

Abstract
Our lab recently showed that N-methyl-D-aspartate (NMDA) evokes ATP-sensitive K+ (K-ATP) currents in subthalamic nucleus (STN) neurons in slices of rat brain. Both K-ATP channels and 5′-adenosine monophosphate-activated protein kinase (AMPK) are considered cellular energy sensors because their activities are influenced by the phosphorylation state of adenosine nucleotides. Moreover, AMPK has been shown to regulate K-ATP function in a variety of tissues including pancreas, cardiac myocytes, and hypothalamus. We used whole-cell patch clamp recordings to study the effect of AMPK activation on K-ATP channel function in STN neurons in slices of rat brain. We found that bath or intracellular application of the AMPK activators A769662 and PT1 augmented tolbutamide-sensitive K-ATP currents evoked by NMDA receptor stimulation. The effect of AMPK activators was blocked by the AMPK inhibitor dorsomorphin (compound C), and by STO609, an inhibitor of the upstream AMPK activator CaMKKβ. AMPK augmentation of NMDA-induced K-ATP current was also blocked by intracellular BAPTA and by inhibitors of nitric oxide synthase and guanylyl cyclase. However, A769662 did not augment currents evoked by the K-ATP channel opener diazoxide. In the presence of NMDA, A769662 inhibited depolarizing plateau potentials and burst firing, both of which could be antagonized by tolbutamide or dorsomorphin. These studies show that AMPK augments NMDA-induced K-ATP currents by a Ca2+-dependent process that involves nitric oxide and cGMP. By augmenting K-ATP currents, AMPK activation would be expected to dampen the excitatory effect of glutamate-mediated transmission in the STN.
Keywords: AMP kinase, NMDA, subthalamic nucleus, brain slice, K-ATP, patch-clamp
1. INTRODUCTION
Glutamate-containing neurons of the subthalamic nucleus (STN) exert a powerful excitatory influence on basal ganglia output (Parent and Hazrati, 1995). Dopamine deficiency, such as occurs in Parkinson’s disease, has been shown to increase burst firing in STN neurons (Ni et al., 2001; Hollerman and Grace, 1992; Wichmann and Dostrovsky, 2011). Moreover, increased burst firing in the STN has been shown to correlate with symptoms of Parkinson’s disease (Wichmann et al., 1994; Benedetti et al., 2009). Experimentally-induced parkinsonism in animals is improved by lesioning the STN (Bergman et al., 1990), and deep brain stimulation of the STN also improves many symptoms of Parkinson’s disease, most likely by interrupting the abnormal firing pattern in basal ganglia output neurons (Maltete et al., 2007). Intrinsic membrane properties enable burst firing (Beurrier et al., 1999; Bevan and Wilson, 1999; Do and Bean, 2003), whereas synaptic inputs control the timing of burst discharges (Bevan et al., 2002). The so-called hyperdirect pathway of glutamate input that arises from cerebral cortex has long been known to induce bursts of action potentials in STN neurons (Kitai and Deniau, 1981; Rouzaire-Dubois and Scarnati, 1987). Moreover, glutamate-mediated EPSPs have been shown to trigger plateau potentials that induce burst discharges in STN neurons (Kass and Mintz, 2006). Thus, glutamate-containing inputs play important roles in regulating firing pattern in the STN.
Concerning the glutamate receptor subtype that triggers bursts, we and others have shown that burst firing can be evoked in STN neurons by NMDA but not AMPA receptor stimulation in slices of rodent brain (Zhu et al., 2004; Loucif et al., 2005; Chu et al., 2012). But in addition to the traditional NMDA-gated current, subsequent studies in our lab showed that NMDA also evoked a tolbutamide-sensitive ATP-sensitive K+ (K-ATP) current. The K-ATP channel is widely expressed in brain (Dunn-Meynell et al., 1998) and is often referred to as an energy-sensing channel because it is closed by ATP and opened by Mg2+-ADP (Ashcroft, 1988). However, we found that NMDA did not evoke K-ATP current in adjacent neurons in the substantia nigra (Shen and Johnson, 2010). Our studies showed that the NMDA-evoked K-ATP current in STN neurons was Ca2+-dependent and also required activation of nitric oxide (NO)-and cGMP-dependent pathways (Shen and Johnson, 2010). Although Ca2+ influx through NMDA-gated channels can activate K-ATP channels, we showed that release of Ca2+ from intracellular stores could also activate K-ATP current in STN neurons (Shen and Johnson, 2013). Moreover, we showed that block of K-ATP current greatly potentiated the excitatory effect of NMDA on burst firing, which suggests that concomitant activation of K-ATP currents tends to dampen the excitatory influence of glutamate in the STN. Because burst firing is associated with symptoms of Parkinson’s disease, it is possible that augmentation of K-ATP current in the STN might reduce symptoms of this disease.
