

Abstract
A major mechanism regulating the accessibility and function of eukaryotic genomes are the covalent modifications to DNA and histone proteins that dependably package our genetic information inside the nucleus of every cell. Formally postulated over a decade ago, it is becoming increasingly clear that post-translational modifications (PTMs) on histones act singly and in combination to form a language or ‘code’ that is read by specialized proteins to facilitate downstream functions in chromatin. Underappreciated at the time was the level of complexity harbored both within histone PTMs and their combinations, as well as within the proteins that read and interpret the language. In addition to histone PTMs, newly-identified DNA modifications that can recruit specific effector proteins has raised further awareness that histone PTMs operate within a broader language of epigenetic modifications to orchestrate the dynamic functions associated with chromatin. Here, we highlight key recent advances in our understanding of the epigenetic language encompassing histone and DNA modifications and foreshadow challenges that lie ahead as we continue our quest to decipher the fundamental mechanisms of chromatin regulation.
Keywords: histones, post-translational modifications, DNA methylation, chromatin, epigenetics, histone code
1. Introduction
1.1. Chromatin balances DNA packaging and accessibility
Eukaryotic DNA is tightly packaged within the nucleus of each cell and must be faithfully regulated, copied, and transmitted during cell division. DNA packaging amounts to an amazing feat, often requiring several meters of DNA to be compacted into the confines of a 2–10 micron nucleus. This high level of compaction presents a potential problem, as the underlying DNA must remain accessible to the vast protein machineries that utilize it for critical biological functions. Thus, a fundamental question being addressed by many labs has been how these diverse genomic functions, such as gene transcription, DNA repair, replication, and recombination, occur at the appropriate place and time to promote cellular growth, differentiation, and proper organismal development.
Key contributors to DNA packaging are the highly basic histone proteins, which wrap ~147 base pairs of DNA around an octamer of histones (2 copies each of H3, H4, H2A and H2B) to form thenucleosome core particle [1, 2]. This repeating nucleosomal subunit is the fundamental building block of chromatin. Chromatin is organized into distinct domains, such as euchromatin and heterochromatin, which are defined by the level of compaction and associated genomic functions. For example, euchromatin has relatively loose compaction and is typically transcriptionally permissive, whereas heterochromatin (facultative or constitutive) is more condensed and typically transcriptionally repressive [3–5]. The degree to which chromatin is organized and packaged is highly influenced by numerous factors, including the actions of linker histone H1 that regulates the formation of higher-order chromatin states [6], histone variants that can be substituted for canonical histones in the nucleosome core particle [7], chromatin remodelers that use the power of ATP hydrolysis to slide and evict histones [8], histone chaperones that facilitate deposition and eviction of histones [9, 10], and small chemical modifications to histones and DNA [11–15], whose interwoven functions are the primary focus of this review.
1.2. Histone PTMs and the ‘histone code’:a rich and vibrant new language has emerged
A major mechanism by which chromatin structure and function is regulated is through the actions of histone post-translational modifications (PTMs). An astonishing number of PTMs, including lysine acetylation, lysine and arginine methylation, arginine citrullination, lysine ubiquitination, lysine sumoylation, ADP-ribosylation, proline isomerization, and serine/threonine/tyrosine phosphorylation occur on histones [11, 15] (Table 1). While the majority are found in the flexible N- and C-terminal ‘tail’ domains that protrude away from the nucleosome core particle, a significant number also occur in the histone fold or globular domains that regulate histone-histone and histone-DNA interactions [16, 17].
Table 1.
Reading and interpreting histone post-translational modifications
| Modification Type | Structure | Associated Functions | Reader Domains |
|---|---|---|---|
| Lysine | 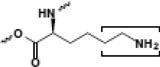 |
gene regulation | PHD, ADD, WD40 |
| mettiylation | 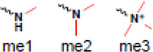 |
gene regulation DMA repair, DNA replication, heterochromatin | chromo, PHD, Tudor, MBT, ZF-CW, PWWP, ADD Ankyrin Repeats, WD4D, BAH |
| acetylation |  |
gene regulation, chromatin structure | bnomo, PHD |
| fomnylation | ? | ? | |
| proprionylation | ? | ? | |
| butyrylation | 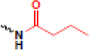 |
? | ? |
| crotonylation | 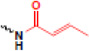 |
gene regulation, chromatin structure | ? |
| malonylation | 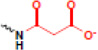 |
? | ? |
| succinylation | 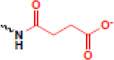 |
? | ? |
| 5-hydroxylation | ? | ? | |
| ubiqurtination | 8.5 kDa polypeptide, C-term Gly isopeptide bonded wilti Lys ε-amine | gene regulation DNA repair, heterochromatin | ? |
| sumoylation | 11 kDa polypeptide, 2° structure similar to ub, C-term Gly isopeptide bonded wilti Lys ε-amine | gene regulation | ? |
| ADP-ribosylation | 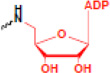 |
gene regulation DNA repair, DNA replication | PAR |
| Arginine |  |
gene regulation | PHD |
| methylation |  |
gene regulation | Tudor, WD4D, ADD |
| citrullination | gene regulation, chromatin structure | ? | |
| Serine |  |
||
| phosphorylation | gene regulation, DNA repair, mitosis | BRCT, 14-3-3 | |
| acetylation | ? | ? | |
| glycosylation | β-N-aceryiglucosamine (O-GlcNAc) | gene regulation | ? |
| Threonine |  |
||
| phosphorylation | 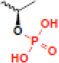 |
mitosis | HIR |
| acetylation | 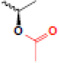 |
? | ? |
| glycosylation | β-N-acery1glucosarnine (O-GlcNAc) | gene regulation | ? |
| Tyrosine | 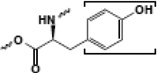 |
||
| phosphorylation | gene regulation | ? | |
| acetylation | ? | ? | |
| hydroxylation | 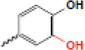 |
gene regulation | ? |
| Histidine | 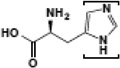 |
||
| phosphorylation | 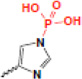 |
gene regulation | ? |
A long-standing question in the field has been how histone PTMs function in chromatin regulation. While it has been half a century since Vincent Allfrey first described the presence of acetylation and methylation on histones [18], and Lubomir Hnilica documented histone phosphorylation [19], the functional significance of these modifications remained elusive for many years. Fundamental breakthroughs in our understanding of histone PTM function (many of which occurred in recent years) have been made through the identification of the protein machineries that incorporate (write), remove (erase), and bind (read) histone PTMs. Two landmark discoveries in this regard were the identifications in 1996 of p55/Gcn5 and HDAC1/Rpd3 as transcription-associated histone acetyltransferases[20]and deacetylases[21], respectively – thereby linking dynamic histone modification activity directly to the transcription process. These findings changed the landscape of how the transcription and chromatin fields viewed histone PTMs and have resulted in a fast-paced and exciting field that shows no signs of slowing down.
In 2000, the concept of a‘histone code’ emerged as a hypothesis to stimulate new thinking about how histone PTMs might function [22, 23]. This postulate grew out of the observation that histone H3 serine 10 (H3S10) phosphorylation could be associated with seemingly opposite functions in chromatin (i.e., chromatin decompaction in transcription and chromatin condensation in mitosis) [24–28]. From analysis of this and other PTMs where the associated functions were known, it was possible to infer that PTMs might work singly as well as in combination (on one or more histone tails) to mediate the distinct functions associated with them. It was envisioned that, in addition to histone PTMs having a direct physical effect on chromatin structure (as is the case for lysine acetylation negating the positive charge of this residue) [29], they might also function through the selective recruitment of effector proteins or readers that ‘dock’ onto histone PTMs to direct specific downstream events in chromatin. One of the major landmarks in deciphering this aspect of the hypothesis was the discovery of the bromodomain module as an acetyl-lysine reader motif [30]. This result paved the way for subsequent characterization of this important family of chromatin effectors [31, 32], and further suggested the existence of other, yet-to-be identified protein domains, that read histone PTMs.
A large body of data now supports the notion that histone PTMs function, at least in part, through the recruitment of effector proteins harboring specialized reader domains [33, 34]. While the ‘histone code’ hypothesis provided a retrospectively simplistic explanation of how histone PTMs function, recent advances have revealed that the context in which histone PTMs operate is much more complex than originally envisioned. In addition to combinatorial PTMs that function together both synergistically and antagonistically, there is now an appreciation for PTM asymmetry within individual nucleosomes [35], novel types of PTMs with unique functions [36], nucleosomes bearing histone variants [7], and nuclear compartmentalization events [5] that are all contributing to the final output of chromatin organization and function. Furthermore, fueled in part by the recent discovery of an active DNA demethylation pathway and proteins that read these pathway intermediates [14, 37], a new appreciation for the function of histone PTMs in coordination with DNA modifications is emerging. This review will focus on several key findings from the last few years that have helped to expand our current understanding of how histone PTMs and DNA modifications function in the context of a more integrated epigenetic language [22, 38]. Posing several key questions to the field, we also foreshadow where studies into chromatin organization and function are headed and highlight several new technologies that are taking us there.
2. Increasing complexity of the histone PTM landscape
2.1. Identifying and mapping histone PTMs beyond the histone tails
Technological advances in mass spectrometric (MS)-based analyses of histones have greatly facilitated our understanding of the types of PTMs that occur on histones, as well as their abundance and co-occurrence [39–43]. Perhaps one of the most surprising findings from early MS studies using the high resolving power of Fourier transform ion cyclotron resonance MS was the significant number of previously unknown acetylation, methylation and phosphorylation events that were detected on the N- and C-terminal tail domains as well as on the globular regions of histones [44]. From this analysis and additional studies [16, 45–47], a vast and mostly unexplored PTM landscape emerged that has greatly expanded the potential complexity of the histone modification language (Table 1). Underappreciated at the time of our original postulate was the potential for a significant number of PTMs to occur along or near the histone-DNA interface where the histone octamer contacts wrapped DNA – thus providing an additional means by which nucleosome stability and/or DNA interactions on the surface can be regulated [16]. Histone-DNA interface PTMs (in contrast to those located on the histone tail domains) largely act in a physical manner by tweaking or fine-tuning histone-DNA interactions that in turn affect nucleosome stability and/or mobility. Evidence for this effect came from elegant in vitro studies employing semi-synthetic or genetically modified nucleosomes to show that histone fold acetylation at H3K56, H3K155 and H3K122, in addition to phosphorylation of H3T118, resulted in weakened histone-DNA interactions, increased nucleosome mobility, and DNA unwrapping [48–53]. Significantly, H3K122 was recently identified as a substrate for the p300/CBP histone acetyltransferase [54]. H3K122ac was found to be enriched at gene promoters and is required for proper transcriptional activation, but how p300/CBP targets H3K122, which is in a largely inaccessible and buried region in the nucleosome core particle, is not yet understood.
While PTMs located at the histone-DNA interface regulate the intrinsic properties of the nucleosome core particle, it is noteworthy that these modifications can also regulate effector protein association similar to those located in the tail domains. For example, JAK2-mediated phosphorylation of H3Y41, a site located at the DNA entry/exit point [55], was found to be refractory to HP1α chromatin association by perturbing an interaction between the HP1α chromoshadow domain and H3 in this region. While H3Y41 was shown to relieve HP1-mediated gene repression, it remains to be determined whether H3Y41 phosphorylation negates HP1α interaction in the context of H3K9me3 [56, 57]. Interestingly, neighboring H3R42 was recently identified as a substrate for CARM1 and PRMT6-mediated asymmetric dimethylation (H3R42me2a) [58]. H3R42me2a was linked to active transcription in vitro, suggesting this PTM alters chromatin structure by removing a potential hydrogen bond donor contributing to histone-DNA interaction. It is intriguing to also speculate that H3R42me2a may elicit positive transcriptional effects in the cell by negating HP1 binding.
2.2. An expanding dictionary of histone PTMs
Along with the identification of new PTM sites on histones, recent MS analyses have also focused on the identification of novel types of histone PTMs. From these studies, a surprising number of newly-identified modifications have been uncovered (Table 1), including lysine crotonylation, lysine butyrylation, lysine propionylation, lysine succinylation,lysine malonylation (all of which are derived from distinct co-enzyme A molecules), lysine 5-hydroxylation, lysine N-formylation, tyrosine hydroxylation, serine/threonine/tyrosine acetylation, and serine/threonine O-GlcNAcylation (for an in depth review, see [36]). It is important note that a number of these above-mentioned PTMs are low in abundance as compared to more well-studied modifications, suggesting their roles in physical and functional cross-talk may be discreet. While the functions of these newly described PTMs are largely awaiting discovery, exciting recent studies are revealing that they can play key roles in genome function. Several examples are highlighted below.
Lysine crotonylation is a highly conserved PTM that is causally linked to gene activation and nucleosome disruption [59]. Crotonylation is a four-carbon moiety, and (like lysine acetylation) neutralizes the positive charge of this residue. Using pan-lysine crotonylation-specific antibodies that detect each core histone in addition to H1, this novel modification was shown to be enriched at gene promoters, predicted enhancers, and transcriptionally active X/Y sex-linked genes that escape repression in male post-meiotic spermatids [59–61]. While these studies suggest a role for lysine crotonylation in active transcription, it remains unclear which specific crotonylated lysine residues are linked to the observations above. Future work will therefore be needed to identify all of the residues in histones that harbor this mark, and how each mark may contribute to gene regulation or other functions associated with DNA. It is noteworthy that histone acetyltransferases capable of utilizing butyryl-CoA and propionyl-CoA are unable to utilize the crotonyl-CoA cofactor due to its flattened, extended, and more rigid carbon-carbon π-bond[59]. Thus, it will be exciting to identify the class of histone-modifying enzymes that write and erase this PTM.
Intriguingly, two sites of lysine crotonylation have recently been identified to occur at H3K122 (a site also found to be acetylated, see above) and H4K77 [62]. These lysines lie near the dyad axis on opposite sides of the nucleosome, and thus, are in a position to directly regulate histone-DNA interactions and nucleosome stability. This study found that defective incorporation of the H2B variant TH2B, normally required for the disruption and replacement of histones with protamines during spermatogenesis, resulted in increased lysine crotonylation (in addition to increased arginine methylation) in the histone fold domains of H4 and H2B. This appears to be a compensatory mechanism for not having TH2B present.
Another example where significant insight has been revealed for a novel histone PTM is with the serine/threonine sugar modification O-linked β-N-acetylglucosamine (O-GlcNAc). Studies have found that O-GlcNAc transferase (OGT) and the hydrolase O-GlcNAcase (OGA) are the enzymes that write and erase this PTM, both on histones and non-histone proteins [63–66]. The nucleotide sugar donor used by OGT (UDP-GlcNAc)is highly regulated by nutrient levels and is rate limiting in the cell [67, 68]. Studies therefore suggest O-GlcNAcis a sensor of nutrient status, controlling gene expression programs in response to changing nutrient conditions and environmental stress. In C. elegans, OGT has been mapped to gene promoters that are connected to metabolism, growth, aging, and the stress response, and is important for the proper transcriptional regulation of these genes [69]. In Drosophila, OGT has been identified as the protein product of the super sex combs (sxc) gene, a member of the polycomb group (PcG) proteins required for the PcG-mediated repression of developmentally regulated genes [70, 71]. Given O-GlcNAcylation is intimately associated with nutrient sensing and metabolism, this finding suggests that there is a nutrient responsive and/or metabolic component to PcG repression and organismal development. Important to note, however, is that O-GlcNAcylation is well known to occur on non-histone proteins involved in transcription (e.g., MLL5, polymerase II (RNAPII), and the PcG factor Polyhomeotic) [70, 72–74]. Therefore, it will be important to resolve whether O-GlcNAcylation that occurs on histones is indeed contributing to the gene regulation programs identified above.
