

Abstract
The increasing incidence of infectious diseases caused by fungi in immunocompromised patients has encouraged researchers to develop rapid and accurate diagnosis methods. Identification of the causative fungal species is critical in deciding the appropriate treatment, but it is not easy to get satisfactory results due to the difficulty of fungal cultivation and morphological identification from clinical samples. In this study, we established a microarray system that can identify 42 species from 24 genera of clinically important fungal pathogens by using a chemical color reaction in the detection process. The array uses the internal transcribed spacer region of the rRNA gene for identification of fungal DNA at the species level. The specificity of this array was tested against a total of 355 target and nontarget fungal species. The fungal detection was succeeded directly from 103 CFU/ml for whole blood samples, and 50 fg DNA per 1 ml of serum samples indicating that the array system we established is sensitive to identify infecting fungi from clinical sample. Furthermore, we conducted isothermal amplification in place of PCR amplification and labeling. The successful identification with PCR-amplified as well as isothermally amplified target genes demonstrated that our microarray system is an efficient and robust method for identifying a variety of fungal species in a sample.
Electronic supplementary material
The online version of this article (doi:10.1007/s11046-014-9756-2) contains supplementary material, which is available to authorized users.
Keywords: Diagnosis, Microarray, Fungal infection, Isothermal amplification
Introduction
Systemic fungal infections with high morbidity and mortality rates in immunocompromised patients are growing. Besides the increasing incidence, recent epidemiology of fungal infection shows the expanding variety of fungal pathogens [1].
Identification of the causative pathogen is a fundamental step for appropriate treatment of infectious diseases, and early initiation of antifungal therapy is crucial for reducing the mortality rate in infected patients. Despite efforts by many researchers, however, early and rapid diagnosis of systemic fungal infection remains limited. Conventional diagnostic procedures, such as cultivation of fungi from clinical samples, are time-consuming and suffer from low sensitivity. Furthermore, sufficient technique and experience are required at the identification step. In recent years, other methods, such as PCR and serological tests, have been established for rapid and sensitive detection of fungi from clinical samples [2, 3]. However, these methods are difficult to use to identify a variety of fungal species at a time. Although multiplex PCR can be used to identify several species in one test, its applicability is limited by the primer sets used because specific primer sets are needed for each species.
A variety of DNA array systems have been developed to identify several bacteria and/or fungi simultaneously with high sensitivity and specificity [4–8]. Fluorescent labels are widely used to detect the signal in DNA microarrays due to their high sensitivity. However, the low stability of most fluorescent dyes and the necessity of expensive scanning equipment call for the development of alternative labeling systems that are inexpensive and robust.
To facilitate the diagnosis of fungal infectious disease, we established a rapid and specific DNA microarray system for identifying a variety of causative fungal species simultaneously. We applied the chemical color reaction of biotin-peroxidase and its substrate as the signal detection for the microarray system, enabling examination of the spot pattern with the naked eyes, without the need for expensive scanning equipment. To evaluate the specificity and sensitivity of this visible DNA microarray system, we tested it on several kinds of samples, such as reference fungal strains, blood samples containing a certain number of fungal cells, serum samples with serial dilutions of fungal DNA, and blood culture samples from patients.
Conventional PCR methods have been used for labeling and amplifying DNA from pathogen in microarray identification systems, but this method could not be used for bedside analysis and therefore difficult to be widely adopted. Recently, several isothermal amplification methods that do not require expensive thermal cyclers, such as loop-mediated isothermal amplification (LAMP), helicase-dependent amplification (HAD), and isothermal and chimeric primer-initiated amplification of nucleic acids (ICAN), have been developed to replace PCR amplification [9–12]. However, the maximum amplifiable length of the products in these isothermal methods is too small (100–500 bp) for our purposes, making primer design difficult. Hence, in this study, we applied the recombinase polymerase amplification cycle (RPA) for labeling and amplification of DNA products from the pathogen for microarray detection [13]. The RPA technology is based on a combination of polymerases and DNA recombinases. These enzyme mixtures are active at low temperature (optimum around 37 °C) and recognize template target sites by oligonucleotide primers, followed by strand-displacing DNA synthesis. Thus, exponential DNA amplification of the target region is proceeded under the isothermal condition.
