product generated by PCR primers designed for the additional RAPD PCR band, which discriminated the first and the last two strains in this study. Hybridization was performed at 60°C.
PFGE band electroelution for BAC cloning.
Agarose plugs were prepared as described above with the following exceptions: 2.5 × 109 cells/ml−1 were used, 5 times more units of proteinase K and lysozyme were used for digestion, and 80 units of SpeI were used per plug. PFGE was performed (6 replicates per strain) for the first two isolates as described in “PFGE and Southern hybridization analysis” above. Partial gel staining with ethidium bromide was performed to prevent UV exposure to the DNA bands of interest and guide electroelution of the appropriate agarose strips, which was performed as previously described (19). The doublet of bands exhibiting a putative genetic rearrangement or deletion event in strain 2 and their corresponding homologs from strain 1 were cloned into a bacterial artificial chromosome (BAC) vector, pEpiFOS-5 (Epicentre FOS0901). Briefly, the vector was digested with the BamHI and HindIII restriction enzymes and ligated to a linker sequence containing the BamHI-AvrII-ZraI-NheI-HindIII restriction sites. The modified vector was then cut with NheI to create a cohesive overhang of CTAG to match the SpeI ends of the electroeluted DNA. The resulted BAC clones were then subjected to sequencing.
Shotgun sequencing and gene island annotation.
Pyrosequencing was performed on the isolated BAC DNA using the genome sequencer FLX system (454 Life Sciences) according to the manufacturer's protocols. These BAC clones were derived from strains 1 and 2, representing the upper and lower bands of a doublet that exhibited homology to the Southern hybridization probe and evidence of a rearrangement event. Following quality filtering of sequence reads, contigs were assembled using Newbler 2.3 (454 Life Sciences). Open reading frames (ORFs) were identified using Glimmer 3.02 (20), and BLAST searches were performed (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
Whole-genome sequencing of P. aeruginosa strains.
Strains 1, 2, and 3 were also sequenced using the Illumina genome analyzer II system (Illumina) according to the published Joint Genome Institute (JGI) protocol (http://www.jgi.doe.gov/sequencing/protocols/prots_production.html) and produced 1.9, 2.2, and 2.4 million reads per strain, representing approximately 12×, 14×, and 15× coverage, respectively. Strain 3 was also resequenced using 454 FLX technology, which produced 80 Mb of reads (∼12× coverage). Newbler assembly was again carried out on 312,000 reads, which produced 6.1 Mb in contigs of >100 bp. Comparative genomics was performed using the VISTA package on the JGI's integrated microbial genome (IMG) system (https://img.jgi.doe.gov/cgi-bin/w/main.cgi). Illumina read data were mapped to P. aeruginosa C3719 using BLAST and were analyzed to identify single nucleotide polymorphisms (SNPs) and indels.
Antibiotic resistance testing.
Antibiotic resistance to aminoglycosides, penicillins, penicillin-beta-lactam inhibitor combinations, cephalosporins, quinolones, aztreonam, and imipenem was tested using Dade Behring panels by the Microscan system (Dade Behring Inc., CA). Further testing by the MIC dilution method was performed on all strains for clindamycin and piperacillin-tazobactam using a doubling-dilution technique with a range of concentrations from 0 to 1,024 μg ml−1.
Quantitative reverse transcriptase PCR (qRT-PCR) analysis of gene island ORF expression in response to clinically administered antibiotics.
Following overnight culture in LB broth, each strain was diluted in fresh LB medium to an optical density at 600 nm (OD600) of 0.1 and reincubated at 37°C with shaking for 4 h. Cultures were split into two equal volumes of 10 ml each. One 10-ml culture was exposed to antibiotic (concentrations of 384 μg ml−1 clindamycin and 896 μg ml−1 of piperacillin-tazobactam were chosen as they were the MICs at which strain 1 plateaued at ∼25% viability); the parallel 10-ml culture served as the unexposed control. Cultures were reincubated for 1 h at 37°C with shaking prior to centrifugation at 4,500 × g at 4°C for 30 min. Cell pellets were immediately resuspended in TRIzol (Invitrogen, CA), and RNA was extracted according to the manufacturer's protocol.
DNA contamination was removed from extracted RNA using the Turbo DNA-free kit (Ambion, CA) followed by RNA NanoDrop quantification and RNA integrity assessed by a bioanalyzer (Agilent, CA). One microgram of PCR-confirmed, DNA-free RNA per sample was converted to cDNA using SuperScript III RT (Invitrogen, CA) according to the manufacturer's instructions. Triplicate qRT-PCRs were performed per sample to quantify expression of copA, copB, copC, and copD for the clindamycin exposure experiment. Separate triplicate qRT-PCRs were performed to quantify β-lactamase gene expression from strains exposed to piperacillin-tazobactam. Each 25-μl qRT-PCR volume comprised 12.5 μl QuantiTec SYBR green PCR master mix, 15 pmol of forward primer, 15 pmol of reverse primer, 6.5 μl of water, and 1 μl of cDNA. Cycling was performed as follows: 1 cycle of 10 min at 95°C and 40 cycles of 30 s at 95°C, 1 min at 58°C, and 30 s at 72°C. Copy number was normalized to 16S rRNA gene expression (also performed in triplicate per sample) using the Liu-Saint method (21). Descriptions of all primer pairs used in this study are provided in Table 1.
