

Abstract
When many protein sequences are available for estimating the time of divergence between two species, it is customary to estimate the time for each protein separately and then use the average for all proteins as the final estimate. However, it can be shown that this estimate generally has an upward bias, and that an unbiased estimate is obtained by using distances based on concatenated sequences. We have shown that two concatenation-based distances, i.e., average gamma distance weighted with sequence length (d2) and multiprotein gamma distance (d3), generally give more satisfactory results than other concatenation-based distances. Using these two distance measures for 104 protein sequences, we estimated the time of divergence between mice and rats to be approximately 33 million years ago. Similarly, the time of divergence between humans and rodents was estimated to be approximately 96 million years ago. We also investigated the dependency of time estimates on statistical methods and various assumptions made by using sequence data from eubacteria, protists, plants, fungi, and animals. Our best estimates of the times of divergence between eubacteria and eukaryotes, between protists and other eukaryotes, and between plants, fungi, and animals were 3, 1.7, and 1.3 billion years ago, respectively. However, estimates of ancient divergence times are subject to a substantial amount of error caused by uncertainty of the molecular clock, horizontal gene transfer, errors in sequence alignments, etc.
The molecular clock hypothesis asserts that the number of amino acid substitutions in a protein is roughly proportional to the time since divergence of the two species compared (1, 2). Strictly speaking, no gene or protein would evolve at a constant rate for a long evolutionary time, because gene function is likely to change over time, and mutational and DNA repair mechanisms appear to vary among different groups of organisms (3). For this reason, the molecular clock has been controversial for several decades (3–6). However, even if the substitution rate is not strictly constant, it is still possible to obtain rough estimates of divergence times, and these estimates are very useful when there is no reliable fossil record (6, 7). Furthermore, if a gene evolves excessively fast or slow in a few evolutionary lineages, one can eliminate these lineages and then estimate divergence times for the rest of the species (8). The accuracy of time estimates is expected to increase as the number of genes or proteins used increases, and in recent years, many authors have used multiple genes or proteins for this purpose (9–11).
There are several statistical methods for estimating divergence times, but the theoretical basis of the methods is not well understood when multiple genes are used. For this reason, different authors obtained widely different estimates for the same set of species by using different methods (10, 12–16). We have therefore examined the reliability of different methods for estimating divergence times and have developed new methods that are likely to give more reasonable estimates than previous ones. The purpose of this paper is to discuss statistical problems related to this subject and to present new methods. These new methods will then be used to estimate the divergence times between mice and rats and between humans and rodents, which have been controversial for the last few decades. We also consider the divergence times of animals, plants, fungi, protists, and bacteria to show the dependency of time estimates on the assumptions made and the statistical methods used. In this paper, we consider only protein sequences, because they usually give more satisfactory results than DNA sequences when long-term evolution is considered. We also consider only distance methods of time estimation, because most recent time estimates have been obtained by these methods.
Theoretical Basis of Estimation of Divergence Times
Individual Protein (IP) Approach.
In the past, most investigators have used a method that may be called the IP approach (11, 13, 14, 17, 18). In this approach, the estimate of divergence time is computed for each protein, and the average of the estimates over all proteins is used as the final estimate. Consider Fig. 1A, in which a phylogenetic tree for five species is given. Here, a–f stand for the least-squares estimates of branch lengths (numbers of amino acid substitutions) obtained from a pairwise distance matrix for a protein. Species 5 is used as an outgroup to determine the root of the tree for the remaining sequences, and therefore the branch length estimate for this branch is not given. Here we assume that the topology of the tree for the five species has been established from other information. To estimate divergence times between species, it is convenient to construct a linearized tree (8), in which the branch lengths are reestimated under the assumption of a constant-rate evolution (Fig. 1B). When this linearized tree is constructed, a timescale for the tree is produced to estimate divergence times (t1and t2). This timescale can be obtained by computing the rate of amino acid substitution per year (r) by using the known divergence time and the corresponding branch-length estimate for a pair of species or species clusters.
Figure 1.

Phylogeny of the five species used. (A) NJ tree constructed by using distance d2 for 104 protein sequences. (B) Linearized tree of the above NJ tree. R, root of the four species under consideration.
For example, if T is the calibration point in Fig. 1B, the rate of amino acid substitution can be estimated by r̂ = b̂3/T, where b̂3 is the branch-length estimate for species 4 after divergence from species 1, 2, and 3 in the linearized tree (Fig. 1B). When this rate is obtained, we can estimate t1 by
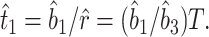 |
1 |
Here, the estimates b̂1, b̂2, and b̂3 can be obtained from pairwise distances by Takezaki et al.'s (8) method. Similarly, the estimate of t2 is given by t̂2 = b̂2/r̂ = (b̂2/b̂3)T. The variances of t̂1 or t̂2 can be obtained by Takezaki et al.'s method or by the bootstrap method (19). For certain data sets (18, 20), it is possible to use several calibration points (paleontological dates for different pairs of species). In this case, r may be estimated by fitting a regression line to the relationship between branch length estimates and paleontological dates.