Materials and Methods
Microorganisms and Growth Conditions
A total of 355 strains were obtained from the Medical Mycology Research Center IFM Collection (Chiba university, Japan) (Table S1). All fungal strains were cultivated on PDA medium at appropriate temperatures. Small amount of fungal cells were picked by toothpick and suspended in distilled water to become little bit cloudy solution and used as template for PCR amplification.
Design of Capture Probes
The fungal oligonucleotide probes were designed based on the whole internal transcribed spacer (ITS) sequences regions available in the GenBank database and from our own sequencing data. The alignment was prepared by BioEdit using several objective and nonobjective fungal ITS sequences as listed in Table S2. After sequence alignment, species- or genus-specific oligonucleotide sequences were selected to be unique to each species/genus. To evaluate the specificities against other organisms, we performed additional BLASTN searches of the GenBank database. The designed probes were consisted of 14–21 species/genus-specific oligonucleotides and a poly-T anchor at the end of the oligonucleotides [14]. Detailed sequences of the capture probes are given in Table 1.
Table 1.
Oligonucleotide sequence of probes
| Organism | Probe name | Probe sequence (5′-3′) | Length (bp) |
|---|---|---|---|
| Common for all fungi | 50-17 | GATGAAGAACGCAGCGATTTTTTTTTT | 27 |
| 50-19 | CGATGAAGAACGCAGCGAATTTTTTTTTT | 29 | |
| 51-17 | GAGTCTTTGAACGCACATTTTTTTTTT | 27 | |
| 51-19 | CGAGTCTTTGAACGCACATTTTTTTTTTT | 29 | |
| 52-17R | TTTTTTTTTTACCAAGAGATCCGTTGT | 27 | |
| 52-19R | TTTTTTTTTTAACCAAGAGATCCGTTGTT | 29 | |
| Absidia corymbifera | Ab3-19t | CCGGATGGAGACTCTAGAGTTTTTTTTTT | 29 |
| Ab2-18Rt | ATTTAAGGCCATGACAGCTTTTTTTTTT | 28 | |
| Ab2-t18R | TTTTTTTTTTATTTAAGGCCATGACAGC | 28 | |
| Alternaria sp. | AlA-17Rt | GAAGTACGCAAAAGACATTTTTTTTTT | 27 |
| AlA-t17R | TTTTTTTTTTGAAGTACGCAAAAGACA | 27 | |
| AlD-16Rt | ACGCCCAACACCAAGCTTTTTTTTTT | 26 | |
| AlE-16t | TCGGAGCGCAGCACAATTTTTTTTTT | 26 | |
| Aspergillus flavus | 60B 1 | TTTTTTTTTTTGATCTAGTGAAGTCTGAG | 29 |
| 60B 1R | TTTTTTTTTTCTCAGACTTCACTAGATCA | 29 | |
| 60B 17R | TCAGACTTCACTAGATCTTTTTTTTTT | 27 | |
| 60C 1R | TTTTTTTTTTTAACTGATTGCGATACAAT | 29 | |
| 60C 2R | TTTTTTTTTTACTGATTGCGATACAAT | 27 | |
| 60C-19R | TAACTGATTGCGATACAATTTTTTTTTTT | 29 | |
| Aspergillus fumigatus | 33B-1R | TTTTTTTTTTTAACTGATTACGATAATCAA | 30 |
| 33B-2R | TTTTTTTTTTTAACTGATTACGATAATCA | 29 | |
| 33B-4R | TAACTGATTACGATAATCAATTTTTTTTTT | 30 | |
| 33C 1R | TTTTTTTTTTTAACTGATTACGATAATCAAC | 31 | |
| 33C 2R | TTTTTTTTTTACTGATTACGATAATCAAC | 29 | |
| 33C 3R | TTTTTTTTTTCTGATTACGATAATCAAC | 28 | |
| 34A-8 | TTGTCACCTGCTCTGTTTTTTTTTTT | 26 | |
| 34A-14 | TTGTCACCTGCTCTTTTTTTTTTT | 24 | |
| 34A-17 | GTCACCTGCTCTGTTTTTTTTTT | 23 | |
| 34A-20 | TTTTTTTTTTTTTGTCACCTGCTC | 24 | |
| Aspergillus nidulans | 64B 8 | TTTTTTTTTTAGTTCAGTGGTCCCCGGC | 28 |
| 64B 9 | TTTTTTTTTTAGTTCAGTGGTCCCCG | 26 | |
| 65A 15 | GGCGTCTCCAACCTTTTTTTTTTTT | 25 | |
| 65A 17 | CGGCGTCTCCAACCTTATTTTTTTTTT | 27 | |
| 65A 19 | CCGGCGTCTCCAACCTTATTTTTTTTTTT | 29 | |
| Aspergillus niger | 62A 4 | TTTTTTTTTTATAGACACGGATG | 23 |
| 63A 15 | TTTTTTTTTTCCAACCATTCTTTCCA | 26 | |
| 63A 17 | TTTTTTTTTTTCCAACCATTCTTTCCA | 27 | |
| 63A 19 | TTTTTTTTTTTTCCAACCATTCTTTCCAG | 29 | |
| Aspergillus terreus | 35A 17R | GCAAAGAATCACACTCATTTTTTTTTT | 27 |
| 35A 19 | TGAGTGTGATTCTTTGCAATTTTTTTTTT | 29 | |
| 35A 19R | TTGCAAAGAATCACACTCATTTTTTTTTT | 29 | |
| 36A 1 | TTTTTTTTTTGGCTTCGTCTTCCGCTCCG | 29 | |
| 36A 2 | TTTTTTTTTTGCTTCGTCTTCCGCTCC | 27 | |
| 36A 19 | GGCTTCGTCTTCCGCTCCGTTTTTTTTTT | 29 | |
| 36B 15 | CGACGCATTTATTTGTTTTTTTTTT | 25 | |
| 36B 17 | GCCGACGCATTTATTTGTTTTTTTTTT | 27 | |
| 36B 19 | CGCCGACGCATTTATTTGCTTTTTTTTTT | 29 | |
| Blastomyces dermatitidis | 41A 17R | GTTCCTCCGGTCTAGGATTTTTTTTTT | 27 |
| 41A 19R | GGTTCCTCCGGTCTAGGAGTTTTTTTTTT | 29 | |
| 42A 15 | CCGGCCCCATCTCAATTTTTTTTTT | 25 | |
| 42A 17 | TCCGGCCCCATCTCAAATTTTTTTTTT | 27 | |
| Candida albicans | 14A 15 | CGGAGATGCTTGACTTTTTTTTTTT | 25 |
| 14A 17 | CGGAGATGCTTGACAATTTTTTTTTTT | 27 | |
| 1A 17R | TTTTTTTTTTAAGTTTAGACCTCTGGC | 27 | |
| 1A 19 | CCGCCAGAGGTCTAAACTTTTTTTTTTTT | 29 | |
| 1B 15R | TTTTTTTTTTATCTGGTGTGACAAG | 25 | |
| 1B 17R | TTTTTTTTTTTAATCTGGTGTGACAAG | 27 | |
| 1B 19 | ACTTGTCACACCAGATTATTTTTTTTTTT | 29 | |
| 2A 15 | CGTCCACCACGTATATTTTTTTTTT | 25 | |