TABLE 1.
Primer pairs used in this study
| Primer name | Forward primer sequence (5′–3′) | Reverse primer sequence (5′–3′) | Source or reference |
|---|---|---|---|
| Cop A | 5′-CCAGGAAGCTGATGCGTTGT-3′ | 5′-GATGACCCATCCCATGCACC-3′ | This study |
| Cop B | 5′-TCAATGGTTGGCTTTCGGCA-3′ | 5′TCCAGAATCAGTCGCTGGGT-3′ | This study |
| Cop C | 5′-GCATCCTTCGCTGGTGTCTT-3′ | 5′-GCCATTCGACGCGATAGTCA-3′ | This study |
| Cop D | 5′-CGTCGGATATCACGCACCTC-3′ | 5′-ACGCACGATCGCTGTACCTA-3′ | This study |
| ORF 110 β-lactamase | 5′-TGCTCTTCTCCGGCGACATC-3′ | 5′-CTTCATGCTTGCGGTTGCC-3′ | This study |
| 16S rRNA | 5′-ACTCCTACGGGAGGCAGCAG-3′ | 5′-ATTACCGCGGCTGCTGG-3′ | 37 |
| PA0976.1 tRNAlys site (SpF1f and SpF3r) | 5′-ATCCCGTTCCCGATGAACAAACCCG-3′ | 5′-GACAGAGCTTGCTGAACCAAGAGAGC-3′ | 22 |
Screen of gene island integration sites.
A total of 76 randomly chosen P. aeruginosa strains isolated from 76 ETAs of patients with VAP were cultured overnight in 5 ml of LB at 37°C with shaking. DNA was extracted from each strain using the Wizard genomic DNA purification kit (Promega, WI) according to the manufacturer's protocol. The tRNAlys site was screened for an integration event using PCR primers designed on the genomic region flanking this recombination site (SpF1f and SpF3r) (22) (Table 1). PCRs were performed in a final volume of 50 μl under the following conditions: 95°C for 5 min, followed by 30 cycles of 95°C for 1 min, 55°C for 30 s, and 72°C for 1 min and a final extension of 72°C for 7 min. Products were visualized on a 1% agarose gel by UV light and scored integration negative if a visible band was present.
Nucleotide sequence accession numbers strains.
The whole-genome sequence data for P. aeruginosa strains 1, 2, and 3 are deposited in the NCBI Sequence Read Archive under accession numbers SRX099642, SRX099643, and SRX099644.
RESULTS
P. aeruginosa strain isogenicity and gene island location.
P. aeruginosa strains were isolated from sequential ETAs obtained over a 3-week period from an intubated patient who developed P. aeruginosa VAP and was treated with various classes of antimicrobials (Fig. 1A). To determine the dominant P. aeruginosa genotype in the ETAs, 10 colonies isolated from each ETA were cultured and profiled by RAPD PCR analysis. Isolates examined at each individual time point exhibited an identical banding pattern (Fig. 1B), indicating that the populations of Pseudomonas aeruginosa strains at each of these time points were dominated by a single strain type, permitting us to choose a representative strain for each time point for further analysis. However, minor differences in the banding pattern were evident between the time points sampled, suggesting that subtle alterations in the P. aeruginosa genome existed across the time points sampled. Standard PFGE analysis of these representative strains indicated broad-level clonality (Fig. 1C) and that the decline in patient health was not due to superinfection or acquisition of a very distinct P. aeruginosa strain. However, we noted that while the RAPD PCR profiles of each of the representative strains exhibited similar banding patterns, the last isolate (strain 3) demonstrated an additional RAPD PCR product not observed in the first 2 strains. Cloning and sequence analysis of this band revealed 100% homology with two genes, C60 and C61, encoding hypothetical proteins, in the previously described PAGI-2(C) gene island of P. aeruginosa strain C (11).
Although the initial PFGE analysis, performed under standard conditions, did not reveal any gross differences among isolate genomes, the RAPD PCR analysis led us to hypothesize that discrete chromosomal differences between these strains, involving a gene island, may exist. We therefore performed PFGE analysis under a variety of conditions to resolve various-sized chromosomal fractions. This approach revealed a small, reproducible size variation between a doublet of bands in the first isolate compared to the last two strains (Fig. 2A). Southern blotting of this PFGE gel with a probe generated from the sequenced RAPD band demonstrated hybridization to the lower of these two bands in all isolates (Fig. 2B), suggesting genome remodeling via deletion or inversion in an ∼150-kb stretch (based on PFGE band size) in the last two strains, in a region that exhibited homology to the previously described PAGI-2(C) gene island.
