

Abstract
Total syntheses of the dimeric tetrahydroxanthone natural products secalonic acids A and D are described. Key steps involve kinetic resolution of the tetrahydroxanthone core structure using homobenzotetramisole (HBTM) catalysis and late-stage copper (I)-mediated homodimerization of complex aryl stannane monomers.
Keywords: secalonic acids, dimeric tetrahydroxanthone, stannane coupling, kinetic resolution
Dimeric tetrahydroxanthones belong to a class of mycotoxins[1] which connect tetrahydroxanthone monomers via a 2,2′-biphenol linkage (Figure 1). Among these compounds, the secalonic acids[2] were first isolated in 1960 and were found to exhibit interesting bioactivities. Secalonic acid B (1) has antitumor activity and its diastereomer secalonic acid D (2) shows potent cytotoxicity to HL60/K562 cells by down-regulation of c-Myc.[3a] Compound 2 has also been reported to inhibit DNA topoisomerase I.[3b] The enantiomer of 2, secalonic acid A (not shown, ent-2), has antitumor properties and also reduces colchicine toxicity in rat cortical neurons.[4] Related natural products include gonytolide A[5] (3) and rugulotrosin A[6] (4), both of which possess axial chirality. Recently, our laboratory[7] as well as the Bräse,[8] Nicolaou,[9] and Tietze[10] groups have accomplished syntheses of monomeric tetrahydroxanthone natural products. Herein, we describe the first total syntheses of the dimeric natural products secalonic acids A and D utilizing copper (I)-mediated dimerization of complex aryl stannane monomers to construct the requisite 2,2′-biphenol linkage.
Figure 1.
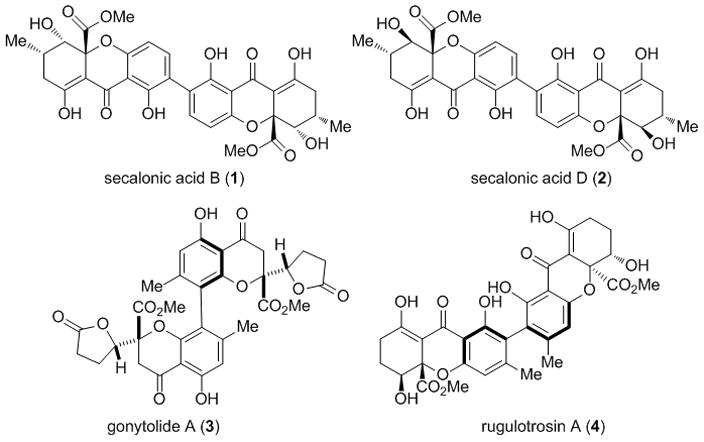
Representative dimeric tetrahydroxanthone natural products
Based on the proposed biosynthetic relationship[11] between tetrahydroxanthones and chromone lactones and our previous synthetic studies,[7] we anticipated that secalonic acid D (2) could be obtained from the dimeric chromone lactone 5[12] via double Dieckmann cyclization (Figure 2). We initially envisioned that oxidative coupling of chromone lactone 6 could be used to access dimeric precursor 5. Following our developed methodology,[7] substrate 6 may be obtained by vinylogous addition of 2-(trimethylsiloxy)furan to a siloxybenzopyrylium species derived from chromone ester 7.
Figure 2.