In mammals, O-GlcNAcylation is a modification that occurs on each of the core histones and is regulated in a cell cycle-dependent manner [75]. Peak levels of O-GlcNAc occur during G1 and drop in S-phase, followed by a rise occurring through G2 and M phase. O-GlcNAc was found to occur at H3S10, posing an interesting question as to whether O-GlcNAcylation is functionally antagonistic with H3S10 phosphorylation. Since O-GlcNAc levels are highest in G1, it is plausible that H3S10 O-GlcNAcylation acts as a molecular ‘switch’ to negatively regulate gene expression normally controlled by H3S10 phosphorylation. Intriguingly, O-GlcNAc has also been found to crosstalk with other histone PTMs, notably H2BK120 monoubiquitination [76]. Specifically, O-GlcNAcylation of H2BS112 in Drosophila was found to promote H2BK120Ub1 through the recruitment of the ubiquitination machinery [76]. These studies indicate a positive role for O-GlcNAcylation in transcription. In agreement with this notion, it was shown that OGT recruitment by the DNA methylcytosine dioxygenase TET2 (see section 3.3) facilitates H2BS112 O-GlcNAcylation during gene reactivation [77]. As OGT was found to be required with TET2 for gene reactivation, the study suggests that DNA methylcytosine oxidation is coordinated with other histone marks to facilitate reactivation of TET2-regulated genes. This serves as an important example of how our understanding of histone PTM function has evolved over the years to encompass an intimate DNA component (a topic discussed in detail below).
The studies described above have provided important insights into the function of newly discovered PTMs that occur on histones – yet fundamental questions remain about the role of these and other aforementioned PTMs in chromatin regulation. For example, what are the writers, erasers, and potential readers of these modifications? In the case of lysine butyrylation and propionylation, which use distinct co-enzyme A cofactors (butyryl-CoA and propionyl-CoA), studies have shown that they can be catalyzed and removed by histone acetyltransferases and deacetylases [78, 79]. Given that these different Co-A forms are created by distinct metabolic pathways, and thus can be regulated by metabolic flux, it may be that these distinct lysine modification forms play specialized roles in fine-tuning gene expression programs in response to nutrient imbalance.Significantly, this principle of metabolic flux regulating epigenetic signatures is a fundamental aspect of chromatin regulation, recently reviewed in detail elsewhere [80, 81].
It is also intriguing to speculate that, like lysine acetylation, distinct Co-A forms are read by bromodomain-containing proteins. Indeed, structures of the first bromodomain of Brd4 solved in complex with proprionylated H3K23 and butyrylated H3K14 demonstrate that these moieties are coordinated similarly to acetyllysine [82]. For these and other newly identified PTMs, it will be important to determine where they occur across the genome, how they show crosstalk with other PTMs, and what functions are mediated downstream of these modifications. Given what has been discovered so far with lysine crotonylation and O-GlcNAcylation, it is likely that these other novel modifications will also have important and exciting roles in chromatin function.
2.3. PTM combinations and asymmetry add increasing diversity to histone function
In addition to expanded mapping of individual histone PTMs, the co-existence of histone modifications has gained new appreciation based on recent studiesemploying high resolution tandem MS and improved MS approaches to detect the short and long-range histone PTM combinations that occur. These studies, examining the N-termini of H31–50 and H41–23, have identified a staggering number of diversely modified H3.2 and H4 N-terminal forms (> 200 each for H3.2 and H4) in mammalian histones [40, 83, 84]. The H3 N-terminus was found to be the most highly modified histone N-terminus compared to the other core histones, and can contain up to 7 PTM combinations in a single N-terminal fragment. These studies also confirm several previously observed findings, including the co-occurrence of H3K4 methylation with neighboring acetylation [85, 86]. Given that the C-terminal regions of histones have not been examined to this same extent, along with the inclusion of phosphorylation events that were not readily observed from the asynchronous cells used in the above studies (but would exist during mitosis), there appears be an astonishing number of potential PTM combinations that add diversity to the histone language. Key to our understanding of how histone PTMs function is to elucidate how the readers, writers, and erasers behave in this combinatorial context. Along these lines, a number of high-throughput technologies, including peptide microarray-based screening approaches, have recently been employed to begin characterizing these complex interactions and substrate specificities [87–92].
While much progress has been made in identifying the co-occurrence and mutual exclusivity of histone PTM combinations, it has been technically challenging to determine whether polypeptides within a histone homodimer are modified in the same way. Further challenges have been faced in determining whether identified combinations occur along the same histone polypeptide (symmetrically) or on adjacent polypeptides within a histone homodimer (asymmetrically). Major advances in our understanding of histone PTM patterning within a single nucleosome came from a recent study by Reinberg, Garcia, and colleagues [35]. Histone PTM-specific antibodies were used to enrich for mononucleosomes bearing a PTM of interest, and the abundance of these marks was quantified following tryptic digestion using liquid chromatography-coupled MS (LC/MS). Assuming the histone PTM antibody was specific and did not enrich for mononucleosomes lacking the mark of interest (a key assumption for accurate interpretation of these data) [93], the relative abundance of unmodified vs. modified histone peptide in the MS pool provided insight into the degree of symmetry of a specific PTM. Applying this approach to H3K27me2 and H3K27me3 (marks of facultative heterochromatin written by the EZH2 subunit of the PRC2 complex [94]) as well as H4K20me1 (written by PR-Set7 and involved in the DNA damage response and mitotic chromatin condensation [95]), populations of symmetrically and asymmetrically modified nucleosomes were identified. These experiments provided the first evidence that histone H3 and H4 homodimers are not necessarily modified identically within a single nucleosome.
A potentially important role for asymmetry has now been realized in the context of development. Genome-wide mapping studies have documented the co-occurrence of H3K4me3 (associated with active transcription) and H3K27me3 (a dominant mark of transcriptional repression) at developmental gene promoters in embryonic stem cells (defined as ‘bivalency’) [96] – the notion being that these two marks might reside on the same H3 tail to transcriptionally ‘poise’ these loci for rapid transcriptional activation in response to cellular cues leading to demethylation of H3K27me3. Surprisingly, high-resolution MS studies found that these marks do not co-exist on the same H3 tail [83]. This seemingly opposing result was clarified by the finding that H3K4me3 and H3K27me3 exist within a single nucleosome, but occur on adjacent histones within a homodimer [35]. Consistent with this observation, it has been demonstrated that PRC2 cannot methylate H3K27 when H3K4me3 is present on the same tail [35, 97].
The identification of asymmetrically modified nucleosomes greatly increases our awareness for how histone PTMs operate. It will be important to extend these studies to other histone modifications to determine the degree of symmetry and asymmetry across the spectrum of histone PTM combinations. Furthermore, the enzymology and biochemistry directing symmetric and asymmetric PTM combinations will surely be exciting new avenues of discovery. Given our recent appreciation for the ability of histone effector proteins to sense PTMs on adjacent histone tails (see below and Fig. 1), it is intriguing to also speculate that the functional outcomes of symmetric and asymmetric PTM combinations may be distinct.
Figure 1. Writing, erasing, and reading the histone and DNA modification landscape.

Isolated and linked protein domains coordinate the addition (writing), removal (erasing), and association (reading) of DNA modifications (black circles) and histone PTMs (blue circles and red triangles), creating a dynamic and variable chromatin environment. Cis refers to multivalent events occurring on the same histone. Trans refers to multivalent events occurring on adjacent histones or spanning histones and DNA, either within the same nucleosome or on neighboring nucleosomes. Trans interactionswith distant nucleosomes (which may be in 3-dimensional proximity) are not depicted. Multivalent interactions facilitated by membership in macromolecular complexes are also not depicted.
2.4. Multivalency in histone PTM recognition
Much of our understanding of histone PTM function has come from the identification that unique folds or domains within chromatin-associated proteins can ‘read’ histone PTMs and their combinations to drive distinct functions in chromatin, a central tenet in the original ‘histone code’ postulate. A structurally diverse family of histone reader domains has been discovered over the last decade, including bromodomains that bind acetyllysine, chromodomains that bind methyllysine, plant homeodomain (PHD) fingers that bind acetyllysine, methyllysine, and even unmodified lysine, Tudor domains that bind methyllysine and methylarginine, and BRCT domains that bind phosphoserine (Table 1). Recent comprehensive reviews detail the structural properties of these and other identified histone reader domains and highlight the diverse biochemical implications of their interactions [33, 34].
It has become increasingly apparent that many chromatin-associated factors harbor multiple known or predicted histone binding domains, both within a single protein, and within distinct subunits of macromolecular complexes. This offers a diverse and exciting potential for multivalent histone engagement [98] (Fig. 1) that adds a previously underappreciated layer of specificity to histone PTM recognition. A number of closely spaced histone binding domains within single proteins have been shown to coordinately engage histone PTM signatures on a single histone tail, termed cis interactions [99–106]. A recent example is the tandem Tudor domain (TTD) and PHD finger of UHRF1, which together, coordinate an interaction with H3K9me3 and the N-terminus of an H3 tail, respectively [100, 107, 108]. Importantly, disrupting the reader function of either domain individually, or perturbing the ability for this dual domain to engage the H3 tail in cis, was shown to inhibit the UHRF1 chromatin interaction and prevent UHRF1 from facilitating the epigenetic inheritance of DNA methylation [104] (Fig.3).
Figure 3. Distinct combinatorial epigenetic modifications play key roles in the erasure, establishment, and maintenance of DNA methylation patterns through organismal development.

(Left) PGC7 protects the maternal genome from active DNA demethylation. PGC7 binding to H3K9me2 (blue circles) protects maternal DNA methylation (black circles) from TET3-dependent oxidative demethylation by an unknown mechanism. The paternal genome lacks histones, and TET3 demethylation is therefore active in these cells. (Center) DNMT3A/B and DNMT3L re-establish DNA methylation patterns in early development. DNMT3A/B interacts with the H3 N-terminus through an ADD domain and H3K36me3 (blue circles) through a PWWP domain. DNMT3L also interacts with the H3 N-terminus through an ADD domain and physically associates with the methyltransferase domain of DNMT3A/B to allosterically stimulate de novo DNA methyltransferase activity. (Right) UHRF1 facilitates the DNMT1-mediated maintenance of established DNA methylation patterns through embryonic and somatic cell divisions. UHRF1 physically associates with chromatin in a trivalent manner through its TTD and PHD domains that engage a single histone H3 tail (cis interaction) that is tri-methylated at lysine 9 (H3K9me3; blue circles) and unmodified at the N-terminus, respectively, and through its SRA domain that engages hemi-methylated DNA (black circles), a DNA replication intermediate. The UHRF1 RING domain aids in the catalysis of H3K23 ubiquitination (H3K23ub; green triangle), which serves as a binding platform for DNMT1. DNMT1 also physically interacts with DNA (see text) and with the SRA domain of UHRF1. Cartoon representations of nucleosomal interactions are depicted based on biochemical and structural studies, but may not be accurate in regards to orientation within the nucleosome.
Interestingly, crystal structures of the UHRF1 TTD-PHD bound to H31–11K9me3 revealed that H3 residues lysine 4 through alanine 7 adopt a single turn of an α-helix [108]. Since the H3 tail peptide in complex with the isolated PHD finger is in an extended conformation, it is assumed that multivalent TTD-PHD engagement induces conformational change in the H3 tail. It is notable that tail domains of histone proteins are rich in positively charged lysines and arginines and lack detectable secondary structure when observed as nucleosomes, free histones, and/or peptides. These findings have led to the perception that tail domains are intrinsically disordered. However, classic studies from Grunstein and colleagues predicted that regions within the H3 and H4 tails have the propensity to adopt amphipathic α-helices, β-sheets, and β-turns [109]. More recent studies using advanced molecular modeling algorithms have reached similar conclusions, suggesting that the H4 tail can form β-hairpins, and the H3 and H2B tails may adopt α-helices [110, 111]. Interestingly, it appears that PTMs may influence the departure from extended tail conformations, either directly by stabilizing secondary structures [111] or indirectly (as described above with UHRF1) through the recruitment of effector proteins that appear to induce conformational change in histone tails [57, 100, 101]. Indeed, H31–13in complex with the double PHD finger of the MOZ lysine acetyltransferase adopts an extensive α-helix, spanning H3 residues lysine 4 through threonine 11, and, unlike the helix adopted by UHRF1, several residues spanning the helix make hydrogen bonds with MOZ [101]. This helical conformation, and effector protein interaction, is maintained when lysines 9 or 14 are acetylated. Importantly, the acetyl group on lysine 14 forms a hydrogen bond with the main chain amine of isoleucine 260 on MOZ, and the proximity of this interaction appears to be facilitated by the helical turn.
The potential for histone PTMs to either induce or stabilize secondary structures suggests that histone PTMs distant in sequence may in fact be in proximity for combinatorial influence. This possibility adds an exciting new level of regulation that may underlie how histone PTMs function. Future structural work will reveal the dynamics of induced secondary structures upon effector protein binding, the propensity for other effector protein interactions to induce conformational change in histone tails, and the direct effects (or lack-there-of) on PTM-induced conformational change. It is important to note that these recent instances of histone tails with stabilized secondary conformations were observed with peptides. The propensity for histone tail structure in the physiological context of chromatin may be more challenging, both energetically and physically. Furthermore, it will be interesting to question whether effector proteins are able to recognize histone tails that have already adopted secondary structure, or whether the ground state of histone tails is indeed unstructured.
Multivalent interactions spanning adjacent histone tails within a single nucleosome or on adjacent nucleosomes (Fig. 1), termed trans interactions, have been more difficult to study, largely due to technological challenges in generating optimal templates for biophysical assays. However, a number of recent studies provide compelling evidence for trans interactions in chromatin [112–114]. The BPTF subunit of the NURF chromatin-remodeling complex harbors a PHD finger adjacent to a bromodomain, a common reader domain combination found in chromatin-associated proteins [98]. Biophysical studies using modified histone peptides connected by rigid DNA linkers, as well as semi-synthetic mono- and di-nucleosomes, demonstrated that PHD engagement of H3K4me3 and bromodomain engagement of H4K16ac within a single nucleosome provided an optimal scaffold for BPTF recruitment [114]. This important finding was supported by structural analysis, which suggested that in nature, a rigid, helical linker connecting the domains provides the optimal spacing to facilitate this trans interaction. While disruption of either reader domain in BPTF was demonstrated by chromatin immunoprecipitation (ChIP) to affect HOX gene targeting of the NURF complex, in what way this multivalent trans interaction impacts NURF chromatin remodeling function at these loci and others remains to be determined.
These and other recent studies have conveyed awareness that histone binding domains within proteins and protein complexes are often functioning in a coordinate manner. Notably, a number of paired histone binding domains characterized to engage the chromatin template in a multivalent manner harbor single domain reader functions that are broadly shared among chromatin-associated factors. For instance, the PHD finger of BPTF shares the ability to read H3K4me3 with a growing list of chromatin-associated factors [33, 115–117], and the TTD of UHRF1 is one of a host of identified H3K9me3 readers [57, 102, 118–121]. Importantly, in vitro affinity measurements for these interactions are often similar, suggesting the possibility that competition for histone PTMs exists at defined regions of chromatin. It is intriguing to speculate that multivalency may in fact provide a mechanism to circumvent this potential competition problem, as these dual interactions are often more energetically favorable than single domain interactions [122]. Decoding the energetics and interaction networks of paired or multidomain complexes will certainly be of fundamental importance moving forward. Importantly, these studies indicate that it may be insufficient to characterize isolated histone binding domains without taking into account the potential function or influence of adjacent regions. In addition, the discovery of asymmetry within histone tails in a single nucleosome [35] suggests that trans-tail interactions may be more prevalent than initially thought. Technological advances, such as more straightforward routes to facilitate the construction of defined semi-synthetic nucleosomes [123], will also be needed to aid future discoveries of trans-tail histone interactions.