Before the construction of a linearized tree, it is customary to conduct a statistical test of the molecular clock and eliminate species that evolved excessively fast or slow, although this is not always necessary (see below). A number of authors (7, 11) have used the relative rate tests (21–23) for this purpose. In these tests, three sequences (or three sequence clusters) are used, and the equality of evolutionary rate for two evolutionary lineages is tested by using a third sequence as an outgroup. For example, the equality of the expectations of branch lengths a (for mice) and b (for rats) in Fig. 1A can be tested by using the chicken sequence as the outgroup. Theoretically, however, this test is not appropriate for estimating the divergence time t1, because it does not test the equal rates for sequences 1, 2, and 3. For this purpose, we should test the null hypotheses E(a) = E(b) and E(a/2 + b/2 + c + e) = E(f), where E is the expectation operator. These null hypotheses can be tested by Takezaki et al.'s (8) U statistic, which approximately follows the χ2 distribution and tests the equality of the branch lengths from the root (R) to tips (1, 2, 3, and 4) for all species. In this test, at least four sequences are necessary.
When there are data from many different proteins, the divergence time t1 is often estimated by the simple average of t̂1s for all the proteins used, as mentioned above. That is,
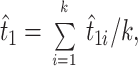 |
2 |
where t̂1i is the estimate of t1 obtained by the ith protein, and k is the total number of proteins used. Theoretically, however, t̂1 obtained in this way is not an unbiased estimator of t̂1, even if the branch length estimates (numbers of amino acid substitutions) for each protein are unbiased. Let b̂1i and b̂3i be unbiased estimates of b1 and b3 for the ith protein, so that t̂1i = (b̂1i/b̂3i)T. In this case, an unbiased estimator of t1 is given by [(∑ib̂1i)/(∑ib̂3i)]T rather than by t̂1 = ∑i(b̂1i/b̂3i)T/k. If we assume that b̂1 and b̂3 are random variables (subscript i dropped), the expectation of b̂1/b̂3 is approximately given by
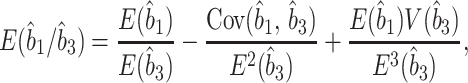 |
3 |
where V(b̂3) is the variance of b̂3, and Cov(b̂1, b̂3) is the covariance of b̂1 and b̂3 (24). Numerical evaluation of the second and third terms in Eq. 3 suggests that t̂1 in Eq. 2 often gives overestimates of divergence times. This is particularly so when the calibration point is smaller than the divergence time to be estimated. Suppose we know t1 instead of T in Fig. 1B and want to estimate T by T̂ = ∑i(b̂3i/b̂1i)t1/k. In this case, T̂i (b̂3i/b̂1i)t1 may become very large if b̂1i happens to be close to 0 by chance (the upper limit being infinity). If b̂1i happens to be large relative to b̂3i, T̂i becomes small but never smaller than t1. Therefore, T̂ tends to be an overestimate when b̂1i varies extensively. To avoid this overestimation, we should use concatenation-based distances (CDs) mentioned below.
Distance Measures to Be Used.
When all protein sequences used are closely related, the Poisson correction (PC) distance appears to give sufficiently accurate estimates of divergence times (25). This distance is given by d = −ln(1 − p), where p is the proportion of sites at which the amino acids of the two sequences compared are different. However, the PC distance is obtained under the assumption that the rate of amino acid substitution per year (r) is the same for all amino acid sites. In practice, this assumption rarely holds, and empirical data have suggested that the rate varies from site to site approximately following the gamma distribution (26). In this case, the evolutionary distance between two sequences can be measured by the following PC gamma distance
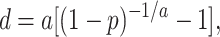 |
4 |
where a is the shape parameter of the gamma distribution (gamma parameter) and decreases as the variation of r among sites increases (27, 28).
If we assume that a is a constant, the variance of d is given by V1(d) = p[(1 − p)−(1+2/a)]/n (28), but if we take into account the sampling variance [V(â)] of the estimate (â) of a, the total variance is approximately given by
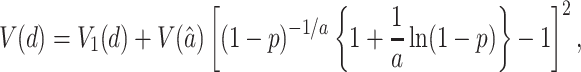 |
5 |
where V(â) = [2a(a + 1)(p + a)2]/(np2), and n is the number of amino acids used. This equation was obtained by the delta method by using Anscombe's (29) formula for V(â).
One might question the applicability of Eq. 4 to actual data, because it does not take into account higher rates of substitution between similar amino acids than between dissimilar amino acids (30). Grishin (31) developed a complex distance measure by taking into account variation in substitution rate among different amino acid sites as well as among different pairs of amino acids. However, this distance can also be approximated very well by a PC gamma distance with a = 0.65 (32). Therefore, for most practical purposes, we may use PC gamma distance.
CD Approach.