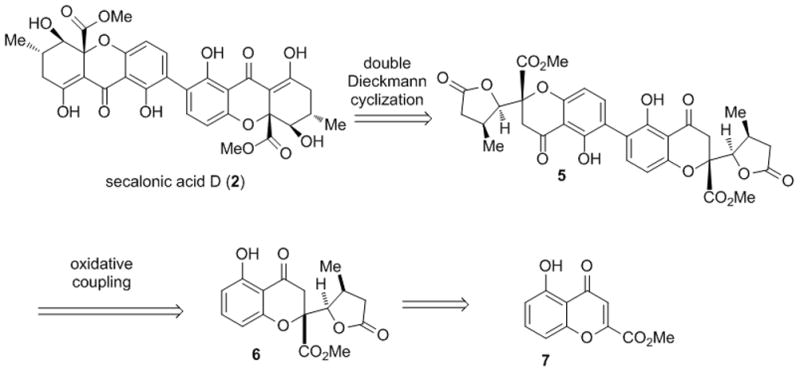
Initial retrosynthetic analysis for secalonic acid D
We first evaluated the possibility for oxidative coupling of a chromone lactone monomer due to the likely instability of the tetrahydroxanthone core structure. Accordingly, we prepared model chromone lactone 8[7] (Scheme 1) on a gram-scale and investigated a number of oxidative coupling conditions[13] (e.g. VOCl3, Mn(acac)3, FeCl3). However, we found that oxidative conditions afforded 2,2′-linked-chromone lactone dimers in very low yield and, in the case of VOCl3, chlorinated products. We then changed our focus to prefunctionalize the monomers to achieve dimerization of the chromone lactone which we anticipated could provide selectivity for biaryl coupling. Both thallium acetate-directed[14] or Me3NBnICl2-directed iodination[15,16] of 8 afforded a major ortho-iodinated product which was followed by base-mediated O-methylation to provide aryl iodide 9 (Scheme 1). Noteably, traditional copper-mediated Ullmann coupling of 9 (10 equiv Cu, 160 °C) led to deiodination and decomposition products. Stannylation of 9 was mediated by Pd(0) catalysis in which case nBu4NI[17] was found to be an essential additive to obtain full conversion to stannane 10. With both 9 and 10 in hand, a number of Pd sources, ligands, and solvents were investigated for biaryl couplings. However, in these experiments the desired dimer was obtained in trace amounts. Intriguingly, we found that stannane 10 was converted into an inseparable mixture of C2 dimer 11 and Cs dimer 12 when the commercial reagent CuCl[18,19] was used as additive for Pd(0)-mediated cross coupling. Surprisingly, this pathway was totally suppressed using freshly prepared CuCl and degassed solvent.
Scheme 1.
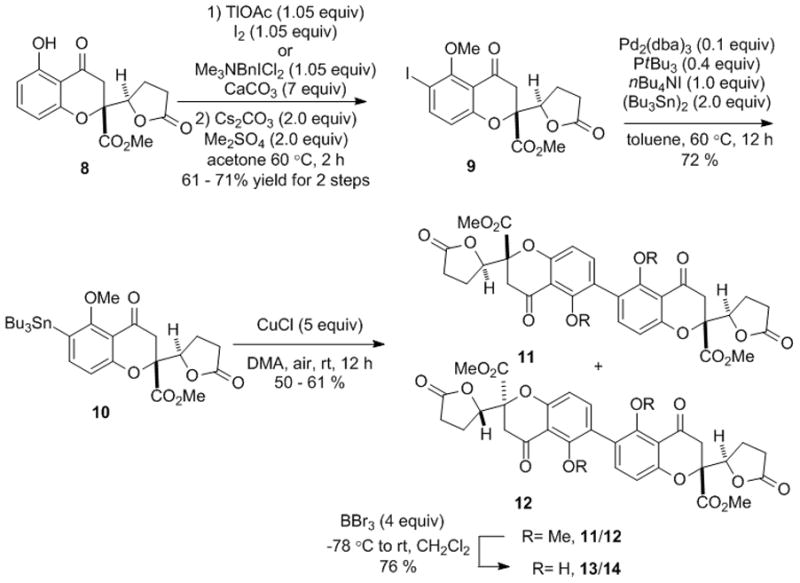
Synthesis of a racemic, model 2,2′-linked chromanone dimer
We found that using ambient air as oxidant[20,21] was critical to enable reproducible dimerization chemistry and that the polar aprotic solvent DMA enhanced the rate of transmetallation of tributyltin to copper(I). Treatment of the resulting dimers 11/12 with BBr3 cleanly afforded the deprotected dimers 13/14.
