

Abstract
Conventional analyses distinguish between antimicrobial peptides by differences in amino acid sequence. Yet structural paradigms common to broader classes of these molecules have not been established. The current analyses examined the potential conservation of structural themes in antimicrobial peptides from evolutionarily diverse organisms. Using proteomics, an antimicrobial peptide signature was discovered to integrate stereospecific sequence patterns and a hallmark three-dimensional motif. This striking multidimensional signature is conserved among disulfide-containing antimicrobial peptides spanning biological kingdoms, and it transcends motifs previously limited to defined peptide subclasses. Experimental data validating this model enabled the identification of previously unrecognized antimicrobial activity in peptides of known identity. The multidimensional signature model provides a unifying structural theme in broad classes of antimicrobial peptides, will facilitate discovery of antimicrobial peptides as yet unknown, and offers insights into the evolution of molecular determinants in these and related host defense effector molecules.
Nature provides a context in which hosts across the phylogenetic spectrum are confronted by potential microbial pathogens. In turn, natural selection provides a corresponding requirement for rapid and effective molecular stratagems of host defense. Antimicrobial peptides represent a pivotal result of this coevolutionary relationship. While higher organisms have evolved complex and adaptive immune systems, virtually all organisms rely on innate immune mechanisms that involve antimicrobial peptides that are rapidly deployed to ward off microbial invasion.
Antimicrobial peptides may be categorized as those with or without disulfide bridges. Members of both classes share a number of features that likely contribute to their antimicrobial actions, including (i) small size, typically <10 kDa; (ii) cationic charge, often ranging from +2 to +7 at pH 7; and (iii) amphipathic stereogeometry, conferring relatively polarized hydrophilic and hydrophobic facets (1). The general recurrence of such themes in antimicrobial peptides from diverse organisms suggests further structural relationships yet to be defined. The limited size of these polypeptides places restrictions on the structural repertoire available to meet these requirements. Despite this limitation, antimicrobial peptides display a high degree of variability at nonconserved sites, with amino acid substitution rates on the order of those associated with positive selection (2). These observations are consistent with the hypothesis that coevolutionary selective pressures drive hostpathogen interactions (3).
Amino acid sequence motifs have been identified in certain antimicrobial peptide subclasses [e.g., cysteine array in mammalian defensins (4)], and prior studies suggested that structural themes may occur within distinct peptide subsets (5, 6). Yet comparatively little is known about potentially more comprehensive relationships common to broader classes of antimicrobial peptides. The current investigation integrated proteomic and microbiologic techniques to explore structure-function commonalities among antimicrobial peptides from phylogenetically diverse organisms.
Materials and Methods
Antimicrobial Peptide Dataset. Prototypic representatives from virtually all classes of disulfide-containing antimicrobial peptides were included to generate a diverse primary dataset by using the following criteria: (i) mature primary sequence, (ii) cysteine-containing, (iii) published antimicrobial activity, and (iv) up to 75 aa in length. The initial dataset contained over 500 known antimicrobial peptides (see supporting information, which is published on the PNAS web site). From this initial cohort, representatives of specific classes [e.g., α-defensins, β-defensins, insect defensins, plant defensins, cysteine-stabilized (CS)-αβ peptides] were identified on the basis of their being well characterized in the literature, having a known 3D structure, and being prototypic of their respective subclass. No cysteine-containing antimicrobial peptide subclasses were specifically excluded from this algorithm. Thus, the primary dataset was broadly encompassing of antimicrobial peptides with diverse sequence and secondary structures known at the time of this investigation. Amino acid sequence data were analyzed, because not all nucleotide sequences have been characterized, and saturation of nucleotides occurs within nonmitochondrial sequences over evolutionary time scales.