FIG 2.
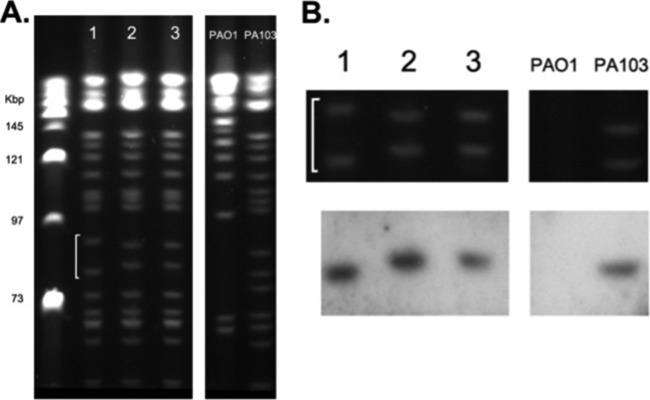
(A) PFGE run under nonstandard conditions to resolve various fractions of the P. aeruginosa genome reveals a repeatable shift in banding pattern (white line) between isolates 1 and 2 that persists in the latter strain. (B) Southern blot of PFGE gel using a probe designed on the PAGI-2 sequence identified by RAPD PCR analysis exhibits hybridization with the lower band in all P. aeruginosa isolates (the region of interest from the PFGE gel is enlarged for clarity).
Sequence characterization of a novel 124-kb P. aeruginosa gene island.
The doublet of bands exhibiting size differences were electroeluted from strains 1 and 2 and cloned into a BAC vector prior to shotgun sequencing. Annotation of sequences from these two bands in strain 1 revealed the existence of a novel 124-kb gene island, designated PAGI-12, characteristically flanked by a split tRNAlys site, which permitted orientation of the island. This commonly known recombination hot spot also serves as an integration site for the PAPI-1, PAPI-2, pKLK106, and PAGI-4 gene islands (3, 6). The G+C content of the island was 64.3%, not significantly different from that of the P. aeruginosa core genome (G+C content = 66% [23]), putatively due to the fact that genes present in this island originated from phylogenetically related species. Sequence homology-based annotation of PAGI-12 revealed that the island included 137 predicted ORFs (see Table S1 in the supplemental material and Fig. 3), which exhibit highest homology with genes from Cupriavidus spp., Parvibaculum spp., Acidovorax spp., Achromobacter spp., Bordetella spp., Delftia spp., Rhodopseudomonas spp., and Xanthomonas spp. Retrospective examination of previously published culture-independent airway microbiota of the ETAs from which these P. aeruginosa strains were isolated (17) revealed that several Cupriavidus and Xanthomonas species were detected in these samples (data not shown), demonstrating cooccurrence of potential donor species and P. aeruginosa in the airways of this patient.
FIG 3.

Gene annotation of a novel gene island identified in clinical isolates of P. aeruginosa (strains 1 and 2). Gene function is indicated by color, and the inversion event is denoted by a hatched line.
The majority of genes on the island, identified through BLAST homology searches, were broadly distributed into eight groups: (i) antibiotic and heavy metal resistance; (ii) transcriptional regulation; (iii) DNA recombination, repair, and modification; (iv) metabolism; (v) two-component response regulator systems, e.g., GacS-like system; (vi) pathogenesis, e.g., type IV secretion components; (vii) hypothetical protein; and (viii) other (Fig. 3). A large number of ORFs (44%) encoded either hypothetical proteins or proteins of unknown function. Toward the 3′ end of the gene island, several stretches of DNA included ORFs with no significant homologs in the NCBI BLAST database, putatively representing novel genes.
Further comparative sequence analyses revealed substantial homology between the gene islands in strains 1 and 2. Alignment of the entire sequences, including the flanking chromosomal regions, demonstrated the existence of identical islands, except for a region at the 3′ end. As predicted by the PFGE analysis, strain 2 exhibited rearrangement of a 36-kb fragment encoding ORFs 82 to 122 (Fig. 3) and deletions resulting in the loss of 3.3 kb of DNA including ORF 98 (d-xylulose 5-phosphate/d-fructose 6-phosphate phosphoketolase) and ORF 99 (no known homolog). Genes borne on the inverted segment included those encoding two heavy metal translocating P-type ATPases (ORFs 88 and 114), a predicted two-component heavy metal response transcriptional regulator (ORF 83), and a putative beta-lactamase domain protein of the metallo-beta-lactamase family (ORF 109), in addition to several copper resistance genes (ORFs 86, a copper resistance B precursor protein [copB] and ORF 87, copper resistance protein [copA]).