With the chromone lactone dimers in hand, we anticipated that double Dieckmann cyclization[7] could provide access to the dimeric tetrahydroxanthone. However, in contrast to monomeric substrates,[7] treatment of dimers 13/14 under basic conditions (e.g. NaH/THF, NaH/Mg(OTf)2, Et3N/TMSOTf), did not lead to significant production of dimeric tetrahydroxanthone products or mono-rearranged products and only led to decomposition of starting material (Scheme 2). Accordingly, we investigated dimerization of the tetrahydroxanthone core structure. Protection of the enol moiety was necessary due to its high reactivity. Tetrahydroxanthone 15[7] was successfully methylated with (trimethylsilyl)diazomethane to afford the desired enol ether 17 in 57 % yield along with isomer 16 (20 %) (Scheme 3). Strict control of reaction temperature and time was required to prevent double O-methylation. Employing the previous ortho-iodination conditions, iodide 18 was obtained in 80 % yield. Utilizing the homobenzotetramisole (HBTM) catalyst 19 developed by Birman and coworkers,[22] kinetic resolution[23] proceeded smoothly to afford (−)-18 in 48 % yield (96 % ee) and (+)-20 in 49 % yield (96 % ee) (s= 146). Furthermore, we were able to obtain (−)-18 in 99 % ee using a longer reaction time (13 h). The absolute stereochemistry of kinetic resolution products was verified by single crystal analysis of the deiodinated congener (−)-17.[16,24]
Scheme 2.
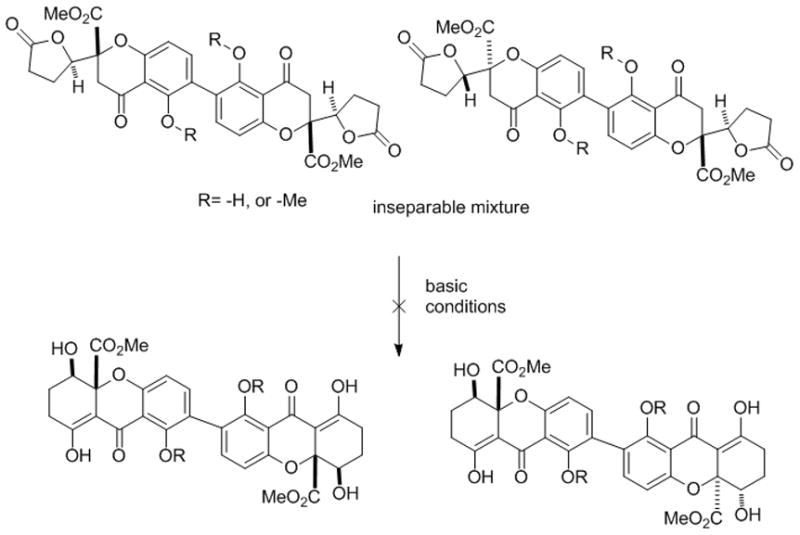
Attempted double Dieckmann cyclization
Scheme 3.
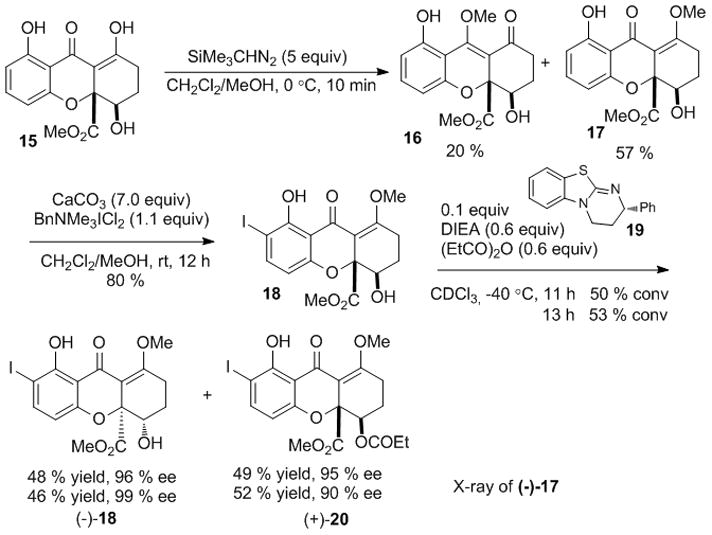
Kinetic resolution on a model system
We propose that the observed high selectivity for the kinetic resolution results from π-stacking between the tetrahydroxanthone substrate and the HBTM catalyst[22] leading to an assembly which minimizes steric repulsion (Scheme 4, A). Steric repulsion between the phenyl group and the tetrahydroxanthone in B renders the latter assembly less favorable.
Scheme 4.