5′-Adenosine monophosphate-activated protein kinase (AMPK) is also frequently described as a sensor of cellular energy because it is activated by AMP and inhibited by ATP. AMPK is a heterotrimeric enzyme complex composed of an alpha catalytic subunit and beta and gamma regulatory subunits. With rising levels of AMP, this molecule displaces ATP at the gamma subunit and produces a conformational change that enables phosphorylation of the alpha subunit at Thr-172 (Oakhill et al., 2010). Phosphorylation, which is accomplished by the upstream kinases liver kinase B1 (LKB1) and Ca2+-calmodulin-dependent kinase kinase beta (CaM-KKβ), increases AMPK activity 50-100 fold (Amato and Man, 2011). Once activated, AMPK up-regulates those processes that increase ATP, such as increasing glucose uptake, glycolysis, and mitochondrial biogenesis (Hallows, 2005). At the same time, AMPK switches off pathways that consume ATP, such as protein and glycogen synthesis (Russo et al., 2013). Thus, AMPK can be described as a master regulator that is designed to maintain cellular energy homeostasis (Hardie et al., 2003).
Because both K-ATP and AMPK respond to changes in energy supply, it is not surprising that their activities are frequently linked. In pancreatic β-cells, small molecule activators of AMPK have been reported to increase insulin release by reducing K-ATP channel activity (Wang et al., 2005; Langelueddecke et al., 2012; Düfer et al., 2010). However, others have reported that AMPK activation increases the surface expression of K-ATP channels in islet cells (Lim et al., 2009; Smith et al., 2006), which would seem to oppose the hypothesis that AMPK augments insulin release. Nevertheless, AMPK activation has also been reported to increase the surface expression of K-ATP channels in cardiac myocytes (Sukhodub et al., 2007; Yoshida et al., 2012). In hypothalamic neurons, hypoglycemia causes hyperpolarization in glucose-sensing neurons by activation of K-ATP channels, and this response is regulated by AMPK (Beall et al., 2012). Thus, there is considerable evidence to suggest that AMPK and KATP channels have interrelated functions, although controversy remains concerning some aspects of their functional relationship.
The purpose of the present study was to identify and characterize effects of AMPK activation on K-ATP channel function in the STN in slices of rat brain. We used small molecule activators of AMPK to study effects on K-ATP currents evoked by NMDA and by the K-ATP channel opener diazoxide. Our studies show that acute application of AMPK activators augment NMDA-evoked K-ATP current but not K-ATP current evoked by diazoxide. Furthermore, we show that augmentation of NMDA-evoked K-ATP current is mediated by Ca2+-dependent NO-and cGMP-coupled pathways. By augmenting K-ATP current, we found that NMDA-induced burst firing and depolarizing plateau potentials are suppressed by AMPK activation. We suggest that AMPK activation in the STN could be explored as a therapeutic strategy in the management of Parkinson’s disease.
