

Abstract
Chiral saturated oxygen heterocycles are important components of bioactive compounds. Cyclization of alcohols onto pendant alkenes is a direct route to their synthesis, but few catalytic enantioselective methods enabling cyclization onto unactivated alkenes exist. Herein is reported a highly efficient copper-catalyzed cyclization of γ-unsaturated pentenols that terminates in C-C bond formation, a net alkene carboetherification. Both intra- and intermolecular C-C bond formations are demonstrated, yielding functionalized chiral tetrahydrofurans as well as fused-ring and bridged-ring oxabicyclic products. Transition state calculations support a cis-oxycupration stereochemistry-determining step.
Keywords: copper-catalyzed, carboetherification, alkene, tetrahydrofuran, enantioselective
Oxygen heterocycles, such as tetrahydrofurans, can be found in numerous bioactive compounds.[1,2] Metal-catalyzed carboetherification/cyclization of unsaturated alcohols is a powerful complexity-building strategy for the synthesis of functionalized saturated oxygen heterocycles from simple unactivated alkenols. Significant effort in this field has resulted in the synthesis of diverse oxygen heterocycle products with predictable regio- and diastereoselectivity.[3–12] While the related synthesis of enantiomerically enriched nitrogen heterocycles via Pd- or Cu-catalyzed enantioselective alkene carboamination methodologies has been substantially developed in recent years,[13,14] advances in reaction technology for catalytic enantioselective carboetherification/cyclization of unactivated alkenes have largely remained elusive.[15] Notable exceptions include Pd-catalyzed reactions of ortho-vinyl phenols that proceed through quinone methide intermediates[16] and Pd-catalyzed cyclizations of γ- and δ-unsaturated phenols that terminate in carbonylation or olefin insertion (whose substituted intermediates are unable to undergo β-hydride elimination).[17] More recently, an enantioselective copper-catalyzed oxytrifluoromethylation of 4-aryl-4-pentenoic acids where carbon radical-initiated C-C bond formation occurs prior to C-O bond formation, and copper-catalyzed C-O bond-formation appears to be the enantioselectivity-determining step has been demonstrated.[18] In 2012 we reported a new copper-catalyzed doubly intramolecular alkene carboetherification that produced bicyclic tetrahydrofurans with promising (up to 75% ee) enantioselectivity using the (R,R)-Ph-box ligand (Scheme 1).[19] At that time, we tentatively assigned the absolute configuration of the product as shown (Scheme 1). We report herein optimization of the enantioselective carboetherification reaction (up to >95% ee) and a revision of the absolute stereochemical assignment. Importantly, we show herein expansion of the method to involve intermolecular C-C bond formation via alkyl Heck-type couplings with vinyl arenes (Scheme 1).[13b,20] This method is complementary in substrate scope to existing catalytic enantioselective carboetherifications[15–18,21–23] as aliphatic alcohols with unactivated terminal alkenes are excellent substrates, and various vinylarenes undergo coupling with them (vide infra).
Scheme 1.

Copper-catalyzed enantioselective alkene carboetherification.
The promising level of enantioselectivity (75% ee) obtained with 1,1-diphenyl-3,3-dibenzyl-4-pentenol (1a) using the (R,R)-Ph-box ligand shown in Scheme 1 prompted us to screen additional ligands (Table 1). It should be noted that these oxidative cyclizations are net C-H functionalizations (arene functionalization) where the stoichiometric oxidant, hypothesized to turnover [Cu(I)] to [Cu(II)] (vide infra), is readily available and inexpensive activated MnO2 (ca. 85%, <5 μM). We were fortunate to quickly find that the commercially available (S,S)-t-Bu-box ligand gave the opposite enantiomer (by chiral HPLC and optical rotation) of tetrahydrobenzofuran 2a in >95% ee (Table 1, entry 3). Interestingly, the (S)-i-Pr-quinox ligand, though not highly selective (20% ee), provides a product enantiomeric to that of (S,S)-t-Bu-Box (compare Table 1 entries 3 and 4). Reaction of the chloro-substituted pentenol 1b provided the corresponding tetrahydrobenzofuran adduct 2b in >95% ee as well, and we were able to obtain an X-ray crystal structure of 2b that definitively assigned its absolute stereochemistry (using its heavy atom) as (R,R), as shown (Figure 1). This result indicated our original, tentative assignment of the absolute stereochemistry with the (R,R)-Ph-box ligand (Scheme 1) was incorrect.[19]
Table 1.