3. Emergence of a DNA methylation language
Classic studies have unquestionably shown that DNA sequence is a major driving force in genome function through the direct recruitment of sequence-specific binding factors to cognate DNA elements (Fig. 2). Perhaps the clearest examples lie in the control of gene expression, where many landmark studies have identified an expanding list of activators and repressors that target DNA elements to drive transcriptional states [124]. These studies have also shaped our view of how the transcription regulatory machinery, via DNA binding, regulates histone modification states and histone variant patterns [125]. In addition to DNA sequence, it has become increasingly apparent that DNA modifications, much like histone PTMs, can regulate the association and downstream functions of factors that directly bind DNA (Fig. 2). High-resolution mapping of DNA modification patterns and systematic characterization of factors that interact specifically with these marks are revealing important new insights into how DNA modifications play an active role in chromatin regulation. Below, we highlight recent advances in our understanding of DNA modifications and how these marks integrate with histone modifications to create a diverse epigenetic landscape that functions coordinately with the underlying DNA sequence to dynamically regulate chromatin function.
Figure 2. Coordinate genetic and epigenetic mechanisms regulate transcription factor binding.

(Left) Transcription factor (TF) binding to cognate DNA sequence motifs (yellow box) regulates transcriptional output (orange box). (Right) TF binding to methylated DNA sequence motifs (black circles) both positively and negatively regulates transcriptional output.
3.1. Reading DNA methylation
DNA methylation in eukaryotes, which occurs primarily at the 5-position of cytosine residues (5mC) in the context of CpG dinucleotides (Table 2), is a heritable chromatin modification long-studied for its key role in repressive chromatin regulatory processes like X-chromosome inactivation, genomic imprinting, and retrotransposon silencing [13]. It is estimated that 80% of all CpG sites in the genome are methylated, accounting for roughly 1% of the total nucleotide pool [126]. DNA methylation has classically been shown to function in gene silencing through direct occlusion of the interaction of transcription factors with DNA [127](Fig. 2) and by serving as a binding site for methyl-binding domain (MBD) proteins often associated with chromatin remodelers and transcriptional co-repressor complexes [13]. Methyl-CpG binding protein 2 (MeCP2) was the first MBD identified to specifically read a symmetrically methylated CpG dinucleotide [128], and mutations in this domain are causally linked to the X-linked neurodevelopmental disorder Rett Syndrome [129, 130]. More recently, a second mutational cluster identified outside of the MBD domain of MeCP2 [131] was shown to disrupt the interaction with the NCoR/SMRT co-repressor complexes [132], suggesting a key role for MeCP2 as a bridging molecule between 5mC and NCoR/SMRT. Subsequent members of the MBD family (MBD1-MBD4) have been classified based on primary sequence similarity to the minimal region of MeCP2 required to bind symmetrically methylated DNA [128, 133]. Potential functional redundancy of MBD1, MBD2, andMBD4, but not MBD3, is suggested from mouse knockout studies [134–137]. Interestingly, MBD3 is unable to read methylated DNA due to two amino acid substitutions in its MBD domain [138]. A recent study suggests this protein may in fact read 5-hydroxymethylcytosine (5hmC) [139], a proposed intermediate of oxidative DNA demethylation [14] (see below). Genome-wide analysis demonstrated that MBD1, MBD2, MBD4, and MeCP2 occupancy is directly proportional to CpG density [140]. However, in vitro affinities for methylated CpG sites vary considerably among this protein family [141], suggesting that other factors, such as additional known or putative chromatin-binding domains within these proteins or unique interacting partners, may also contribute to their cellular specificity.
Table 2.
Reading and interpreting DNA modifications
| Modification Type | Structure | Associated Functions | Reader Domains |
|---|---|---|---|
| Cytosine (CpG) | 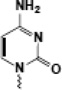 |
gene regulation | CxxC |
| methylaion (5mC) | 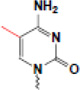 |
X-inactivation, imprinting, long-term silencing, development, gene regulation | MBD, ZF, SRA (others?) |
| hydroxymethylation (5hmC) | 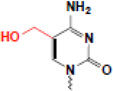 |
? | MBD, SRA (others?) |
| fomnylation (5fC) | 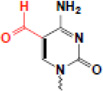 |
? | ? |
| caiboxylation (ScaC) | 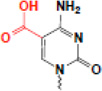 |
? | SRA (others?) |
Several other 5mC reader families have been recently identified, including a family of zinc finger-containing proteins and SET and RING-associated (SRA) domain-containing proteins. Kaiso, the flagship member of the zinc finger family, reads symmetrically modified 5mC (occurring on both DNA strands) in the context of tandem CpG dinucleotides [142]. Similar to MeCP2, Kaiso may be involved in the recruitment of the NCoR co-repressor complex to mediate silencing of target loci [143]. Interestingly, Kaiso also reads with high affinity a specific 5 base-pair unmethylated DNA sequence found at promoters of target genes regulated by the Wnt signaling pathway [144, 145]. As structural analysis of Kaiso binding to methylated and unmethylated DNA templates reveals similar molecular interactions [146], a challenge for future study will be to determine the extent to which this unique bimodal DNA recognition contributes to Kaiso-mediated gene regulation.
The SRA domain was first described as a bona-fide 5mC-binding motif in Arabidopsis thaliana [147] and is restricted to the UHRF family of chromatin-interacting proteins in mammals [148]. Evidence for an interaction of the UHRF1 SRA domain with hemi-methylated DNA was initially shown in crude nuclear extracts [149]. Structural studies determined that the UHRF1 SRA domain binds hemi-methylated CpG sites [150–153], similar to nucleotide-modifying enzymes [154, 155], by flipping the methyl-cytosine nucleotide out of the DNA double helix into a cage-like hydrophobic pocket [150–153]. Importantly, this is the first example of a sequence-specific non-enzymatic DNA binding domain to harbor base-flipping activity. Hemi-methylated CpG dinucleotides are a DNA synthesis intermediate found at replication forks, and the faithful copying of the parental DNA methylation pattern on the daughter strand by DNA methyltransferase 1 (DNMT1) is central to the epigenetic inheritance model of DNA methylation [156, 157] (Fig.3). The interaction of UHRF1 with both DNMT1 and hemi-methylated DNA suggests UHRF1 is involved in this process [149, 158–160]. Indeed, deletion of Uhrf1 results in a pronounced reduction in cellular DNA methylation levels [159, 160], and the SRA interaction with DNA is required for DNMT1 chromatin targeting and subsequent DNA methylation maintenance [161].
3.2. Breaking the dogma of DNA methylation silencing
Large-scale mass spectrometry-based screening approaches are revealing what appears to be an expanded repertoire of proteins that read 5mC either directly or indirectly through membership in macromolecular complexes [37, 162–164]. Paradoxical to the silencing function of DNA methylation, a number of DNA-binding transcription factors, notably, three Kruppel-like zinc fingers (KLF2, KLF4, and KLF5), bind specific DNA sequences in a methylation-dependent manner [37, 165](Fig. 2). Interestingly, the Yamanaka reprogramming factor KLF4 associates with both methylated and unmethylated DNA binding motifs in cells, and biochemical studies suggest these non-competitive reading activities are harbored in distinct domains of the protein [165]. Importantly, 5mC recognition by KLF4 plays a stimulatory role in KLF4-mediated transcription. However, the precise role of 5mC recognition in the stem-cell reprogramming function of KLF4 remains to be determined.
Taken together, these intriguing findings expand the functional role of 5mC in gene regulation, thus underscoring the potential for a DNA methylation language to regulate active and repressive gene states in a site-specific manner. One of the most exciting concepts to emerge from these studies is the realization that a methylated DNA sequence motif can take on a new function by creating a novel DNA binding site for transcriptional activators that could not be predicted from sequence information alone (Fig. 2). Thus, the complexity of DNA modifications regulating genomic function is perhaps as complex now as understanding the dynamics of histone PTM regulation. This complexity is further typified by the recent findings that factors read the newly discovered oxidized derivatives of 5mC, described further below.
3.3. Reading DNA hydroxymethylation and oxidized derivatives
Mechanisms facilitating the active removal of DNA methylation have been the subject of much debate [14]. The recent discovery that the ten-eleven translocation (TET) family of enzymes catalyze iterative 5mC oxidation to 5-hmC [166–168], 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC) (the latter two being substrates for thymine DNA glycosylase-mediated base excision repair) [169, 170], significantly advanced our understanding of the active DNA demethylation process. Importantly, recent identification of proteins that read these oxidized derivatives of 5mC [37, 139, 171, 172] suggests that DNA-demethylation independent functions of these DNA modifications may exist.
In addition to its interaction with 5mC, it has been demonstrated that UHRF1 can also read 5hmC [37, 171, 173]. While the significance of this interaction remains to be determined, it was recently shown that the structurally homologous protein UHRF2 reads 5hmC and 5caC, but not 5mC or 5fC [37]. Importantly, UHRF2 is unable to restore DNA methylation in cells devoid of UHRF1 [174, 175], suggesting that while these two proteins share structural identity, they may function in distinct biological pathways. Indeed, oxidative demethylation by TET1 in HEK293T cells was enhanced in the presence of ectopically expressed UHRF2 [37]. Clearly, a relationship is emerging between the DNA demethylation pathway and UHRF1/UHRF2, and future biochemical and biological studies are warranted to gain mechanistic insight into how these discreet modifications to the cytosine ring are helping orchestrate the complex recruitment and function of these factors.
Recent studies have also suggested that MBD proteins are capable of reading 5hmC [139, 172]. Interestingly, MBD3 localizes to hydroxymethylated gene promoters in a TET1-dependent manner, and an identified physical interaction of MBD3 (unable to read 5mC; see above) with 5hmC suggests TET1-mediated 5hmC may be a recruitment mechanism for MBD3/NuRD-dependent gene regulation [139]. Additionally, MeCP2 was identified as a reader of 5hmC [172], and a common mutation of MeCP2 found in Rett Syndrome [176] disrupts the interaction with 5hmC while maintaining the ability of MeCP2 to read 5mC [172]. The strong correlation between 5hmC and gene activation provides yet another paradigm whereby a dual modification reader like MeCP2 may function as both a co-activator and a co-repressor of transcription depending on the chromatin mark it reads.
Perhaps the most intriguing findings from the above studies are the wide range of potential reader domains capable of interacting with the distinct methylation states on CpG dinucleotides. This finding creates a new hierarchy of regulation and will lead to many important studies into the functional significance of these interactions.
3.4. Reading unmodified CpG dinucleotides
While most CpG dinucleotides across the genome are methylated, repetitive clusters known as CpG islands are found at more than half of transcriptionally active mammalian gene promoters, and these islands are largely hypomethylated [177]. The identification of cysteine-rich zinc-finger CxxC-containing proteins as readers of unmethylated CpG dinucleotides suggests that CpG islands can play an active role in transcription [178]. Indeed, CxxC-finger protein 1 (CFP1), a subunit of the SET domain 1 (SETD1) H3K4 methyltransferase complex [179], associates with a large fraction of non-methylated CpG sites in the mouse brain, most of which are also enriched for H3K4me3 [180]. Importantly, integration of artificial CpG islands into promoter-free regions of the mouse genome is sufficient to establish new H3K4me3 domains, suggesting a key role for CFP1 in directing H3K4me3-dependent transcription.
The presence of CxxC domains in TET1 and TET3, but not TET2, suggests unmethylated CpG sites may constitute, at least in part, a targeting mechanism and nucleation point for subsequent DNA demethylation. However, efforts to characterize the CxxC domains of TET1 and TET3 as bona-fide CpG readers in vitro have been difficult, presumably due to the positive charge of these domains promoting non-specific DNA interactions [181–184]. Interestingly, the CxxC domain of IDAX, an ancestral gene product of TET2, now transcribed from a divergent promoter [185], is necessary to promote caspase-dependent TET2 degradation [186]. TET3, through the action of its own CxxC domain, is also regulated in a similar manner. These unexpected consequences of TET2 and TET3 chromatin recruitment are suggestive of interesting auto-regulatory loops to ensure the maintenance of hypomethylated genomic states.
4. Coordinate function of histone and DNA modifications in chromatin regulation
4.1. Multivalency in DNA and histone recognition
Significant insight into chromatin regulation has come from recent studies revealing that DNA and histone modifications are functioning in a cooperative manner to re-shape chromatin organization and facilitate the recruitment of effector proteins and their macromolecular complexes to discreet locations throughout the genome. Significantly, pioneering work of Kouzarides, Mann, and colleagues using a semi-synthetic mononucleosome assembly strategy coupled with quantitative MS has begun defining an interaction network of proteins and protein complexes that read both histone and DNA modification states [163]. CpG methylated or unmethylated DNAs wrapped around recombinant histone octamers containing uniformly modified H3K4me3, H3K9me3, or H3K27me3 were used as baits to enrich for interacting proteins from HeLa nuclear extracts. From these proteomic experiments, a rich list of factors whose interaction with chromatin may be directed by modifications on both histones and DNA has emerged [163]. For example, UHRF1 binding to H3K9me3-containing nucleosomes was enhanced by CpG methylation, consistent with the identified functions of its TTD-PHD histone binding module [104] and SRA domain [150–153]. In contrast, KDM2A binding to H3K9me3-containing nucleosomes was perturbed by CpG methylation, consistent with the identified function of its CxxC domain as a reader of unmethylated CpG dinucleotides [187]. Importantly, several protein complexes, including ORC and PRC2, were identified that appear to read simultaneous modification states on both histones and DNA, likely through distinct subunits. Indeed, the Mi-2/NuRD complex can sense methylated DNA through its MBD2 subunit and unmodified H3 N-terminal tails through its CHD4 [112] and CHD5 subunits [113], and the repressive function of the MBD2/CHD4-containing NuRD complex appears to be dependent on the coordinate function of these two interactions [188].
Another example of DNA and histone multivalency is the pre-initiation complex for RNAPII transcription. Here, the largest multi-subunit complex, TFIID, harbors the TATA-box binding protein (TBP) in addition to the TAF1 and TAF3 subunits that contain paired bromodomains and a PHD finger, respectively. A landmark study by Tjian and colleagues in 2000 identified the bromodomains of TAF1 to be dual histone acetyllysine readers [32] – thus providing an expanded mechanism by which TFIID may be recruited and/or stabilized at gene promoters. This mechanism may be particularly important in proper positioning of TFIID at TATA-less promoters that do not utilize TBP. In addition to the TAF1 interaction with histone acetylation, recent SILAC coupled with MS analysis revealed that the PHD finger of TAF3 is a specific reader of H3K4me3 [189] – a mark found at the +1 nucleosome along with H3/H4 acetylation. Significantly, recent studies have shown that TAF3 interaction with H3K4me3 is required for proper gene activation [190]. Taken together, these data underscore the role of multivalent histone and DNA interactions in creating a precise epigenetic signature that is read by the RNAPII transcription machinery.
4.2. Multivalency in the generation of defined epigenetic landscapes
Combinatorial DNA and histone modifications also contribute to the diversity of epigenetic landscapes and chromatin states by influencing the generation and removal of distant marks. Several recent examples of multivalent mechanisms underlying the creation of defined chromatin signatures on histones and DNA are discussed below.
The Jumonji C (JmjC) domain-containing histone lysine demethylase KDM2A catalyzes the removal of H3K36me2 [191]. The identified function of its CxxC domain as a reader of unmethylated CpG dinucleotides [187] suggests that DNA recognition may be important for cellular KDM2A catalytic activity. Indeed, this was shown to be the case [187], providing a likely explanation for why CpG island promoters are devoid of H3K36me2. Likewise, CxxC-mediated recognition of unmethylated CpG islands by SET1-associated CFP1 is necessary to prevent the spread of H3K4me3 to distal regulatory elements [192].