2. EXPERIMENTAL PROCEDURES
2.1 Tissue Preparation
All procedures were carried out in accordance with a protocol approved by Institutional Animal Care and Use Committee at the Portland Veterans Affairs Medical Center. Every precaution was taken to minimize animal stress and the number of animals used. Horizontal slices containing diencephalon and rostral midbrain were prepared from male Sprague-Dawley rats (120–180 g, Harlan, Indianapolis, IN) as described previously (Shen and Johnson, 2000). Briefly, rats were anesthetized with isoflurane and euthanized by severing major thoracic vessels. Brains were rapidly removed and slices were cut with a vibratome in an ice-cold sucrose buffer solution of the following composition (in mM): sucrose, 196; KCl, 2.5; MgCl2, 3.5; CaCl2, 0.5; NaH2PO4, 1.2; glucose, 20; and NaHCO3, 26, equilibrated with 95% O2 and 5% CO2. A slice containing the STN was then placed on a supporting net and submerged in a continuously flowing solution (2 ml/min) of the following composition (in mM): NaCl, 126; KCl, 2.5; CaCl2, 2.4; MgCl2, 1.2; NaH2PO4, 1.2; NaHCO3, 19; glucose, 11, gassed with 95% O2 and 5% CO2 (pH 7.4) at 36°C. By using a microscope for visual guidance, the STN was located as gray matter approximately 2.7 mm lateral to the midline and 2 mm rostral to the center of the substantia nigra zona reticulata.
2.2 Electrophysiological Recordings
Whole-cell recordings were made with pipettes containing (in mM): potassium gluconate, 138; MgCl2, 2; CaCl2, 1; EGTA, 11; HEPES, 10; ATP, 1.5; GTP, 0.3 (pH 7.3). In some experiments, EGTA was replaced by 10 mM BAPTA. Pipette resistance ranged from 3 to 8 MΩ, whereas series resistance typically ranged from 15 to 40 MΩ. Membrane currents or potentials were amplified and recorded with an Axopatch-1D amplifier with 5-kHz low-pass filter (Molecular Devices, Foster City, CA, USA). Data were acquired using a personal computer with a Digidata analog/digital interface and analyzed using pCLAMP software (Molecular Devices/Axon Instruments, Foster City, CA). Membrane potentials for whole-cell recordings were corrected for the liquid junction potential (10 mV).
2.3 Current-Voltage Studies
Current-voltage (I-V) relationships were constructed by measuring currents during a series of hyperpolarizing voltage steps (400 ms duration) with 10 mV increments from a holding potential of −70 mV. Currents were measured immediately after capacitive transients to minimize the influence of hyperpolarization-activated cation currents. When measuring the voltage dependence of NMDA currents, I-V plots were always obtained at the end of the 3-5 min application of NMDA. Slope conductance was calculated for each cell as the slope of a straight line in the I-V plot at voltages between −70 and −100 mV. Mean slope conductance and S.E.M. were obtained by averaging slope conductance from all cells in each experimental group. In some experiments, I-V plots are displayed as “subtracted” currents in which currents recorded during the experimental treatment were subtracted from those currents recorded in the control condition. Therefore, these subtracted currents represent “net” currents that were produced or blocked by an experimental treatment.
2.4 Drugs and Chemicals
All drugs were dissolved in aqueous or dimethyl sulfoxide stock solutions. Most drugs were added to the slice perfusate. Stock solutions were diluted at least 1:1000 to the desired concentration in superfusate immediately prior to use. Approximately 30 seconds were required for the drug solution to enter the recording chamber; this delay was due to passage of the superfusate through a heat exchanger. In some experiments, drugs were added to the internal pipette solution and delivered by passive intracellular dialysis. Diazoxide, penitrem A, dorsomorphin, 6,7-dihydro-4-hydroxy-3-(2′-hydroxy[1,1′-biphenyl]-4-yl)-6-oxo-thieno[2,3-b]pyridine-5-carbonitrile (A769662), 2-chloro-5-[[5-[[5-(4,5-dimethyl-2-nitrophenyl)-2-furanyl]methylene]-4,5-dihydro-4-oxo-2-thiazolyl]amino]benzoic acid (PT1), N1-(β-D-ribofuranosyl)-5-aminoimidazole-4-carboxamide (AICAR), CGS9343B, STO609 acetate, and 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) were obtained from Tocris Cookson Inc. (Bristol, UK). Apamin, barium chloride, N-methyl-D-aspartic acid (NMDA), NG-nitro-l-arginine methyl ester hydrochloride (L-NAME), and tolbutamide were obtained from Sigma-Aldrich (St. Louis, MO).