Ligand Screening.[a]
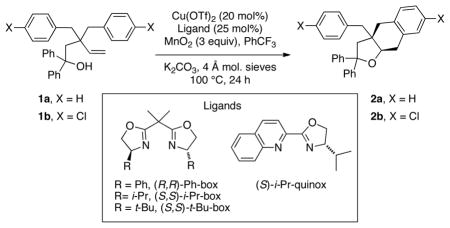
| ||||
|---|---|---|---|---|
| Entry | Substrate | Ligand | Yield (%)[b] | ee (%)[c] |
| 1 | 1a | (R,R)-Ph-box | 89 | −75 |
| 2 | 1a | (S,S)-i-Pr-box | 96 | 93 |
| 3 | 1a | (S,S)-t-Bu-box | 95 | >95 |
| 4 | 1a | (S)-i-Pr-quinox | 90 | −20[d] |
| 5 | 1b | (S,S)-t-Bu-box | 95 | >95 |
All reactions were run under anhydrous conditions at 0.1 M under Ar in sealed tubes with ca. 0.139 mmol 1 and 1 equiv K2CO3.
Isolated yield after flash chromatography on silica gel.
Enantiomeric excess was determined by chiral HPLC.
Enantiomers not baseline resolved in this HPLC trace.
Figure 1.
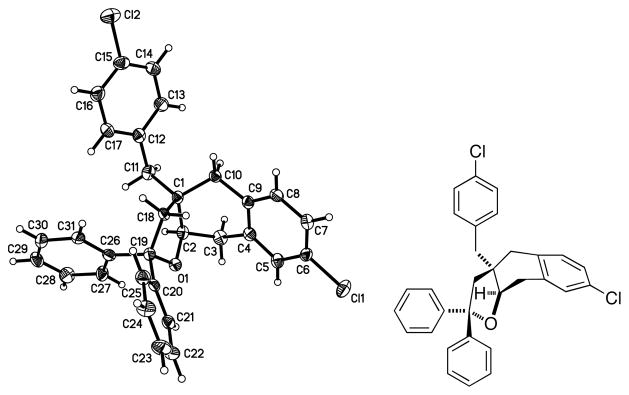
X-ray structure of 2b showing (R, R) absolute stereochemistry.
The scope of the copper-catalyzed enantioselective doubly intramolecular alkene carboetherification was briefly explored where both substrate backbone and alkene substitution were varied using the optimized (Table 1, entry 3) conditions (Table 2). Reaction of the less substituted pentenol 3 gave tetrahydrobenzofuran 4 in 70% ee, substantially better than previously observed (Scheme 1), but not as high as observed with 1,1-diaryl substrates 1 (Table 1). Pentenols 5 and 7, containing 2,2-diaryl substitution provided oxabicyclo[3.2.1]octanes 6 and 8 with good selectivity (82% and 84% ee, respectively, Table 2, entries 2 and 3). (E)-2,2-Diphenyl-4-hexenol (9) provided a 1:1 diastereomeric mixture of oxabicyclo[3.2.1]octanes 10 and 11 with surprisingly excellent enantioselectivity (94% ee for both, Table 2, entry 4).
Table 2.
Affect of Substrate Structure on Enantioselective Intramolecular Carboetherification[a]
| Entry | Substrate | Product | Yield (%)[b] | ee (%)[c] |
|---|---|---|---|---|
| 1[d] |
 3 |
 4 |
71 | 70 |
| 2 |
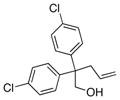 5 |
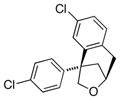 6 |
77 | 82 |
| 3 |
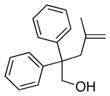 7 |
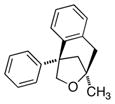 8 |
99 | 84 |
| 4 |
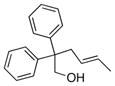 9 |
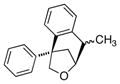 10/11 (dr = 1:1) |
96 | 94/94 |
Same reaction conditions as Table 1, entry 3.
Isolated yield.
Enantiomeric excess was determined by chiral HPLC.