Recent studies also demonstrate that the establishment, maintenance, and removal of DNA methylation is intricately linked to the recognition of histone PTMs. Parental DNA methylation patterns, largely erased between fertilization and the blastocyst stage, are re-established during early embryogenesis by the de novo DNA methyltransferases DNMT3A and DNMT3B (with the associated, but non-catalytic DNMT3L) [193], and are stably maintained through somatic cell divisions [194] primarily by the maintenance methyltransferase DNMT1 [195] (Fig. 3). Waves of DNA methylation reprogramming also occur through germ cell development and carcinogenesis, recently reviewed elsewhere [196, 197]. DNMT3A, DNMT3B and DNMT3L share a cysteine-rich ADD domain, aptly named for the four proteins identified to harbor this motif (ATRX-DNMT3A/B-DNMT3L), that binds the unmodified N-terminus of histone H3 [198– 200]. Furthermore, in vitro catalysis of DNA methylation by DNMT3A/DNMT3L on linker DNA of semi-synthetic nucleosomal arrays was disrupted when nucleosomes harbored H3K4me2 or H3K4me3, suggesting a role for the ADD domain in DNMT3A/DNMT3L recruitment [201] (Fig. 3). While these data are consistent with the anti-correlative genome-wide distribution of DNA methylation and H3K4me3, thebiologicalsignificance of these putative recruitment mechanisms remains to be determined. Interestingly, DNMT3A and DNMT3B also harbor PWWP domains essential for chromatin targeting in cells [202–204], and biochemical studies suggest the PWWP domain of DNMT3A reads H3K36me3 [205]. Given that H3K36me3 is a histone PTM associated with transcription elongation and is found within the bodies of RNAPII-regulated genes, it has been speculated that DNMT3A/B contributes to gene-body DNA methylation that widely occurs but is poorly understood [206]. Indeed, it was recently shown in mouse ESCs that DNMT3L is a positive regulator of gene body DNA methylation within active genes [207]. Interestingly, this same study showed that a physical interaction between DNMT3L and the EZH2 subunit of the PRC2 complex ensures that poised bivalent promoters remain hypomethylated.
Unlike the de novo methyltransferases, mechanisms facilitating the recruitment of the maintenance methyltransferase DNMT1 to chromatin have been somewhat elusive. A replication foci targeting sequence (RFTS) facilitates the interaction of DNMT1 with proliferating cell nuclear antigen (PCNA) at replication forks [208]. However, disruption of this interaction has a minimal effect on cellular DNA methylation [209]. The identification of a CxxC domain in DNMT1 that binds unmethylated CpG dinucleotides and is necessary in cells for enzymatic function [210] suggests an interesting interplay between the ability of DNMT1 to sense both unmethylated and hemi-methylated DNA, the latter of which appears to be an inherent property of its catalytic domain [208, 211, 212]. Interestingly, recent structural studies suggest that these unbound domains function as auto-regulators of DNMT1 activity, as both an acidic loop C-terminal to the CxxC domain and the RFTS domain limit catalytic function of the enzyme by inserting into the active site [213, 214]. Recent studies linking UHRF1 genetically and physically to DNMT1 chromatin targeting and activity [150–153, 159, 160] strongly suggest that UHRF1 modulates DNMT1 chromatin function. Excitingly, it was recently shown that the UHRF1 RING domain has E3 ubiquitin ligase activity towards H3K23 [161]. H3K23 monoubiquitination was identified as being read by DNMT1 in an RFTS-dependent manner, providing an unexpected mechanism by which a histone modification facilitates DNA methylation maintenance (Fig. 3). This is one of the few examples of how ubiquitination on a histone may be driving recruitment of a chromatin modifier, and further, underscores the more general potential for how histone monoubiquitination may serve as a binding platform, as is found for monoubiquitinated proteins in cell signaling [215].
Aided by important technological advances in both sample collection and genome-wide mapping capabilities, the dynamics of epigenetic reprogramming in the early stages of development have been re-examined extensively over the past several years (for recent in-depth reviews, see [193, 197]). Intriguingly, chromatin-templated mechanisms regulating these impressive dynamics of DNA methylation during embryogenesis are being uncovered. The time-scale in which DNA methylation is lost in the paternal and maternal nuclei suggests that both active and passive removal mechanisms, respectively, are in place (Fig. 3). Indeed, recognition of H3K9me2 by the maternal factor PGC7 is necessary to protect maternal 5mC from TET3-mediated oxidative demethylation [216, 217] (Fig. 3). Although paternal nuclei also express PGC7, sperm DNA is packaged with protamines rather than histones, and therefore lack the ability to recruit PGC7, resulting in active TET3. In addition, Dnmt1 and Np95 (mouse UHRF1) are excluded from maternal nuclei and primordial germ cells, accounting for the passive loss of DNA methylation in these cells [197, 218]. Understanding the relationship between PGC7 and TET3, the structural basis for PGC7 recognition of H3K9me2, and the signals controlling the subcellular localization of Dnmt1 and Np95 at specific cell stages will be required to fully appreciate these key developmental regulatory mechanisms.
5. Future challenges towards understanding chromatin regulation
Similar to histone PTMs, we are still just scratching the surface of understanding how key players in the generation, removal, and interpretation of DNA modifications are regulated in the context of the epigenetic landscape. To fully grasp the depth and breadth of complexity underlying how histone and DNA modifications function together, many remaining unanswered questions must be tested experimentally. A number of immediate future challenges towards interpreting the complex epigenetic language are discussed below.
5.1. Determine the influence ofsequence context on the recognition of DNA modifications
Compared to histone residues, the sequence context surrounding CpG dinucleotides throughout the genome is enormous. This diversity alone suggests the rules governing DNA modification interactions will be more challenging to decipher than histone PTM interactions. Indeed, several recent studies suggest that, similar to histone PTM recognition and transcription factor binding, DNA readers have the ability to differentially sense the sequence surrounding a modified CpG. For example, in the above-described semi-synthetic nucleosome strategy for identifying dual readers of histone and DNA modifications, two DNA sequences with similar nucleosome forming properties but differing sequences [219] were used to assemble nucleosomes [163] for MS enrichment. Interestingly, factors were identified that bound in a sequence-dependent and independent manner, suggesting that some, but not all CpG readers are influenced by sequence context. Moreover, recent analysis of transcription factor binding to 154 unique CpG methylated DNA sequences demonstrated that most of these newly-identified CpG readers bound methylated DNA in a sequence-dependent fashion [165]. Differing sequence preferences have also been documented for several MBD proteins [220, 221], a property to consider when using these domains as methylated CpG affinity enrichment tools. This poses intriguing questions regarding the rules that dictate whether specific neighboring nucleotides (or combinations thereof) attract or repel CpG readers. The already realized level of apparent sequence preference suggests that there is minimal redundancy in the DNA modification language. Rather, in parallel to the original postulate of the ‘histone code,’ CpG modifications at defined genomic locations may also direct distinct biological outcomes.
5.2. Definethe structural families of DNA modification readers
Our understanding of functions associated with histone PTMs has been facilitated, in part, by detailed structural and biochemical characterization of the protein domains that bind to histone modifications [33, 34]. Undoubtedly, atomic resolution structural mapping of binding interactions for newly identified DNA modification readers will facilitate our understanding of how DNA modifications are read and interpreted. While a number of these proteins harbor DNA binding domains commonly found in transcription factors, including zinc fingers, homeoboxes, and helix-loop-helix domains [37, 165], it is likely that many newly discovered DNA modification readers will engage their mark through novel motifs. Excitingly, a number of identified DNA modification readers also harbor known or putative histone binding domains. For example, death inducer obliterator (DIDO) isoforms contain PHD fingers shown to read H3K4me3 [116, 117, 163]. The recent identification of DIDO1 as a sequence-specific reader of CpG methylation [165] poses an interesting question regarding the contributions of these DNA and histone reading activities to DIDO functions. Indeed, determining how proteins interact with CpG modifications, as well as cytosine modifications in other, lesser-studied, sequence contexts [222], will ultimately lead to a deeper understanding of known and novel biological functions of these marks.
5.3. Complete the dictionary of chromatin modifications
Drawing the analogy of DNA and histone modifications being akin to letters of the alphabet, we would be unable to form words and sentences like transcriptional activation or DNA repair without first learning the identity of all of the letters in the alphabet as well as the order in which to place them. Therefore, crucial to our future understanding of chromatin function will be the identification of the full complement of DNA and histone modifications and their combinations, as well as their precise locations throughout the genome.
While much of the PTM landscape on the N-termini of histones have been defined, we surprisingly still know very little about the identity and combinations of PTMs present on histone C-termini, globular domains, and variant histones. Indeed, important functions associated with modifications to these less-studied regions of histones are continually emerging. For example, Zhang and colleagues recently used an elegant combination of yeast genetics and biochemistry to identify a conserved mechanism by which H3K56ac promotes Rtt101-dependent ubiquitination of three lysines in the H3 C-terminal tail (K121, K122, and K125) [223]. This ubiquitination promotes the necessary transfer of H3-H4 dimers between the histone chaperones Asf1 and RTT106/CAF-1 for nucleosome assembly and reveals novel crosstalk between the histone ubiquitination and acetylation machinery beyond competition for the same residue.
Proteomic technologies are also advancing to the point where comprehensive analysis of all core histones and their variants is now possible[36, 39, 42]. As new types of histone PTMs will likely continue to be identified, a full understanding of epigenetic regulation will require knowledge of how these PTMs function, and how they contribute antagonistically or synergistically with known PTMs to regulate the distinct functions associated with them. Likewise, the recent discovery of oxidized derivatives of CpG methylation suggests that the dictionary of DNA modifications is also incomplete. For example, the existence of CpA, CpT, and CpC methylation has been known for some time now [222], but it is unclear whether these modifications are also susceptible to oxidative demethylation.
5.4. Define the modification landscape at individual genomic loci
Another significant set of challenges we face is to develop new technologies that enable characterization of the PTM landscape at defined genomic loci and to determine how this landscape contributes to the architecture and function at these regions, through specific cell stages, cycles, and developmental contexts. From a practical standpoint, knowing this information will be key to understanding of how deregulation of the epigenetic machinery contributes to the re-wiring of the epigenetic landscape in human disease.
Several recent studies have indeed begun addressing this challenge using distinct approaches. Notably, Tackett and colleagues recently developed a technique they call Chromatin Affinity Purification with Mass Spectrometry (ChAP-MS) that allows for enrichment of native ~ 1 kilobase sections of chromatin for site-specific identification of chromatin-associated proteins and histone PTMs [224]. While powerful, this technique requires insertion of a LexA DNA binding site into the genomic region of interest for site-specific targeting of the LexA protein, and is therefore limited to routine use in genetically tractable systems. An important modification to this technique circumvents this problem by using transcription activator-like (TAL) effector proteins as targeting molecules [225]. TALs can easily be designed to target any ~ 20-nucleotide genomic locus of interest without genetic manipulation, and are therefore a powerful tool for site-specific high-resolution genomic targeting. This modified technique was used to define the chromatin architecture of the GAL1 gene in Sacchromyces cerevisae, which was found to be enriched for a number of single histone PTMs, including H3K14ac, H3K56ac, all three states of H3K79 methylation, H2BK17ac, and H2AK7ac [225]. A number of di-acetyl histone PTM combinations were also found at this locus. Furthermore, over 50 unique proteins were identified to reside at the GAL1 gene, including a number previously identified transcriptional regulators like the Spt16 subunit of the FACT transcription elongation complex and Gal3.
Recent studies have also begun defining with high-resolution the nucleosomal landscapes occupied by chromatin-associated proteins in an unbiased manner [226, 227]. One notable study coupled the powerful approaches of mononucleosome-resolution ChIP coupled with next-generation DNA sequencing (Mnase-ChIP-seq), RNA sequencing, and MS to simultaneously reveal 1) the combinations of histone PTMs associated with HP1 isoforms and several members of the bromodomain and extraterminal family (BET), 2) the genomic loci harboring these nucleosomes, and 3) the transcriptional state of these nucleosomes [226]. BET family proteins (Brd2, Brd3, and Brd4) were enriched at active gene promoters predominantly harboring H3K4me3 in combination with K9ac, K14ac, and K23ac, as well as H4K5ac in combination with K12ac and K16ac. Gene bodies were also enriched for BET family proteins, and histones in these regions were predominantly marked with H3K4me1 in combination with K14ac, K23ac, and K79me1 as well as H4K8ac in combination with K12ac, K16ac, and K20me1. Interestingly, a large number of gene promoters occupied by BET family proteins were the key developmental HOX genes, although the functional significance of this intriguing finding is not understood. Approaches like this are an important step towards understanding how chromatin-associated proteins read and translate the epigenetic language, and future studies incorporating DNA modification analyses will give an even more complete picture of how histone and DNA modification states coordinate functional outcomes in defined regions of chromatin.
5.5. Assign function to chromatin modifications
As we continue to discover new chromatin modifications, their combinations, genomic locations, and protein readers, a key challenge moving forward is to define how any particular modification, on DNA or histones, tips the balance towards distinct biological outcomes. Our ability to now site-specifically modify the chromatin landscape in complex eukaryotic systems using re-purposed genome editing tools like zinc-finger (ZF) nuclease, transcription activatorlike effector (TALE) nuclease, and clustered regulatory interspaced short palindromic repeat (CRISPR) technologies [228–231] is an important step towards tackling this central question. In addition, chemical-induced [56] and light-induced [232] chromatin targeting approaches also allow the direct examination of the functional consequences of DNA and histone modifications. For example, Crabtree and colleagues elegantly adapted a chemical-induced proximity system for precision targeting of chromatin modifiers [56], whereby stimulation by a bi-functional small molecule causes the rapid, reversible association of two different peptide tags fused to proteins of interest in the cell. They then engineered the mouse Oct4 promoter to harbor ZFHD1 and GAL4 DNA binding sites in-frame with a GFP reporter. By tethering ZFHD1 to FKBP, and the chromoshadow domain of HP1α (which directly interacts with the H3K9 methyltransferases SUV39h1/2 and SETDB1) to Frb, they were able to use rapamycin to promote an interaction between FKBP and Frb, which resulted in the recruitment of HP1α and H3K9 methylation to the engineered ZFHD1 binding site of Oct4. Excitingly, this resulted in a cascade of events, including the spread of H3K9me3, the loss of H3K4me3, and the gradual increase in DNA methylation that collectively created a new, stable domain of transcriptionally repressed heterochromatin even in the absence of continued HP1α targeting. Importantly, they showed that the kinetics of DNA methylation at this locus trailed the kinetics of H3K9 methylation and discovered that the maintenance of the repressed domain in the absence of further HP1α targeting was dependent on DNA methylation at this locus. Interestingly, by tethering the transcriptional activator VP16 to PYL1, and GAL4 to ABI1, they were similarly able to recruit VP16 to the GAL4 DNA binding site at this repressed Oct4 locus by addition of abscisic acid, which resulted in reactivation of silenced GFP. This result suggests that robust transcriptional activation can outcompete the signals of heterochromatin, and the authors propose that DNA methylation at the Oct4 promoter may function to maintain this heterochromatin domain by directly preventing the binding of transcription factors.