Proteomic Analysis of Structural Patterns. Primary sequences were analyzed for conserved motifs by multiple sequence alignment [MSA (7, 8)]. Stereospecific sequence analyses used PROSITE to probe forward and reverse Swiss-Prot protein databases. Peptides with known structures [Protein Data Bank (PDB)] were used in conformation analyses. Selection criteria for comparative nonantimicrobial peptides followed a systematic algorithm paralleling that used for the antimicrobial peptides examined. A Boolean argument was used to identify appropriate controls from the group of cysteine-containing peptides with known mature primary sequences and 3D structures: chain length 10-75 residues [AND] sequence containing a GXC motif [NOT] keywords antimicrobial, toxin, or protease inhibitor. Peptide fragments, nonnatural variants, and duplicates were removed from search outcomes. A cohort of >50 comparators with no evidence of antimicrobial activity in the literature or to our knowledge were identified in the Protein Data Bank. Specific comparators were identified to represent diverse phylogenetic sources, including mammalian, plant, fungal, and prokaryotic. Three-dimensional alignments of peptides were carried out by using combinatorial extension (9), and quantified by rms deviation (rmsd). As a control for alignment specificity, analyses included a prototypic α-helical antimicrobial peptide that lacks disulfide-bridging (magainin, from Xenopus).
Assay for Antimicrobial Activity. Antimicrobial assays were performed by using a well established radial diffusion method modified to pH 5.5 or 7.5 (10). Peptides were obtained from commercial sources, with the exception of recombinant brazzein, kindly provided by J. L. Markley and F. M. Assadi-Porter (University of Wisconsin) (11). Defensin HNP-1 was included in each assay as a control. A panel of microorganisms was tested: Gram-positive Staphylococcus aureus (ATCC 27217) and Bacillus subtilis (ATCC 6633), Gram-negative Escherichia coli (strain ML-35), and the fungus Candida albicans (ATCC 36082). Logarithmic-phase organisms were inoculated (106 colony-forming units/ml) into buffered agarose, and poured into plates. Peptides (10 μg) were introduced into wells in the seeded matrix and incubated for 3 h at 37°C. Nutrient overlay medium was applied, and assays were incubated at 37°C for bacteria or 30°C for fungi. After 24 h, zones of inhibition were measured. Independent experiments were repeated a minimum of two times.
Results
Identification of Multidimensional Signatures in Antimicrobial Peptides. The primary dataset included antimicrobial peptides from taxa encompassing broad biological diversity spanning an evolutionary distance of 2.6 billion years [estimated divergence of fungi and plants from higher organisms (12)]. This dataset included prototypes of all major classes of disulfide-containing antimicrobial peptides, including distinct conformation groups such as defensin, CS-αβ, ranabox, and β-hairpin.
Conventional multiple sequence alignment (MSA; N- to C-terminal; dextromeric) revealed no clear consensus patterns among primary sequences of the antimicrobial peptide study set. However, visual inspection revealed an absolutely conserved GXC motif, oriented in reverse in some peptides. We hypothesized that conventional MSA failed to recognize this inverted consensus pattern. Therefore, peptides containing inverted GXC motifs were aligned in their C- to N-terminal (levomeric) orientation. This stereospecific MSA revealed a striking sequence pattern common to all disulfide-containing antimicrobial peptide classes (Fig. 1). The consensus patterns, defined herein as the enantiomeric sequence signature, adhere to the following formulae:
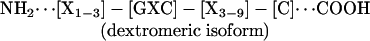 |
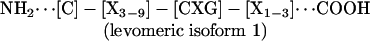 |
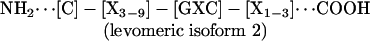 |
Fig. 1.

Enantiomeric sequence patterns in cysteine-containing antimicrobial peptides. Representatives are, in descending order (peptide name, GenBank GI accession code): α-defensins (HNP-3, 229858); β-defensins (mBD-8, 15826276); insect defensins (phormicin, 118432); insect CS-αβ peptides (drosomycin, 2780893); plant CS-αβ peptides (Ah-AMP-1, 6730111); crustacean CS-αβ peptides (MGD-1, 12084380); protegrins (protegrin-1, 3212589); big defensin (2493577); gaegurins (gaegurin-1, 1169813); tachyplesins (tachyplesin-1, 84665); polyphemusins (polyphemusin-1, 130777); mytilins (mytilin A, 6225740); gomesin (20664097); thanatin (6730068); and AFP-1 (1421258). Sequences are shown in their conventional dextromeric orientations unless indicated as levomeric (levo). The GXC or CXG-C motif is highlighted [glycine (G), orange; cysteine (C), yellow] within primary sequences corresponding to the γ-core motif (outlined). Conserved cysteine residues beyond the γ-core are shaded gray.