This substrate gives 37% ee using (S)-i-Pr-box and −37% ee using (S)-i-Pr-quinox.
We next turned our attention to intermolecular C-C bond forming reactions, whose development of required consideration of the carboetherification reaction mechanism. We have provided evidence for a reaction mechanism involving oxycupration across the alkene with concommittant generation of an unstable organocopper(II) intermediate that undergoes homolysis to generate [Cu(I)] and a carbon radical intermediate (Scheme 2).[19] In that study, evidence for a primary carbon radical intermediate was provided by 1) H-atom abstraction from 1,4-cyclohexadiene and 2) an isotopic labeling study which gave a 1:1 diastereomeric product mixture from a (Z)-deuterated alkene.[19] In the case of the doubly intramolecular carboetherification, the radical adds to a pendant arene, and subsequent re-aromatization under the oxidizing reaction conditions provides the observed bicyclic products (Scheme 2). Intramolecularity in the arene addition step seems important as we have not yet observed intermolecular additions to arenes. We hypothesized that intermolecular C-C bond formation could occur if the carbon radical intermediate were intercepted with superior radical acceptors, namely, vinylarenes. Subsequent oxidation of the resulting benzylic radicals would provide new vinyl arenes. We were optimistic the vinyl arene additions would be feasible as these radical acceptors worked in our related copper-catalyzed enantioselective alkene carboamination/alkyl-Heck type reactions.[13b]
Scheme 2.
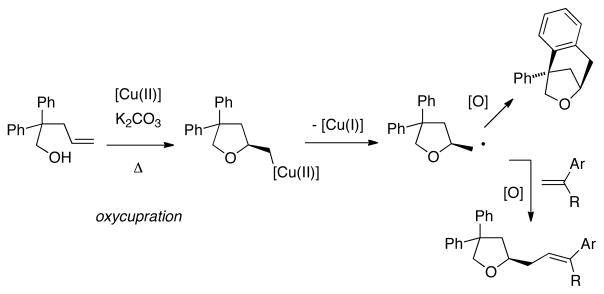
Proposed intra- and intermolecular carboetherification mechanism.
Table 3 summarizes the results of enantioselective copper-catalyzed coupling of variously substituted γ-unsaturated alcohols with various vinylarenes. 4-Pentenol (12) underwent coupling with diphenylethylene to provide tetrahydrofuran 14 in excellent yield and good enantioselectivity (Table 3, entry 1). The absolute stereochemistry of 14 was assigned as (S) by its conversion to a known tetrahydrofuran and optical rotation comparison (see Supporting Information for details). The absolute stereochemistry of all other products was assigned by analogy to 2b and 14.
Table 3.
Intermolecular carboetherification/Heck-type couplings[a]
| Entry | Substrate | Alkene | Product | Yield (%)[b] | ee (%)[c] |
|---|---|---|---|---|---|
| 1[d] |
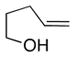 12 |
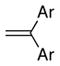 13a Ar = Ph |
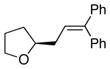 14 |
92 | 82 |
| 2 |
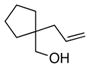 15 |
13a |
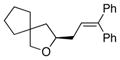 16 |
90 | 82 |
| 3 |
 17 |
13a |
 18a, Ar = Ph |
90 | >95 |
| 4 | 5 | 13a |
 19, Ar = 4-ClC6H4 |
80 | 80 |
| 5 |
 20 |
13a |
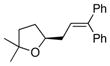 21 |
84 | >95 |
| 6 |
22 |
13a |
23/24 (dr = 6:1) |
90 | 86 (maj) >95 (min) |
| 7 | 17 |
13b Ar = 4-MeOC6H4 |
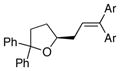 18b Ar = 4-MeOC6H4 |
88 | >95 |
| 8 | 17 | 13c, Ar = 4-F-C6H4 | 18c, Ar = 4-F-C6H4 | 70 | >95 |
| 9 | 17 |
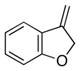 25 |
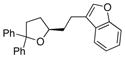 26 |
88 | 94 |
| 10 | 17 |
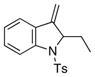 27 |
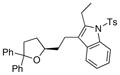 28 |
81 | nd[e] |
| 11 | 17 |
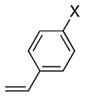 29a X = OMe |
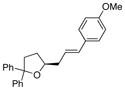 30, E:Z = >20:1 |
46 | >95 |
| 12 | 20 |
29b X = t-Bu |
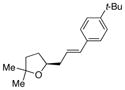 31, E:Z = >20:1 |
42 | 95 |
Reactions were run under anhydrous conditions under Ar in a sealed tube. 20 mol% of Cu(OTf)2 was complexed with 25 mol% (S,S)-t-Bu-box (60 °C for 2 h in 1 mL PhCF3) then ca. 0.145 mmol alkenol substrate in PhCF3 (0.1 M total), 3 equiv vinylarene, 1 equiv K2CO3, 3 equiv MnO2 and ca. 36 mg 4 Å mol sieves were added and the reaction stirred at 100 °C for 16 h unless otherwise noted.