6. Concluding remarks
Progress over the past several decades has ushered in a new wave of appreciation for the role of histone modifications in diverse chromatin functions. The complexity of histone modifications in regards to the sheer number of identified PTMs, alone and in combination, is astonishing in itself. The discovery of proteins harboring multiple reader domains that interpret these combinatorial PTMs on histones to perform their chromatin functions suggests our original one modification-one domain model underestimates the real situation and complexity of histone function. The discovery of new protein families that read DNA modification states and patterns of histone and DNA modification embedded in nucleosomes places the ‘histone code’ within the framework of a much broader language of epigenetic modifications. Taking into account known single PTMs on the four core histone tail domains, occurring symmetrically or asymmetrically, in conjunction with the known single modification states of CpG dinucleotides, regardless of DNA sequence context, would imply the potential for an enormous number of unique chromatin signatures throughout the genome. Even if only a fraction of these combinations contribute to biological functions of chromatin, we will surely stay busy decoding the astonishing language of histone and DNA modifications for years to come.
HIGHLIGHTS.
Histone PTMs and DNA modifications coordinately regulate chromatin function
Newly revealed modifications add distinct elements of regulatory control on the chromatin template
Aided by new technologies, a newfound appreciation for the complexities of DNA and histone recognition is emerging
Acknowledgements
We thank C. David Allis for his insightful discussions, suggestions, and critical reading of the manuscript. We also thank members of the Strahl Lab, Zu-Wen Sun, Jean Cook, and Lindsey Rizzardi for their helpful comments and suggestions, and Alexey Soshnev from the Allis lab for sharing his cartoon rendering that served as the basis for Fig. 2. We apologize to authors whose contributions could not be acknowledged due to space limitations. S.B.R. is supported by a Pathway to Independence Award from the NIH (K99CA181343). B.D.S. is supported by grants from the NIH (R01DA036877 and R01DA036897)and the WM Keck Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. 1999;98:285–294. doi: 10.1016/s0092-8674(00)81958-3. [DOI] [PubMed] [Google Scholar]
- 2.van Holde KE. Chromatin. New York: Springer-Verlag; 1989. [Google Scholar]
- 3.Li G, Reinberg D. Chromatin higher-order structures and gene regulation. Curr Opin Genet Dev. 2011;21:175–186. doi: 10.1016/j.gde.2011.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luger K, Hansen JC. Nucleosome and chromatin fiber dynamics. Curr Opin Struct Biol. 2005;15:188–196. doi: 10.1016/j.sbi.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 5.Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12:7–18. doi: 10.1038/nrg2905. [DOI] [PubMed] [Google Scholar]
- 6.Harshman SW, Young NL, Parthun MR, Freitas MA. H1 histones: current perspectives and challenges. Nucleic Acids Res. 2013;41:9593–9609. doi: 10.1093/nar/gkt700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Talbert PB, Henikoff S. Histone variants--ancient wrap artists of the epigenome. Nat Rev Mol Cell Biol. 2010;11:264–275. doi: 10.1038/nrm2861. [DOI] [PubMed] [Google Scholar]
- 8.Narlikar GJ, Sundaramoorthy R, Owen-Hughes T. Mechanisms and functions of ATP-dependent chromatin-remodeling enzymes. Cell. 2013;154:490–503. doi: 10.1016/j.cell.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Avvakumov N, Nourani A, Cote J. Histone chaperones: modulators of chromatin marks. Mol Cell. 2011;41:502–514. doi: 10.1016/j.molcel.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 10.Burgess RJ, Zhang Z. Histone chaperones in nucleosome assembly and human disease. Nat Struct Mol Biol. 2013;20:14–22. doi: 10.1038/nsmb.2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13:343–357. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 14.Kohli RM, Zhang Y, TET enzymes. TDG and the dynamics of DNA demethylation. Nature. 2013;502:472–479. doi: 10.1038/nature12750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 16.Cosgrove MS, Boeke JD, Wolberger C. Regulated nucleosome mobility and the histone code. Nat Struct Mol Biol. 2004;11:1037–1043. doi: 10.1038/nsmb851. [DOI] [PubMed] [Google Scholar]
- 17.Cosgrove MS, Wolberger C. How does the histone code work? Biochem Cell Biol. 2005;83:468–476. doi: 10.1139/o05-137. [DOI] [PubMed] [Google Scholar]
- 18.Allfrey VG, Faulkner R, Mirsky AE. Acetylation and Methylation of Histones and Their Possible Role in the Regulation of Rna Synthesis. Proc Natl Acad Sci U S A. 1964;51:786–794. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gutierrez RM, Hnilica LS. Tissue specificity of histone phosphorylation. Science. 1967;157:1324–1325. doi: 10.1126/science.157.3794.1324. [DOI] [PubMed] [Google Scholar]
- 20.Brownell JE, Zhou J, Ranalli T, Kobayashi R, Edmondson DG, Roth SY, Allis CD. Tetrahymena histone acetyltransferase A: a homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell. 1996;84:843–851. doi: 10.1016/s0092-8674(00)81063-6. [DOI] [PubMed] [Google Scholar]
- 21.Taunton J, Hassig CA, Schreiber SL. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science. 1996;272:408–411. doi: 10.1126/science.272.5260.408. [DOI] [PubMed] [Google Scholar]
- 22.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 23.Turner BM. Histone acetylation and an epigenetic code. Bioessays. 2000;22:836–845. doi: 10.1002/1521-1878(200009)22:9<836::AID-BIES9>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 24.Bradbury EM. Reversible histone modifications and the chromosome cell cycle. Bioessays. 1992;14:9–16. doi: 10.1002/bies.950140103. [DOI] [PubMed] [Google Scholar]
- 25.Koshland D, Strunnikov A. Mitotic chromosome condensation. Annu Rev Cell Dev Biol. 1996;12:305–333. doi: 10.1146/annurev.cellbio.12.1.305. [DOI] [PubMed] [Google Scholar]
- 26.Mahadevan LC, Willis AC, Barratt MJ. Rapid histone H3 phosphorylation in response to growth factors, phorbol esters, okadaic acid, and protein synthesis inhibitors. Cell. 1991;65:775–783. doi: 10.1016/0092-8674(91)90385-c. [DOI] [PubMed] [Google Scholar]
- 27.Chadee DN, Hendzel MJ, Tylipski CP, Allis CD, Bazett-Jones DP, Wright JA, Davie JR. Increased Ser-10 phosphorylation of histone H3 in mitogen-stimulated and oncogene-transformed mouse fibroblasts. J Biol Chem. 1999;274:24914–24920. doi: 10.1074/jbc.274.35.24914. [DOI] [PubMed] [Google Scholar]
- 28.De Cesare D, Jacquot S, Hanauer A, Sassone-Corsi P. Rsk-2 activity is necessary for epidermal growth factor-induced phosphorylation of CREB protein and transcription of c-fos gene. Proc Natl Acad Sci U S A. 1998;95:12202–12207. doi: 10.1073/pnas.95.21.12202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 30.Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou MM. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999;399:491–496. doi: 10.1038/20974. [DOI] [PubMed] [Google Scholar]
- 31.Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Muller S, Pawson T, Gingras AC, Arrowsmith CH, Knapp S. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149:214–231. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jacobson RH, Ladurner AG, King DS, Tjian R. Structure and function of a human TAFII250 double bromodomain module. Science. 2000;288:1422–1425. doi: 10.1126/science.288.5470.1422. [DOI] [PubMed] [Google Scholar]
- 33.Musselman CA, Lalonde ME, Cote J, Kutateladze TG. Perceiving the epigenetic landscape through histone readers. Nat Struct Mol Biol. 2012;19:1218–1227. doi: 10.1038/nsmb.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patel DJ, Wang Z. Readout of epigenetic modifications. Annu Rev Biochem. 2013;82:81–118. doi: 10.1146/annurev-biochem-072711-165700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Voigt P, LeRoy G, Drury WJ, 3rd, Zee BM, Son J, Beck DB, Young NL, Garcia BA, Reinberg D. Asymmetrically modified nucleosomes. Cell. 2012;151:181–193. doi: 10.1016/j.cell.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arnaudo AM, Garcia BA. Proteomic characterization of novel histone post-translational modifications. Epigenetics Chromatin. 2013;6:24. doi: 10.1186/1756-8935-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spruijt CG, Gnerlich F, Smits AH, Pfaffeneder T, Jansen PW, Bauer C, Munzel M, Wagner M, Muller M, Khan F, Eberl HC, Mensinga A, Brinkman AB, Lephikov K, Muller U, Walter J, Boelens R, van Ingen H, Leonhardt H, Carell T, Vermeulen M. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152:1146–1159. doi: 10.1016/j.cell.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 38.Hake SB, Xiao A, Allis CD. Linking the epigenetic 'language' of covalent histone modifications to cancer. Br J Cancer. 2004;90:761–769. doi: 10.1038/sj.bjc.6601575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bartke T, Borgel J, DiMaggio PA. Proteomics in epigenetics: new perspectives for cancer research. Brief Funct Genomics. 2013;12:205–218. doi: 10.1093/bfgp/elt002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garcia BA, Pesavento JJ, Mizzen CA, Kelleher NL. Pervasive combinatorial modification of histone H3 in human cells. Nat Methods. 2007;4:487–489. doi: 10.1038/nmeth1052. [DOI] [PubMed] [Google Scholar]
- 41.Han Y, Garcia BA. Combining genomic and proteomic approaches for epigenetics research. Epigenomics. 2013;5:439–452. doi: 10.2217/epi.13.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Soldi M, Cuomo A, Bremang M, Bonaldi T. Mass spectrometry-based proteomics for the analysis of chromatin structure and dynamics. Int J Mol Sci. 2013;14:5402–5431. doi: 10.3390/ijms14035402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vermeulen M, Eberl HC, Matarese F, Marks H, Denissov S, Butter F, Lee KK, Olsen JV, Hyman AA, Stunnenberg HG, Mann M. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell. 2010;142:967–980. doi: 10.1016/j.cell.2010.08.020. [DOI] [PubMed] [Google Scholar]
- 44.Zhang L, Eugeni EE, Parthun MR, Freitas MA. Identification of novel histone post-translational modifications by peptide mass fingerprinting. Chromosoma. 2003;112:77–86. doi: 10.1007/s00412-003-0244-6. [DOI] [PubMed] [Google Scholar]
- 45.Zhang K, Tang H, Huang L, Blankenship JW, Jones PR, Xiang F, Yau PM, Burlingame AL. Identification of acetylation and methylation sites of histone H3 from chicken erythrocytes by highaccuracy matrix-assisted laser desorption ionization-time-of-flight, matrix-assisted laser desorption ionization-postsource decay, and nanoelectrospray ionization tandem mass spectrometry. Anal Biochem. 2002;306:259–269. doi: 10.1006/abio.2002.5719. [DOI] [PubMed] [Google Scholar]
- 46.Cosgrove MS. Histone proteomics and the epigenetic regulation of nucleosome mobility. Expert Rev Proteomics. 2007;4:465–478. doi: 10.1586/14789450.4.4.465. [DOI] [PubMed] [Google Scholar]
- 47.Cocklin RR, Wang M. Identification of methylation and acetylation sites on mouse histone H3 using matrix-assisted laser desorption/ionization time-of-flight and nanoelectrospray ionization tandem mass spectrometry. J Protein Chem. 2003;22:327–334. doi: 10.1023/a:1025334006014. [DOI] [PubMed] [Google Scholar]
- 48.Neumann H, Hancock SM, Buning R, Routh A, Chapman L, Somers J, Owen-Hughes T, van Noort J, Rhodes D, Chin JW. A method for genetically installing site-specific acetylation in recombinant histones defines the effects of H3 K56 acetylation. Mol Cell. 2009;36:153–163. doi: 10.1016/j.molcel.2009.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Manohar M, Mooney AM, North JA, Nakkula RJ, Picking JW, Edon A, Fishel R, Poirier MG, Ottesen JJ. Acetylation of histone H3 at the nucleosome dyad alters DNA-histone binding. J Biol Chem. 2009;284:23312–23321. doi: 10.1074/jbc.M109.003202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.North JA, Javaid S, Ferdinand MB, Chatterjee N, Picking JW, Shoffner M, Nakkula RJ, Bartholomew B, Ottesen JJ, Fishel R, Poirier MG. Phosphorylation of histone H3(T118) alters nucleosome dynamics and remodeling. Nucleic Acids Res. 2011;39:6465–6474. doi: 10.1093/nar/gkr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Simon M, North JA, Shimko JC, Forties RA, Ferdinand MB, Manohar M, Zhang M, Fishel R, Ottesen JJ, Poirier MG. Histone fold modifications control nucleosome unwrapping and disassembly. Proc Natl Acad Sci U S A. 2011;108:12711–12716. doi: 10.1073/pnas.1106264108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shimko JC, North JA, Bruns AN, Poirier MG, Ottesen JJ. Preparation of fully synthetic histone H3 reveals that acetyl-lysine 56 facilitates protein binding within nucleosomes. J Mol Biol. 2011;408:187–204. doi: 10.1016/j.jmb.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Watanabe S, Resch M, Lilyestrom W, Clark N, Hansen JC, Peterson C, Luger K. Structural characterization of H3K56Q nucleosomes and nucleosomal arrays. Biochim Biophys Acta. 2010;1799:480–486. doi: 10.1016/j.bbagrm.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tropberger P, Pott S, Keller C, Kamieniarz-Gdula K, Caron M, Richter F, Li G, Mittler G, Liu ET, Buhler M, Margueron R, Schneider R. Regulation of transcription through acetylation of H3K122 on the lateral surface of the histone octamer. Cell. 2013;152:859–872. doi: 10.1016/j.cell.2013.01.032. [DOI] [PubMed] [Google Scholar]
- 55.Dawson MA, Bannister AJ, Gottgens B, Foster SD, Bartke T, Green AR, Kouzarides T. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature. 2009;461:819–822. doi: 10.1038/nature08448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hathaway NA, Bell O, Hodges C, Miller EL, Neel DS, Crabtree GR. Dynamics and memory of heterochromatin in living cells. Cell. 2012;149:1447–1460. doi: 10.1016/j.cell.2012.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jacobs SA, Khorasanizadeh S. Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail. Science. 2002;295:2080–2083. doi: 10.1126/science.1069473. [DOI] [PubMed] [Google Scholar]
- 58.Casadio F, Lu X, Pollock SB, LeRoy G, Garcia BA, Muir TW, Roeder RG, Allis CD. H3R42me2a is a histone modification with positive transcriptional effects. Proc Natl Acad Sci U S A. 2013;110:14894–14899. doi: 10.1073/pnas.1312925110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, Lu Z, Ye Z, Zhu Q, Wysocka J, Ye Y, Khochbin S, Ren B, Zhao Y. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146:1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Montellier E, Rousseaux S, Zhao Y, Khochbin S. Histone crotonylation specifically marks the haploid male germ cell gene expression program: post-meiotic male-specific gene expression. Bioessays. 2012;34:187–193. doi: 10.1002/bies.201100141. [DOI] [PubMed] [Google Scholar]