| 2A 17 | AACGTCCACCACGTATATTTTTTTTTT | 27 | |
| 2A 19 | GTAACGTCCACCACGTATATTTTTTTTTT | 29 | |
| 2B 15 | TTTTTTTTTTATTGCTTGCGGCGGT | 25 | |
| 2B 17 | TTTTTTTTTTACATTGCTTGCGGCGGT | 27 | |
| Candida dubliniensis | 13A-2R | TTTTTTTTTTAACAAAACACATGTGGT | 27 |
| 13A-3R | TTTTTTTTTTAACAAAACACATGTGG | 26 | |
| 13B 15 | TTTTTTTTTTTATAAACTTGTCACG | 25 | |
| 13B 17 | TTTTTTTTTTTATAAACTTGTCACGAG | 27 | |
| Candida famata | 80B-1 | TGGTCTGGACTAGAAATATTTTTTTTT | 28 |
| 80B-1R | TTTTTTTTTTATTTCTAGTCCAGACCA | 28 | |
| 81A-1 | TTTTTTTTTTTAGTGCTATATGACTTTC | 28 | |
| 81A-3 | TTTTTTTTTTAGTGCTATATGACTTTC | 27 | |
| Candida glabrata | 7A 15R | TTTTTTTTTTTGTCTCTCTCCGAGC | 25 |
| 7A 17R | TTTTTTTTTTATGTCTCTCTCCGAGCT | 27 | |
| 7A 19R | TTTTTTTTTTGATGTCTCTCTCCGAGCTC | 29 | |
| 7B 17 | CTCCTGCCTGCGCTTAATTTTTTTTTT | 27 | |
| 7B 19 | TTCTCCTGCCTGCGCTTAATTTTTTTTTT | 29 | |
| 7B 19R | TTAAGCGCAGGCAGGAGAATTTTTTTTTT | 29 | |
| 8A 17 | TTTTTTTTTTAACTTGAAATTGTAGGC | 27 | |
| 8A 19 | TTTTTTTTTTAACTTGAAATTGTAGGCCA | 29 | |
| 8B 15 | TTTTTTTTTTTGCTGCTCGTTTGCG | 25 | |
| 8B 17 | TTTTTTTTTTTTGCTGCTCGTTTGCGC | 27 | |
| 8B 19 | TTTTTTTTTTTCTGCTGCTCGTTTGCGCG | 29 | |
| Candida guilliermondii | 55A 17R | TTTTTTTTTTAAAATTTGACTAACTGT | 27 |
| 55A 19 | TTTACAGTTAGTCAAATTTTTTTTTTTTT | 29 | |
| 55A 19R | TTTTTTTTTTCAAAATTTGACTAACTGTA | 29 | |
| 55B 15 | GTCGACCTCTCAATGTTTTTTTTTT | 25 | |
| 55B 17 | TGTCGACCTCTCAATGTTTTTTTTTTT | 27 | |
| 55B 19 | CTGTCGACCTCTCAATGTATTTTTTTTTT | 29 | |
| Candida kefyr | Ck1-t16R | TTTTTTTTTTGTCAGACGATTCCCCC | 26 |
| Ck2-20Rt | TAGCAGAGAATCAAGAACTGTTTTTTTTTT | 30 | |
| Ck2-t20R | TTTTTTTTTTTAGCAGAGAATCAAGAACTG | 30 | |
| Ck4-t17 | TTTTTTTTTTCGTCTCGGGTTAACTTG | 27 | |
| Ck4-17Rt | CAAGTTAACCCGAGACGTTTTTTTTTT | 27 | |
| Ck4-t17R | TTTTTTTTTTCAAGTTAACCCGAGACG | 27 | |
| Ck6-18Rt | GCAAGAGTCGAGTCCATATTTTTTTTTT | 28 | |
| Candida krusei | 9B 17R | GCTATATTCCACATTTTTTTTTTTTTT | 27 |
| 9B 19R | ATGCTATATTCCACATTTTTTTTTTTTTT | 29 | |
| 9C-1R | TTTTTTTTTTTCGACTATATGCTATATTC | 29 | |
| 9C-2R | CGACTATATGCTATATTCCTTTTTTTTTT | 29 | |
| 9C-3R | TTTTTTTTTTCGACTATATGCTATATTCC | 29 | |
| 10A 15 | GCGGACGACGTGTAATTTTTTTTTT | 25 | |
| 10A 17 | GCGGACGACGTGTAAAGTTTTTTTTTT | 27 | |
| 10A 19 | GAGCGGACGACGTGTAAAGTTTTTTTTTT | 29 | |
| 10B 15 | TTTTTTTTTTGAGCGAAGCTGGCCG | 25 | |
| 10B 17 | TTTTTTTTTTAGCGAAGCTGGCCGAGC | 27 | |
| 10B 19 | TTTTTTTTTTGAGCGAAGCTGGCCGAGCG | 29 | |
| Candida lusitaniae | 11C 14R | TGTTCGCAAAAACATTTTTTTTTT | 24 |
| 11C 15R | TGTTCGCAAAAACAATTTTTTTTTT | 25 | |
| 11C 16R | TGTTCGCAAAAACAATTTTTTTTTTT | 26 | |
| 11B 19 | TTCGAATTTCTTAATATCATTTTTTTTTT | 29 | |
| 11B 19R | TTGATATTAAGAAATTCGATTTTTTTTTT | 29 | |
| 12A 17R | TTTCGGAGCAACGCCTATTTTTTTTTT | 27 | |
| 12A 19 | TTAGGCGTTGCTCCGAAATTTTTTTTTT | 29 | |
| 12A 19R | TTTCGGAGCAACGCCTAACTTTTTTTTTT | 29 | |
| 12B 17 | CGTTTACAGCACGACATTTTTTTTTTT | 27 | |
| 12B 19 | CACGTTTACAGCACGACATTTTTTTTTTT | 29 | |
| Candida rugosa | 17B 15R | GATCGGTACTTGAAGTTTTTTTTTT | 25 |
| 18B 15R | TTTTTTTTTTAGACGGTCGCGTTTC | 25 | |
| Candida parapsilosis | 5A 17 | CTGCCAGAGATTAAACTTTTTTTTTTT | 27 |
| 5A 18 | CTGCCAGAGATTAAACTCTTTTTTTTTT | 28 | |
| 5A 18R | GAGTTTAATCTCTGGCAGTTTTTTTTTT | 28 | |
| 6A 17 | TTTTTTTTTTCCAAAACTTCTTCCATT | 27 | |
| 6A 19 | TTTTTTTTTTCTCCAAAACTTCTTCCATT | 29 | |
| 6A 19R | TTTTTTTTTTAATGGAAGAAGTTTTGGAG | 29 | |
| 6B 17 | TTTTTTTTTTACTCCAAAACTTCTTCC | 27 | |
| 6B 18 | TTTTTTTTTTACTCCAAAACTTCTTCCA | 28 | |
| 6B 18R | TTTTTTTTTTTGGAAGAAGTTTTGGAGT | 28 | |
| Candida tropicalis | 3A 16R | TTTTTTTTTTGGATTGCTCCCGCCAC | 26 |
| 3A 17R | TTTTTTTTTTGGATTGCTCCCGCCACC | 27 | |
| 3B 15R | TTTTTTTTTTATCAAGTTTGACTGT | 25 | |
| 3B 17R | TTTTTTTTTTAAATCAAGTTTGACTGT | 27 | |
| 3B 19R | TTTTTTTTTTAAATCAAGTTTGACTGTAA | 29 | |
| 4A 15 | TTTTTTTTTTATACGCTAGGTTTGT | 25 | |
| 4A 17 | TTTTTTTTTTATACGCTAGGTTTGTTT | 27 | |
| 4A 19 | TTTTTTTTTTCAATACGCTAGGTTTGTTT | 29 | |
| 4B 17 | GCTAGTGGCCACCACTTTTTTTTTTTT | 27 | |
| 4B 19 | GCTAGTGGCCACCACAATTTTTTTTTTTT | 29 | |
| Candida zeylanoides | 15A 19 | GTTTTATACTAAAACTTCATTTTTTTTTT | 29 |
| 15A 1 | GGTTTTATACTAAAACTTCATTTTTTTTTT | 30 | |
| 15B 1 | TTTTTTTTTTATTGAATTGTTAATTAATTA | 30 | |
| 15B 1R | TTTTTTTTTTTAATTAATTAACAATTCAAT | 30 | |
| 16A 19 | TTTTTTTTTTGACCAGTATAGTATTTGT | 28 | |
| 16A 17 | TTTTTTTTTTACCAGTATAGTATTTG | 26 | |
| Coccidioides posadasii | 37C 15R | GGAGGTGCGCAGCCGTTTTTTTTTT | 25 |
| 37C 17R | GGGAGGTGCGCAGCCGGTTTTTTTTTT | 27 | |
| 37C 19R | GGGGAGGTGCGCAGCCGGATTTTTTTTTT | 29 | |
| 37E 15R | TTTTTGCTATGATGCTTTTTTTTTT | 25 | |
| 37E 17R | GATTTTTGCTATGATGCTTTTTTTTTT | 27 | |
| 37E 18R | GATTTTTGCTATGATGCTTTTTTTTTTT | 28 | |
| 38D-1 | TTATATCCGGTTTGACCTCTTTTTTTTTT | 29 | |
| 38D-2 | ATATCCGGTTTGACCTCTTTTTTTTTT | 27 | |
| 38D-3 | TATCCGGTTTGACCTTTTTTTTTTT | 25 | |
| 38E 15 | TTTTTTTTTTACCCGATCGGGGCCG | 25 | |
| 38E 17 | TTTTTTTTTTGACCCGATCGGGGCCGA | 27 | |
| 38E 19 | TTTTTTTTTTAGACCCGATCGGGGCCGAT | 29 | |