We also performed whole-genome shotgun sequencing on the 3 sequential isolates to determine the level of genome sequence homology between strains, outside the gene island region, and to determine their homology to existing P. aeruginosa reference genomes. Comparative analysis of draft genomes from these strains identified a total of 33 deletions at various positions around the genome in strain 3, compared to strains 1 and 2, in genes encoding a variety of functional proteins such as transporters, regulatory genes, and protein secretion system genes (see Table S2 in the supplemental material). Though these SNPs may represent bona fide point mutations in strain 3, it should be acknowledged that these findings are within the range of sequencing error, particularly at the draft levels described here. Nonetheless, these data suggested that the primary source of genomic variation and remodeling in these strains existed within the tRNAlys-flanked gene island rather than the genomic backbone of these isolates. All strains exhibited high homology with the sequenced strain P. aeruginosa C3719.
Expression of genes on the island in response to antibiotics administered to the patient.
We hypothesized that the gene island inversion event represented rapid evolution in response to selective pressures from antimicrobials administered to the patient, which thus provided these strains with a fitness advantage upon such exposures. The inversion event coincided with administration of clindamycin and piperacillin-tazobactam to the patient (Fig. 1A), and the inverted DNA segment carried a putative gene (ORF 109) that exhibited high homology with an Acidovorax species β-lactamase. MIC testing of all three strains against doubling dilutions of piperacillin-tazobactam revealed that all three strains were considered resistant (based on the CLSI breakpoint), although at higher antibiotic concentrations (896 μg ml−1) strains 2 and 3 exhibited greater viability (see Fig. S1A in the supplemental material). We hypothesized that this was due to altered regulation of the island-borne putative β-lactamase gene in strains in which inversion of the island had occurred. To examine this, we cultured all 3 strains in the presence or absence of piperacillin-tazobactam and performed qRT-PCR expression analysis of this gene, normalized to total 16S rRNA copy number to account for changes in viability that may impact relative expression over these strains. Compared to P. aeruginosa C3719 as a negative control, we demonstrated that all of the clinical isolates expressed this β-lactamase gene at appreciable levels even in the absence of piperacillin-tazobactam. However, upon exposure of the strains to this antibiotic, the relative expression of this gene increased significantly only in strains 2 and 3, which housed the inverted island genotype (Fig. 4), providing evidence for an association between island remodeling and increased expression of this gene. This increased expression in strains 2 and 3 was associated with significantly increased resistance of these strains to piperacillin/tazobactam (see Fig. S1A in the supplemental material). However, the clinical implications of this increased resistance are unclear, since the concentrations of antibiotic at which significantly increased resistance occurred is not clinically relevant and all three strains are deemed resistant to piperacillin-tazobactam based on the CLSI breakpoint.
FIG 4.

qRT-PCR relative gene expression of the gene island-borne β-lactamase gene by all three clinical isolates in the absence (white bars) or presence (black bars) of piperacillin-tazobactam. P. aeruginosa C3719 acts as a negative control.
We noted that the inversion event also appeared to reconstitute an Achromobacter xylosoxidans copper resistance operon, placing the genes copA and copB proximal to copC and copD. Though clindamycin is not bactericidal to P. aeruginosa, it is known to impact production of alginic acid and exopolysaccharide (24), suggesting that it represents a stress for this species. Given the well-documented relationship between bacterial heavy metal and antibiotic resistance (25–27), we hypothesized that expression of these genes involved in copper sequestration (copA and copC) and efflux (copB and copD) (28, 29) would be increased in response to clindamycin exposure. Clindamycin MIC analyses of these three strains indicated that strains 2 and 3 exhibited increased viability compared to strain 1, and this was particularly evident at a concentration of 384 μg/ml (see Fig. S1B in the supplemental material). Therefore, all three strains were incubated with and without 384 μg ml−1 of clindamycin as described in Materials and Methods. All three P. aeruginosa strains expressed the four cop genes, and exposure to clindamycin resulted in significantly higher gene expression of all four genes across the three strains (with the exception of copA expression by strain 2; Fig. 5A to D).
FIG 5.

qRT-PCR-based relative expression of copA (A), copB (B), copC (C), and copD (D) heavy metal resistance genes in the absence (white bars) or presence (black bars) of clindamycin.
Prevalence of chromosomal integration at tRNAlys among P. aeruginosa VAP isolates.