- 61.Sin HS, Barski A, Zhang F, Kartashov AV, Nussenzweig A, Chen J, Andreassen PR, Namekawa SH. RNF8 regulates active epigenetic modifications and escape gene activation from inactive sex chromosomes in post-meiotic spermatids. Genes Dev. 2012;26:2737–2748. doi: 10.1101/gad.202713.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Montellier E, Boussouar F, Rousseaux S, Zhang K, Buchou T, Fenaille F, Shiota H, Debernardi A, Hery P, Curtet S, Jamshidikia M, Barral S, Holota H, Bergon A, Lopez F, Guardiola P, Pernet K, Imbert J, Petosa C, Tan M, Zhao Y, Gerard M, Khochbin S. Chromatin-to-nucleoprotamine transition is controlled by the histone H2B variant TH2B. Genes Dev. 2013;27:1680–1692. doi: 10.1101/gad.220095.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kreppel LK, Blomberg MA, Hart GW. Dynamic glycosylation of nuclear and cytosolic proteins. Cloning and characterization of a unique O-GlcNAc transferase with multiple tetratricopeptide repeats. J Biol Chem. 1997;272:9308–9315. doi: 10.1074/jbc.272.14.9308. [DOI] [PubMed] [Google Scholar]
- 64.Lubas WA, Frank DW, Krause M, Hanover JA. O-Linked GlcNAc transferase is a conserved nucleocytoplasmic protein containing tetratricopeptide repeats. J Biol Chem. 1997;272:9316–9324. doi: 10.1074/jbc.272.14.9316. [DOI] [PubMed] [Google Scholar]
- 65.Gao Y, Wells L, Comer FI, Parker GJ, Hart GW. Dynamic O-glycosylation of nuclear and cytosolic proteins: cloning and characterization of a neutral, cytosolic beta-N-acetylglucosaminidase from human brain. J Biol Chem. 2001;276:9838–9845. doi: 10.1074/jbc.M010420200. [DOI] [PubMed] [Google Scholar]
- 66.Comtesse N, Maldener E, Meese E. Identification of a nuclear variant of MGEA5, a cytoplasmic hyaluronidase and a beta-N-acetylglucosaminidase. Biochem Biophys Res Commun. 2001;283:634–640. doi: 10.1006/bbrc.2001.4815. [DOI] [PubMed] [Google Scholar]
- 67.Bond MR, Hanover JA. O-GlcNAc cycling: a link between metabolism and chronic disease. Annu Rev Nutr. 2013;33:205–229. doi: 10.1146/annurev-nutr-071812-161240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hanover JA, Krause MW, Love DC. Bittersweet memories: linking metabolism to epigenetics through O-GlcNAcylation. Nat Rev Mol Cell Biol. 2012;13:312–321. doi: 10.1038/nrm3334. [DOI] [PubMed] [Google Scholar]
- 69.Love DC, Ghosh S, Mondoux MA, Fukushige T, Wang P, Wilson MA, Iser WB, Wolkow CA, Krause MW, Hanover JA. Dynamic O-GlcNAc cycling at promoters of Caenorhabditis elegans genes regulating longevity, stress, and immunity. Proc Natl Acad Sci U S A. 2010;107:7413–7418. doi: 10.1073/pnas.0911857107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gambetta MC, Oktaba K, Muller J. Essential role of the glycosyltransferase sxc/Ogt in polycomb repression. Science. 2009;325:93–96. doi: 10.1126/science.1169727. [DOI] [PubMed] [Google Scholar]
- 71.Sinclair DA, Syrzycka M, Macauley MS, Rastgardani T, Komljenovic I, Vocadlo DJ, Brock HW, Honda BM. Drosophila O-GlcNAc transferase (OGT) is encoded by the Polycomb group (PcG) gene, super sex combs (sxc) Proc Natl Acad Sci U S A. 2009;106:13427–13432. doi: 10.1073/pnas.0904638106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kelly WG, Dahmus ME, Hart GW. RNA polymerase II is a glycoprotein. Modification of the COOH-terminal domain by O-GlcNAc. J Biol Chem. 1993;268:10416–10424. [PubMed] [Google Scholar]
- 73.Comer FI, Hart GW. Reciprocity between O-GlcNAc and O-phosphate on the carboxyl terminal domain of RNA polymerase II. Biochemistry. 2001;40:7845–7852. doi: 10.1021/bi0027480. [DOI] [PubMed] [Google Scholar]
- 74.Fujiki R, Chikanishi T, Hashiba W, Ito H, Takada I, Roeder RG, Kitagawa H, Kato S. GlcNAcylation of a histone methyltransferase in retinoic-acid-induced granulopoiesis. Nature. 2009;459:455–459. doi: 10.1038/nature07954. [DOI] [PubMed] [Google Scholar]
- 75.Zhang S, Roche K, Nasheuer HP, Lowndes NF. Modification of histones by sugar beta-Nacetylglucosamine (GlcNAc) occurs on multiple residues, including histone H3 serine 10, and is cell cycleregulated. J Biol Chem. 2011;286:37483–37495. doi: 10.1074/jbc.M111.284885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fujiki R, Hashiba W, Sekine H, Yokoyama A, Chikanishi T, Ito S, Imai Y, Kim J, He HH, Igarashi K, Kanno J, Ohtake F, Kitagawa H, Roeder RG, Brown M, Kato S. GlcNAcylation of histone H2B facilitates its monoubiquitination. Nature. 2011;480:557–560. doi: 10.1038/nature10656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen Q, Chen Y, Bian C, Fujiki R, Yu X. TET2 promotes histone O-GlcNAcylation during gene transcription. Nature. 2013;493:561–564. doi: 10.1038/nature11742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen Y, Sprung R, Tang Y, Ball H, Sangras B, Kim SC, Falck JR, Peng J, Gu W, Zhao Y. Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol Cell Proteomics. 2007;6:812–819. doi: 10.1074/mcp.M700021-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang K, Chen Y, Zhang Z, Zhao Y. Identification and verification of lysine propionylation and butyrylation in yeast core histones using PTMap software. J Proteome Res. 2009;8:900–906. doi: 10.1021/pr8005155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kaelin WG, Jr., McKnight SL. Influence of metabolism on epigenetics and disease. Cell. 2013;153:56–69. doi: 10.1016/j.cell.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gut P, Verdin E. The nexus of chromatin regulation and intermediary metabolism. Nature. 2013;502:489–498. doi: 10.1038/nature12752. [DOI] [PubMed] [Google Scholar]
- 82.Vollmuth F, Geyer M. Interaction of propionylated and butyrylated histone H3 lysine marks with Brd4 bromodomains. Angew Chem Int Ed Engl. 2010;49:6768–6772. doi: 10.1002/anie.201002724. [DOI] [PubMed] [Google Scholar]
- 83.Young NL, DiMaggio PA, Plazas-Mayorca MD, Baliban RC, Floudas CA, Garcia BA. High throughput characterization of combinatorial histone codes. Mol Cell Proteomics. 2009;8:2266–2284. doi: 10.1074/mcp.M900238-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tweedie-Cullen RY, Brunner AM, Grossmann J, Mohanna S, Sichau D, Nanni P, Panse C, Mansuy IM. Identification of combinatorial patterns of post-translational modifications on individual histones in the mouse brain. PLoS One. 2012;7:e36980. doi: 10.1371/journal.pone.0036980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Taverna SD, Ueberheide BM, Liu Y, Tackett AJ, Diaz RL, Shabanowitz J, Chait BT, Hunt DF, Allis CD. Long-distance combinatorial linkage between methylation and acetylation on histone H3 N termini. Proc Natl Acad Sci U S A. 2007;104:2086–2091. doi: 10.1073/pnas.0610993104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hazzalin CA, Mahadevan LC. Dynamic acetylation of all lysine 4-methylated histone H3 in the mouse nucleus: analysis at c-fos and c-jun. PLoS Biol. 2005;3:e393. doi: 10.1371/journal.pbio.0030393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bock I, Kudithipudi S, Tamas R, Kungulovski G, Dhayalan A, Jeltsch A. Application of Celluspots peptide arrays for the analysis of the binding specificity of epigenetic reading domains to modified histone tails. BMC Biochem. 2011;12:48. doi: 10.1186/1471-2091-12-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bua DJ, Kuo AJ, Cheung P, Liu CL, Migliori V, Espejo A, Casadio F, Bassi C, Amati B, Bedford MT, Guccione E, Gozani O. Epigenome microarray platform for proteome-wide dissection of chromatin-signaling networks. PLoS One. 2009;4:e6789. doi: 10.1371/journal.pone.0006789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fuchs SM, Krajewski K, Baker RW, Miller VL, Strahl BD. Influence of combinatorial histone modifications on antibody and effector protein recognition. Curr Biol. 2011;21:53–58. doi: 10.1016/j.cub.2010.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nady N, Min J, Kareta MS, Chedin F, Arrowsmith CH. A SPOT on the chromatin landscape? Histone peptide arrays as a tool for epigenetic research. Trends Biochem Sci. 2008;33:305–313. doi: 10.1016/j.tibs.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 91.Rathert P, Dhayalan A, Murakami M, Zhang X, Tamas R, Jurkowska R, Komatsu Y, Shinkai Y, Cheng X, Jeltsch A. Protein lysine methyltransferase G9a acts on non-histone targets. Nat Chem Biol. 2008;4:344–346. doi: 10.1038/nchembio.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rothbart SB, Krajewski K, Strahl BD, Fuchs SM. Peptide microarrays to interrogate the “histone code”. Methods Enzymol. 2012;512:107–135. doi: 10.1016/B978-0-12-391940-3.00006-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fuchs SM, Strahl BD. Antibody recognition of histone post-translational modifications: emerging issues and future prospects. Epigenomics. 2011;3:247–249. doi: 10.2217/epi.11.23. [DOI] [PubMed] [Google Scholar]
- 94.Di Croce L, Helin K. Transcriptional regulation by Polycomb group proteins. Nat Struct Mol Biol. 2013;20:1147–1155. doi: 10.1038/nsmb.2669. [DOI] [PubMed] [Google Scholar]
- 95.Beck DB, Oda H, Shen SS, Reinberg D. PR-Set7 and H4K20me1: at the crossroads of genome integrity, cell cycle, chromosome condensation, and transcription. Genes Dev. 2012;26:325–337. doi: 10.1101/gad.177444.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Voigt P, Tee WW, Reinberg D. A double take on bivalent promoters. Genes Dev. 2013;27:1318–1338. doi: 10.1101/gad.219626.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schmitges FW, Prusty AB, Faty M, Stutzer A, Lingaraju GM, Aiwazian J, Sack R, Hess D, Li L, Zhou S, Bunker RD, Wirth U, Bouwmeester T, Bauer A, Ly-Hartig N, Zhao K, Chan H, Gu J, Gut H, Fischle W, Muller J, Thoma NH. Histone methylation by PRC2 is inhibited by active chromatin marks. Mol Cell. 2011;42:330–341. doi: 10.1016/j.molcel.2011.03.025. [DOI] [PubMed] [Google Scholar]
- 98.Ruthenburg AJ, Li H, Patel DJ, Allis CD. Multivalent engagement of chromatin modifications by linked binding modules. Nat Rev Mol Cell Biol. 2007;8:983–994. doi: 10.1038/nrm2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ali M, Yan K, Lalonde ME, Degerny C, Rothbart SB, Strahl BD, Cote J, Yang XJ, Kutateladze TG. Tandem PHD fingers of MORF/MOZ acetyltransferases display selectivity for acetylated histone H3 and are required for the association with chromatin. J Mol Biol. 2012;424:328–338. doi: 10.1016/j.jmb.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Arita K, Isogai S, Oda T, Unoki M, Sugita K, Sekiyama N, Kuwata K, Hamamoto R, Tochio H, Sato M, Ariyoshi M, Shirakawa M. Recognition of modification status on a histone H3 tail by linked histone reader modules of the epigenetic regulator UHRF1. Proc Natl Acad Sci U S A. 2012;109:12950–12955. doi: 10.1073/pnas.1203701109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dreveny I, Deeves SE, Fulton J, Yue B, Messmer M, Bhattacharya A, Collins HM, Heery DM. The double PHD finger domain of MOZ/MYST3 induces alpha-helical structure of the histone H3 tail to facilitate acetylation and methylation sampling and modification. Nucleic Acids Res. 2013 doi: 10.1093/nar/gkt931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Eustermann S, Yang JC, Law MJ, Amos R, Chapman LM, Jelinska C, Garrick D, Clynes D, Gibbons RJ, Rhodes D, Higgs DR, Neuhaus D. Combinatorial readout of histone H3 modifications specifies localization of ATRX to heterochromatin. Nat Struct Mol Biol. 2011;18:777–782. doi: 10.1038/nsmb.2070. [DOI] [PubMed] [Google Scholar]
- 103.Qiu Y, Liu L, Zhao C, Han C, Li F, Zhang J, Wang Y, Li G, Mei Y, Wu M, Wu J, Shi Y. Combinatorial readout of unmodified H3R2 and acetylated H3K14 by the tandem PHD finger of MOZ reveals a regulatory mechanism for HOXA9 transcription. Genes Dev. 2012;26:1376–1391. doi: 10.1101/gad.188359.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rothbart SB, Dickson BM, Ong MS, Krajewski K, Houliston S, Kireev DB, Arrowsmith CH, Strahl BD. Multivalent histone engagement by the linked tandem Tudor and PHD domains of UHRF1 is required for the epigenetic inheritance of DNA methylation. Genes Dev. 2013;27:1288–1298. doi: 10.1101/gad.220467.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tsai WW, Wang Z, Yiu TT, Akdemir KC, Xia W, Winter S, Tsai CY, Shi X, Schwarzer D, Plunkett W, Aronow B, Gozani O, Fischle W, Hung MC, Patel DJ, Barton MC. TRIM24 links a non-canonical histone signature to breast cancer. Nature. 2010;468:927–932. doi: 10.1038/nature09542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Xi Q, Wang Z, Zaromytidou AI, Zhang XH, Chow-Tsang LF, Liu JX, Kim H, Barlas A, Manova-Todorova K, Kaartinen V, Studer L, Mark W, Patel DJ, Massague J. A poised chromatin platform for TGF-beta access to master regulators. Cell. 2011;147:1511–1524. doi: 10.1016/j.cell.2011.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nady N, Lemak A, Walker JR, Avvakumov GV, Kareta MS, Achour M, Xue S, Duan S, Allali-Hassani A, Zuo X, Wang YX, Bronner C, Chedin F, Arrowsmith CH, Dhe-Paganon S. Recognition of multivalent histone states associated with heterochromatin by UHRF1 protein. J Biol Chem. 2011;286:24300–24311. doi: 10.1074/jbc.M111.234104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rajakumara E, Wang Z, Ma H, Hu L, Chen H, Lin Y, Guo R, Wu F, Li H, Lan F, Shi YG, Xu Y, Patel DJ, Shi Y. PHD finger recognition of unmodified histone H3R2 links UHRF1 to regulation of euchromatic gene expression. Mol Cell. 2011;43:275–284. doi: 10.1016/j.molcel.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Johnson LM, Fisher-Adams G, Grunstein M. Identification of a non-basic domain in the histone H4 N-terminus required for repression of the yeast silent mating loci. Embo J. 1992;11:2201–2209. doi: 10.1002/j.1460-2075.1992.tb05279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Potoyan DA, Papoian GA. Energy landscape analyses of disordered histone tails reveal special organization of their conformational dynamics. J Am Chem Soc. 2011;133:7405–7415. doi: 10.1021/ja1111964. [DOI] [PubMed] [Google Scholar]