| Cryptococcus neoformans var. neoformans, grubii, gattii | 22A-8 | GTTTATGTGCTTCGGCACTTTTTTTTTT | 28 |
| 22A 17 | TTTTTTTTTTGTTTATGTGCTTCGGCA | 27 | |
| 23A 17 | TTTTTTTTTTGAAGGTGATTACCTGTC | 27 | |
| 23A 19 | TTTTTTTTTTGGAAGGTGATTACCTGTCA | 29 | |
| 23B 1 | TTTTTTTTTTTTTCGCTGGGCCTATGG | 27 | |
| 23B 2 | TTTTTTTTTTGTTTCGCTGGGCCTATGGG | 29 | |
| Cryptococcus gattii | 20-2R | TTTTTTTTTTTGGACCGAAGCCCAGTATT | 29 |
| 20-5R | TTTTTTTTTTGACCGAAGCCCAGTATT | 27 | |
| 20-6R | TTTTTTTTTTGGACCGAAGCCCAGTAT | 27 | |
| Cunninghamella bertholletiae | 70A-1R | CCCAAAGATCCCTTGATCTATTTTTTTTTT | 30 |
| 70A-2R | CCCAAAGATCCCTTGATCTTTTTTTTTTT | 29 | |
| 71A 19 | TAGTCGGCTTTAATAGATTTTTTTTTTTT | 29 | |
| 71A 17 | TAGTCGGCTTTAATAGATTTTTTTTTT | 27 | |
| 71A 15 | AGTCGGCTTTAATAGTTTTTTTTTT | 25 | |
| 71B-1 | TTTTTTTTTTTAAATACAAGGCTCGACTTT | 30 | |
| 71B-2 | TTTTTTTTTTAATACAAGGCTCGACT | 26 | |
| 71B-3 | TTTTTTTTTTTAATACAAGGCTCGACTTT | 29 | |
| Epidermophyton floccosum | 76B 19R | CTCAGACTGAACCACCTATTTTTTTTTTT | 29 |
| 76B 17R | TCAGACTGAACCACCTATTTTTTTTTT | 27 | |
| 76B 15R | CAGACTGAACCACCTTTTTTTTTTT | 25 | |
| 77A 19 | TTTTTTTTTTAGTTTCCGTCGGGAGGACG | 29 | |
| 77A 17 | TTTTTTTTTTGTTTCCGTCGGGAGGAC | 27 | |
| Fusarium sp. | 7-16t | GGCCACGCCGTTAAACTTTTTTTTTT | 26 |
| 7-18t | CTTCTGAATGTTGACCTCTTTTTTTTTT | 28 | |
| 7-19t | CGCGGCCACGCCGTTAAACTTTTTTTTTT | 29 | |
| 7B-19t | CAACTTCTGAATGTTGACCTTTTTTTTTT | 29 | |
| 7C-18t | ACCCCAACTTCTGAATGTTTTTTTTTT | 28 | |
| 7C-19t | CCGTAAACCCCAACTTCTGTTTTTTTTTT | 29 | |
| 10B-16Rt | GTATGTTCACAGGGGTTTTTTTTTTT | 26 | |
| 10B-18Rt | GTATGTTCACAGGGGTTGTTTTTTTTTT | 28 | |
| Fusarium solani complex (FSSC) | 1-16Rt | CCGTCTGTTCCCGCCGTTTTTTTTTT | 26 |
| 1-18Rt | GCCGTCTGTTCCCGCCGATTTTTTTTTT | 28 | |
| 1-19Rt | CCGTCTGTTCCCGCCGAAGTTTTTTTTTT | 29 | |
| 2-19Rt | GCCGATCCCCAACGCCAGGTTTTTTTTTT | 29 | |
| 4-18t | CACCTCGCAACTGGAGAGTTTTTTTTTT | 28 | |
| 4-19t | GCTAACACCTCGCAACTGGATTTTTTTTTT | 29 | |
| 4-20t | GTAGCTAACACCTCGCAACTTTTTTTTTTT | 30 | |
| 6B-17Rt | CAGAGTTAGGGGTCCTCTTTTTTTTTT | 27 | |
| 9-17t | ACGTTGCTTCGGCGGGATTTTTTTTTT | 27 | |
| Histoplasma capsulatum | 39B-22 | TTTTTTTTTTCGTTCACCGACGGTTCTT | 28 |
| 39B-24 | TTTTTTTTTTGTTCACCGACGGTTCT | 26 | |
| 39B-25 | TTTTTTTTTTGTTCACCGACGGTTC | 25 | |
| 39C 15R | AGGTCCGGTAGACAATTTTTTTTTT | 25 | |
| 39C 17R | CAGGTCCGGTAGACAAGTTTTTTTTTT | 27 | |
| 39C 19R | ACAGGTCCGGTAGACAAGGTTTTTTTTTT | 29 | |
| Malassezia furfur | 48A 15R | TTTTTTTTTTCCAAACGGTGCACAC | 25 |
| 48A 17R | TTTTTTTTTTTCCAAACGGTGCACACG | 27 | |
| 48A 19R | GATTTCCACGTTCATACAATTTTTTTTTT | 29 | |
| 48B 15R | TTTCCACGTTCATACTTTTTTTTTT | 25 | |
| 48B 17R | ATTTCCACGTTCATACATTTTTTTTTT | 27 | |
| 48B 19R | GATTTCCACGTTCATACAATTTTTTTTTT | 29 | |
| 49A 7 | TGCGATTGCACTGCTTTGTTTTTTTTTT | 28 | |
| 49A 8 | GCGATTGCACTGCTTTGTTTTTTTTTT | 27 | |
| 49A 9 | CGATTGCACTGCTTTGTTTTTTTTTT | 26 | |
| 49B 15 | TTTTTTTTTTGCATTAGCGCCTTTG | 25 | |
| 49B 17 | TTTTTTTTTTTGCATTAGCGCCTTTGG | 27 | |
| 49B 19 | TTTTTTTTTTATGCATTAGCGCCTTTGGG | 29 | |
| Microsporum canis | 73A 6 | TTTTTTTTTTGTAACCACCCACCGCTTA | 28 |
| 73A 7 | GTAACCACCCACCGCTTAGTTTTTTTTTT | 29 | |
| 73A 9 | GTAACCACCCACCGCTTATTTTTTTTTT | 28 | |
| 73B 19 | CGCACCATGTATTATTCAGTTTTTTTTTT | 29 | |
| 73B 17 | GCACCATGTATTATTCATTTTTTTTTT | 27 | |
| 73B 1 | TTTTTTTTTTCGCACCATGTATTATTCAG | 29 | |
| Microsporum gypseum | 74A 2R | GATTTTACTTGCTAACGTTTTTTTTTT | 27 |
| 74B 1 | CGGAACAGTATTCATGGATTTTTTTTTTT | 29 | |
| 74B 2 | GGAACAGTATTCATGGATTTTTTTTTT | 27 | |
| 74B 4 | TTTTTTTTTTCGGAACAGTATTCATGGAT | 29 | |
| Mucor sp. | M1-t15R | TTTTTTTTTTTAATACAGTTCACAG | 25 |
| M1-16Rt | AATAATACAGTTCACATTTTTTTTTT | 26 | |
| M1-t16R | TTTTTTTTTTAATAATACAGTTCACA | 26 | |
| M3-20Rt | GGTAAATAATAATAGGATACTTTTTTTTTT | 30 | |
| M3-t20R | TTTTTTTTTTGGTAAATAATAATAGGATAC | 30 | |
| M4-t15R | TTTTTTTTTTGGTCTATGTTACAAT | 25 | |
| Paracoccidioides brasiliensis | 45A 15R | CCCCGTCCCCCCACGTTTTTTTTTT | 25 |
| 45A 17R | GCCCCGTCCCCCCACGGTTTTTTTTTT | 27 | |
| 45A 18R | GGCCCCGTCCCCCCACGGTTTTTTTTTT | 28 | |
| 45B 15R | TTTTTTTTTTTCAAAGCTCCGAACC | 25 | |
| 45B 17R | TTTTTTTTTTGTCAAAGCTCCGAACCA | 27 | |
| 45B 19R | TTTTTTTTTTCGTCAAAGCTCCGAACCAG | 29 | |
| 46A 15 | CCCCACTCATCGACCTTTTTTTTTT | 25 | |
| 46A 17 | GCCCCACTCATCGACCCTTTTTTTTTT | 27 | |
| 46A 19 | GGCCCCACTCATCGACCCCTTTTTTTTTT | 29 | |
| Penicillium marneffei | 43B 15R | TTTTTTTTTTTCAGACAGTCCATCT | 25 |
| 43B 17R | TTTTTTTTTTCTCAGACAGTCCATCTT | 27 | |
| 43B 19R | TTTTTTTTTTACTCAGACAGTCCATCTTC | 29 | |
| 44A 17 | TTTTTTTTTTCCACCATATTTACCACG | 27 | |
| 44A 19 | TTTTTTTTTTACCACCATATTTACCACGG | 29 | |
| Pichia anomala | Pa2-16Rt | GACTATTGGTTAAAGGTTTTTTTTTT | 26 |
| Pa3-17t | AGCAGTCTTTCTGAAATTTTTTTTTT | 27 | |
| Pa3-t17 | TTTTTTTTTTAGCAGTCTTTCTGAAAT | 27 | |
| Pa4-20Rt | CTTCTAAACCTGCCTAGCTGTTTTTTTTTT | 30 | |
| Pa4-t20R | TTTTTTTTTTCTTCTAAACCTGCCTAGCTG | 30 | |
| Pichia norvegensis | Pin2-20t | CACGAATAACCATGTCACCCTTTTTTTTTT | 30 |