Isolated yield following chromatography on silica gel.
Enantiomeric excess determined by chiral HPLC.
Reaction concentration of 0.08 M used.
Not determined. Enantiomers would not separate on several chiral HPLC columns.
As illustrated in Table 3, entries 1–6, various alcohol substitution patterns were tolerated although α,α-disubstituted alcohols 17 and 20 provided the highest level of enantioselectivity (>95% ee). The meso dieneol 22 provides a 6:1 ratio of diastereomers favoring the trans-substituted tetrahydrofuran 23, albeit the minor, cis diastereomer 24 is formed with higher enantioselectivity (>95% ee versus 86% ee, Table 3, entry 6). Both electron-rich (p-MeO) and electron poor (p-F) 1,1-diaryl ethylenes can be used in the coupling (Table 3, entries 7 and 8). It should be noted that conversion of the product’s alkene to an aldehyde should be facile[13b] and would extend their utility beyond diarylalkenes. Net benzofuran and indole coupling adducts 26 and 28 were obtained by reaction of the alkenols with heterocyclic vinylarenes 25 and 27,[24] respectively (Table 3, entries 9 and 10). Additionally, both 4-methoxy- and 4-tert-butyl styrene served as vinylarene component in this coupling reaction, giving homoallylic ethers 30 and 31 in moderate yield and high enantioselectivity (Table 3, entries 11 and 12).
The high enantioselectivity was used to further probe the mechanism of this novel reaction. Involvement of the chiral copper catalyst in the alkene addition step is evident from the observed reaction enantioselectivity. This step is thought to occur via enantioselective cis-oxycupration.[19] Cis-oxycupration presupposes that the alcohol moiety of the substrate first coordinates to the copper(II) center in preference to the alkene. This is supported by reported coordination preferences of copper(II) complexes.[25] An alternative trans-oxycupration mechanism was thus considered less likely. To further probe the enantiodetermining alkene addition step, the pro-S (major) and pro-R (minor) cis-oxycupration transition states were modeled using density functional theory calculations.
Unrestricted density functional theory calculations were performed at the B3LYP/6-31+G(d)[26–30] level of theory using the Gaussian 09 software suite.[31] Single-point calculations using the Polarizable Continuum Model[32] were carried out to determine the solvation free energy, but CH2Cl2 (ε = 8.93) was used instead of PhCF3 (ε = 9.18) as the program does not contain parameters for the latter. Calculations of the pro-S (major) and pro-R (minor) cis-oxycupration transition states at 100 °C for substrate 15 are illustrated in Figure 2. The major transition state is 1.59 kcal/mol lower in Gibbs free energy, translating to a calculated % ee of 79.1% (82% is experimentally observed, Table 3, entry 2). While no steric interactions of 2.2 Å or less were observed in the major transition state, two were observed in the minor transition state, one between a substrate terminal alkene H and a ligand backbone H, and another between a substrate alpha carbon H and a ligand backbone H (see Figure 2). The supporting information contains full details of the computational methodology employed, as well as in-depth analysis of the most important computational results.
Figure 2.

(a) Calculated major transition state; (b) Calculated minor transition state. ΔΔG‡ = 1.59 kcal/mol (79.1% ee).