Gene islands described to date are characteristically recombined into the bacterial genome at several well-known recombination hot spots, including the tRNAlys site, which was identified as the site of integration in this study (3, 6). Since gene islands provide bacteria with a diversity of auxiliary genes and functions, including virulence factors (15), we hypothesized that integration of DNA at a tRNAlys site is correlated with poorer clinical outcomes. We therefore designed a simple PCR-based assay using primers designed to flank the tRNAlys site, which was performed at a relatively permissive annealing temperature (to permit PCR amplification of nonidentical tRNAlys sequence). A total of 76 P. aeruginosa strains isolated from ETAs of 76 patients with ventilator-associated pneumonia were screened using this assay. Isolates were obtained from a range of adult patients, including those with high and low burdens of P. aeruginosa and those with and without signs of clinical infection (i.e., fever, leukocytosis, purulent secretions, or consolidations on chest radiograph). Isolates that produced a PCR product were considered integration negative, while those that did not were considered integration positive. A total of 40 of these isolates (52.6%) were considered integration positive, while 36 (47.4%) were integration negative. We next sought to determine whether possessing a putatively integration-positive P. aeruginosa genotype was associated with clinical outcome, by stratifying patients based on integration status (positive or negative) and examining 28-day mortality across these groupings. Overall patient mortality was 26.3% (20 of 76 patients). Of the 20 P. aeruginosa strains isolated from the deceased patients, 15 (75%) demonstrated integration at the tRNAlys site examined. Of the isolates from the patients who survived, only 25 of the 56 strains (44.6%) exhibited putative integration at this site. These preliminary data indicate that recombination events at this single integration site are associated with significantly increased patient mortality in this cohort (P < 0.035; Fisher's exact test). Though preliminary, this observation indicates that genome-based pathogen population studies may permit enhanced insights into variable outcomes in patient populations.
DISCUSSION
Gene island-mediated chromosomal plasticity in P. aeruginosa is a well-documented phenomenon thought to confer a competitive advantage to this species in a variety of environments (3–5, 7, 10, 30). Identification of these islands has revealed the remarkable potential that these accessory elements confer upon bacterial species to evolve and expand beyond their relatively conserved genotypes. Multiple distinct gene islands have been identified in a number of clinical isolates, and in certain cases, it has been demonstrated that these ancillary genetic elements provide P. aeruginosa with the means to better adapt to and compete in a changing host environment (7).
Though the strains in this study were deemed clonal by standard pulsed-field electrophoresis, RAPD PCR analysis suggested otherwise, and more-exhaustive efforts to resolve the genomes revealed a very subtle but reproducible difference in the genome at a site exhibiting homology to elements from a known gene island [PAGI-2(C)]. Subsequent sequencing of this region revealed what is, to our knowledge, the largest documented gene island to date in P. aeruginosa. Many genes in this island are involved in metabolism and in heavy metal and antibiotic resistance, again suggesting that, as for many other described gene islands, these mosaics of exogenous genomic DNA expand the functional repertoire of this pathogen and likely enhance its ability to survive in the human host. For example, ORF 112, which lies within the inverted region, exhibits homology to GacS, a membrane-bound sensor kinase that forms part of a two-component regulatory system in Pseudomonas species regulating genes involved in virulence, secondary metabolism, and biofilm formation (31). Multiple two-component response regulators, which commonly possess a histidine kinase and response regulator domain, have been shown to facilitate rapid responses to various environmental conditions, including coordinated expression of specific virulence determinants (32). Increased ability to sense and rapidly respond to changing environmental conditions clearly confers a fitness advantage; therefore, accumulation of additional two-component systems that putatively expand this repertoire represents an attractive strategy for improving bacterial strain versatility, particularly in human host niches.
The conjugation subfamily of the type IV secretion system (T4SS) has been implicated as a major mechanism of transmission of virulence factors, antibiotic resistance elements, and other fitness traits among human-pathogenic bacteria. Other T4SS subfamilies are employed by bacteria such as Legionella pneumophila and Bordetella pertussis to deliver proteins and molecules to eukaryotic target cells facilitating host invasion and colonization (33). ORFs 46 and 64 of the gene island encode component proteins VirD4/VirB4 of the T4SS. The identification of T4SS genes within this island that facilitate the transfer of DNA from cells prompted speculation that these islands encode essential elements that support their transmission across strains or possibly even species. Carter and colleagues have demonstrated transfer of PAPI-1 by a specific type IV pilus apparatus encoded by elements within this gene island in P. aeruginosa (34). The authors further suggest that PAPI-1 may have evolved by acquisition of this pilus-based conjugation system, further supporting the hypothesis that these mobile genetic mosaics commonly carry genes capable of facilitating their transfer to other bacterial cells.
The PAGI-12 island described here contains several genes for copper resistance, which are transcribed in the same direction as adjacent copper regulatory genes, including copC and the CzcA family heavy metal efflux pump gene, located in the 3′ chromosomal area just outside the inverted segment in the last strains. Studies have demonstrated that environmentally sourced, heavy metal-resistant P. aeruginosa strains have a higher incidence of antibiotic resistance (25–27). We hypothesized that genes involved in copper resistance may also play a role in clindamycin resistance of these strains. Though clindamycin, a lincosamide class antimicrobial, is not used to treat P. aeruginosa infection, it was the first empirical antimicrobial administered to the patient in this study and the first antimicrobial encountered by P. aeruginosa in this setting. All three strains demonstrated clindamycin-responsive cop gene expression in vitro, implying that these genes may also be expressed in vivo upon such exposures.