| Pin2-t20 | TTTTTTTTTTCACGAATAACCATGTCACCC | 30 | |
| Pin2-20Rt | GGGTGACATGGTTATTCGTGTTTTTTTTTT | 30 | |
| Pin2-t20R | TTTTTTTTTTGGGTGACATGGTTATTCGTG | 30 | |
| Pin4-17t | GGCAGCGGGACTGAGCGTTTTTTTTTT | 27 | |
| Pin4-t17 | TTTTTTTTTTGGCAGCGGGACTGAGCG | 27 | |
| Pin4-t17R | TTTTTTTTTTCGCTCAGTCCCGCTGCC | 27 | |
| Pin5-20t | CACTCGCGCTTGGCCCGCCGTTTTTTTTTT | 30 | |
| Pin5-t20 | TTTTTTTTTTCACTCGCGCTTGGCCCGCCG | 30 | |
| Pin5-20Rt | CGGCGGGCCAAGCGCGAGTGTTTTTTTTTT | 30 | |
| Rhizomucor sp. | Rm1-17t | AGGGATTGCTCCAGATCTTTTTTTTTT | 27 |
| Rm1-t17R | TTTTTTTTTTGATCTGGAGCAATCCCT | 27 | |
| Rm2-17t | CTTTGGATTTGCGGTGCTTTTTTTTTT | 27 | |
| Rm2-17Rt | GCACCGCAAATCCAAAGTTTTTTTTTT | 27 | |
| Rm3-19t | GGGCTTGCTTGGTATCTATTTTTTTTTT | 29 | |
| Rm3-19Rt | TAGATACCAAGCAAGCCCTTTTTTTTTT | 29 | |
| Rm4-19t | GATCTGAACTTAGACGGGATTTTTTTTTT | 29 | |
| Rm4-t19R | TTTTTTTTTTTCCCGTCTAAGTTCAGATC | 29 | |
| Rhizopus microspores* | Rizm1-19Rt | CTGAGAAGTAAATCCCAGTTTTTTTTTT | 29 |
| Rizm1-t19R | TTTTTTTTTTCTGAGAAGTAAATCCCAGT | 29 | |
| Rizm2-t20 | TTTTTTTTTTCTGGCGATGAAGGTCGTAAC | 30 | |
| Rizm2-20Rt | GTTACGACCTTCATCGCCAGTTTTTTTTTT | 30 | |
| Rizm2-t20R | TTTTTTTTTTGTTACGACCTTCATCGCCAG | 30 | |
| Rizm3-19t | CTTCCTTGGGAAGGAAGGTTTTTTTTTT | 29 | |
| Rizm3-t19 | TTTTTTTTTTCTTCCTTTGGGAAGGAAGG | 29 | |
| Rizm3-19Rt | CCTTCCTTCCCAAAGGAAGTTTTTTTTTT | 29 | |
| Rizm4B-17Rt | GCACGATGGCTAGGTAGTTTTTTTTTT | 27 | |
| Rizm4B-t17R | TTTTTTTTTTGCACGATGGCTAGGTAG | 27 | |
| Rhizopus oryzae | Rizo1-19Rt | TACCCCAGAGGAAACCCTATTTTTTTTTT | 29 |
| Rizo1-t19R | TTTTTTTTTTTACCCCAGAGGAAACCCTA | 29 | |
| Rizo2-t18R | TTTTTTTTTTCTCCTGAAACCAGGAGTG | 28 | |
| Rizo3A-19t | ACAGTGAGCACCTAAAATGTTTTTTTTTT | 29 | |
| Rizo3A-t19 | TTTTTTTTTTACAGTGAGCACCTAAAATG | 29 | |
| Rizo3B-19t | GCTAGGCAGGAATATTACGTTTTTTTTTT | 29 | |
| Rizo3B-t19 | TTTTTTTTTTGCTAGGCAGGAATATTACG | 29 | |
| Rhodotorula mucilaginosa | Rho2-19Rt | CACCTCCTTCAATCATTAAGTTTTTTTTTT | 29 |
| Rho2-t19R | TTTTTTTTTTCACCTCTTCAATCATTAAG | 29 | |
| Rho5-18Rt | CTAGACCGTAAAGGCCAGTTTTTTTTTT | 28 | |
| Rho5-17Rt | CGAGCTAGACCGTAAAGTTTTTTTTTT | 27 | |
| Rho5-t17R | TTTTTTTTTTCGAGCTAGACCGTAAAG | 27 | |
| Scedosporium prolificans | Scp2-t15R | TTTTTTTTTTGTATTGTATTCAGAA | 25 |
| ScpP-19Rt | GGCTTGTAAAAACCTAGGCTTTTTTTTTT | 29 | |
| ScP-t19R | TTTTTTTTTTGGCTTGTAAAAACCTAGGC | 29 | |
| Sporothrix schenckii* | Sps2-t20R | TTTTTTTTTTGTAGGGCCCGCCGCCCCTGG | 30 |
| Sps4-20t | CACAACTCCCAACCCTTGCTTTTTTTTTT | 30 | |
| Sps4-20Rt | GCAAGGGTTGGGAGTTGTGTTTTTTTTTT | 30 | |
| Sps4-17t | GCGAACCGTACCCAATCTTTTTTTTTT | 27 | |
| Trichophyton mentagrophytes | 68A 1 | TTTTTTTTTTGTTTAGCCACTAAAGAGAG | 29 |
| 68A 2 | TTTTTTTTTTGTTTAGCCACTAAAGAGA | 28 | |
| 68A 4R | TTTTTTTTTTGTTTAGCCACTAAAGAGAGG | 30 | |
| 69A-10 | GCCCCCGTCTTTGGGGGTTTTTTTTTTT | 28 | |
| Trichophyton rubrum | 66B 6R | TTTTTTTTTTGCTCGAGGCTCCCAGAAGG | 29 |
| 66B 13R | TTTTTTTTTTCTCGAGGCTCCCAGAAGG | 28 | |
| 66B 14R | TTTTTTTTTTGCTCGAGGCTCCCAGAAG | 28 | |
| 67A 1 | TTTTTTTTTTCAGCCAATCCAGCGCCCTCA | 30 | |
| 67A 7 | TTTTTTTTTTCAGCCAATCCAGCGCCCTC | 29 | |
| 67A 8 | TTTTTTTTTTAGCCAATCCAGCGCCCTCA | 29 | |
| 67B 17 | AGCCAATTCAGCGCCCTTTTTTTTTTT | 27 | |
| 67B 19 | CAGCCAATTCAGCGCCCTCTTTTTTTTTT | 29 | |
| Trichophyton tonsurans | 47A 6 | CCTATCCTGGGGGGCCTTTTTTTTTT | 26 |
| 47A 7 | TTTTTTTTTTCCTATCCTGGGGGGCC | 26 | |
| 47A 19R | TTTTTTTTTTTATCCTGGGGGGCCGGCCT | 29 | |
| 47B 1 | TTTTTTTTTTGAGCCGCTATAAAGAGAGG | 29 | |
| 47B 4 | TTTTTTTTTTGAGCCGCTATAAAGAGAGGC | 30 | |
| 47B 19R | GAGCCGCTATAAAGAGAGGTTTTTTTTTT | 29 | |
| Trichosporon sp. | 78A-3 | TTTTTTTTTTCTTGCGCTCTCTGGTA | 26 |
| 78C-1 | TTTTTTTTTTGCTCGCCTTAAAAGAGTT | 28 | |
| Trichosporon asahii | 79A-5 | TTTTTTTTTTGCGTCTGCGATTTCT | 25 |
| 79A-6a | TTTTTTTTTTGGGCGTCTGCAATTTC | 26 | |
| Trichosporon cutaneum | 31A-2 | TTTTTTTTTTCGGTCAATTGATTTTACAAA | 30 |
| 31A-4R | TTTGTAAAATCAATTGACCGTTTTTTTTTT | 30 | |
| 32A 17 | TTTTTTTTTTAACTTGTCTTATCTGGC | 27 |
The probes with asterisk (*) shows cross-hybridization within the same genus
The ex-type classification name was used in some of the fungi
Preparation of DNA Microarray Slides
The synthetic oligonucleotides were diluted to 20 pmol/μl in TE buffer and mixed with an equal volume of 6× SSC [20× SSC is 3 M NaCl, 0.3 M sodium citrate (pH7.0)] to make a final concentration of 10 pmol/μl oligonucleotides in 3× SSC. The probe solutions were spotted on NGK plastic slides (NGK insulators LTD, Aichi, Japan) using a KCS-mini microarray printer (Kubota Comps Corporation, Hyogo, Japan). After spotting, the slides were irradiated with UV at 0.6 J/cm2 using a UV cross-linker (model CL-1000; UVP, San Gabriel, Ca) to fix the probes on plastic slides. The slides were then gently shaken in blocking buffer [3 % BSA, 0.2 M NaCl, 0.1 M Tris–HCl (pH 8.0), and 0.05 % Triton X-100] for 5 min and washed with TE buffer for 10 min. The array slides were air-dried and stably stored at room temperature at least 3 years.