These consensus patterns transcend defensin-specific motifs identified previously (4, 13). Specific characteristics of the enantiomeric sequence signatures include (i) a length of 8-16 amino acid residues, and (ii) conserved GXC or CXG motifs within the sequence isoforms. Interestingly, levomeric isoform 2 peptides retain a dextromeric GXC motif within the levomeric sequence signature (Fig. 1).
Identification of the conserved enantiomeric signature suggested that a corresponding motif would also be present in the 3D structures of disulfide-stabilized antimicrobial peptides. Conformation alignments revealed a core motif that was absolutely conserved across all classes of disulfide-stabilized antimicrobial peptides (Fig. 2 and Table 1). This 3D archetype, termed herein as the γ-core motif, is composed of two antiparallel β-sheets, with an interposed short turn region (Figs. 3 and 4). All three isoforms of the enantiomeric sequence signature conform to the γ-core motif, reflecting their 3D convergence (Fig. 4). Additional features that characterize the γ-core include the following: (i) net cationic charge (+0.5 to +7) with basic residues typically polarized along its axis; (ii) periodic charge and hydrophobicity yielding amphipathic stereogeometry; and (iii) participation in one to four disulfide bonds. This motif may constitute the entire peptide, or it may link to adjacent structural domains.
Fig. 2.

(A-H) Conservation of 3D structure among disulfide-stabilized antimicrobial peptides. Comparisons (peptide name, PDB ID code, source genus, common name; rmsd) are between Ah-AMP-1 (1BK8, Aesculus, horse chestnut tree) and protegrin-1 (PRO-1,1PG1, Sus, domestic pig; rmsd 1.2 Å; A and B); drosomycin (DRO, 1MYN, Drosophila, fruit fly; rmsd 1.2 Å; C and D); HNP-3 (1DFN, Homo, human; rmsd 3.2 Å; E and F); and magainin-2 (MAG-2, 2MAG, Xenopus, frog; rmsd 2.6 Å; G and H). A, C, E, and G use clustal secondary structure coloration (gold, β-sheet; red, α-helix; blue, turn). B, D, F, and H use the clustal polarity-2 color scheme (hydrophobic, gray; hydrophilic, purple). Oxidized cysteine residues (cystine) are colored gray, indicating hydrophobicity. Disulfide bonds are indicated as dotted yellow lines in A-H. Proteins were visualized by using protein explorer (26). (I-L) Absence of the γ-core signature in nonantimicrobial peptides. Representative comparisons are between the antimicrobial peptide Ah-AMP-1 and nonantimicrobial peptides: allergen-5 (2BBG, Ambrosia, ragweed; rmsd 6.5 Å; I); metallothionein II (1AOO, Saccharomyces, yeast; rmsd 5.3 Å; J); transforming growth factor α (3TGF, Homo, human; rmsd 4.7 Å; K); and ferredoxin (2FDN, Clostridium, bacterium; rmsd 7.4 Å; L). Nonantimicrobial peptides (blue) are shown in maximal alignment to Ah-AMP-1 (gray). Formatting is same as in A-H.
Table 1. Quantification of 3D congruence among disulfide-containing antimicrobial peptides.