- 111.Feng Y, Wang J, Asher S, Hoang L, Guardiani C, Ivanov I, Zheng YG. Histone H4 acetylation differentially modulates arginine methylation by an in Cis mechanism. J Biol Chem. 2011;286:20323–20334. doi: 10.1074/jbc.M110.207258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Musselman CA, Ramirez J, Sims JK, Mansfield RE, Oliver SS, Denu JM, Mackay JP, Wade PA, Hagman J, Kutateladze TG. Bivalent recognition of nucleosomes by the tandem PHD fingers of the CHD4 ATPase is required for CHD4-mediated repression. Proc Natl Acad Sci U S A. 2012;109:787–792. doi: 10.1073/pnas.1113655109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Oliver SS, Musselman CA, Srinivasan R, Svaren JP, Kutateladze TG, Denu JM. Multivalent recognition of histone tails by the PHD fingers of CHD5. Biochemistry. 2012;51:6534–6544. doi: 10.1021/bi3006972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ruthenburg AJ, Li H, Milne TA, Dewell S, McGinty RK, Yuen M, Ueberheide B, Dou Y, Muir TW, Patel DJ, Allis CD. Recognition of a mononucleosomal histone modification pattern by BPTF via multivalent interactions. Cell. 2011;145:692–706. doi: 10.1016/j.cell.2011.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ali M, Rincon-Arano H, Zhao W, Rothbart SB, Tong Q, Parkhurst SM, Strahl BD, Deng LW, Groudine M, Kutateladze TG. Molecular basis for chromatin binding and regulation of MLL5. Proc Natl Acad Sci U S A. 2013;110:11296–11301. doi: 10.1073/pnas.1310156110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gatchalian J, Futterer A, Rothbart SB, Tong Q, Rincon-Arano H, Sanchez de Diego A, Groudine M, Strahl BD, Martinez AC, van Wely KH, Kutateladze TG. Dido3 PHD modulates cell differentiation and division. Cell Rep. 2013;4:148–158. doi: 10.1016/j.celrep.2013.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kinkelin K, Wozniak GG, Rothbart SB, Lidschreiber M, Strahl BD, Cramer P. Structures of RNA polymerase II complexes with Bye1, a chromatin-binding PHF3/DIDO homologue. Proc Natl Acad Sci U S A. 2013;110:15277–15282. doi: 10.1073/pnas.1311010110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rothbart SB, Krajewski K, Nady N, Tempel W, Xue S, Badeaux AI, Barsyte-Lovejoy D, Martinez JY, Bedford MT, Fuchs SM, Arrowsmith CH, Strahl BD. Association of UHRF1 with methylated H3K9 directs the maintenance of DNA methylation. Nat Struct Mol Biol. 2012;19:1155–1160. doi: 10.1038/nsmb.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kokura K, Sun L, Bedford MT, Fang J. Methyl-H3K9-binding protein MPP8 mediates E-cadherin gene silencing and promotes tumour cell motility and invasion. Embo J. 2010;29:3673–3687. doi: 10.1038/emboj.2010.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Escamilla-Del-Arenal M, da Rocha ST, Spruijt CG, Masui O, Renaud O, Smits AH, Margueron R, Vermeulen M, Heard E. Cdyl a New Partner of the Inactive X Chromosome and Potential Reader of H3K27me3 and H3K9me2. Mol Cell Biol. 2013;33:5005–5020. doi: 10.1128/MCB.00866-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Collins RE, Northrop JP, Horton JR, Lee DY, Zhang X, Stallcup MR, Cheng X. The ankyrin repeats of G9a and GLP histone methyltransferases are mono- and dimethyllysine binding modules. Nat Struct Mol Biol. 2008;15:245–250. doi: 10.1038/nsmb.1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fasting C, Schalley CA, Weber M, Seitz O, Hecht S, Koksch B, Dernedde J, Graf C, Knapp EW, Haag R. Multivalency as a chemical organization and action principle. Angew Chem Int Ed Engl. 2012;51:10472–10498. doi: 10.1002/anie.201201114. [DOI] [PubMed] [Google Scholar]
- 123.Fierz B, Muir TW. Chromatin as an expansive canvas for chemical biology. Nat Chem Biol. 2012;8:417–427. doi: 10.1038/nchembio.938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ptashne M. Regulation of transcription: from lambda to eukaryotes. Trends Biochem Sci. 2005;30:275–279. doi: 10.1016/j.tibs.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 125.Henikoff S, Furuyama T, Ahmad K, Histone variants. nucleosome assembly and epigenetic inheritance. Trends Genet. 2004;20:320–326. doi: 10.1016/j.tig.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 126.Bird AP. DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res. 1980;8:1499–1504. doi: 10.1093/nar/8.7.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Watt F, Molloy PL. Cytosine methylation prevents binding to DNA of a HeLa cell transcription factor required for optimal expression of the adenovirus major late promoter. Genes Dev. 1988;2:1136–1143. doi: 10.1101/gad.2.9.1136. [DOI] [PubMed] [Google Scholar]
- 128.Nan X, Meehan RR, Bird A. Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res. 1993;21:4886–4892. doi: 10.1093/nar/21.21.4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 130.Guy J, Cheval H, Selfridge J, Bird A. The role of MeCP2 in the brain. Annu Rev Cell Dev Biol. 2011;27:631–652. doi: 10.1146/annurev-cellbio-092910-154121. [DOI] [PubMed] [Google Scholar]
- 131.Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell. 1997;88:471–481. doi: 10.1016/s0092-8674(00)81887-5. [DOI] [PubMed] [Google Scholar]
- 132.Lyst MJ, Ekiert R, Ebert DH, Merusi C, Nowak J, Selfridge J, Guy J, Kastan NR, Robinson ND, de Lima Alve F, Rappsilber J, Greenberg ME, Bird A. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat Neurosci. 2013;16:898–902. doi: 10.1038/nn.3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hendrich B, Bird A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol Cell Biol. 1998;18:6538–6547. doi: 10.1128/mcb.18.11.6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hendrich B, Guy J, Ramsahoye B, Wilson VA, Bird A. Closely related proteins MBD2 and MBD3 play distinctive but interacting roles in mouse development. Genes Dev. 2001;15:710–723. doi: 10.1101/gad.194101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Martin Caballero I, Hansen J, Leaford D, Pollard S, Hendrich BD. The methyl-CpG binding proteins Mecp2, Mbd2 and Kaiso are dispensable for mouse embryogenesis. but play a redundant function in neural differentiation, PLoS One. 2009;4:e4315. doi: 10.1371/journal.pone.0004315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hutchins AS, Mullen AC, Lee HW, Sykes KJ, High FA, Hendrich BD, Bird AP, Reiner SL. Gene silencing quantitatively controls the function of a developmental trans-activator. Mol Cell. 2002;10:81–91. doi: 10.1016/s1097-2765(02)00564-6. [DOI] [PubMed] [Google Scholar]
- 137.Zhao X, Ueba T, Christie BR, Barkho B, McConnell MJ, Nakashima K, Lein ES, Eadie BD, Willhoite AR, Muotri AR, Summers RG, Chun J, Lee KF, Gage FH. Mice lacking methyl-CpG binding protein 1 have deficits in adult neurogenesis and hippocampal function. Proc Natl Acad Sci U S A. 2003;100:6777–6782. doi: 10.1073/pnas.1131928100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Saito M, Ishikawa F. The mCpG-binding domain of human MBD3 does not bind to mCpG but interacts with NuRD/Mi2 components HDAC1 and MTA2. J Biol Chem. 2002;277:35434–35439. doi: 10.1074/jbc.M203455200. [DOI] [PubMed] [Google Scholar]
- 139.Yildirim O, Li R, Hung JH, Chen PB, Dong X, Ee LS, Weng Z, Rando OJ, Fazzio TG. Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell. 2011;147:1498–1510. doi: 10.1016/j.cell.2011.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Baubec T, Ivanek R, Lienert F, Schubeler D. Methylation-dependent and -independent genomic targeting principles of the MBD protein family. Cell. 2013;153:480–492. doi: 10.1016/j.cell.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 141.Hashimoto H, Liu Y, Upadhyay AK, Chang Y, Howerton SB, Vertino PM, Zhang X, Cheng X. Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic Acids Res. 2012;40:4841–4849. doi: 10.1093/nar/gks155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Prokhortchouk A, Hendrich B, Jorgensen H, Ruzov A, Wilm M, Georgiev G, Bird A, Prokhortchouk E. The p120 catenin partner Kaiso is a DNA methylation-dependent transcriptional repressor. Genes Dev. 2001;15:1613–1618. doi: 10.1101/gad.198501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yoon HG, Chan DW, Reynolds AB, Qin J, Wong J. N-CoR mediates DNA methylation-dependent repression through a methyl CpG binding protein Kaiso. Mol Cell. 2003;12:723–734. doi: 10.1016/j.molcel.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 144.Kim SW, Park JI, Spring CM, Sater AK, Ji H, Otchere AA, Daniel JM, McCrea PD. Non-canonical Wnt signals are modulated by the Kaiso transcriptional repressor and p120-catenin. Nat Cell Biol. 2004;6:1212–1220. doi: 10.1038/ncb1191. [DOI] [PubMed] [Google Scholar]
- 145.Park JI, Kim SW, Lyons JP, Ji H, Nguyen TT, Cho K, Barton MC, Deroo T, Vleminckx K, Moon RT, McCrea PD. Kaiso/p120-catenin and TCF/beta-catenin complexes coordinately regulate canonical Wnt gene targets. Dev Cell. 2005;8:843–854. doi: 10.1016/j.devcel.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 146.Buck-Koehntop BA, Stanfield RL, Ekiert DC, Martinez-Yamout MA, Dyson HJ, Wilson IA, Wright PE. Molecular basis for recognition of methylated and specific DNA sequences by the zinc finger protein Kaiso. Proc Natl Acad Sci U S A. 2012;109:15229–15234. doi: 10.1073/pnas.1213726109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Johnson LM, Bostick M, Zhang X, Kraft E, Henderson I, Callis J, Jacobsen SE. The SRA methyl-cytosine-binding domain links DNA and histone methylation. Curr Biol. 2007;17:379–384. doi: 10.1016/j.cub.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bronner C, Achour M, Arima Y, Chataigneau T, Saya H, Schini-Kerth VB. The UHRF family: oncogenes that are drugable targets for cancer therapy in the near future? Pharmacol Ther. 2007;115:419–434. doi: 10.1016/j.pharmthera.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 149.Unoki M, Nishidate T, Nakamura Y. ICBP90, an E2F-1 target, recruits HDAC1 and binds to methyl-CpG through its SRA domain. Oncogene. 2004;23:7601–7610. doi: 10.1038/sj.onc.1208053. [DOI] [PubMed] [Google Scholar]
- 150.Qian C, Li S, Jakoncic J, Zeng L, Walsh MJ, Zhou MM. Structure and hemimethylated CpG binding of the SRA domain from human UHRF1. J Biol Chem. 2008;283:34490–34494. doi: 10.1074/jbc.C800169200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Arita K, Ariyoshi M, Tochio H, Nakamura Y, Shirakawa M. Recognition of hemi-methylated DNA by the SRA protein UHRF1 by a base-flipping mechanism. Nature. 2008;455:818–821. doi: 10.1038/nature07249. [DOI] [PubMed] [Google Scholar]
- 152.Avvakumov GV, Walker JR, Xue S, Li Y, Duan S, Bronner C, Arrowsmith CH, Dhe-Paganon S. Structural basis for recognition of hemi-methylated DNA by the SRA domain of human UHRF1. Nature. 2008;455:822–825. doi: 10.1038/nature07273. [DOI] [PubMed] [Google Scholar]
- 153.Hashimoto H, Horton JR, Zhang X, Bostick M, Jacobsen SE, Cheng X. The SRA domain of UHRF1 flips 5-methylcytosine out of the DNA helix. Nature. 2008;455:826–829. doi: 10.1038/nature07280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Klimasauskas S, Kumar S, Roberts RJ, Cheng X. HhaI methyltransferase flips its target base out of the DNA helix. Cell. 1994;76:357–369. doi: 10.1016/0092-8674(94)90342-5. [DOI] [PubMed] [Google Scholar]
- 155.Song J, Teplova M, Ishibe-Murakami S, Patel DJ. Structure-based mechanistic insights into DNMT1-mediated maintenance DNA methylation. Science. 2012;335:709–712. doi: 10.1126/science.1214453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Holliday R, Pugh JE. DNA modification mechanisms and gene activity during development. Science. 1975;187:226–232. [PubMed] [Google Scholar]
- 157.Razin A, Riggs AD. DNA methylation and gene function. Science. 1980;210:604–610. doi: 10.1126/science.6254144. [DOI] [PubMed] [Google Scholar]
- 158.Achour M, Jacq X, Ronde P, Alhosin M, Charlot C, Chataigneau T, Jeanblanc M, Macaluso M, Giordano A, Hughes AD, Schini-Kerth VB, Bronner C. The interaction of the SRA domain of ICBP90 with a novel domain of DNMT1 is involved in the regulation of VEGF gene expression. Oncogene. 2008;27:2187–2197. doi: 10.1038/sj.onc.1210855. [DOI] [PubMed] [Google Scholar]
- 159.Bostick M, Kim JK, Esteve PO, Clark A, Pradhan S, Jacobsen SE. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science. 2007;317:1760–1764. doi: 10.1126/science.1147939. [DOI] [PubMed] [Google Scholar]
- 160.Sharif J, Muto M, Takebayashi S, Suetake I, Iwamatsu A, Endo TA, Shinga J, Mizutani-Koseki Y, Toyoda T, Okamura K, Tajima S, Mitsuya K, Okano M, Koseki H. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature. 2007;450:908–912. doi: 10.1038/nature06397. [DOI] [PubMed] [Google Scholar]
- 161.Nishiyama A, Yamaguchi L, Sharif J, Johmura Y, Kawamura T, Nakanishi K, Shimamura S, Arita K, Kodama T, Ishikawa F, Koseki H, Nakanishi M. Uhrf1-dependent H3K23 ubiquitylation couples maintenance DNA methylation and replication. Nature. 2013;502:249–253. doi: 10.1038/nature12488. [DOI] [PubMed] [Google Scholar]
- 162.Mittler G, Butter F, Mann M. A SILAC-based DNA protein interaction screen that identifies candidate binding proteins to functional DNA elements. Genome Res. 2009;19:284–293. doi: 10.1101/gr.081711.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Bartke T, Vermeulen M, Xhemalce B, Robson SC, Mann M, Kouzarides T. Nucleosome-interacting proteins regulated by DNA and histone methylation. Cell. 2010;143:470–484. doi: 10.1016/j.cell.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Bartels SJ, Spruijt CG, Brinkman AB, Jansen PW, Vermeulen M, Stunnenberg HG. A SILAC-based screen for Methyl-CpG binding proteins identifies RBP-J as a DNA methylation and sequence-specific binding protein. PLoS One. 2011;6:e25884. doi: 10.1371/journal.pone.0025884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hu S, Wan J, Su Y, Song Q, Zeng Y, Nguyen HN, Shin J, Cox E, Rho HS, Woodard C, Xia S, Liu S, Lyu H, Ming GL, Wade H, Song H, Qian J, Zhu H. DNA methylation presents distinct binding sites for human transcription factors. Elife. 2013;2:e00726. doi: 10.7554/eLife.00726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ito S, D'Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–1133. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, Sun Y, Li X, Dai Q, Song CX, Zhang K, He C, Xu GL. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Iurlaro M, Ficz G, Oxley D, Raiber EA, Bachman M, Booth MJ, Andrews S, Balasubramanian S, Reik W. A screen for hydroxymethylcytosine and formylcytosine binding proteins suggests functions in transcription and chromatin regulation. Genome Biol. 2013;14:R119. doi: 10.1186/gb-2013-14-10-r119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Mellen M, Ayata P, Dewell S, Kriaucionis S, Heintz N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell. 2012;151:1417–1430. doi: 10.1016/j.cell.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Frauer C, Hoffmann T, Bultmann S, Casa V, Cardoso MC, Antes I, Leonhardt H. Recognition of 5-hydroxymethylcytosine by the Uhrf1 SRA domain. PLoS One. 2011;6:e21306. doi: 10.1371/journal.pone.0021306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Pichler G, Wolf P, Schmidt CS, Meilinger D, Schneider K, Frauer C, Fellinger K, Rottach A, Leonhardt H. Cooperative DNA and histone binding by Uhrf2 links the two major repressive epigenetic pathways. J Cell Biochem. 2011;112:2585–2593. doi: 10.1002/jcb.23185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zhang J, Gao Q, Li P, Liu X, Jia Y, Wu W, Li J, Dong S, Koseki H, Wong J. S phase-dependent interaction with DNMT1 dictates the role of UHRF1 but not UHRF2 in DNA methylation maintenance. Cell Res. 2011;21:1723–1739. doi: 10.1038/cr.2011.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kudo S, Nomura Y, Segawa M, Fujita N, Nakao M, Schanen C, Tamura M. Heterogeneity in residual function of MeCP2 carrying missense mutations in the methyl CpG binding domain. J Med Genet. 2003;40:487–493. doi: 10.1136/jmg.40.7.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Bird A, Taggart M, Frommer M, Miller OJ, Macleod D. A fraction of the mouse genome that is derived from islands of nonmethylated, CpG-rich DNA. Cell. 1985;40:91–99. doi: 10.1016/0092-8674(85)90312-5. [DOI] [PubMed] [Google Scholar]