Infectious Mouse Model
As an infection model, male ICR mice (Charles River Laboratories) were infected intravenously with Aspergillus fumigatus Af293 or Fusarium solani complex IFM40718 (FSSC) conidia (1 × 106 conidia/mouse) in a 200 µl volume of saline. Three mice were used in each fungal species. One hour after infection, mice were killed, and blood was collected from the heart tissues under sterilized conditions. The CFU was determined by inoculating 100 µl of collected blood on a PDA with Chloramphenicol plate, and colonies were counted after 24 h of cultivation at 30 °C. Blood samples were used directly as template for PCR.
Blood Culture
As the routine diagnosis of blood infection, blood samples were taken from patients and cultivated using the BD BACTEC FX system (BD, Tokyo, Japan) for 7 days, following the ethics of Chiba University Hospital. After cultivation, growth positive samples were inoculated onto several kinds of agar to identify bacteria and/or fungi. For the microarray identification, one growth positive and one growth negative blood culture samples were used directly as a template for PCR.
DNA Extraction
Fungal DNA was extracted as normal phenol–chloroform method. The conidia of A. fumigatus were inoculated in PDB medium and cultivated for 2 days at 37 °C. The mycelium was collected by filtration and ground by mortar using liquid N2. The ground cells were suspended in DNA extraction buffer (200 mM Tris, 25 mM NaCl, 25 mM EDTA, 0.5 % SDS, pH 8.5) and extracted with phenol/chloroform/isoamyl alcohol. After that, RNase treatment and ethanol precipitation were conducted. The cells of Candida albicans were cultivated in PDB medium 1 day at 30 °C. The cells were collected by centrifugation, and the DNA was extracted using GenTorukun (TaKaRa Bio Inc., Shiga, Japan).
PCR Amplification and Labeling
The 5′-biotin-labeled fungus-specific universal primers ITS1-bio (5′-TCCGTAGGTGAACCTGCGG-3′) and ITS4-bio (5′-TCCTCCGCTTATTGATATGC-3′) [15] were used for amplifying the entire ITS region and biotin labeling. The amplified fragment ranged from 426 to 930 bp depending on the fungal species. PCR was performed using MightyAmp DNA polymerase ver. 2 (TaKaRa Bio Inc.) in a total reaction volume of 10 μl containing 1 μl of template (fungal cell suspension, whole blood, serum, blood culture, and extracted DNAs). Amplification was carried out as follows, 2 min of initial denaturation at 98 °C, 40 cycles of DNA denaturation at 98 °C for 10 s, primer annealing at 55 °C for 15 s and elongation at 68 °C for 45 s, and a final elongation step at 68 °C for 5 min. After the PCR reaction, amplification was verified by electrophoresis. In case of low content of fungal cells or DNA, nested PCR was performed using ITS1-n (5′-GAGGAAGTAAAAGTCG-3′) and ITS4-n (5′-TTCACTCGCCGTTACT-3′) as the first-round PCR primer set. One µl of first-round PCR sample was then used as template in the second round of PCR performed in a total reaction volume of 10 μl.
Isothermal Amplification
Isothermal amplification was performed with a TwistAmp basic kit (TwistDx Limited, Cambridge, UK). Amplification was carried out at 37 °C for 40 min according to the manufacturer’s protocol using 1.5 μl of fungal cell suspension or DNA as template.
Microarray Hybridization and Signal Detection
Before microarray hybridization, amplified and labeled PCR samples were denatured at 95 °C for 2 min and chilled on ice for 2 min. Four μl of a denatured sample was then mixed with 16 μl of hybridization buffer (0.2 g tetramethylammonium chloride, 0.5 % SDS, and 1.9 mg EDTA in 1 ml of 6 × SSC). Samples were applied to the array slide, covered with a cover-film to prevent sample evaporation, and incubated at 37 °C for 1 h in a moist-chamber. Array slides were then washed with PBS buffer at 37 °C for 5 min. A color development reaction was performed on the slide in accordance with the avidin-biotinylated peroxidase complex (ABC) method using a 3, 3′, 5, 5′-tetramethylbenzidine (TMB) solution for visualization [16]. First, the conjugation reaction was performed with streptavidin and biotin-HRP for 30 min, and the array was washed twice with PBST buffer (PBS buffer with 0.1 % Tween 20) for 5 min. After washing, color development was performed with 0.02 % TMB, 0.015 % H2O2, and 0.5 mg/ml alginic acid in 0.2 M acetate buffer (pH 3.3). Color development was terminated after 30 min by washing the array slides with distilled water. The results were evaluated by visual observation.
Results
Design of Probes and DNA Microarray Slides
Because the ribosomal RNA gene, especially the 28S rRNA gene, is highly conserved among species, sequences of the ITS region are widely used for identification of fungal species. We designed species- and/or genus-specific probes within the ITS region and tested the specificity of selected sequences using 355 reference strains (Table S1). Genus-specific probes were designed for some fungi (Alternaria sp., Rhizomucor sp., Mucor sp., Trichosporon sp.); because they have highly conserved ITS region sequences within the genus, we could not design species-specific probes. We successfully designed 319 probes of species/genus-specific oligonucleotides ranging from 13 to 21 bp with a poly-T anchor at the 5′ or 3′ end for the identification of 42 species from 24 genera of fungal pathogens (Table 1). Three to twelve different specific capture probes were designed and spotted on the array slides for each fungal species/genus to ensure hybridization reaction for proper identification. Among the 319 probes, six universal probes for fungi were designed, so that the array would give a positive signal at universal probe even if fungal species in the tested sample was not listed on the Table 1. In other words, 6 universal probes could detect any fungi other than listed objective fungi without specific signal.
All designed probes and the positive control marker (biotinylated-poly-T) were spotted on one plastic slide. Figure 1a shows an example of the spot pattern of the microarray slide.
Fig. 1.