| Peptide | Length, aa | rmsd, Å | Identity, % | Align/gap |
|---|---|---|---|---|
| Antimicrobial | ||||
| Ah-AMP-1 (Aesculus, tree, 1BK8) | 50 | 0.0 | 100 | 50/0 |
| Sapecin (Sarcophaga, fly, 1L4V) | 40 | 0.9 | 25.0 | 38/0 |
| Protegrin-1 (Sus, Pig, 1PG1) | 19 | 1.2 | 18.8 | 16/0 |
| Defensin (Raphanus, radish, 1AYJ) | 51 | 1.3 | 47.6 | 49/0 |
| Drosomycin (Drosophila, fruit fly, 1MYN) | 44 | 1.4 | 29.3 | 41/6 |
| Thionin (Triticalis, wheat, 1GPS) | 47 | 1.8 | 26.1 | 46/3 |
| MGD-1 (Mytilus, mussel, 1FJN) | 39 | 2.0 | 26.5 | 34/1 |
| Thanatin (Podisus, soldier bug, 8TFV) | 21 | 2.2 | 12.5 | 16/0 |
| HNP-3 (Homo, human, 1DFN) | 34 | 3.2 | 8.3 | 24/17 |
| MBD-8 (Mus, mouse, 1E4R) | 35 | 3.4 | 0.0 | 24/13 |
| AFP-1 (Aspergillus, fungus, 1AFP) | 51 | 4.8 | 6.2 | 32/7 |
| Mean ± SD | 2.2 ± 1.2* | |||
| Model-predicted antimicrobial | ||||
| Brazzein (Pentadiplandra, j'oublie berry, 1BRZ) | 54 | 1.9 | 34.4 | 32/4 |
| Charybdotoxin (Leiurus, scorpion, 2CRD) | 37 | 2.0 | 17.4 | 46/2 |
| Mean ± SD | 1.9 ± 0.7* | |||
| Nonantimicrobial | ||||
| Transforming growth factor α (Homo, human, 3TGF) | 50 | 4.7 | 3.1 | 32/7 |
| Metallothionein (Saccharomyces, yeast, 1AOO) | 40 | 5.3 | 18.8 | 32/16 |
| Allergen-5 (Ambrosia, ragweed, 2BBG) | 40 | 6.5 | 18.8 | 32/7 |
| Ferredoxin (Clostridium, Bacterium, 2FDN) | 55 | 7.4 | 5.0 | 40/6 |
| Mean ± SD | 6.0 ± 1.2 | |||
Conformation alignments of antimicrobial and nonantimicrobial peptide structures were analyzed by pairwise comparison with Ah-AMP-1 (Aesculus, horse chestnut tree, 1BK8) by using combinatorial extension. Peptide formatting: peptide (source genus, common name, PDB ID code). Control peptides (Fig. 2 I-L) were equivalent to Ah-AMP-1 in disulfide content, sequence length, and mass. Representative data are shown. All rmsd values were determined for distances between α-carbon atoms over the length of the alignment. Identity is percentage sequence identity between comparators. Align/gap indicates the number of residues aligned, and number of gaps inserted. *, rmsd values significantly different from nonantimicrobial peptides (two-tailed t test; P < 0.01).
Fig. 3.

Conservation of the γ-core motif among disulfide-containing antimicrobial peptides. The conserved γ-core motif (red) is indicated with corresponding sequences (GXC or CXG-C motifs are denoted in red text). Example molecules, peptide (source), are organized into four structural groups relative to the γ-core. γ Group: protegrin-1 (1PG1), gomesin (1KFP), tachyplesin-1 (1MA2), RTD-1 (1HVZ), thanatin (8TFV), and hepcidin (1M4F). γ-α Group: sapecin (1L4V), insect defensin A (1ICA), heliomycin (1I2U), drosomycin (1MYN), MGD-1 (1FJN), and charybdotoxin (2CRD). β-γ Group: HNP-3 (1DFN), RK-1 (1EWS), BNBD-12 (1BNB), HBD-1 (1E4S), HBD-2 (1E4Q), and mBD-8 (1E4R). β-γ-α Group: Ah-AMP-1 (1BK8), Rs-AFP-1 (1AYJ), Ps-Def-1 (1JKZ), γ-1-H-thionin (1GPT), γ-1-P-thionin (1GPS), and brazzein (1BRZ). Protegrin, gomesin, tachyplesin, RTD-1, and thanatin γ-core sequences (γ Group) are depicted in levomeric orientation. Other formatting is as in Fig. 2.
Fig. 4.

Iterations of the 3D γ-core motif. Amino acid consensus patterns of the three γ-core sequence isoforms are shown. Coloration represents the most common residue (>50% frequency) at a given position, as adapted from the RASMOL (raster-pixel array molecule) schema: cysteine (C), yellow; glycine (G), orange; lysine or arginine, royal blue; serine or threonine, peach; leucine, isoleucine, alanine, or valine, dark green; aromatic, aqua; and variable positions (<50% consensus), gray.