Previously, we mentioned that to obtain an unbiased estimate of t1, pairwise CDs for all proteins should be computed and b1 and b3 be estimated from these distances. There are several ways of concatenating pairwise distances (ds) for different proteins to obtain unbiased estimates of b1 and b3.
(i) Simple average distance (d1).
In this method, a PC or PC gamma distance is computed for each protein, and the simple average of the distances for all proteins is used.
(ii) Average distance weighted by sequence length (d2).
One disadvantage of distance d1 is that the average of ds over loci is computed without regard to the number of amino acids (sequence length). Because a protein distance based on many amino acids would be more reliable than d1, it would be better to use the average distance weighted with sequence length.
(iii) Multiprotein gamma distance (d3).
As mentioned earlier, PC gamma distance is very flexible and can be applied to most amino acid sequence data (32). However, the gamma parameter a is expected to vary from protein to protein, and it has been shown that the rate of amino acid substitution per protein roughly follows the gamma distribution (25). This suggests that if we consider many protein sequences simultaneously, the rate of amino acid substitution per site approximately follows the gamma distribution when the entire set of amino acids for all proteins is considered. We can therefore estimate the gamma parameter a for the entire set of amino acids and compute the gamma distance using Eq. 4. We call this distance the multiprotein gamma distance in this paper. The standard errors of the estimates (d̂1, d̂2, and d̂3) of the above three distances may be computed by the bootstrap or the jackknife method by using individual proteins as units of resampling. For distance estimate d̂3, the variance can also be computed by Eq. 5, but the jackknife variance appears to be more appropriate, because the unit of evolution is a gene or protein rather than an amino acid.
A standard way of concatenating different statistical estimators is to use the inverse of variance as the weight. Lynch (15) suggested that the average of the variances of all pairwise distances for each protein be used as the weight. Although the CD obtained in this way does not give unbiased estimates of b1 and b2, it may be useful for the estimation of divergence times. The CD obtained by this method will be denoted by dL. We used Eqs. 1 and 2 in his paper to compute the (gamma) distance and its variance (with a = 0.5) for each protein, following his suggestion.
Divergence Times Between Mice and Rats and Between Humans and Rodents
A large number of authors have estimated the times of divergence between different groups of mammals by using molecular data (11, 33, 34), but the results obtained are conflicting and controversial (35–37). Of special interest in this regard are the divergence times between mice and rats and between humans and rodents. Molecular estimates of these divergence times have been controversial, because the fossil record is poor (38, 39), and rodent genes appear to have evolved faster than primate genes (22). In this paper, we therefore focus our attention primarily on these divergence times. We use five vertebrate species, i.e., mice, rats, humans, chickens, and Xenopus laevis, of which the evolutionary relationships are well established and for which many shared protein sequences are available. Xenopus is used as an outgroup species (Fig. 1).
Protein Sequence Data.
We obtained protein sequence data from the December 1999 edition of the hovergen database (40) and used only sequences that are available for all five species. In this database, orthologous and paralogous genes are not always distinguished, and we attempted to exclude paralogous genes as much as possible by eliminating multigene families such as major histocompatibility complex and immunoglobulin genes. We also constructed a neighbor-joining (NJ) tree using p distance (41) for each gene and eliminated all genes that produced a topology different from the known tree for the five species.
Using the above procedure, we obtained 104 putative orthologous proteins (see Table 3, which is published as supplemental data at www.pnas.org). We used both PC and PC gamma distances in this paper. The gamma parameter a was estimated by Gu and Zhang's (42) method (the computer program available from the web site http://mep.bio.psu.edu) for each protein separately. Fig. 2 shows the estimates (â) of a for the 104 proteins in relation to the extents of sequence divergence (average root-to-tip branch length; bR). The â value varies extensively from protein to protein, and it is positively correlated with bR (43) or the proportion (pv) of variable sites among the five sequences. Because PC gamma distance is disproportionately large compared with PC distance when â is small, the relationships in Fig. 2 suggest that the extent of sequence divergence as measured by PC gamma is less heterogeneous among proteins than that obtained by PC. As mentioned earlier, the multiprotein gamma distance is computed by using the â value obtained from the entire set of amino acids. This value was 0.57, whereas the average (ā) of âs for all proteins was 0.76.
Figure 2.

(A) Relationship between estimated gamma parameter (â) and the average root-to-tip distance (bR) for 104 nuclear proteins from the five species used. (B) Relationships between â and the proportion of variable sites (pv) among the five sequences.
Time Estimation.
Theoretically, it is better to eliminate sequence data that do not pass the molecular clock test. In practice, however, proteins that violate the molecular clock hypothesis do not necessarily give unreasonable estimates of divergence times (11). We therefore examined the relationships between U statistic values and time estimates for individual proteins (Fig. 3). In the case of PC distance, 25 proteins did not pass the molecular clock test and showed a U value of 11.3 (1% significance level of χ2 with 3 degrees of freedoms) or higher. However, the estimates of the mouse-rat and the human-rodent divergence times were nearly the same whether these 25 deviant proteins were included or not. In the case of PC gamma distance, only 7 proteins did not pass the molecular clock test, and the mean estimates of divergence times were again virtually unaffected by inclusion or exclusion of these proteins. For this reason, we used the average time estimates for all proteins as the final estimates disregarding the U values in the independent protein (IP) approach. However, note that the extent of variation in t̂1 and t̂2 is so enormous that the average estimates based on a few proteins are quite unreliable. In this paper, we used T = 310 million year (MY) (divergence time between birds and mammals) as the calibration point (11, 38).