2.5 Western immunoblot
Horizontal slices of ventral midbrain (300 μm) were cut on a vibratome and placed in glass vials containing normal aCSF that was equilibrated with 95% O2 and 5% CO2 at 36 oC. After one hour, A769662 was added to the media for an additional hour. Concentration-response curves were constructed by equilibrating slices with different concentrations of A769662 (0-100 μM). Some slices were equilibrated in dorsomorphin (30 μM) 45 min prior to A769662 treatment. All slices were removed from the media 2 hours after slice preparation and snap frozen in liquid nitrogen then stored at −80°C until processed for Western immunoblot. Midbrain slices were sonicated in lysis buffer that contained protease and phosphatase inhibitors (137 mM NaCl, 15 mM Tris, 5 mM EDTA, 1% Triton-X 100, 1% Halt™ Protease and Phosphatase Inhibitor Cocktail, Thermo Scientific). Soluble protein fractions were decanted and pellets discarded after centrifugation at 22,000g for 20 min. A BCA protein assay (Thermo) was used to facilitate equal sample loading. Samples were denatured with B-mercaptoethanol 2X Laemmli Buffer (1:1; Sigma) and heated for 10 min at 95°C, then electrophoresed in a 7.5% Tris–HCl SDS polyacrylamide gel (TGX Criterion™, Bio-Rad). Protein standards (Precision Plus Kaleidescope™, Bio-Rad) were run along-side samples to locate approximate positions of expected bands. Separated proteins were then transferred to polyvinylidene difluoride (PVDF) membranes (Immun-Blot, Bio-Rad), which were blocked in 5% BSA in Tris–buffered saline with Tween-20 (TBST) for 60 min. Membranes were then probed with primary antibodies for Thr-172 phosphorylated (rabbit, 1:2000) and total AMPK (rabbit; 1:1500, Santa Cruz Bio-tech). After three 5 min washes in TBST, membranes were probed with alkaline phosphatase conjugated secondary antibody (1:40000, goat anti-rabbit) for 1 hr, then washed again in TBST and incubated in ECF substrate (GE Healthcare Life Sciences) for 5 min. Blots were dried briefly, excited with UV light and imaged through a 565nm filter (Ultra-Lum Omega™). Quantification of protein was done with densitometry using protein band illumination (summation of pixel intensities) using Image Studio Lite (LI-COR) software. Average background signal was subtracted from each band individually. To allow interblot analysis, raw intensity for each band was divided by the sum intensity of all lanes of a blot. Statistics were done on these normalized values grouped from replicated blots.
2.6 Data Analysis
Numerical data in the text and error bars in figures are expressed as mean ± S.E.M. I-V plots and concentration-response curves were analyzed by analysis of variance (ANOVA) followed by a Holm-Sidak pairwise comparison test unless stated otherwise (SigmaStat; Jandel Scientific, San Rafael, CA). An F-test was used to compare differences between two datasets. Averaged data in each set were fitted with a third degree polynomial to obtain sum-of-squares for error and degrees of freedom. The null hypothesis was that one curve fits all data points from both data sets, whereas the alternative hypothesis was that each set of data was better fit separately. The F value was calculated as the relative difference in sum-of-squares divided by the relative difference in degrees of freedom. A P value <0.05 was considered significant.
3. RESULTS
3.1 AMPK activation augments the ability of NMDA to increase membrane conductance
We reported previously that activation of either NMDA or metabotropic glutamate receptors evoke Ca2+-dependent K-ATP current in STN neurons (Shen and Johnson, 2010; Shen and Johnson, 2013). These previous experiments used whole-cell pipette solutions that contained a low level of EGTA (0.2 mM) in order to facilitate Ca2+-dependent mechanisms. In the present study, we used whole-cell pipette solutions that contained 11 mM EGTA in order to dampen Ca2+-dependent activation of K-ATP channels. Under these conditions, NMDA only evoked inward currents at −70 mV (Fig.1A1) and did not increase membrane conductance as shown in the voltage-dependent current traces in Fig. 1A2. On average, the first application of NMDA (10 μM) evoked 28 ± 6 pA of inward current at −70 mV (n = 5), whereas this current increased to 38 ± 7 pA after 30 min of recording (P < 0.01, paired t test). In order to study effects of AMPK on NMDA currents, we superfused slices with A769662 or PT1, which have been shown to activate AMPK by stabilizing phosphorylation at Thr-172 (Göransson et al., 2007; Pang et al., 2008). NMDA (10 μM) evoked a small inward current that was followed by an outward current when slices were superfused with either A769662 (10 μM, Fig. 1B1) or PT1 (10 μM, Fig. 1C1) at −70 mV. Moreover, amplitudes of NMDA-evoked outward currents became progressively larger during 30 min of superfusion with either AMPK activator. As shown in the voltage-dependent current traces in Fig. 1B2 and C2, NMDA increased membrane conductance in the presence of A769662 and PT1.