Relative to the γ-core, disulfide-stabilized antimicrobial peptides of evolutionarily distant organisms exhibited a striking conservation in conformation, which was essentially isomeric, or at a minimum, highly homologous (Fig. 2 A-F). This 3D correspondence encompassed overall conformations or was localized to specific domains in comparative peptides. For example, the structures of Ah-AMP-1 (horse chestnut tree, Aesculus) and drosomycin (fruit fly, Drosophila) are essentially superimposable over their entire backbone trajectories (Fig. 2 C and D). Alternatively, protegrin-1 (domestic pig, Sus) and Ah-AMP-1 share conformational homology corresponding to their γ-core motifs (Fig. 2 A and B). As anticipated, magainin aligned to the α-helical motif in Ah-AMP-1 (Fig. 2 G and H), verifying the specificity of conformational alignments.
To test the significance of this structural homology, comparative alignments of disulfide-containing antimicrobial and nonantimicrobial peptides of equivalent mass and cysteine composition were performed. Quantitative rmsd outcomes demonstrated significant differences between these groups (Table 1). Moreover, nonantimicrobial peptides failed to achieve the 3D γ-core motif seen in antimicrobial peptides (Fig. 2 I-L). These data substantiate the concept that the multidimensional signature differentiates disulfide-stabilized antimicrobial and nonantimicrobial peptides.
Validation of the Multidimensional Signature Model. The multidimensional signature model predicted that peptides integrating an enantiomeric sequence pattern with the 3D γ-core would exert antimicrobial activity, even if such activity had not yet been recognized. Complementary approaches were used to challenge the model in this regard.
To test the hypothesis that the primary sequence patterns of the multidimensional signature are relevant to all classes of disulfide-containing antimicrobial peptides, Swiss-Prot forward and reverse databases (14) were queried with the enantiomeric sequence formulae. Representatives of all major disulfide-containing antimicrobial peptide classes were retrieved (Table 2). Searches also retrieved members of other peptide subclasses: (i) neurotoxins, particularly the charybdotoxin class of the family Buthidae (scorpion); (ii) protease inhibitor or related peptides (e.g., brazzein) from plants; (iii) ferredoxins; and (iv) metallothioneins. Prototypes with known 3D structures, but no known antimicrobial activity, were analyzed for the presence of the γ-core signature. Of these, the peptides brazzein and charybdotoxin were selected to test for antimicrobial activity on the basis of two criteria: (i) their quantitative rmsd values reflected greatest homology to the comparator γ-core motif; and (ii) they represented diverse nonmammalian (plant or scorpion) host sources and distinct structure classes not previously known to have antimicrobial activity. Thus, brazzein and charybdotoxin exemplified peptides that fulfilled the enantiomeric sequence and γ-core criteria required for the multidimensional signature (Fig. 5 G and H). These peptides were predicted to have direct antimicrobial activity. In contrast, prototype metallothioneins and ferredoxins did not contain γ-core motifs (Fig. 2 J and L, Table 1). Thus, metallothionein II was selected as an example comparator predicted to lack antimicrobial activity.
Table 2. Recognition of diverse classes of antimicrobial peptides by the enantiomeric sequence formulae.
| Sequence isoform, no. found
|
||||||
|---|---|---|---|---|---|---|
| Antimicrobial peptide class | Phylogeny | Dextro | Levo-1 | Levo-2 | Total | % of total |
| α-Defensin | Chordata | 24 | 42 | 6 | 72 | 15.3 |
| β-Defensin | Chordata | 52 | 65 | 31 | 148 | 31.4 |
| θ-Defensin | Chordata | 1 | 1 | 0 | 2 | 0.4 |
| Insect defensin/CS-αβ | Insectae | 21 | 23 | 12 | 56 | 11.9 |
| Plant defensin/CS-αβ | Plantae | 51 | 67 | 20 | 138 | 29.3 |
| Invertebrate defensin/CS-αβ | Mollusca | 3 | 4 | 4 | 11 | 2.3 |
| Protegrins/gomesins | Chordata/Arthropoda | 0 | 0 | 6 | 6 | 1.3 |
| Tachyplesins/Polyphemusins | Arthropoda | 6 | 5 | 2 | 13 | 2.8 |
| Thanatin | Arthropoda | 0 | 1 | 0 | 1 | 0.2 |
| Mytilins/Big Defensin | Mollusca | 3 | 3 | 2 | 8 | 1.7 |
| AFP-1 | Ascomycota | 1 | 0 | 0 | 1 | 0.2 |
| Lantibiotics/microcins | Proteobacteria | 3 | 3 | 9 | 15 | 3.2 |
| Total | 165 | 214 | 92 | 471 | ||
Forward or reverse Swiss-Prot databases (release 42.4; November 14, 2003; 138,347 entries) were probed with formulae containing the dextromeric or levomeric motifs of the antimicrobial peptide signature by using prosite (14). Data indicate the proportionate distribution of a nonredundant cohort of retrieval sets; in some cases, peptides were retrieved by more than one formula isoform. Note that search results include members of the lantibiotic superfamily of antimicrobial peptides, which lack conventional disulfide bridges but have thioether stabilization.