The electronic structure of the transition states was further analyzed. (These data were obtained in the gas phase since the inclusion of solvent effects, via single point calculations on gas phase geometries, did not have a notable effect on these parameters.) Spin analysis of the major transition state indicates that the unpaired electron resides mainly on copper(II) (46%) but the emergent terminal carbon (C1-Cu bond) also picks up spin (26%) as does the oxygen adding to the alkene (21%). Mulliken charge analysis indicates the internal alkene carbon (C2) increases in positive charge from 0.16 in the substrate to 0.95 at the major transiton state, and Wiberg bond indices[33–35] analysis indicates the C1-Cu bond is 68% formed while the C2-O bond is 45% formed in the major transition state. Taken together, these data support an alkene addition with substantial polar character, though radical contributions cannot be discounted. Tetrahedral twist measurements indicate the major transition state is distorted tetrahedral (16.5° less than a perfect tetrahedron) about the copper center, while the minor transition state is almost perfectly tetrahedral. Natural Bond Order (NBO) analysis of the two transition states indicate there are more favorable bonding interactions in the major transition state than in the minor.
In conclusion, we have rendered the copper-catalyzed carboetherification of 4-alkenols highly enantioselective. We have developed a new intra/intermolecular coupling reaction of 4-alkenols with vinyl arenes that results in net alkyl Heck-type products where the alkyl component consists of an enantiomerically enriched tetrahydrofuran. This reaction extends the scope of what is possible via polar/radical reaction cascades.[36] The enantioselectivity of the reaction can be >95% ee. The absolute stereochemistry of the major products was definitively assigned. DFT transition state calculations are consistent with a cis-oxycupration stereodetermining transition state and there is good agreement between experimental and calculated levels of enantiomeric excess. Theoretical studies to fully understand the entire reaction mechanism are on-going.
Supplementary Material
Footnotes
The National Institutes of Health (NIGMS RO1 078383), the donors of the American Chemical Society Petroleum Research Fund (51672DNI6 EZ) and the Center for Computational Research at SUNY Buffalo are acknowledged for support of this research. We thank William W. Brennessel and the Crystallographic Facility at the University of Rochester for obtaining the X-ray structure of 2b (CCDC 985356).
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201xxxxxx.
Contributor Information
Prof. Eva Zurek, Email: ezurek@buffalo.edu.
Sherry R. Chemler, Prof., Email: schemler@buffalo.edu.
References
- 1.Wolfe JP, Hay MB. Tetrahedron. 2007;63:261. doi: 10.1016/j.tet.2006.08.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li N, Shi Z, Tang Y, Chen J, Li X. Beilstein J Org Chem. 2008;4:48. doi: 10.3762/bjoc.4.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hay MB, Hardin AR, Wolfe JP. J Org Chem. 2005;70:3099. doi: 10.1021/jo050022+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hay MB, Wolfe JP. J Am Chem Soc. 2005;127:16468. doi: 10.1021/ja054754v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ward AF, Wolfe JP. Org Lett. 2010;12:1268. doi: 10.1021/ol1001472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ward AF, Xu Y, Wolfe JP. Chem Commun. 2012;48:609. doi: 10.1039/c1cc15880e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fries P, Halter D, Kleinschek A, Hartung J. J Am Chem Soc. 2011;133:3906. doi: 10.1021/ja108403s. [DOI] [PubMed] [Google Scholar]
- 8.Zhang G, Cui L, Wang Y, Zhang L. J Am Chem Soc. 2010;132:1474. doi: 10.1021/ja909555d. [DOI] [PubMed] [Google Scholar]
- 9.Zhu C, Falck JR. Angew Chem Int Ed. 2011;50:6626. doi: 10.1002/anie.201101857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Protti S, Dondi D, Fagnoni M, Albini A. Eur J Org Chem. 2008:2240. [Google Scholar]
- 11.Nicolai S, Waser J. Org Lett. 2011;13:6324. doi: 10.1021/ol2029383. [DOI] [PubMed] [Google Scholar]
- 12.Nicolai S, Erard S, Gonzalez DF, Waser J. Org Lett. 2010;12:384. doi: 10.1021/ol9027286. [DOI] [PubMed] [Google Scholar]
- 13.Selected Cu-catalyzed enantioselective alkene carboaminations: Zeng W, Chemler SR. J Am Chem Soc. 2007;129:12948. doi: 10.1021/ja0762240.Liwosz TW, Chemler SR. J Am Chem Soc. 2012;134:2020. doi: 10.1021/ja211272v.