Figure 3.

Relationships between estimated divergence times and U statistic values for each of 104 proteins. The molecular clock hypothesis was rejected for the proteins indicated with the × symbol.
In the CD approach, CDs with a large number of amino acids (48,092 in the present case) are used, so that even small differences in evolutionary rate among species become statistically significant. For example, the tree in Fig. 1A shows the branch length estimates obtained when distance d2 was used. The average branch length (0.053) for the mouse and rat lineages after their separation from the human lineage is about 1.2 times longer than that (0.043) of the human lineage, and the difference is highly significant (at the 0.01% level). Similarly, the branch length (0.17) for the rodent lineage after separation from the chicken lineage is 1.3 times greater than that (0.12) of the chicken lineage, and the difference is again significant at the 0.01% level.
However, this extent of variation in evolutionary rate among lineages does not seem to affect time estimates seriously (32). In Fig. 1A, it is unclear whether the evolutionary rate was accelerated in the rodent lineage compared with the chicken lineage or was decelerated in the chicken lineage. In either case, however, it is possible to estimate the divergence times t1 and t2 by considering the times of separation of chicken and humans from the rodent lineage. For example, t1 and t2 can be estimated by [(a + b)/(a + b + 2c + 2e)]T and [(a + b + 2c)/(a + b + 2c + 2e)]T, respectively, where T = 310 MY. The estimates of t1 and t2 obtained in this way are 31 MY and 97 MY, respectively. These estimates are close to those obtained by the linearized tree method (Table 1). For this reason, we decided to use all 104 protein data in all the methods of the CD approach.
Table 1.
Estimates (± standard errors) of divergence times (MY) between mice and rats and between humans and rodents
| Method | Mouse-rat
(t1)
|
Human-rodent
(t2)
|
||
|---|---|---|---|---|
| PC | PC Gamma | PC | PC Gamma | |
| IP approach | ||||
| 44.0 ± 3.4 | 38.5 ± 3.2 | 113.4 ± 5.0 | 102.9 ± 5.0 | |
| CD approach | ||||
| d1 (ā = 0.76) | 40.7 ± 3.0 | 34.4 ± 2.8 | 112.3 ± 5.4 | 99.9 ± 5.3 |
| d2 (ā = 0.76) | 39.1 ± 2.3 | 33.0 ± 2.0 | 110.0 ± 4.4 | 97.6 ± 4.4 |
| d3 (a = 0.57) | 32.9 ± 2.3 | 95.5 ± 4.2 | ||
| d3 (a = 0.28) | (25.2 ± 2.0) | (82.0 ± 4.0) | ||
| dL (a = 0.5) | 32.0 ± 5.0 | 90.0 ± 10.0 | ||
d1, unweighted average distance; d2, average distance weighted with sequence length; d3, multiprotein gamma distance; dL, Lynch's distance. The standard errors for the CD approach were computed by the jackknife method. The standard errors of t̂1 and t̂2 obtained by using Eq. 5 for d3 with a = 0.57 were 2.0 and 4.0, respectively.
Estimates of divergence times between mice and rats and between humans and rodents (t1 and t2 in Fig. 1B) obtained by all methods are presented in Table 1. When the IP approach is used, the estimates of t1 and t2 obtained with PC distance are 44 and 113 MY, respectively. When PC gamma distances are used, the estimates of t1 and t2 are both considerably smaller than those obtained by using PC distances.
In the case of the CD approach, PC distances d1 and d2 with a = ∞ give similar estimates of t1 (39∼41 MY) and t2 (110∼112 MY). When PC gamma is used, the time estimates are somewhat smaller than those obtained by the IP approach for both t1 and t2. In general, distance d2 with ā = 0.76 and d3 with a = 0.57 give very close estimates. Distance dL gives t̂1 = 32 MY, which is similar to t̂1 obtained by d3, but t̂2 obtained by dL is considerably smaller than the values obtained by the other methods. Note also that the standard errors of the estimates obtained by dL are greater than those obtained by other methods.
Previously we mentioned that the IP approach tends to give overestimates of divergence times, particularly when the times are estimated from recent calibration points. Assuming that t1 is known to be 33 MY but T is unknown, we can estimate T for the present data set. In this case, we obtain T̂ = ∞, because there are four proteins for which b̂1i is 0. If we eliminate these four proteins, the average estimate is still 450 MY. By contrast, if we use d3 with a = 0.57, we obtain T̂ = 310 MY, as expected. If we use t2 (=96 MY) as the calibration point, the IP approach gives T̂ = 353 MY, but the CD with d3 gives T̂ = 310 MY.