Fig. 1.

AMPK activators augment the ability of NMDA (10 μM) to evoke outward currents in STN neurons. (A1) Current trace shows that repeated applications of NMDA (10 μM) consistently evoke inward currents (at – 70 mV) in an STN neuron. Truncated deflections in these and subsequent current records are artifacts caused by voltage steps that were used to measure series resistance or membrane conductance for the construction of I-V plots. (A2) Current traces recorded during a series of hyperpolarizing voltage steps (from −70 to −140 mV) in the absence and presence of NMDA. Wash indicates recording after NMDA was washed from the slice. Dashed line indicates zero current. (B1) Current trace shows that NMDA evokes outward currents with increasing amplitudes when the slice is superfused with the AMPK activator A769662 (10 μM). (B2) Current traces recorded during a series of hyperpolarizing voltage steps (from −70 to −140 mV) show that NMDA increases membrane conductance in an STN neuron when recorded in the presence of A769662 (10 μM). (C1) Current trace shows that NMDA evokes outward currents with increasing amplitudes when the slice is superfused with the AMPK activator PT1 (10 μM). (C2) Current traces recorded during a series of hyperpolarizing voltage steps (from −70 to −140 mV) show that NMDA increases membrane conductance in an STN neuron when recorded in the presence of PT1 (10 μM).
We obtained similar results with intracellular dialysis of AMPK activators, as shown in Fig. 2. NMDA was bath applied at 15- to 20-minute intervals while recording with pipettes that contained either normal internal solution (control) or solutions containing an AMPK activating agent. The current trace in Fig. 2A1 shows that intracellular dialysis of A769662 (5 μM) caused NMDA to evoke increasing amounts of outward current with repeated applications. In nine STN neurons dialyzed with A769662, the initial application of NMDA (10 μM) evoked an inward current of 9 ± 12 pA at −70 mV. However, NMDA evoked 31 ± 16 pA of outward current after more than 30 minutes of dialysis (n = 9; P < 0.01, paired t test). Figure 2A2 shows that the voltage dependence of NMDA current recorded with A769662 in pipettes was significantly different from that recorded under control conditions (P < 0.0001; F4,104 = 15.17). I-V plots were constructed at least 30 min after beginning whole-cell recordings. In the presence of A769662 (5 μM), NMDA current was associated with a positive slope conductance of 3.9 ± 0.8 nS when measured between −70 and −100 mV (n = 9), which was significantly different from the negative slope conductance of 0.29 ± 0.18 nS (n = 5) in the control group (P < 0.001, t test). Similarly, intracellular dialysis with PT1 (10 μM) also caused increasing amplitudes of outward current evoked by repeated applications of NMDA (Fig. 2B1). In the presence of PT1, the initial application of NMDA (10 μM) evoked an inward current of 3 ± 27 pA at −70 mV. However, NMDA evoked 82 ± 33 pA of outward current after more than 30 minutes of dialysis (n = 4; P < 0.05, paired t test). Figure 2B2 shows IV plots for NMDA currents evoked with PT1 versus the control condition. The presence of PT1 caused a significant shift in voltage dependence compared to NMDA control (P < 0.0001, F4,64 = 14.94). In the presence of PT1, NMDA current was associated with a positive slope conductance of 4.5 ± 1.4 nS when measured between −70 and −100 mV (n = 4), which was significantly different from the negative slope conductance evoked by NMDA under control conditions (P < 0.05, t test). These data show that both A769662 and PT1 cause NMDA to evoke outward currents with subsequent alteration of I-V plots with increasing positive slope conductance.
Fig. 2.