Fig. 5.

Structural validation of the multidimensional signature model. Structures: brazzein [1BRZ, Pentadiplandra,j'oblie berry (11); A, D, and G], charybdotoxin [2CRD, Leiurus, scorpion (27); B, E, and H)], tachyplesin I (1MA2, Tachypleus, horseshoe crab; C, F, and I). Respective γ-core motifs are highlighted in red (G-I; as in Fig. 3). Formatting is as in Fig. 2.
As predicted by the signature model, brazzein and charybdotoxin exerted direct antimicrobial activity against bacteria and Candida albicans (Fig. 6). Notably, these peptides exhibited pH-specific antimicrobial activities, which in some conditions exceeded the activity of HNP-1. These results demonstrate for the first time to our knowledge the direct antimicrobial activities of brazzein and charybdotoxin. In contrast, metallothionein II failed to exert antimicrobial activity against any organism tested under any condition, as predicted by the model.
Fig. 6.

Functional validation of the multidimensional signature model. Radial diffusion assays (10) were conducted by using defensin HNP-1 (HNP); brazzein (BRZ); charybdotoxin (CTX); or metallothionein II (MTL). Histograms express mean (±SD; minimum n = 2) zones of complete (blue) or incomplete (yellow) growth inhibition. Note differences in scale.
An alternative approach was also used to validate the multidimensional signature model. Tachyplesins are known cysteine-containing antimicrobial peptides from the horseshoe crab Tachypleus. Tachyplesins were retrieved from protein database searches employing the levomeric signature isoforms (Table 2). The model predicted that, because they have known antimicrobial activity and fulfill the enantiomeric sequence criteria, tachyplesins would possess a γ-core motif. The 3D structure of tachyplesin I became available subsequent to development of the model (15), and as predicted, exhibits a γ-core motif integral to the multidimensional signature (see Fig. 5I).
Discussion
Conventional analyses have yielded limited sequence conservation and have failed to uncover comprehensive structural motifs encompassing diverse classes of antimicrobial peptides. If present, consensus motifs relating broad classes of antimicrobial peptides would provide insights into their shared or distinct structure activity relationships and mechanisms of action, reveal their evolutionary origins, and allow prediction of antimicrobial activity in molecules recognized to have other functions.
Stereospecific sequence and 3D conformation analyses were integrated and reduced to reveal a multidimensional signature common to all classes of disulfide-stabilized antimicrobial peptides. This signature represents a unifying structural paradigm in antimicrobial peptides from organisms separated by profound evolutionary distances. The γ-core motif integrates hallmark physicochemical properties, including amino acid sequence, composition, and 3D structure, that likely facilitate antimicrobial functions (1, 16). Remarkably, the γ-core can be derived from dextromeric or levomeric sequence patterns in distinct antimicrobial peptides (Fig. 4). Thus, this motif has been preserved despite amino acid site- or orientation-specific variations in primary sequence.
Conservation of the γ-core motif across the phylogenetic spectrum suggests it is an antimicrobial peptide archetype. Yet the γ-core is not necessarily an exclusive structural determinant of antimicrobial activity. In some cases, the γ-core alone is sufficient for antimicrobial activity (e.g., protegrins, tachyplesins, RTD-1). However, this motif also appears to serve as a scaffold, to which complementary antimicrobial determinants (e.g., α-helices or β-sheets) are attached as modules coordinated in various configurations relative to the γ-core. Examples of this theme are abundant in diverse antimicrobial peptides ranging in source from microbes to humans (Fig. 3). It is likely that specific configurations of such structural mosaics optimize the functions of antimicrobial peptides against relevant pathogens in respective environmental, physiologic, or anatomic contexts.