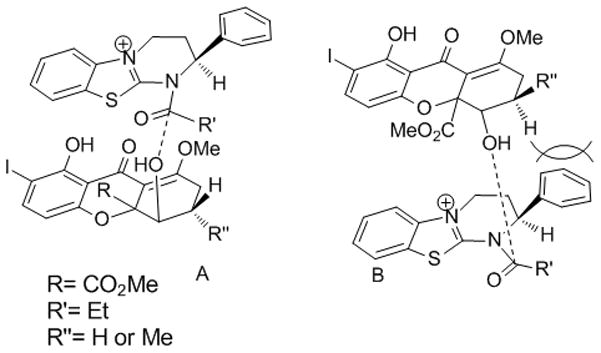
Proposed assemblies for kinetic resolution
With monomer (−)-18 in hand, and after evaluating several protecting groups, we prepared MOM-protected (+)-21 in 81 % yield which was stannylated to afford the key intermediate (+)-22 (Scheme 5). As before, the additive nBu4NI was found to be crucial for Pd(0)-catalyzed stannylation. Furthermore, longer reaction times or increased amounts of additives led to enol ether deprotection. Gratifyingly, copper-mediated coupling of (+)-22 proceeded smoothly to afford the 2,2′-linked dimer (−)-23 in 60 % yield. Finally, treatment of 23 with 3M HCl led to the fully deprotected tetrahydroxanthone dimer (−)-24 in 89 % yield.
Scheme 5.
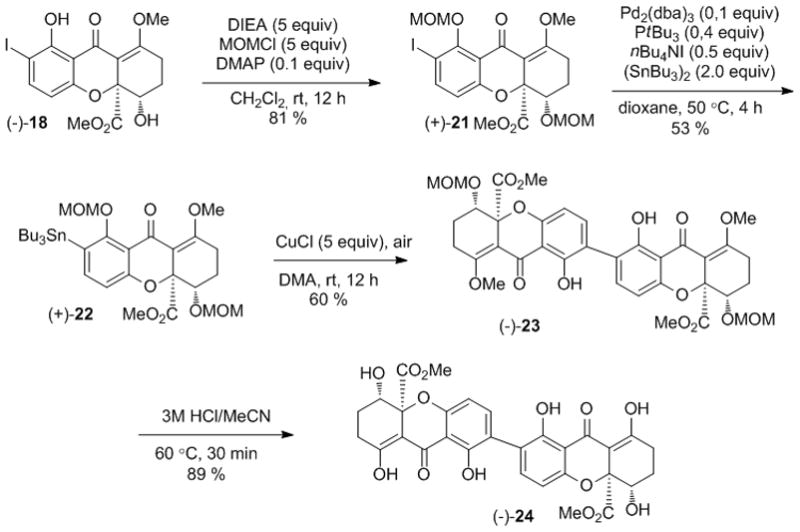
Synthesis of a model 2,2′-linked tetrahydroxanthone dimer
Encouraged by our model studies, we initiated syntheses of secalonic acids A and D using racemic blennolide B (25)[7]. Following previously optimized conditions, methyl enol ether protection and ortho-iodination afforded blennolide derivative 28 (Scheme 6). Due to the presence of an additional methyl group on the C ring, acylative kinetic resolution was much slower in comparison to the model substrate (25 h, 0 °C). However, a longer reaction time and higher temperature provided both (−)-28 and (+)-29 in excellent yield and enantioselectivity. Fortunately, after recrystallization, both (+)-29 and (−)-28 could be obtained in >99 % ee.
Scheme 6.