Of further interest, the inverted gene island segment also encoded a putative beta-lactamase domain protein of the metallo-beta-lactamase family, associated with resistance to beta-lactam antimicrobials. Piperacillin-tazobactam (a beta-lactam antimicrobial) is frequently used to treat P. aeruginosa infections and was specifically administered to the patient whose isolates were used in this study. A statistically significant increase in expression of this putative beta-lactamase gene in the presence of this antimicrobial was observed only in strains exhibiting the gene island inversion event, suggesting that gene island remodeling events alter regulation of such genes. Accounts of chromosomal inversion impacting gene expression in bacterial species have been described previously; for example, inversions in the genome of Bacteroides fragilis, a common gastrointestinal inhabitant, are known to control variable gene expression (35).
Germane to this discussion is the relatively large number of ORFs located in regions flanking the site of inversion that displayed no known homology to known genes in publicly available databases. This observation raises the possibility that remodeling events, through coding region loss and genomic inversion, may represent sites of novel ORF evolution. From the bacterial perspective, evolution within confined regions of what essentially amounts to accessory genes may provide a low-risk and efficient mechanism of adaptation to changes in environmental conditions, while maintaining a relatively stable genomic backbone. In support of this theory is the observation that despite exposure to the selective pressures of antimicrobials, the genomic sequence outside the gene island in each of the strains remained relatively stable over the sampling period, while the gene island itself underwent substantial remodeling. This suggests that P. aeruginosa has developed a strategy involving evolution of accessory DNA to rapidly customize its fitness under antimicrobial selective pressure in the infected human host. Given that a large number of P. aeruginosa clinical isolates with gene islands originate from mixed-species microbiotas that are frequently exposed to antimicrobials, e.g., cystic fibrosis airways (7, 36), it is not entirely unexpected that such a strategy exists in this highly successful respiratory pathogen.
Pilot data from this study suggest that integration at one of the well-known recombination hot spots, tRNAlys, is associated with mortality of patients with ventilator-associated pneumonia. There are some caveats to our observation, which include the possibility that some of the integration-positive strains may simply encode very distinct genome sequences not targeted by the PCR primers designed for this assay or that patients who died were infected by a very different P. aeruginosa strain, which cannot be ascertained by the PCR assay used in this preliminary survey. Nonetheless, these data suggest that further studies involving whole-genome sequences of populations of pathogen isolates are necessary to improve our understanding of both the prevalence and the role of gene islands in patient outcome.
The rapidly evolving field of human microbiome research has demonstrated mixed-species microbial populations associated with a number of respiratory diseases and infections, which undoubtedly leads to transfer of genetic material between species within these communities. While interspecies interactions play a key role in community function, there is an equally great need to comprehend the fundamentals of strain population dynamics and genomic evolution of individual species within these consortia.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by research grants from the National Institutes of Health (HLO74005, HL69809, and AI075410PO1) and the Pathways to Careers in Clinical and Translational Research.
We thank Judith Flanagan and Eoin Brodie for their advice and Yoriko Sawa and Amua Camargo for technical support. We also thank Steven Lory for the kind provision of P. aeruginosa C3719 to us for this study.
Footnotes
Published ahead of print 30 April 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JCM.01626-13.
REFERENCES
- 1. Chastre J, Fagon JY. 2002. Ventilator-associated pneumonia. Am. J. Respir. Crit. Care Med. 165:867–903. 10.1164/ajrccm.165.7.2105078 [DOI] [PubMed] [Google Scholar]
- 2. Kollef MH, Shorr A, Tabak YP, Gupta V, Liu LZ, Johannes RS. 2005. Epidemiology and outcomes of health-care-associated pneumonia: results from a large US database of culture-positive pneumonia. Chest 128:3854–3862. 10.1378/chest.128.6.3854 [DOI] [PubMed] [Google Scholar]