Estimates of Gamma Parameter a and Divergence Times.
In the above computation of divergence times for humans and rodents, we estimated gamma parameter a from the entire set of species. In practice, the estimate of a tends to be smaller when closely related species are used than when distantly related species are used (43). This occurs because multiple substitutions at the same amino acid sites can be detected more easily in the former group of species than in the latter. Because the estimate obtained from closely related species should be closer to the true a value in the absence of sampling bias (43), one might argue that this estimate should be used for computing pairwise distances for all species. We therefore estimated the a value for d3 using the mammalian sequences only and obtained â = 0.28. When this estimate was used, we obtained t̂1 = 25 MY and t̂2 = 82 MY, respectively (Table 1).
However, our computer simulation (G.G. and M.N., unpublished data) has shown that when the a value estimated from closely related species is used for computing pairwise distances, divergence times smaller than the calibration point tend to be underestimated, whereas divergence times greater than the calibration point tend to be overestimated. This happens because for distantly related species the amino acid sites that evolve very fast have little effect on overall sequence divergence and further divergence is primarily determined by slowly evolving sites, which show a rather large a value. Therefore, if we use a small a value obtained from closely related species, it will give unduly large pairwise distances for distantly related species and consequently give overestimates of divergence times for them but may give underestimates for closely related species. Therefore, time estimates of 82 and 25 MY for the human-rodent and the mouse-rat divergence time appear to be underestimates. In practice, this problem is rather complex, and detailed aspects will be discussed elsewhere.
Estimation of Ancient Divergence Times
Estimation of early divergence times such as those between animals, fungi, and plants is much more difficult than that of divergence times of mammalian species, because the timescale for a linearized tree for these species has to be produced from the fossil record for vertebrates (16, 18, 33), and there is no assurance of constant-rate evolution for a long evolutionary time (12, 44). Furthermore, different statistical methods often give different time estimates even if the same calibration time is used. Here we would like to examine only statistical problems considering protein sequence data from eubacteria (mostly Escherichia coli), protists (mostly Plasmodium), plants (Arabidopsis), fungi (yeast), and five species of animals (nematode, Drosophila, chicken, rat, and human). Although a large number of genes have been sequenced in some of these organisms, we could find only 11 orthologous genes that are shared by the above nine species and show relatively few alignment gaps (see supplemental Table 4 at www.pnas.org). All of these proteins were considered to be of eubacterial rather than archebacterial origin (45).
We tested the molecular clock hypothesis for each protein using PC gamma distance, but none of the proteins except one violated the clock hypothesis. This hypothesis was not rejected even when the multiprotein gamma distance for all proteins (3,310 amino acids) was used. The a value obtained for the latter set of proteins was 1.24, and the NJ and the linearized trees constructed from the multiprotein gamma distances are presented in Fig. 4. The timescale for this tree was obtained by using the calibration point of 310 MY between chicken and mammals. The time estimates obtained by this and other methods are presented in Table 2. The divergence time between the E. coli genes and their homologues from the eukaryotes used here was obtained under the assumption of a molecular clock, because there was no outgroup for this species group.
Figure 4.

(A) NJ tree constructed by using distance d3 with a = 1.24 for 11 protein sequences. The numbers given for this tree stand for the bootstrap values (500 replications). (B) Linearized tree of the above NJ tree. The numbers given for this tree represent the estimates of divergent times.
Table 2.
Estimates (± standard errors) of divergence times (MY) of various organisms from the human lineage
| Method | Rats | Chicken | Drosophila | Nematodes | Fungi | Plants | Protists | Eubacteria |
|---|---|---|---|---|---|---|---|---|
| IP approach | ||||||||
| PC gamma | 124 ± 28 | 310 | 962 ± 132 | 1,225 ± 211 | 1,768 ± 311 | 1,715 ± 257 | 2,282 ± 557 | 3,557 ± 649 |
| CD approach | ||||||||
| d1 (ā = 1.53) | 113 ± 38 | 310 | 745 ± 196 | 930 ± 274 | 1,229 ± 402 | 1,343 ± 394 | 1,578 ± 485 | 2,600 ± 568 |
| d2 (ā = 1.53) | 128 ± 38 | 310 | 798 ± 121 | 951 ± 168 | 1,372 ± 275 | 1,372 ± 272 | 1,707 ± 379 | 3,000 ± 476 |
| d3 (a = 1.24) | 120 ± 36 | 310 | 833 ± 114 | 970 ± 160 | 1,392 ± 256 | 1,392 ± 252 | 1,717 ± 349 | 3,036 ± 470 |
| d3 (a = 0.54) | 115 ± 35 | 310 | 931 ± 153 | 1,115 ± 229 | 1,740 ± 422 | 1,740 ± 424 | 2,276 ± 667 | 5,010 ± 1,060 |
| dL (a = 0.50) | 62 ± 10 | 310 | 798 ± 274 | 881 ± 354 | 1,779 ± 651 | 1,557 ± 970 | 1,834 ± 1,034 | 6,468 ± 5045 |
d1, unweighted average PC gamma; d2, average PC gamma weighted with sequence length; d3, multiprotein gamma; dL, Lynch's distance. The divergence time (310 MY) between mammals and birds was used as the calibration point. Evolutionary relationships among animals, fungi, and plants varied with distance measure.