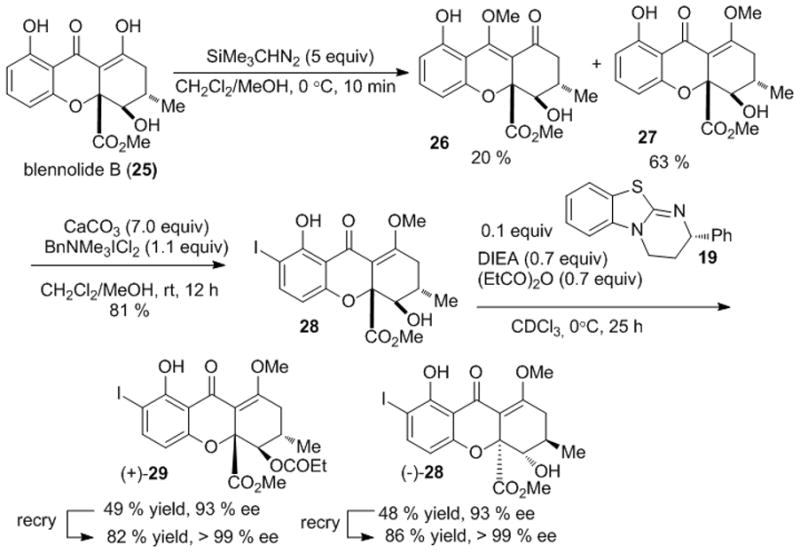
Kinetic resolution of a blennolide derivative
With (+)-29 in hand, we conducted MOM protection followed by stannane formation to obtain the enantioenriched tetrahydroxanthone 30 (Scheme 7). Copper-mediated oxidative coupling provided a single dimeric product 31 whose absolute and relative stereochemistry were unambiguously verified by X-ray crystal structure analysis (Figure 3).[24] Global deprotection under acidic conditions provided secalonic acid D (2) in 80 % yield. NMR spectra, UPLC retention times, and αD values for 2 were found to be identical to those reported for the natural material.[16]
Scheme 7.
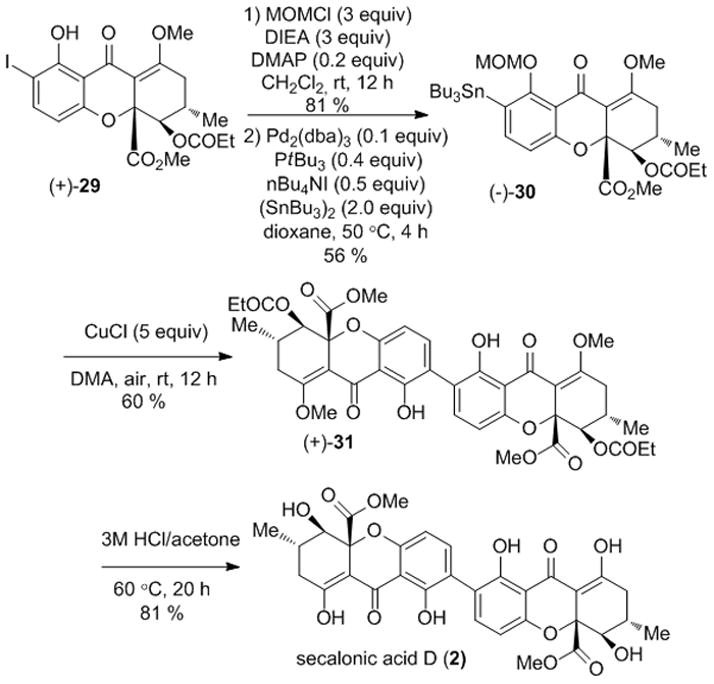
Synthesis of secalonic acid D
Figure 3.
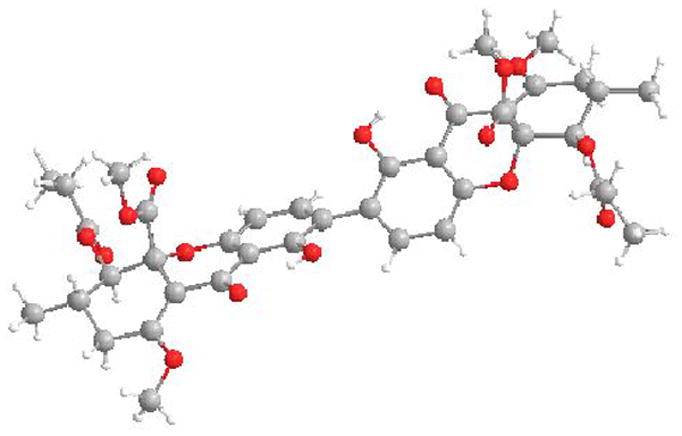
X-ray crystal structure of (+)-31
In a similar manner, the other compound accessed from kinetic resolution, (−)-28, was advanced to secalonic acid A (ent-2) (Scheme 8). After MOM protection and stannane synthesis, (+)-32 was obtained in 49 % yield after two steps. Treatment of (+)-32 with CuCl and air led to the production of dimer (−)-33 in 65 % yield. Final deprotection using 3M HCl/acetonitrile afforded secalonic acid A (ent-2), the enantiomer of secalonic acid D, in 85 % yield.