- 14.Selected Pd-catalyzed enantioselective alkene carboaminations: Yip KT, Yang M, Law KL, Zhu NY, Yang D. J Am Chem Soc. 2006;128:3130. doi: 10.1021/ja060291x.Mai DN, Wolfe JP. J Am Chem Soc. 2010;132:12157. doi: 10.1021/ja106989h.Hopkins BA, Wolfe JP. Angew Chem Int Ed. 2012;51:9886. doi: 10.1002/anie.201205233.
- 15.For reviews of palladium-catalyzed enantioselective alkene nucleopalladation reactions see: McDonald RI, Liu G, Stahl SS. Chem Rev. 2011;111:2981. doi: 10.1021/cr100371y.Dohanosova J, Gracza T. Molecules. 2013;18:6173. doi: 10.3390/molecules18066173.
- 16.Pathak TP, Gligorich KM, Welm BE, Sigman MS. J Am Chem Soc. 2010;132:7870. doi: 10.1021/ja103472a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a) Tietze LF, Sommer KM, Zinngrebe J, Stecker F. Angew Chem Int Ed. 2005;44:257. doi: 10.1002/anie.200461629. [DOI] [PubMed] [Google Scholar]; b) Tietze LF, Spiegl DA, Stecker F, Major J, Raith C, Grobe C. Chem—Eur J. 2008;14:8956. doi: 10.1002/chem.200800967. [DOI] [PubMed] [Google Scholar]
- 18.Zhu R, Buchwald SL. Angew Chem Int Ed. 2013;52:12655. doi: 10.1002/anie.201307790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller Y, Miao L, Hosseini AS, Chemler SR. J Am Chem Soc. 2012;134:12149. doi: 10.1021/ja3034075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.a) Liwosz TW, Chemler SR. Org Lett. 2013;15:3034. doi: 10.1021/ol401220b. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nishikata T, Noda Y, Fujimoto R, Sakashita T. J Am Chem Soc. 2013;135:16372. doi: 10.1021/ja409661n. [DOI] [PubMed] [Google Scholar]
- 21.Nising CF, Brase S. Chem Soc Rev. 2012;41:988. doi: 10.1039/c1cs15167c. [DOI] [PubMed] [Google Scholar]
- 22.McGarraugh PG, Johnston RC, Martinez-Munoz A, Cheong PHY, Brenner-Moyer SE. Chem Eur J. 2012;18:10742. doi: 10.1002/chem.201104029. [DOI] [PubMed] [Google Scholar]
- 23.Corbett MT, Johnson JS. Chem Sci. 2013;4:2828. doi: 10.1039/C3SC51022K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Terada Y, Arisawa M, Nishida A. Angew Chem Int Ed. 2004;43:4063. doi: 10.1002/anie.200454157. [DOI] [PubMed] [Google Scholar]
- 25.Gustafsson B, Hakansson M, Jagner S. New J Chem. 2003;27:459. [Google Scholar]
- 26.Becke AD. Phys Rev A. 1988;38:3098. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 27.Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 28.Clark T, Chandrasekhar J, Spitznagel GW, Schleyer PVR. J Comp Chem. 1983;4:294. [Google Scholar]
- 29.Hariharan PC, Pople JA. Theoret Chimica Acta. 1973;28:213. [Google Scholar]
- 30.Francl MM, Petro WJ, Hehre WJ, Binkley JS, Gordon MS, DeFrees DJ, Pople JA. J Chem Phys. 1982;77:3654. [Google Scholar]
- 31.Frisch MJ, et al. Gaussian ‘09, Revision A.02. Gaussian, Inc; Wallingford CT: 2010. [Google Scholar]
- 32.Tomasi J, Mennucci B, Cances E. J Mol Struct (Theochem) 1999;464:211. [Google Scholar]
- 33.Foster JP, Weinhold F. J Am Chem Soc. 1980;102:7211. [Google Scholar]
- 34.Reed AE, Weinhold F. J Chem Phys. 1983;78:4066. [Google Scholar]
- 35.Reed AE, Weinstock RB, Weinhold F. J Chem Phys. 1985;83:735. [Google Scholar]
- 36.Mondal S, Bertrand MP, Nechab M. Angew Chem Int Ed. 2013;52:809. doi: 10.1002/anie.201207518. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.