

Neuromyelitis optica (NMO) is a demyelinating disease of the CNS that preferentially affects the optic nerve and spinal cord.1 The presence of circulating autoantibodies (NMO–immunoglobulin G [IgG]) having the water channel aquaporin-4 (AQP-4) as their target antigen is associated with NMO.1 Outside the CNS AQP-4 is present in the distal collecting tubes of the kidney, in parietal cells of the stomach,2 and in fast-twitch fibers of skeletal muscle.3 Several findings support the idea that AQP-4 water channels may be associated to the dystrophin-glycoprotein complex (DGC) in skeletal muscle fibers and AQP-4 expression has been found altered in muscle diseases.4
We describe the case of a 13-year-old girl with NMO experiencing recurrent episodes of hyperCKemia.
Case report.
The patient had been well until 2006, when she developed the first acute myelitis presenting with paraparesis, hypoesthesia, and paresthesias below the C7 level. MRI of the spinal cord showed the presence of a hyperintense lesion, extending from C7 to T9. Brain MRI was normal. A CSF analysis showed lymphocytic pleocytosis (85 cells/mm3) and absence of oligoclonal IgG bands. She responded well to IV methylprednisolone (1 g daily for 5 days) and IVIg (0.4 g/kg body weight daily for 5 consecutive days). In 2007, a bilateral optic neuritis occurred. Brain MRI showed the presence of fluid-attenuated inversion recovery signal abnormality around the third ventricle. A repeat CSF analysis showed lymphocytic pleocytosis (10 cells/mm3). Hematologic tests were normal as well as screening for autoimmune and infectious conditions (including testing for antinuclear antibodies, anti PM-Scl antibodies, antineutrophil cytoplasmic antibodies, lupus anticoagulant, anticardiolipin antibody, and anti-Borrelia, Treponema pallidum hemagglutination, and HIV serologies). NMO-IgG (on primate cerebellum) and anti-AQP-4 antibody (on AQP-4-transfected cells; Euroimmun, Lübeck, Germany) testings were positive (anti-AQP-4 antibody titer, 1:500). The patient was diagnosed with NMO. In the following year she experienced other clinical attacks (both optic neuritis and myelitis episodes), requiring IV methylprednisolone. In August 2008, the patient was admitted to the Section of Neurology, Perugia, Italy, because of a cervical myelitis. Laboratory tests demonstrated hyperCKemia (5,465 IU/L, normal values 0–180). In the following days CK rose to 15,818 IU/L. HyperCKemia was accompanied by a concomitant increase of lactic dehydrogenase (1,079 IU/L, normal values 225–450), glutamic-oxaloacetic transaminase (320 IU/L, normal values <45), and myoglobin (677.7 ng/mL, normal values 14.3–65.8). No significant alterations of CK-MB levels were demonstrated. No laboratory evidence of liver dysfunction/disease was found. After 5 days, CK declined to 1,386 IU/L, and then it remained mildly elevated (591 IU/L) in the following weeks. In September 2008, CK rose again to 14,163 IU/L. The patient was asymptomatic with the exception of mild myalgia. Repeat CK 30 days later was 340 IU/L. At the end of September 2008, therapy with azathioprine (2 mg/Kg) was started, with good clinical response. In November 2008, CK rose again to 4,068 UI/L, and then it progressively declined. A retrospective analysis of the patient's medical records also revealed another asymptomatic episode of moderate hyperCKemia in 2007 (1,985 IU/L) that occurred in association with a bilateral optic neuritis treated with methylprednisolone (1 g daily for 5 days). No clinical or laboratory evidence of muscle disease was found before 2007. During the second hyperCKemia episode, EMG was performed twice and it did not demonstrate myopathy or neurogenic changes. Muscle biopsy (taken from the vastus lateralis when CK levels were 7,828 UI/L) failed to show any major histopathologic alteration (figure, A). Immunofluorescent staining showed a normal sarcolemmal reactivity for dystrophin, dysferlin, and caveolin-3. Staining of AQP-4 was performed, ruling out a massive loss of AQP-4 at the surface of type 2 muscle fibers (figure, B and C). No other major causes of hyperCKemia, including medications,5 were found.
Figure. Patient's muscle biopsy.