Scheme 8.
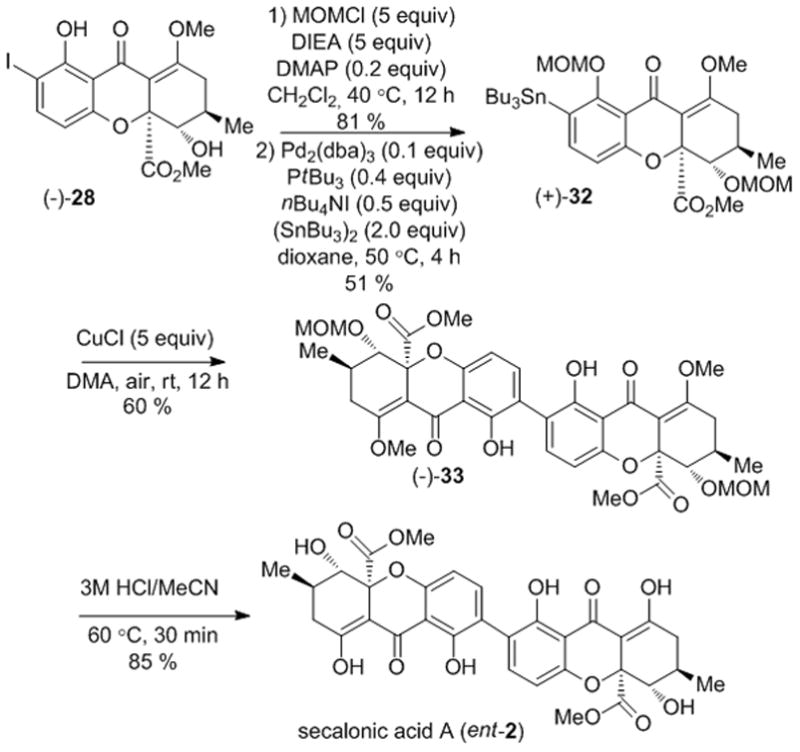
Synthesis of secalonic acid A
In summary, the bioactive tetrahydroxanthone dimers secalonic acids A and D have been synthesized for the first time. Birman’s homobenzotetramisole (HBTM) catalysts were found to be highly effective for kinetic resolution of highly functionlized tetrahydroxanthone monomers. In addition, we found that copper (I) chloride under mild oxidative conditions could be used to access 2,2′ biphenols from complex aryl stannanes. These extremely mild dimerization conditions show excellent functional group tolerance and should be useful for the synthesis of a range of dimeric 2,2′-linked chromone lactone and tetrahydroxanthone natural products. Further studies on the detailed mechanism of copper-mediated coupling of aryl stannanes as well as asymmetric syntheses of additional tetrahydroxanthone natural product targets are currently under investigation and will be reported in due course.
Supplementary Material
Footnotes
Financial support from the NIH (GM-099920 and GM-067041) and Vertex Pharmaceuticals, Inc. (graduate fellowship to T.Q.) is gratefully acknowledged. We thank Dr. Jeffery Bacon (Boston University) for X-ray crystal structure analyses, Prof. Shu-Hua Qi (South China Sea Institute of Oceanology, CAS) for kindly providing a natural sample of secalonic acid D, and Profs. Xiaoguang Lei (National Institute of Biological Sciences, Beijing) and Zhigang She (SunYat-sen University) for helpful information concerning secalonic acid derivatives.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.a) Bräse S, Encinas A, Keck J, Nising CF. Chem Rev. 2009;109:3903–3990. doi: 10.1021/cr050001f. [DOI] [PubMed] [Google Scholar]; b) Masters KS, Bräse S. Chem Rev. 2012;112:3717–3776. doi: 10.1021/cr100446h. [DOI] [PubMed] [Google Scholar]
- 2.For the isolation of secalonic acid A, see: Franck B, Gottschalk EM, Ohnsorge U, Baumann G. Angew Chem. 1964;76:438–439.Angew Chem, Int Ed Engl. 1964;3:441–442.Franck B, Baumann G, Ohnsorge U. Tetrahedron Lett. 1965;6:2031–2037. doi: 10.1016/s0040-4039(00)90148-5.Franck B, Gottschalk EM, Ohnsorge U, Hüper F. Chem Ber. 1966;99:3842–3862.First the isolation of secalonic acid D, see: Steyn PS. Tetrahedron. 1970;26:51–57. doi: 10.1016/0040-4020(70)85006-2.