- 3. Klockgether J, Reva O, Larbig K, Tummler B. 2004. Sequence analysis of the mobile genome island pKLC102 of Pseudomonas aeruginosa C. J. Bacteriol. 186:518–534. 10.1128/JB.186.2.518-534.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Romling U, Schmidt KD, Tummler B. 1997. Large genome rearrangements discovered by the detailed analysis of 21 Pseudomonas aeruginosa clone C isolates found in environment and disease habitats. J. Mol. Biol. 271:386–404. 10.1006/jmbi.1997.1186 [DOI] [PubMed] [Google Scholar]
- 5. Liang X, Pham XQ, Olson MV, Lory S. 2001. Identification of a genomic island present in the majority of pathogenic isolates of Pseudomonas aeruginosa. J. Bacteriol. 183:843–853. 10.1128/JB.183.3.843-853.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Qiu X, Gurkar AU, Lory S. 2006. Interstrain transfer of the large pathogenicity island (PAPI-1) of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 103:19830–19835. 10.1073/pnas.0606810104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Winstanley C, Langille MGI, Fothergill JL, Kukavica-Ibrulj I, Paradis-Bleau C, Sanschagrin F, Thomson NR, Winsor GL, Quail MA, Lennard N, Bignell A, Clarke L, Seeger K, Saunders D, Harris D, Parkhill J, Hancock REW, Brinkman FSL, Levesque RC. 2009. Newly introduced genomic prophage islands are critical determinants of in vivo competitiveness in the Liverpool epidemic strain of Pseudomonas aeruginosa. Genome Res. 19:12–23. 10.1101/gr.086082.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hacker J, Carniel E. 2001. Ecological fitness, genomic islands and bacterial pathogenicity. A Darwinian view of the evolution of microbes. EMBO Rep. 2:376–381. 10.1093/embo-reports/kve097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hacker J, Kaper JB. 2000. Pathogenicity islands and the evolution of microbes. Annu. Rev. Microbiol. 54:641–679. 10.1146/annurev.micro.54.1.641 [DOI] [PubMed] [Google Scholar]
- 10. Harrison EM, Carter ME, Luck S, Ou HY, He X, Deng Z, O'Callaghan C, Kadioglu A, Rajakumar K. 2010. Pathogenicity islands PAPI-1 and PAPI-2 contribute individually and synergistically to the virulence of Pseudomonas aeruginosa strain PA14. Infect. Immun. 78:1437–1446. 10.1128/IAI.00621-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Larbig KD, Christmann A, Johann A, Klockgether J, Hartsch T, Merkl R, Wiehlmann L, Fritz HJ, Tummler B. 2002. Gene islands integrated into tRNAGly genes confer genome diversity on a Pseudomonas aeruginosa clone. J. Bacteriol. 184:6665–6680. 10.1128/JB.184.23.6665-6680.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gal-Mor O, Finlay BB. 2006. Pathogenicity islands: a molecular toolbox for bacterial virulence. Cell. Microbiol. 8:1707–1719. 10.1111/j.1462-5822.2006.00794.x [DOI] [PubMed] [Google Scholar]
- 13. Battle SE, Rello J, Hauser AR. 2009. Genomic islands of Pseudomonas aeruginosa. FEMS Microbiol. Lett. 290:70–78. 10.1111/j.1574-6968.2008.01406.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Klockgether J, Wurdemann D, Reva O, Wiehlmann L, Tummler B. 2007. Diversity of the abundant pKLC102/PAGI-2 family of genomic islands in Pseudomonas aeruginosa. J. Bacteriol. 189:2443–2459. 10.1128/JB.01688-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kulasekara BR, Kulasekara HD, Wolfgang MC, Stevens L, Frank DW, Lory S. 2006. Acquisition and evolution of the exoU locus in Pseudomonas aeruginosa. J. Bacteriol. 188:4037–4050. 10.1128/JB.02000-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Juhas M, Van Der Meer JR, Gaillard M, Harding RM, Hood DW, Crook DW. 2009. Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol. Rev. 33:376–393. 10.1111/j.1574-6976.2008.00136.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Flanagan JL, Brodie EL, Weng L, Lynch SV, Garcia O, Brown R, Hugenholtz P, DeSantis TZ, Andersen GL, Wiener-Kronish JP, Bristow J. 2007. Loss of bacterial diversity during antibiotic treatment of intubated patients colonized with Pseudomonas aeruginosa. J. Clin. Microbiol. 45:1954–1962. 10.1128/JCM.02187-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pellegrino FL, Casali N, Dos Santos KR, Nouer SA, Scheidegger EM, Riley LW, Moreira BM. 2006. Pseudomonas aeruginosa epidemic strain carrying bla(SPM) metallo-beta-lactamase detected in Rio de Janeiro, Brazil. J. Chemother. 18:151–156. 10.1179/joc.2006.18.2.151 [DOI] [PubMed] [Google Scholar]