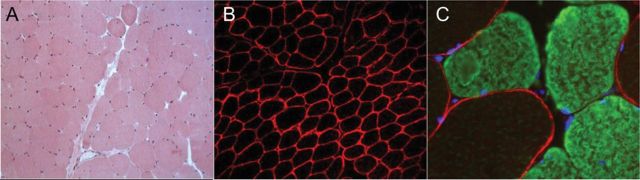
(A) Hematoxylin-eosin staining (×10) shows a normal histologic pattern. (B) A normal localization of AQP-4 (antibody purchased from Sigma-Aldrich, St. Louis, MO) at the surface of type 2 muscle fibers (×10). (C) Double immunofluorescence with anti AQP-4 (red) and anti-myosin heavy chain-slow (green) antibodies shows a preferential localization of AQP-4 around fast myofibers (unstained for myosin). Nuclei were visualized using DAPI-conjugated (blue) mounting medium (×40).
Discussion.
The occurrence of hyperCKemia episodes in 3 anti-AQP-4 antibody-positive NMO female patients has been recently described, suggesting the possibility of an anti-AQP-4 antibody-mediated attack to the sarcolemma.6 This possibility is compatible with what we observed in our patient, particularly with the relapsing behavior of the hyperCKemia episodes. However, muscle biopsy findings excluded major histopathologic muscle alterations. In particular, both histopathologic and AQP-4-specific stainings reciprocally concurred to exclude inflammatory myopathy and significant losses of sarcolemmal AQP-4.
However, the utilized morphologic methods could not detect a partial loss of AQP-4 and the potential multifocality of the inflammatory pattern could have limited our potential to detect histopathologic abnormalities. Moreover, it is known that in about 30% of patients with pauci-asymptomatic hyperCKemia, muscle biopsy, even after comprehensive immunocytochemical and biochemical studies, is normal.5 To date, the real prevalence and pathogenetic meaning of hyperCKemia in NMO remain unknown.
Footnotes
Author contributions: M.D.F., M.D.G., P.S., P.M., and P.C. took care of the patient. R.M., C.T., and E.R. performed and interpreted the muscle biopsy findings. D.F., E.Z., and M.G. performed and interpreted the staining of AQP-4. P.F. interpreted the spinal cord and brain MRI. M.D.F., M.D.G., L.G., A.I., P.S., and P.C. prepared the manuscript draft. All the authors contributed to the final version of the manuscript.
Disclosure: M. Di Filippo received travel grants from Bayer Schering, Biogen Dompé, Biogen Idec, Merck-Serono, Novartis, and Sanofi-Aventis to attend national and international conferences and speaker honoraria from Biogen Idec and Boehringer-Ingelheim. D. Franciotta reports no disclosures. R. Massa received/receives travel grants from Genzyme, Kedrion, and CSL-Behring to attend national and international conferences. M. Di Gregorio, E. Zardini, M. Gastaldi, C. Terracciano, E. Rastelli, L. Gaetani, A. Iannone, P. Menduno, P. Floridi, and P. Sarchielli report no disclosures. P. Calabresi receives/received research support from Bayer Schering, Biogen-Dompé, Boehringer Ingelheim, Eisai, Lundbeck, Merck-Serono, Novartis, Sanofi-Aventis, Sigma-Tau, and UCB Pharma. He also receives/received support from Ricerca Corrente IRCCS, Ricerca Finalizzata IRCCS, European Community Grant REPLACES (restorative plasticity at corticostriatal excitatory synapses), the Italian Minister of Health, and AIFA (Agenzia Italiana del Farmaco). Go to Neurology.org for full disclosures.
References
- 1.Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol 2007;6:805–815 [DOI] [PubMed] [Google Scholar]
- 2.Jarius S, Paul F, Franciotta D, et al. Mechanisms of disease: aquaporin-4 antibodies in neuromyelitis optica. Nat Clin Pract Neurol 2008;4:202–214 [DOI] [PubMed] [Google Scholar]
- 3.Frigeri A, Nicchia GP, Verbavatz JM, Valenti G, Svelto M. Expression of aquaporin-4 in fast-twitch fibers of mammalian skeletal muscle. J Clin Invest 1998;102:695–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frigeri A, Nicchia GP, Repetto S, Bado M, Minetti C, Svelto M. Altered aquaporin-4 expression in human muscular dystrophies: a common feature? FASEB J 2002;16:1120–1122 [DOI] [PubMed] [Google Scholar]
- 5.Kyriakides T, Angelini C, Schaefer J, et al. EFNS guidelines on the diagnostic approach to pauci- or asymptomatic hyperCKemia. Eur J Neurol 2010;17:767–773 [DOI] [PubMed] [Google Scholar]
- 6.Suzuki N, Takahashi T, Aoki M, et al. Neuromyelitis optica preceded by hyperCKemia episode. Neurology 2010;74:1543–1545 [DOI] [PubMed] [Google Scholar]