The present findings emphasize the potential value of the multidimensional signature model in identifying previously unrecognized antimicrobial peptides, as well as peptides that share common structure-activity relationships or mechanisms of action. For example, the sweetener brazzein and the scorpion neurotoxin charybdotoxin were discovered to have direct antimicrobial activity against bacteria and fungi. Certain antimicrobial peptides and neurotoxins share similar mechanisms of action (e.g., cytoplasmic membrane perturbation), paralleling their common γ-core motif. Thus, the presence of the γ-core in antimicrobial peptides, neurotoxins, and other host defense peptides likely reflects an ancient evolutionary relationship.
The current findings also suggest that iterations of the γ-core motif are common to broader classes of disulfide-stabilized host defense effector molecules, including and beyond classical antimicrobial peptides. For example, several peptides originally identified as cytokines are now known to have direct microbicidal activity, including human platelet factor 4 and platelet basic peptide (10, 17), monokine induced by IFN-γ [MIG (18)], IFN-γ-inducible protein of 10 kDa [IP-10 (18)], IFN-inducible T cell α chemoattractant [ITAC (18)], RANTES (10, 17), and other molecules (1, 19). Importantly, such microbicidal cytokines (kinocidins) also contain iterations of the antimicrobial γ-core signature defined herein (unpublished data). The enantiomeric sequence formulae also retrieved plant protease inhibitor and related proteins. The botanical literature indicates many of these peptides are plant defense molecules (20, 21). Moreover, the plant proteinase inhibitor superfamily includes thionins (22) that contain a γ-core motif as defined herein (Table 1). These observations substantiate congruence between the γ-core structural motif and functional correlates in related host defense peptides (17, 23). We anticipate the multidimensional antimicrobial signature will be found in yet other peptides, and the presence of this signature will be associated with otherwise unforeseen antimicrobial activity.
Identification of multidimensional signatures in antimicrobial peptides from phylogenetically distant organisms offers insights into the evolution of host defense effector molecules. The functional role(s) of conserved amino acids integral to the γ-core motif invite further investigation, as do the potential coevolutionary implications of this highly conserved structural motif. The current results support the hypothesis that vertical and horizontal acquisition of genes, along with their recombination, yield mosaic iterations upon the γ-core motif (24, 25). Selective pressures favoring this remarkable degree of structural conservation may include genetic selection against structural variants and convergent evolution of independent ancestral templates. While future studies will resolve their precise phylogenetic lineages, multidimensional signatures in antimicrobial peptides likely reflect fundamental hostpathogen interactions and their coevolution.
Supplementary Material
Acknowledgments
Several colleagues were invaluable in development of this paper, including Drs. Eric Brass, Terry Smith, Arnold Bayer, and John E. Edwards, Jr. We thank Kimberly Gank for excellent technical contributions. Drs. Paul Sullam and Willam Welch provided helpful comments. The contributions of David R. Weaver are acknowledged with gratitude. The efforts of the Robert M. Delzell Foundation are sincerely appreciated. This work was supported by National Institutes of Health Grants AI-39108 and AI-48031.
Abbreviations: CS, cysteine-stabilized; rmsd, rms deviation.