- 3.a) Zhang J, Tao L, Liang Y, Yan Y, Dai C, Xia X, She Z, Lin Y, Fu L. Cell Cycle. 2009;8:2444–2450. doi: 10.4161/cc.8.15.9170. [DOI] [PubMed] [Google Scholar]; b) Hong R. Pharm Biol. 2011;49:796–799. doi: 10.3109/13880209.2010.548817. [DOI] [PubMed] [Google Scholar]
- 4.Zhai A, Zhang Y, Zhu X, Liang J, Wang X, Lin Y, Chen R. Neurochem Int. 2011;58:85–91. doi: 10.1016/j.neuint.2010.10.016. [DOI] [PubMed] [Google Scholar]
- 5.a) Kikuchi H, Isobe M, Sekiya M, Abe Y, Hoshikawa T, Ueda K, Kurata S, Katou Y, Oshima Y. Org Lett. 2011;13:4624–4627. doi: 10.1021/ol2018449. [DOI] [PubMed] [Google Scholar]; b) Kikuchi H, Isobe M, Kurata S, Katou Y, Oshima Y. Tetrahedron. 2012;68:6218–6223. [Google Scholar]
- 6.Stewart M, Capon RJ, White JM, Lacey E, Tennant S, Gill JH, Shaddock MP. J Nat Prod. 2004;67:728–729. doi: 10.1021/np034038b. [DOI] [PubMed] [Google Scholar]
- 7.Qin T, Johnson RP, Porco JA., Jr J Am Chem Soc. 2011;133:1714–1717. doi: 10.1021/ja110698n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Lesch B, Bräse S. Angew Chem. 2004;116:118–120. doi: 10.1002/anie.200352154. [DOI] [PubMed] [Google Scholar]; Angew Chem, Int Ed. 2004;43:115–118. [Google Scholar]; b) Nising CF, Ohnemüller UK, Bräse S. Angew Chem. 2005;118:313–315. doi: 10.1002/anie.200502913. [DOI] [PubMed] [Google Scholar]; Angew Chem, Int Ed. 2006;45:307–309. [Google Scholar]; c) Ohnemüller UK, Nising CF, Nieger M, Bräse S. Eur J Org Chem. 2006:1535–1546. [Google Scholar]; d) Nising CF, Friedrich A, Bräse S. Synlett. 2007:2987–2990. [Google Scholar]; e) Ohnemüller UK, Nising CF, Encinas A, Bräse S. Synthesis. 2007:2175–2185. [Google Scholar]; f) Gérared EMC, Bräse S. Chem Eur J. 2008;14:8086–8089. doi: 10.1002/chem.200801507. [DOI] [PubMed] [Google Scholar]; g) Volz N, Bröhmer MC, Nieger M, Bräse S. Synlett. 2009:550–553. [Google Scholar]; h) Bröhmer MC, Bourcet EB, Nieger M, Bräse S. Chem Eur J. 2011;17:13706–13711. doi: 10.1002/chem.201102192. [DOI] [PubMed] [Google Scholar]; i) Linsenmeier AM, Bräse S. Eur J Org Chem. 2012:6455–6459. [Google Scholar]; j) Meister AC, Nieger M, Bräse S. Chem Eur J. 2013;19:10836–10839. doi: 10.1002/chem.201301358. [DOI] [PubMed] [Google Scholar]
- 9.Nicolaou KC, Li A. Angew Chem. 2008;120:6681–6684. [Google Scholar]; Angew Chem, Int Ed. 2008;47:6579–6582. doi: 10.1002/anie.200802632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Tietze LF, Spiegl DA, Stecker F, Major J, Raith C, Große C. Chem Eur J. 2008;14:8956–8963. doi: 10.1002/chem.200800967. [DOI] [PubMed] [Google Scholar]; b) Tietze LF, Jackenkroll S, Raith C, Spiegl DA, Reiner JR, Claudia M, Campos O. Chem Eur J. 2013;19:4876–4882. doi: 10.1002/chem.201204037. [DOI] [PubMed] [Google Scholar]; c) Tietze LF, Ma L, Reiner JR, Jackenkroll S, Heidemann S. Chem Eur J. 2013;19:8610–8614. doi: 10.1002/chem.201300479. [DOI] [PubMed] [Google Scholar]
- 11.For the biosynthesis of secalonic acid A, see: Kurobane I, Vining LC, McInnes AG, Walter JA, Wright JLC. Tetrahedron Lett. 1978;19:1379–1382.Kurobane I, Vining ILC. J Antibiotics. 1979;32:1256–1266. doi: 10.7164/antibiotics.32.1256.for the possible relationship between tetrahydroxanthones and chromone lactones, see: Tabata N, Tomada H, Matsuzaki K, Ōmura S. J Am Chem Soc. 1993;115:8558–8564.Tabata N, Tomoda H, Iwai Y, Ōmura S. J Antibiotics. 1996;49:267–271. doi: 10.7164/antibiotics.49.267.