CDs d2 with ā = 1.53 and d3 with a = 1.24 gave essentially the same estimates for all the divergence times considered here (Table 2). Distance d1 gave slightly smaller estimates than those obtained by d2 and d3. Table 2 includes the estimates obtained by d3 with a = 0.54, which was obtained by using only animal sequences (five species). This distance again gives a smaller estimate (115 MY) for the human-rat divergence, which is below the calibration point (310 MY). However, it gives rather high estimates for divergence times earlier than the calibration point. In particular, the estimate of the E. coli-eukaryote divergence is unrealistic, because it is older than the age of Earth (ca. 4,500 MY).
Table 2 also includes the time estimates obtained by the IP approach. These estimates are similar to those obtained by Wang et al. (16) with a similar method. However, they are considerably higher than the estimates obtained by d2 and d3 with a = 1.24. Because the IP approach is expected to give overestimates, the values obtained by d2 and d3 with a = 1.24 appear to be more reliable than those obtained by the IP approach. Unlike the case of mammalian data, Lynch's distance (dL) gives the smallest time estimates (62 MY) for the human-rat divergence but give large estimates for ancient divergence times. However, the standard errors of these estimates are very large.
Our estimate of divergence time (about 3,000 MY) between eubacteria and eukaryotes based on d2 and d3 with a = 1.24 is younger than the age (ca. 3,500 MY) of some old microfossils reported (46). If these microfossils are genuine and if the molecular clock hypothesis holds up to ancient bacterial evolution, the difference can be explained by (i) the large standard error of our estimate, (ii) horizontal gene transfer that might have occurred between the ancestors of current eubacteria and eukaryotes, and/or (iii) the possibility that the ancient microfossils reported do not represent the ancestors of current eubacteria and/or eukaryotes.
Discussion
We have examined various methods of estimating divergence times and have shown that the IP approach is expected to give biased estimates, which are usually greater than those obtained by the CD approach. In the latter approach, distances d2 and d3 are expected to give more reliable estimates than distance d1, although the difference is usually small unless ancient divergence times are considered. Distances d2 and d3 usually give similar estimates, but it is easier to compute d3 than d2.
We have seen that molecular estimates of divergence times depend on a number of assumptions, and they are generally very crude. Nevertheless, if we use a large number of protein sequences, the estimates appear to be reasonably good (11, 17, 18). Our estimate (96 MY) of the time of human-rodent divergence from d3 is somewhat smaller than a recent estimate (112 MY) obtained by Kumar and Hedges (11). This difference occurred primarily because we used the CD approach with multiprotein gamma distance, whereas Kumar and Hedges used the IP approach with PC distance. In the case of the mouse-rat divergence, the difference between our estimate (33 MY) and Kumar and Hedges' (41 MY) is substantial.
In the present paper, we did not consider the uncertainty of the calibration point used. In general, the degree of this uncertainty is quite high (12), so that we should always keep in mind that molecular time estimates are very crude, and that the standard errors attached to them merely represent the statistical error associated with molecular data under the substitution model used. Therefore, small standard errors do not necessarily mean a high accuracy of estimates. If we consider uncertainty of the calibration point, the reliability of time estimates is reduced considerably. For example, Lee (12) states that the divergence between birds and mammals probably occurred 288–310 MY ago. In our study, we used T = 310 MY, because the true divergence time is likely to be at the higher end of paleontological estimates. However, if we use 288 MY, all the time estimates in Tables 1 and 2 will be lowered by 7.6%.
Molecular time estimates are usually greater than paleontological estimates. Molecular evolutionists tend to argue that this is mainly caused by incomplete fossil records and that molecular estimates are more accurate (4, 39). By contrast, paleontologists and other critics (12, 36, 37) often ascribe this difference to the inaccuracy of the molecular approach of dating. It is not easy to settle this controversy at this stage. Fortunately, molecular data are now rapidly increasing thanks to the recent genome-sequencing projects for many different organisms, and when more data become available, we will be able to make more reliable phylogenetic trees and more reliable estimates of divergence times. If we can construct consistent phylogenetic trees with time estimates for many species and for many genes, we should be able to reconstruct a reasonably good evolutionary history of different organisms at the molecular level. This history can then be compared with paleontological data to develop a unified view of the tree of life. At the present time, the amount of molecular data used for phylogenetic inference and time estimation are often too small to give reliable results.