References
- 1.Yeaman, M. R. & Yount, N. Y. (2003) Pharmacol. Rev. 55, 27-55. [DOI] [PubMed] [Google Scholar]
- 2.Hughes, A. L. (1999) Cell. Mol. Life Sci. 56, 94-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blaser, M. J. (2002) N. Engl. J. Med. 346, 2083-2085. [DOI] [PubMed] [Google Scholar]
- 4.White, S. H., Wimley, W. C. & Selsted, M. E. (1995) Curr. Opin. Struct. Biol. 5, 521-527. [DOI] [PubMed] [Google Scholar]
- 5.Guder, A., Wiedemann, I. & Sahl, H. G. (2000) Biopolymers 55, 62-73. [DOI] [PubMed] [Google Scholar]
- 6.Mandard, N., Bulet, P., Caille, A., Daffre, S. & Vovelle, F. (2002) Eur. J. Biochem. 269, 1190-1198. [DOI] [PubMed] [Google Scholar]
- 7.Higgins, D. G. & Sharp, P. M. (1988) Gene 73, 237-244. [DOI] [PubMed] [Google Scholar]
- 8.Higgins, D. G. & Sharp, P. M. (1989) Comput. Appl. Biosci. 5, 151-153. [DOI] [PubMed] [Google Scholar]
- 9.Shindyalov, I. N. & Bourne, P. E. (1998) Protein Eng. 11, 739-747. [DOI] [PubMed] [Google Scholar]
- 10.Tang, Y. Q., Yeaman, M. R. & Selsted, M. E. (2002) Infect. Immun. 70, 6524-6533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caldwell, J. E., Abildgaard, F., Dzakula, Z., Ming, D., Hellekant, G. & Markley, J. L. (1998) Nat. Struct. Biol. 5, 427-431. [DOI] [PubMed] [Google Scholar]
- 12.Nei, M., Xu, P. & Glazko, G. (2001) Proc. Natl. Acad. Sci. USA 98, 2497-2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yount, N. Y., Yuan, J., Tarver, A., Castro, T., Diamond, G., Tran, P. A., Levy, J. N., McCullough, C., Cullor, J. S., Bevins, C. L., et al. (1999) J. Biol. Chem. 274, 26249-26258. [DOI] [PubMed] [Google Scholar]
- 14.Gattiker, A., Gasteiger, E. & Bairoch, A. (2002) Appl. Bioinformatics 1, 107-108. [PubMed] [Google Scholar]
- 15.Laederach, A., Andreotti, A. H. & Fulton, D. B. (2002) Biochem. 41, 12359-12368. [DOI] [PubMed] [Google Scholar]
- 16.Hill, C. P., Yee, J., Selsted, M. E. & Eisenberg, D. (1991) Science 251, 1481-1485. [DOI] [PubMed] [Google Scholar]
- 17.Yeaman, M. R. (1997) Clin. Infect. Dis. 25, 951-968. [DOI] [PubMed] [Google Scholar]
- 18.Cole, A. M., Ganz, T., Liese, A. M., Burdick, M. D., Liu, L. & Strieter, R. M. (2001) J. Immunol. 167, 623-627. [DOI] [PubMed] [Google Scholar]
- 19.Wu, Z., Hoover, D. M., Yang, D., Boulegue, C., Santamaria, F., Oppenheim, J. J., Lubkowski, J. & Lu, W. (2003) Proc. Natl. Acad. Sci. USA 100, 8880-8885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sallenave, J. M. (2002) Biochem. Soc. Trans. 30, 111-115. [DOI] [PubMed] [Google Scholar]
- 21.Wijaya, R., Neumann, G. M., Condron, R., Hughes, A. B. & Polya, G. M. (2000) Plant Sci. 159, 243-255. [DOI] [PubMed] [Google Scholar]
- 22.Melo, F. R., Rigden, D. J., Franco, O. L., Mello, L. V., Ary, M. B., Grossi de Sa, M. F. & Bloch, C. Jr. (2002) Proteins 48, 311-319. [DOI] [PubMed] [Google Scholar]
- 23.Ganz, T. (2002) Science 298, 977-979. [DOI] [PubMed] [Google Scholar]
- 24.Bevins, C. L., Jones, D. E., Dutra, A., Schaffzin, J. & Muenke, M. (1996) Genomics 31, 95-106. [DOI] [PubMed] [Google Scholar]
- 25.Gudmundsson, G. H., Magnusson, K. P., Chowdhary, B. P., Johansson, M., Andersson, L. & Boman, H. G. (1995) Proc. Natl. Acad. Sci. USA 92, 7085-7089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martz, E. (2002) Trends Biochem. Sci. 27, 107-109. [DOI] [PubMed] [Google Scholar]
- 27.Bontems, F., Gilquin, B., Roumestand, C., Menez, A. & Toma, F. (1992) Biochemistry 31, 7756-7764. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.