- 12.For synthesis of an isomer of the chromone lactone dimer paecilin A, see: Tietze LF, Ma L, Jackenkroll S, Reiner JR, Hierold J, Gnanaprakasam B, Heidemann S. Heterocycles. 2014;88:1101–1119.
- 13.For a review on aryl-aryl bond formation, see: Hassan J, Sévignon M, Gozzi C, Schulz E, Lemaire M. Chem Rev. 2002;102:1359–1470. doi: 10.1021/cr000664r.
- 14.Cambie RC, Rutledge PS, Smith-Palmer T, Woodgate PD. J Chem Soc, Perkin Trans 1. 1976:1161–1164. [Google Scholar]
- 15.Kajigaeshi S, Kakinami T, Yamasaki H, Fujisaki S, Kondo M, Okamoto T. Chem Lett. 1987:2109–2112. [Google Scholar]
- 16.See the Supporting Information for complete experimental details.
- 17.a) Piber M, Jensen AE, Rottlander M, Knochel P. Org Lett. 1999;1:1323–1326. [Google Scholar]; b) Nguefack JF, Bolitt V, Sinou D. Tetrahedron Lett. 1996;37:5527–5530. [Google Scholar]; c) Powell NA, Rychnovsky SD. Tetrahedron Lett. 1996;37:7901–7904. [Google Scholar]
- 18.Han X, Stoltz BM, Corey EJ. J Am Chem Soc. 1999;121:7600–7605. [Google Scholar]
- 19.For CuCl-mediated coupling of aryl stannanes, see: Piers E, Yee JGK, Gladstone PL. Org Lett. 2000;2:481–484. doi: 10.1021/ol9903988.Nabatame K, Hirama M, Inoue M. Heterocycles. 2008;76:1011–1016.For CuCl-mediated couplings of vinyl stannanes, see: Piers E, Romero MA. J Am Chem Soc. 1996;118:1215–1216.For copper nitrate-mediated arylstannane coupling, see: Ghosal S, Luke GP, Kyler KS. J Org Chem. 1987;52:4296–4298.For catalytic CuCl2-mediated stannane coupling, see: Kang SK, Baik TG, Jiao XH, Lee YT. Tetrahedron Lett. 1999;40:2383–2384.
- 20.For a review on oxidation of organocuprates, see: Surry DS, Spring DR. Chem Soc Rev. 2006;35:218–225. doi: 10.1039/b508391p.
- 21.See the Supporting Information for a details on evaluation of copper sources, solvents, and oxidants.
- 22.a) Birman VB, Li X. Org Lett. 2006;8:1351–1354. doi: 10.1021/ol060065s. [DOI] [PubMed] [Google Scholar]; b) Birman VB, Li X. Org Lett. 2008;10:1115–1118. doi: 10.1021/ol703119n. [DOI] [PubMed] [Google Scholar]; c) Li X, Jiang H, Uffman EW, Guo L, Zhang Y, Yang X, Birman VB. J Org Chem. 2012;77:1722–1737. doi: 10.1021/jo202220x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.For a recent review of organocatalytic, enantioselective acyl transfer, see: Müller CE, Schreiner PR. Angew Chem. 2011;123:6136–6167.Angew Chem, Int Ed. 2011;50:6012–6042. doi: 10.1002/anie.201006128.
- 24.CCDC 961591 ((−)-17), 961692 ((+)-31) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.