Of course, it is important to as much as possible use genes or proteins whose evolution follows the molecular clock hypothesis. In recent years, a number of authors have used mitochondrial genes or proteins for estimating divergence times (47, 48). However, these data appear to be inappropriate for time estimation when different orders or classes of vertebrates are considered, because the evolutionary rate varies extensively from species group to species group. For example, the evolutionary rate appears to be more than two times lower in fish than in mammals (49) and more than two times lower in artiodactyls than in primates (47, 50). In these cases, the linearized tree method would not give reliable time estimates.
Another problem is the absence of reliable fossil records to calibrate ancient divergence times. At present, it is customary to use vertebrate fossil records to infer ancient divergence times such as early metazoan divergence and the divergence between animals, fungi, and plants (e.g., refs. 14, 16, 18). Estimation of ancient divergence times by using recent calibration dates is more error prone than that of recent divergence times. In this case, erroneous sequence alignment often causes a serious problem, and small differences in the gamma parameter value influence the estimates substantially. In the case of bacterial evolution, horizontal gene transfer also plays an important role (51), and this would introduce another source of errors in inferring phylogenies and divergence times. Great caution is necessary in the estimation of ancient evolutionary times.
Supplementary Material
Acknowledgments
We thank Xun Gu, Sudhir Kumar, Bill Martin, Alex Rooney, Naoko Takezaki, and George Zhang for their comments. This study was supported by research grants from the National Institutes of Health (GM-20293) and the National Aeronautics and Space Administration (NCC2-1057) (M.N.).
Abbreviations
- IP
individual protein approach
- CD
concatenation-based distance
- MY
million years
- PC
Poisson correction
- NJ
neighbor joining
References
- 1.Zuckerkandl E, Pauling L. In: Horizons in Biochemistry. Kasha M, Pullman B, editors. New York: Academic; 1962. pp. 189–225. [Google Scholar]
- 2.Margoliash E. Proc Natl Acad Sci USA. 1963;50:672–679. doi: 10.1073/pnas.50.4.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Britten R J. Science. 1986;231:1393–1398. doi: 10.1126/science.3082006. [DOI] [PubMed] [Google Scholar]
- 4.Easteal S, Collet C, Betty D. The Mammalian Molecular Clock. Austin: Landes; 1995. [Google Scholar]
- 5.Li W-H, Ellsworth D L, Krushkal J, Chang B H, Hewett-Emmett D. Mol Phylogenet Evol. 1996;5:182–187. doi: 10.1006/mpev.1996.0012. [DOI] [PubMed] [Google Scholar]
- 6.Nei M. Molecular Population Genetics and Evolution. Amsterdam: North-Holland; 1975. [PubMed] [Google Scholar]
- 7.Wilson A C, Carlson S S, White T J. Annu Rev Biochem. 1977;46:573–639. doi: 10.1146/annurev.bi.46.070177.003041. [DOI] [PubMed] [Google Scholar]
- 8.Takezaki N, Rzhetsky A, Nei M. Mol Biol Evol. 1995;12:823–833. doi: 10.1093/oxfordjournals.molbev.a040259. [DOI] [PubMed] [Google Scholar]
- 9.Doolittle R F, Feng D-F, Tsang S, Cho G, Little E. Science. 1996;271:470–477. doi: 10.1126/science.271.5248.470. [DOI] [PubMed] [Google Scholar]
- 10.Wray G A, Levinton J S, Shapiro L H. Science. 1996;274:568–573. [Google Scholar]
- 11.Kumar S, Hedges B. Nature (London) 1998;392:917–919. doi: 10.1038/31927. [DOI] [PubMed] [Google Scholar]
- 12.Lee M S. J Mol Evol. 1999;49:385–391. doi: 10.1007/pl00006562. [DOI] [PubMed] [Google Scholar]
- 13.Gu X. J Mol Evol. 1998;47:369–371. doi: 10.1007/pl00013150. [DOI] [PubMed] [Google Scholar]
- 14.Ayala F J, Rzhetsky A, Ayala F J. Proc Natl Acad Sci USA. 1998;95:606–611. doi: 10.1073/pnas.95.2.606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lynch M. Evolution (Lawrence, KS) 1999;53:319–325. doi: 10.1111/j.1558-5646.1999.tb03768.x. [DOI] [PubMed] [Google Scholar]
- 16.Wang D Y C, Kumar S, Hedges S B. Proc R Soc London Ser B. 1999;266:163–171. doi: 10.1098/rspb.1999.0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'hUigin C, Li W-H. J Mol Evol. 1992;35:377–384. doi: 10.1007/BF00171816. [DOI] [PubMed] [Google Scholar]