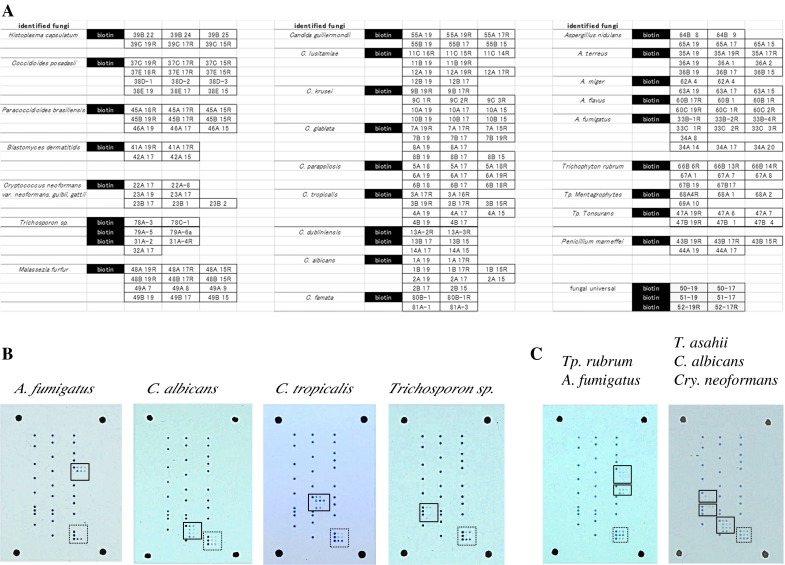
a Example layout of capture probes on microarray slide. Probe names correspond to probe names listed in Table 1. The black column labeled “biotin” indicates the spots for positive control (biotinylated-poly-T) and positional marker. b Typical hybridization patterns using fungal suspension of different fungal species as PCR template. Species-specific signals are enclosed in solid line frames, while universal signals for fungi are enclosed in dotted line frames. These figures show representative results for A. fumigatus, Trichosporon asahii, C. tropicalis, and C. albicans. c Simultaneous hybridization of several species in one array slide. Fungal cell mixtures of C. albicans, Cryptococcus neoformans, and T. asahii or of A. fumigatus and Trichophyton rubrum were directly used as template for PCR amplification and detected on the array slide
Evaluation of the Specificity of DNA Microarray Probes
To evaluate the specificity of the designed capture probes, 66 fungal strains were used (Table S1). All fungal samples tested showed the expected species/genus-specific hybridization patterns as shown in Fig. 1b. Although some probes showed cross-hybridization within the same genus because of their highly conserved sequence (e.g., Rhizopus stlonifer was cross-hybridized to Rhizopus microsporus probes), listed organisms are the major fungi causing infection (Table 1). Moreover, the array system enabled us to identify all the mixed fungi in one test even when several fungal mixtures were used as template (Fig. 1c). Resulted spot number is sometimes varied depending on the sample (e.g., the spot of fungal common probes in Fig. 1b), because the affinity of each designed probes is different.
Sensitivity of the DNA Microarray System
To evaluate microarray detection sensitivity, we used blood samples containing a known number of fungal cells and serum with fungal DNA in place of actual clinical samples. Serial ten-fold dilutions of fungal cells in blood (106–100 CFU/ml) were prepared by adding A. fumigatus conidia or C. albicans cells to rabbit whole blood. The PCR reaction was performed directly using 1 μl of rabbit whole blood with or without fungal cells as template (see Materials and Methods). After the PCR reaction, amplification was verified by electrophoresis, and the samples were used for microarray analysis (Fig. 2a). For both A. fumigatus and C. albicans, 103 CFU/ml was the minimum concentration needed for PCR amplification followed by the microarray detection. Although 102 CFU/ml could be considered as the limit of detection, amplification at this concentration is not reproducible. To increase the sensitivity, we conducted nested PCR. However, it did not enhance the sensitivity.
Fig. 2.

Agarose gel electrophoresis of PCR products. a PCR amplification of ITS region using blood sample spiked with A. fumigatus cells as template. Upper panel shows the results of normal PCR; lower panel shows the results of nested PCR. Lanes: M, molecular marker (Gene Ladder Wide 1: NIPPON GENE co., Tokyo, Japan); 1–7, blood sample spiked with conidia. b PCR amplification of ITS region using serum sample with C. albicans DNA as template. Upper panel shows the results of normal PCR; lower panel shows the results of nested PCR. Lanes: M, molecular marker; 1–8, DNA in 1 ml of serum
We also evaluated detection limits of A. fumigatus and C. albicans DNA in serum. Extracted fungal DNA ranging from 10 ng to 10 fg was separately added to 200 μl of rabbit serum. When 1 μl of the serum sample was used directly as template for PCR, the detection limit was 5 fg per 1 μl of serum. After nested PCR, the minimal amount of DNA required for fungal identification decreased to 0.05 fg per 1 μl of serum (Fig. 2b).
Identification of the Infected Fungi from Mice
Blood samples from infected mice were tested in place of actual human clinical samples. After 1 h of fungal infection, blood was collected and used directly for PCR amplification. Because the first-round PCR did not yield enough amplicon, nested PCR was performed to increase the labeled amplicon, making it possible to detect inoculated fungi in the blood from the infected mouse by microarray. At this moment, the CFU of A. fumigatus and FSSC remained in blood stream were 500 ± 50 (colonies/ml) and 230 ± 30 (colonies/ml), respectively. This detection level is consistent with the result of sensitivity test using rabbit whole blood spiked with fungal cells.
Identification of the Fungi from Blood Culture Sample
Blood from a patient was cultivated in a blood culture bottle for 7 days; one culture positive and one negative sample were subjected to microarray analysis. Samples from blood culture bottle were used directly as a template for nested PCR amplification. The microarray result was consistent with the identification made by the Chiba University Hospital clinical laboratory.
Isothermal Sample DNA Amplification
Because PCR amplification requires a thermal cycler, which is not always available, in small hospitals, or in less developed regions, we attempted to carry out isothermal amplification for biotin labeling of sample DNA using RPA technique [13]. The RPA cycle was performed using 1.5 μl cell suspensions of various fungi as template (Fig. 3). If amplification reaction was successful, the microarray system we developed gave correct identification results in all of the tested samples, even though several amplification bands were observed in some samples.
Fig. 3.

Isothermal amplification. Agarose gel electrophoresis of fungal DNA obtained by isothermal amplification using TwistAmp Basic kit. Lanes: M, molecular marker; Af, A. fumigatus; Ca, C. albicans; Cn, Cryptococcus neoformans; Fs, FSSC; Mf, Malassezia furfur; Ta, T. asahii; Ro, Rhizopus oryzae
Discussion
Fungal infections cause severe morbidity and mortality in immunocompromised patients. Early start of proper treatment is crucial point to achieve better outcome in these patients. Because sensitivity to antifungal drugs differs among different fungal species, identification of the causative fungal agent is important for proper treatment. Rapid detection and identification of pathogens are therefore key points for diagnosis.
Recently, microarray methods have been developed for the identification of a variety of pathogens, including viruses [17], bacteria [4, 8], and fungi [5–7]. These methods are powerful tools to simultaneously detect multiple pathogens. In this study, we developed an easy-to-use, rapid and inexpensive microarray method utilizing biotin-conjugated HRP and color development of the substrate for signal detection. In addition to making the detection system straightforward, we immobilize DNA probes to ordinary plastic slides without any surface modification using UV irradiation via the poly-T anchor of the capture probes [14]. This immobilization system allowed us to use inexpensive, mass-produced, and commercially available ordinary plastic slides as the DNA microarray substrate.
For our DNA microarray, we selected ITS regions of fungal rRNA genes as target because the ITS sequence have been widely used to identify fungi. Although it was difficult to design species-specific probes for some of the fungal genera (Alternaria sp., Rhizomucor sp., Mucor sp., Trichosporon sp.), pathogenic species of these genera have similar MIC values against several antifungal drugs, making designing genus-specific probes useful [18–21]. To our knowledge, the number of fungal species/genera that could be identified by our array system, 42 species from 24 genera, is the largest among microarray identification systems reported to date [5–7]. And the number of identifiable fungal species can be further increased depending on demand of clinical use. Considering the increasing incidence of infectious diseases caused by fungi, this microarray system, which can be used to identify a variety of fungi simultaneously, has great potential.