- 19. Strong SJ, Ohta Y, Litman GW, Amemiya CT. 1997. Marked improvement of PAC and BAC cloning is achieved using electroelution of pulsed-field gel-separated partial digests of genomic DNA. Nucleic Acids Res. 25:3959–3961. 10.1093/nar/25.19.3959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Delcher AL, Bratke KA, Powers EC, Salzberg SL. 2007. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23:673–679. 10.1093/bioinformatics/btm009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu W, Saint DA. 2002. A new quantitative method of real time reverse transcription polymerase chain reaction assay based on simulation of polymerase chain reaction kinetics. Anal. Biochem. 302:52–59. 10.1006/abio.2001.5530 [DOI] [PubMed] [Google Scholar]
- 22. Kiewitz C, Larbig K, Klockgether J, Weinel C, Tummler B. 2000. Monitoring genome evolution ex vivo: reversible chromosomal integration of a 106 kb plasmid at two tRNA(Lys) gene loci in sequential Pseudomonas aeruginosa airway isolates. Microbiology 146(Part 10):2365–2373 [DOI] [PubMed] [Google Scholar]
- 23. Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FS, Hufnagle WO, Kowalik DJ, Lagrou M, Garber RL, Goltry L, Tolentino E, Westbrock-Wadman S, Yuan Y, Brody LL, Coulter SN, Folger KR, Kas A, Larbig K, Lim R, Smith K, Spencer D, Wong GK, Wu Z, Paulsen IT, Reizer J, Saier MH, Hancock RE, Lory S, Olson MV. 2000. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406:959–964. 10.1038/35023079 [DOI] [PubMed] [Google Scholar]
- 24. Ichimiya T, Yamasaki T, Nasu M. 1994. In-vitro effects of antimicrobial agents on Pseudomonas aeruginosa biofilm formation. J. Antimicrob. Chemother. 34:331–341. 10.1093/jac/34.3.331 [DOI] [PubMed] [Google Scholar]
- 25. Upritchard HG, Yang J, Bremer PJ, Lamont IL, McQuillan AJ. 2007. Adsorption to metal oxides of the Pseudomonas aeruginosa siderophore pyoverdine and implications for bacterial biofilm formation on metals. Langmuir 23:7189–7195. 10.1021/la7004024 [DOI] [PubMed] [Google Scholar]
- 26. de Vicente A, Aviles M, Codina JC, Borrego JJ, Romero P. 1990. Resistance to antibiotics and heavy metals of Pseudomonas aeruginosa isolated from natural waters. J. Appl. Bacteriol. 68:625–632. 10.1111/j.1365-2672.1990.tb05228.x [DOI] [PubMed] [Google Scholar]
- 27. Banin E, Vasil ML, Greenberg EP. 2005. Iron and Pseudomonas aeruginosa biofilm formation. Proc. Natl. Acad. Sci. U. S. A. 102:11076–11081. 10.1073/pnas.0504266102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Adaikkalam V, Swarup S. 2005. Characterization of copABCD operon from a copper-sensitive Pseudomonas putida strain. Can. J. Microbiol. 51:209–216. 10.1139/w04-135 [DOI] [PubMed] [Google Scholar]
- 29. Cooksey DA. 1993. Copper uptake and resistance in bacteria. Mol. Microbiol. 7:1–5. 10.1111/j.1365-2958.1993.tb01091.x [DOI] [PubMed] [Google Scholar]
- 30. Wiehlmann L, Wagner G, Cramer N, Siebert B, Gudowius P, Morales G, Kohler T, van Delden C, Weinel C, Slickers P, Tummler B. 2007. Population structure of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 104:8101–8106. 10.1073/pnas.0609213104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Davies JA, Harrison JJ, Marques LL, Foglia GR, Stremick CA, Storey DG, Turner RJ, Olson ME, Ceri H. 2007. The GacS sensor kinase controls phenotypic reversion of small colony variants isolated from biofilms of Pseudomonas aeruginosa PA14. FEMS Microbiol. Ecol. 59:32–46. 10.1111/j.1574-6941.2006.00196.x [DOI] [PubMed] [Google Scholar]
- 32. Krell T, Lacal J, Busch A, Silva-Jiménez H, Guazzaroni M-E, Ramos JL. 2010. Bacterial sensor kinases: diversity in the recognition of environmental signals. Annu. Rev. Microbiol. 64:539–559. 10.1146/annurev.micro.112408.134054 [DOI] [PubMed] [Google Scholar]
- 33. Christie PJ, Atmakuri K, Krishnamoorthy V, Jakubowski S, Cascales E. 2005. Biogenesis, architecture, and function of bacterial type IV secretion systems. Annu. Rev. Microbiol. 59:451–485. 10.1146/annurev.micro.58.030603.123630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Carter MQ, Chen J, Lory S. 2010. The Pseudomonas aeruginosa pathogenicity island PAPI-1 is transferred via a novel type IV pilus. J. Bacteriol. 192:3249–3258. 10.1128/JB.00041-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cerdeno-Tarraga AM. 2005. Extensive DNA inversions in the B. fragilis genome control variable gene expression. Science 307:1463–1465. 10.1126/science.1107008 [DOI] [PubMed] [Google Scholar]
- 36. Mathee K, Narasimhan G, Valdes C, Qiu X, Matewish JM, Koehrsen M, Rokas A, Yandava CN, Engels R, Zeng E, Olavarietta R, Doud M, Smith RS, Montgomery P, White JR, Godfrey PA, Kodira C, Birren B, Galagan JE, Lory S. 2008. Dynamics of Pseudomonas aeruginosa genome evolution. Proc. Natl. Acad. Sci. U. S. A. 105:3100–3105. 10.1073/pnas.0711982105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lane D. 1991. 16S/23S rRNA sequencing, p 115–175 In Stackebrandt E, Goodfellow M. (ed), Nucleic acid techniques in bacterial systematics. John Wiley & Sons, New York, NY [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.