- 18.Feng D-F, Cho G, Doolittle R F. Proc Natl Acad Sci USA. 1997;94:13028–13033. doi: 10.1073/pnas.94.24.13028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Su C, Nei M. Proc Natl Acad Sci USA. 1999;96:9710–9715. doi: 10.1073/pnas.96.17.9710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takahashi K, Rooney A P, Nei M. J Hered. 2000;19:198–204. doi: 10.1093/jhered/91.3.198. [DOI] [PubMed] [Google Scholar]
- 21.Wu C-I, Li W-H. Proc Natl Acad Sci USA. 1985;82:1741–1745. doi: 10.1073/pnas.82.6.1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gu X, Li W-H. Mol Phylogenet Evol. 1992;1:211–214. doi: 10.1016/1055-7903(92)90017-b. [DOI] [PubMed] [Google Scholar]
- 23.Tajima F. Genetics. 1993;135:599–607. doi: 10.1093/genetics/135.2.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nei M, Chakravarti A. Theor Popul Biol. 1977;11:307–325. doi: 10.1016/0040-5809(77)90014-4. [DOI] [PubMed] [Google Scholar]
- 25.Nei M. Molecular Evolutionary Genetics. New York: Columbia Univ. Press; 1987. [Google Scholar]
- 26.Uzzell T, Corbin K. Science. 1971;172:1089–1096. doi: 10.1126/science.172.3988.1089. [DOI] [PubMed] [Google Scholar]
- 27.Nei M, Chakraborty R, Fuerst P A. Proc Natl Acad Sci USA. 1976;73:4164–4168. doi: 10.1073/pnas.73.11.4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ota T, Nei M. J Mol Evol. 1994;38:642–643. [Google Scholar]
- 29.Anscombe F J. Biometrika. 1950;37:358–382. [PubMed] [Google Scholar]
- 30.Dayhoff M O. Atlas of Protein Sequence and Structure. Silver Spring, MD: Natl. Biomed. Res. Found.; 1972. [Google Scholar]
- 31.Grishin N V. J Mol Evol. 1995;41:675–679. doi: 10.1007/BF00175826. [DOI] [PubMed] [Google Scholar]
- 32.Nei M, Kumar S. Molecular Evolution and Phylogenetics. Oxford, U.K.: Oxford Univ. Press; 2000. [Google Scholar]
- 33.Dickerson R E. J Mol Evol. 1971;1:26–45. doi: 10.1007/BF01659392. [DOI] [PubMed] [Google Scholar]
- 34.Li W-H, Gouy M, Sharp P M, O'hUigin C, Yang Y-W. Proc Natl Acad Sci USA. 1990;87:6703–6707. doi: 10.1073/pnas.87.17.6703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arnason U, Gullberg A, Janke A, Xu X. J Mol Evol. 1996;43:650–661. doi: 10.1007/BF02202113. [DOI] [PubMed] [Google Scholar]
- 36.Foote M, Hunter J P, Janis C M, Sepkoski J J., Jr Science. 1999;283:1310–1314. doi: 10.1126/science.283.5406.1310. [DOI] [PubMed] [Google Scholar]
- 37.Bromham L, Penny D, Rambaut A, Hendy M D. J Mol Evol. 2000;50:296–301. doi: 10.1007/s002399910034. [DOI] [PubMed] [Google Scholar]
- 38.Benton M J. The Fossil Record 2. New York: Chapman & Hall; 1993. [Google Scholar]
- 39.Easteal S. BioEssays. 1999;21:1052–1059. doi: 10.1002/(SICI)1521-1878(199912)22:1<1052::AID-BIES9>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 40.Duret L, Mouchiroud D, Gouy M. Nucleic Acids Res. 1994;22:2360–2365. doi: 10.1093/nar/22.12.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saitou N, Nei M. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 42.Gu X, Zhang J. Mol Biol Evol. 1997;15:1106–1113. doi: 10.1093/oxfordjournals.molbev.a025720. [DOI] [PubMed] [Google Scholar]
- 43.Zhang J, Gu X. Genetics. 1998;149:1615–1625. doi: 10.1093/genetics/149.3.1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gogarten J P, Olendzenski L, Hilario E, Simon C, Holsinger K E. Science. 1996;274:1750–1751. [PubMed] [Google Scholar]
- 45.Rivera M C, Jain R, Moore J E, Lake J A. Proc Natl Acad Sci USA. 1998;95:6239–6244. doi: 10.1073/pnas.95.11.6239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schopf J W. Science. 1993;260:640–646. doi: 10.1126/science.260.5108.640. [DOI] [PubMed] [Google Scholar]
- 47.Arnason U, Gullberg A, Janke A. J Mol Evol. 1998;47:718–727. doi: 10.1007/pl00006431. [DOI] [PubMed] [Google Scholar]
- 48.Arnason U, Gullberg A, Gretarsdottir S, Ursing B, Janke A. J Mol Evol. 2000;50:569–578. doi: 10.1007/s002390010060. [DOI] [PubMed] [Google Scholar]
- 49.Nei M. Annu Rev Genet. 1996;30:371–403. doi: 10.1146/annurev.genet.30.1.371. [DOI] [PubMed] [Google Scholar]
- 50.Cao Y, Adachi J, Hasegawa M. Mol Biol Evol. 1998;15:87–89. doi: 10.1093/oxfordjournals.molbev.a025877. [DOI] [PubMed] [Google Scholar]
- 51.Woese C R. Proc Natl Acad Sci USA. 2000;97:8392–8396. doi: 10.1073/pnas.97.15.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.