The sensitivity of our microarray system was evaluated using whole blood spiked with a certain number of fungal cells and serum with fungal DNA. When 1 μl of either sample was used directly as the PCR template, the limit of the detection was 103 cells/ml for blood, and 5 pg/ml of DNA for serum. Nested PCR increased the sensitivity to 50 fg/ml of DNA in serum, but no change in sensitivity was observed in the blood sample. According to calculations, in the 103 cells/ml blood sample, 1 μl of template contains 1 cell, so in case of lower concentration sample, 1 μl of template does not contain any cells. That is why the amplification of 102 cells/ml sample was not constant and sensitivity could not increase by nested PCR. However, DNA extraction or concentration of cells from larger volume of blood or serum sample will have a possibility to decrease detection limit.
When we tested the sample from an infected mouse model and blood culture, it was difficult to get enough intensity in microarray detection with only one step of PCR amplification. This indicated that the amount of DNA in the actual clinical samples is smaller than the detection limit of our microarray system. Nested PCR, however, increased the sensitivity of amplification, and the nested PCR samples successfully gave the expected diagnostic results.
The PCR step in conventional microarray systems has remained as a crucial barrier for wide use in laboratories or hospitals not equipped with a PCR machine. In the present work, we adopted isothermal amplification of DNA samples using the RPA technique as an alternative to PCR to solve this problem. Although, the RPA technique has been used for rapid identification of viruses and bacteria [13], this is the first report of fungal DNA amplification by the RPA technique directly from a fungal cell suspension within 1 h. However, in the present study, the RPA method was found to be less sensitive than the conventional PCR technique. Further optimization of sample preparation and RPA conditions are expected to yield improved results.
In conclusion, we were able to establish a rapid microarray system that can specifically identify a variety of fungal pathogens. Furthermore, we demonstrated that the ABC method could yield enough sensitivity to detect signals from clinical samples, providing an alternative to expensive fluorescence-scanning methods. We also demonstrated that the isothermal amplification technique in combination with this array system has high potential for future applications, such as for bedside diagnosis. This type of assay technique enables simultaneous identification of several agents in a few, relatively simple steps and therefore will become a useful tool in the identification of a wide range of both pathogenic and nonpathogenic microorganisms.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This work was supported by JST/JICA, SATREPS, and the Cooperative Research Grant of NEKKEN 2012, by a research Grant to T.G. (#24659479), by the National BioResource Projects, and SATREPS by JICA/JST, from the Ministry of Education, Culture, Sports, Science, & Technology in Japan. The authors are grateful to Drs. Kizaki and M. Tanaka for critical comments and technical support.
References
- 1.Kume H, Yamazaki T, Togano T. Epidemiology of visceral mycoses in autopsy cases in Japan: comparison of the data from 1989, 1993, 1997, 2001, 2005 and 2007 in annual of pathological autopsy cases in Japan. Med Mycol J. 2011;52:117–127. doi: 10.3314/jjmm.52.117. [DOI] [PubMed] [Google Scholar]
- 2.Fujita S. Serologic diagnosis of fungal infections. Nihon Rinsho. 2008;66:2313–2318. [PubMed] [Google Scholar]
- 3.Su C, Lei L, Duan Y, Zhang KQ, Yang J. Culture-independent methods for studying environmental microorganisms: methods, application, and perspective. Appl Microbiol Biotechnol. 2012;93:993–1003. doi: 10.1007/s00253-011-3800-7. [DOI] [PubMed] [Google Scholar]
- 4.Hong BX, Jiang LF, Hu YS, Fang DY, Guo HY. Application of oligonucleotide array technology for the rapid detection of pathogenic bacteria of foodborne infections. J Microbiol Methods. 2004;58:403–411. doi: 10.1016/j.mimet.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 5.Spiess B, Seifarth W, Hummel M, et al. DNA microarray-based detection and identification of fungal pathogens in clinical samples from neutropenic patients. J Clin Microbiol. 2007;45:3743–3753. doi: 10.1128/JCM.00942-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sato T, Takayanagi A, Nagao K, et al. Simple PCR-based DNA microarray system to identify human pathogenic fungi in skin. J Clin Microbiol. 2010;48:2357–2364. doi: 10.1128/JCM.02185-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hung WT, Su SL, Shiu LY, Chang TC. Rapid identification of allergenic and pathogenic molds in environmental air by an oligonucleotide array. BMC Infect Dis. 2011;11:91–99. doi: 10.1186/1471-2334-11-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sakai T, Kohzaki K, Watanabe A, Tsuneoka H, Shimadzu M. Use of DNA microarray analysis in diagnosis of bacterial and fungal endophthalmitis. Clin Ophthalmol. 2012;6:321–326. doi: 10.2147/OPTH.S29230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vincent M, Xu Y, Kong H. Helicase-dependent isothermal DNA amplification. EMBO Rep. 2004;5:795–800. doi: 10.1038/sj.embor.7400200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gill P, Ghaemi A. Nucleic acid isothermal amplification technologies: a review. Nucleosides Nucleotides Nucleic Acids. 2008;27:224–243. doi: 10.1080/15257770701845204. [DOI] [PubMed] [Google Scholar]
- 11.Mori Y, Notomi T. Loop-mediated isothermal amplification (LAMP): a rapid, accurate, and cost-effective diagnostic method for infectious diseases. J Infect Chemother. 2009;15:62–69. doi: 10.1007/s10156-009-0669-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uemori T, Mukai H, Takeda O, et al. Investigation of the molecular mechanism of ICAN, a novel gene amplification method. J Biochem. 2007;142:283–292. doi: 10.1093/jb/mvm137. [DOI] [PubMed] [Google Scholar]
- 13.Euler M, Wang Y, Otto P, et al. Recombinase polymerase amplification assay for rapid detection of Francisella tularensis. J Clin Microbiol. 2012;50:2234–2238. doi: 10.1128/JCM.06504-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kimura N. One-step immobilization of poly(dT)-modified DNA onto non-modified plastic substrates by UV irradiation for microarrays. Biochem Biophys Res Commun. 2006;347:477–484. doi: 10.1016/j.bbrc.2006.06.130. [DOI] [PubMed] [Google Scholar]
- 15.White TJ, Bruns T, Lee S, Taylor J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ, editors. PCR Protocols : a guide to methods and applications. San Diego: Academic Press Inc.; 1990. pp. 315–322. [Google Scholar]
- 16.Rubtsova MY, Ulyashova MM, Edelstein MV, Egorov AM. Oligonucleotide microarrays with horseradish peroxidase-based detection for the identification of extended-spectrum β-lactamases. Biosens Bioelectron. 2010;26:1252–1260. doi: 10.1016/j.bios.2010.06.053. [DOI] [PubMed] [Google Scholar]
- 17.Huguenin A, Moutte L, Renois F, et al. Broad respiratory virus detection in infants hospitalized for bronchiolitis by use of a multiplex RT-PCR DNA microarray system. J Med Virol. 2012;84:979–985. doi: 10.1002/jmv.23272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hamid B, Sybren GDH, Ilse C, et al. In vitro activities of eight drugs against 70 clinical and environmental isolates of Alternaria species. J Antimicrob Chemother. 2009;63:1295–1297. doi: 10.1093/jac/dkp109. [DOI] [PubMed] [Google Scholar]
- 19.Wang SB, Li LY, Yu J. Identification and susceptibility of Rhizomucor spp. isolated from patients with cutaneous zygomycosis in China. Med Mycol. 2011;49:799–805. doi: 10.3109/13693786.2011.571292. [DOI] [PubMed] [Google Scholar]
- 20.Vitale RG, de Hoog GS, Schwarz P, et al. Antifungal susceptibility and phylogeny of opportunistic members of the order mucorales. J Clin Microbiol. 2012;50:66–75. doi: 10.1128/JCM.06133-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo LN, Xiano M, Kong F, et al. Tree-locus identification, genotyping, and antifungal susceptibilities of medically important Trichosporon species from China. J Clin Microbiol. 2011;49(11):3805–11. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.