- 178.Long HK, Blackledge NP, Klose RJ. ZF-CxxC domain-containing proteins, CpG islands and the chromatin connection. Biochem Soc Trans. 2013;41:727–740. doi: 10.1042/BST20130028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lee JH, Skalnik DG. CpG-binding protein (CXXC finger protein 1) is a component of the mammalian Set1 histone H3-Lys4 methyltransferase complex, the analogue of the yeast Set1/COMPASS complex. J Biol Chem. 2005;280:41725–41731. doi: 10.1074/jbc.M508312200. [DOI] [PubMed] [Google Scholar]
- 180.Thomson JP, Skene PJ, Selfridge J, Clouaire T, Guy J, Webb S, Kerr AR, Deaton A, Andrews R, James KD, Turner DJ, Illingworth R, Bird A. CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature. 2010;464:1082–1086. doi: 10.1038/nature08924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Frauer C, Rottach A, Meilinger D, Bultmann S, Fellinger K, Hasenoder S, Wang M, Qin W, Soding J, Spada F, Leonhardt H. Different binding properties and function of CXXC zinc finger domains in Dnmt1 and Tet1. PLoS One. 2011;6:e16627. doi: 10.1371/journal.pone.0016627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Xu Y, Wu F, Tan L, Kong L, Xiong L, Deng J, Barbera AJ, Zheng L, Zhang H, Huang S, Min J, Nicholson T, Chen T, Xu G, Shi Y, Zhang K, Shi YG. Genome-wide regulation of 5hmC, 5mC, and gene expression by Tet1 hydroxylase in mouse embryonic stem cells. Mol Cell. 2011;42:451–464. doi: 10.1016/j.molcel.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zhang H, Zhang X, Clark E, Mulcahey M, Huang S, Shi YG. TET1 is a DNA-binding protein that modulates DNA methylation and gene transcription via hydroxylation of 5-methylcytosine. Cell Res. 2010;20:1390–1393. doi: 10.1038/cr.2010.156. [DOI] [PubMed] [Google Scholar]
- 184.Xu Y, Xu C, Kato A, Tempel W, Abreu JG, Bian C, Hu Y, Hu D, Zhao B, Cerovina T, Diao J, Wu F, He HH, Cui Q, Clark E, Ma C, Barbara A, Veenstra GJ, Xu G, Kaiser UB, Liu XS, Sugrue SP, He X, Min J, Kato Y, Shi YG. Tet3 CXXC domain and dioxygenase activity cooperatively regulate key genes for Xenopus eye and neural development. Cell. 2012;151:1200–1213. doi: 10.1016/j.cell.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Iyer LM, Abhiman S, Aravind L. Natural history of eukaryotic DNA methylation systems. Prog Mol Biol Transl Sci. 2011;101:25–104. doi: 10.1016/B978-0-12-387685-0.00002-0. [DOI] [PubMed] [Google Scholar]
- 186.Ko M, An J, Bandukwala HS, Chavez L, Aijo T, Pastor WA, Segal MF, Li H, Koh KP, Lahdesmaki H, Hogan PG, Aravind L, Rao A. Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nature. 2013;497:122–126. doi: 10.1038/nature12052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Blackledge NP, Zhou JC, Tolstorukov MY, Farcas AM, Park PJ, Klose RJ. CpG islands recruit a histone H3 lysine 36 demethylase. Mol Cell. 2010;38:179–190. doi: 10.1016/j.molcel.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ramirez J, Dege C, Kutateladze TG, Hagman J. MBD2 and multiple domains of CHD4 are required for transcriptional repression by Mi-2/NuRD complexes. Mol Cell Biol. 2012;32:5078–5088. doi: 10.1128/MCB.00819-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Vermeulen M, Mulder KW, Denissov S, Pijnappel WW, van Schaik FM, Varier RA, Baltissen MP, Stunnenberg HG, Mann M, Timmers HT. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell. 2007;131:58–69. doi: 10.1016/j.cell.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 190.Lauberth SM, Nakayama T, Wu X, Ferris AL, Tang Z, Hughes SH, Roeder RG. H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell. 2013;152:1021–1036. doi: 10.1016/j.cell.2013.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439:811–816. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- 192.Clouaire T, Webb S, Skene P, Illingworth R, Kerr A, Andrews R, Lee JH, Skalnik D, Bird A. Cfp1 integrates both CpG content and gene activity for accurate H3K4me3 deposition in embryonic stem cells. Genes Dev. 2012;26:1714–1728. doi: 10.1101/gad.194209.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14:204–220. doi: 10.1038/nrg3354. [DOI] [PubMed] [Google Scholar]
- 194.Ziller MJ, Gu H, Muller F, Donaghey J, Tsai LT, Kohlbacher O, De Jager PL, Rosen ED, Bennett DA, Bernstein BE, Gnirke A, Meissner A. Charting a dynamic DNA methylation landscape of the human genome. Nature. 2013;500:477–481. doi: 10.1038/nature12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Chen T, Hevi S, Gay F, Tsujimoto N, He T, Zhang B, Ueda Y, Li E. Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nat Genet. 2007;39:391–396. doi: 10.1038/ng1982. [DOI] [PubMed] [Google Scholar]
- 196.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Seisenberger S, Andrews S, Krueger F, Arand J, Walter J, Santos F, Popp C, Thienpont B, Dean W, Reik W. The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Mol Cell. 2012;48:849–862. doi: 10.1016/j.molcel.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Otani J, Nankumo T, Arita K, Inamoto S, Ariyoshi M, Shirakawa M. Structural basis for recognition of H3K4 methylation status by the DNA methyltransferase 3A ATRX-DNMT3-DNMT3L domain. EMBO Rep. 2009;10:1235–1241. doi: 10.1038/embor.2009.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Ooi SK, Qiu C, Bernstein E, Li K, Jia D, Yang Z, Erdjument-Bromage H, Tempst P, Lin SP, Allis CD, Cheng X, Bestor TH. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–717. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Iwase S, Xiang B, Ghosh S, Ren T, Lewis PW, Cochrane JC, Allis CD, Picketts DJ, Patel DJ, Li H, Shi Y. ATRX ADD domain links an atypical histone methylation recognition mechanism to human mental-retardation syndrome. Nat Struct Mol Biol. 2011;18:769–776. doi: 10.1038/nsmb.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Zhang Y, Jurkowska R, Soeroes S, Rajavelu A, Dhayalan A, Bock I, Rathert P, Brandt O, Reinhardt R, Fischle W, Jeltsch A. Chromatin methylation activity of Dnmt3a and Dnmt3a/3L is guided by interaction of the ADD domain with the histone H3 tail. Nucleic Acids Res. 2010;38:4246–4253. doi: 10.1093/nar/gkq147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Ge YZ, Pu MT, Gowher H, Wu HP, Ding JP, Jeltsch A, Xu GL. Chromatin targeting of de novo DNA methyltransferases by the PWWP domain. J Biol Chem. 2004;279:25447–25454. doi: 10.1074/jbc.M312296200. [DOI] [PubMed] [Google Scholar]
- 203.Chen T, Tsujimoto N, Li E. The PWWP domain of Dnmt3a and Dnmt3b is required for directing DNA methylation to the major satellite repeats at pericentric heterochromatin. Mol Cell Biol. 2004;24:9048–9058. doi: 10.1128/MCB.24.20.9048-9058.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Qiu C, Sawada K, Zhang X, Cheng X. The PWWP domain of mammalian DNA methyltransferase Dnmt3b defines a new family of DNA-binding folds. Nat Struct Biol. 2002;9:217–224. doi: 10.1038/nsb759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Dhayalan A, Rajavelu A, Rathert P, Tamas R, Jurkowska RZ, Ragozin S, Jeltsch A. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J Biol Chem. 2010;285:26114–26120. doi: 10.1074/jbc.M109.089433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 207.Neri F, Krepelova A, Incarnato D, Maldotti M, Parlato C, Galvagni F, Matarese F, Stunnenberg HG, Oliviero S. Dnmt3L antagonizes DNA methylation at bivalent promoters and favors DNA methylation at gene bodies in ESCs. Cell. 2013;155:121–134. doi: 10.1016/j.cell.2013.08.056. [DOI] [PubMed] [Google Scholar]
- 208.Chuang LS, Ian HI, Koh TW, Ng HH, Xu G, Li BF. Human DNA-(cytosine-5) methyltransferase-PCNA complex as a target for p21WAF1. Science. 1997;277:1996–2000. doi: 10.1126/science.277.5334.1996. [DOI] [PubMed] [Google Scholar]
- 209.Spada F, Haemmer A, Kuch D, Rothbauer U, Schermelleh L, Kremmer E, Carell T, Langst G, Leonhardt H. DNMT1 but not its interaction with the replication machinery is required for maintenance of DNA methylation in human cells. J Cell Biol. 2007;176:565–571. doi: 10.1083/jcb.200610062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Pradhan M, Esteve PO, Chin HG, Samaranayke M, Kim GD, Pradhan S. CXXC domain of human DNMT1 is essential for enzymatic activity. Biochemistry. 2008;47:10000–10009. doi: 10.1021/bi8011725. [DOI] [PubMed] [Google Scholar]
- 211.Bestor TH, Ingram VM. Two DNA methyltransferases from murine erythroleukemia, cells: purification, sequence specificity, and mode of interaction with DNA. Proc Natl Acad Sci U S A. 1983;80:5559–5563. doi: 10.1073/pnas.80.18.5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Bashtrykov P, Jankevicius G, Smarandache A, Jurkowska RZ, Ragozin S, Jeltsch A. Specificity of Dnmt1 for methylation of hemimethylated CpG sites resides in its catalytic domain. Chem Biol. 2012;19:572–578. doi: 10.1016/j.chembiol.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 213.Takeshita K, Suetake I, Yamashita E, Suga M, Narita H, Nakagawa A, Tajima S. Structural insight into maintenance methylation by mouse DNA methyltransferase 1 (Dnmt1) Proc Natl Acad Sci U S A. 2011;108:9055–9059. doi: 10.1073/pnas.1019629108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Syeda F, Fagan RL, Wean M, Avvakumov GV, Walker JR, Xue S, Dhe-Paganon S, Brenner C. The replication focus targeting sequence (RFTS) domain is a DNA-competitive inhibitor of Dnmt1. J Biol Chem. 2011;286:15344–15351. doi: 10.1074/jbc.M110.209882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Dikic I, Wakatsuki S, Walters KJ. Ubiquitin-binding domains - from structures to functions. Nat Rev Mol Cell Biol. 2009;10:659–671. doi: 10.1038/nrm2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Nakamura T, Liu YJ, Nakashima H, Umehara H, Inoue K, Matoba S, Tachibana M, Ogura A, Shinkai Y, Nakano T. PGC7 binds histone H3K9me2 to protect against conversion of 5mC to 5hmC in early embryos. Nature. 2012;486:415–419. doi: 10.1038/nature11093. [DOI] [PubMed] [Google Scholar]
- 217.Nakamura T, Arai Y, Umehara H, Masuhara M, Kimura T, Taniguchi H, Sekimoto T, Ikawa M, Yoneda Y, Okabe M, Tanaka S, Shiota K, Nakano T. PGC7/Stella protects against DNA demethylation in early embryogenesis. Nat Cell Biol. 2007;9:64–71. doi: 10.1038/ncb1519. [DOI] [PubMed] [Google Scholar]
- 218.Seisenberger S, Peat JR, Reik W. Conceptual links between DNA methylation reprogramming in the early embryo and primordial germ cells. Curr Opin Cell Biol. 2013;25:281–288. doi: 10.1016/j.ceb.2013.02.013. [DOI] [PubMed] [Google Scholar]
- 219.Lowary PT, Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J Mol Biol. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- 220.Clouaire T, de Las Heras JI, Merusi C, Stancheva I. Recruitment of MBD1 to target genes requires sequence-specific interaction of the MBD domain with methylated DNA. Nucleic Acids Res. 2010;38:4620–4634. doi: 10.1093/nar/gkq228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Klose RJ, Sarraf SA, Schmiedeberg L, McDermott SM, Stancheva I, Bird AP. DNA binding selectivity of MeCP2 due to a requirement for A/T sequences adjacent to methyl-CpG. Mol Cell. 2005;19:667–678. doi: 10.1016/j.molcel.2005.07.021. [DOI] [PubMed] [Google Scholar]
- 222.Ramsahoye BH, Biniszkiewicz D, Lyko F, Clark V, Bird AP, Jaenisch R. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc Natl Acad Sci U S A. 2000;97:5237–5242. doi: 10.1073/pnas.97.10.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Han J, Zhang H, Wang Z, Zhou H, Zhang Z. A Cul4 E3 Ubiquitin Ligase Regulates Histone Hand-Off during Nucleosome Assembly. Cell. 2013;155:817–829. doi: 10.1016/j.cell.2013.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Byrum SD, Raman A, Taverna SD, Tackett AJ. ChAP-MS: a method for identification of proteins and histone posttranslational modifications at a single genomic locus. Cell Rep. 2012;2:198–205. doi: 10.1016/j.celrep.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Byrum SD, Taverna SD, Tackett AJ. Purification of a specific native genomic locus for proteomic analysis. Nucleic Acids Res. 2013;41:e195. doi: 10.1093/nar/gkt822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Leroy G, Chepelev I, Dimaggio PA, Blanco MA, Zee BM, Zhao K, Garcia BA. Proteogenomic characterization and mapping of nucleosomes decoded by Brd and HP1 proteins. Genome Biol. 2012;13:R68. doi: 10.1186/gb-2012-13-8-r68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Wang CI, Alekseyenko AA, LeRoy G, Elia AE, Gorchakov AA, Britton LM, Elledge SJ, Kharchenko PV, Garcia BA, Kuroda MI. Chromatin proteins captured by ChIP-mass spectrometry are linked to dosage compensation in Drosophila. Nat Struct Mol Biol. 2013;20:202–209. doi: 10.1038/nsmb.2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Gaj T, Gersbach CA, Barbas CF., 3rd ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, Lim WA, Weissman JS, Qi LS. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Maeder ML, Angstman JF, Richardson ME, Linder SJ, Cascio VM, Tsai SQ, Ho QH, Sander JD, Reyon D, Bernstein BE, Costello JF, Wilkinson MF, Joung JK. Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins. Nat Biotechnol. 2013;31:1137–1142. doi: 10.1038/nbt.2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Mendenhall EM, Williamson KE, Reyon D, Zou JY, Ram O, Joung JK, Bernstein BE. Locus-specific editing of histone modifications at endogenous enhancers. Nat Biotechnol. 2013;31:1133–1136. doi: 10.1038/nbt.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Konermann S, Brigham MD, Trevino AE, Hsu PD, Heidenreich M, Cong L, Platt RJ, Scott DA, Church GM, Zhang F. Optical control of mammalian endogenous transcription and epigenetic states. Nature. 2013;500:472–476. doi: 10.1038/nature12466. [DOI] [PMC free article] [PubMed] [Google Scholar]