AMPK activation potentiates the ability of NMDA (10 μM) to increase conductance in STN neurons. (A1) Trace showing currents evoked by NMDA while recording with a pipette that contained A769662 (5 μM). Bath application of NMDA evoked an inward current during the first application and an outward current in the second application. (A2) Summarized I-V plots showing voltage dependence of NMDA currents evoked in neurons recorded with pipettes that contained either normal internal solution (control; n = 5) or A769662 (n = 9). Note that the region of negative slope conductance seen in the NMDA control plot is replaced by a positive slope conductance when recording with A769662. (B1) Current trace recorded with a pipette that contained PT1 (10 μM); NMDA evoked an inward current during the first application and outward current in the second application. (B2) Summarized I-V plots showing voltage dependence of NMDA currents evoked in neurons recorded with pipettes that contained either normal internal solution (control) or PT1 (n = 4). I-V plots were obtained at least 30 min after starting whole-cell recordings. The I-V plot for NMDA control represents the same data as shown in Fig. 2A2. NMDA control (blue) and NMDA in A769662 (red) are color coded for use in multiple figures. Asterisks: *, P < 0.05; **, P < 0.01.
3.2 Dorsomorphin prevents NMDA-induced membrane conductance increase
To test for the selectivity of AMPK activators, we investigated the ability of the AMPK inhibitor dorsomorphin (also known as compound C) to block the action of A769662 on NMDA currents (Shah et al., 2011; Vucicevic et al., 2011). As shown in the current trace in Fig. 3A, the first application of NMDA (10 μM) evoked an inward current that was followed by an outward current in the presence of dorsomorphin (30 μM). However, subsequent applications of NMDA revealed a progressive loss of the outward current and an increase in inward current. On average, NMDA evoked 16 ± 8 pA of inward current at −70 mV during the initial NMDA application, whereas this current increased to 44 ± 10 pA in later applications in the presence of dorsomorphin (n = 7; P < 0.01, paired t test). Therefore, I-V plots were constructed at least 30 min after starting superfusion with dorsomorphin (30 μM). The summary I-V plots in Fig. 3B show that NMDA-induced outward currents in the presence of dorsomorphin (n = 7) were significantly different from those recorded with A769662 without dorsomorphin (n = 9: P < 0.0001, F4,120 = 30.29). The I-V plot for NMDA currents in the presence of A769662 without dorsomorphin (A769662 control) represent the same data as shown in Fig. 2A2. Slope conductance for NMDA currents, measured between −70 and −100 mV, averaged −1.2 ± 0.2 nS when recorded in dorsomorphin plus A769662, which was significantly different from the positive slope conductance recorded in A769662 alone (P < 0.001, t test). By showing that actions of A769662 are blocked by dorsomorphin, these results support the conclusion that A769662 augments the NMDA-induced membrane conductance increase by a selective activation of AMPK.
Fig. 3.
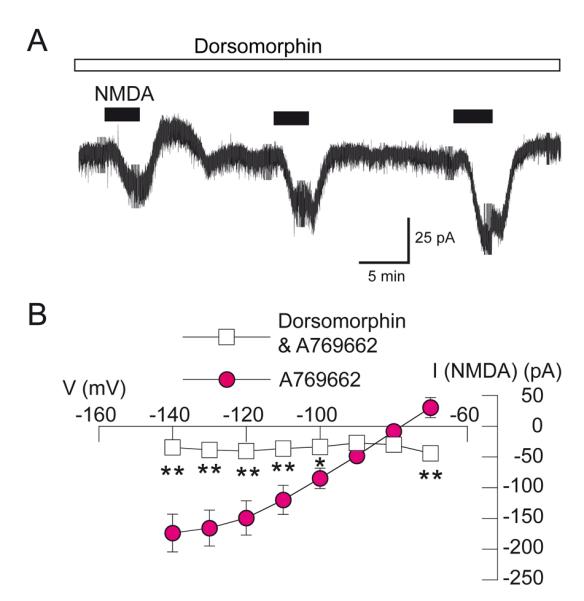
Dorsomorphin prevents the generation of AMPK-dependent NMDA-induced outward current. (A) Current trace shows that bath application of the AMPK inhibitor dorsomorphin (30 μM) blocks NMDA-evoked outward current (at −70 mV) in an STN neuron recorded with internal solution containing A769662 (5 μM). Note that outward current evoked by repeated applications of NMDA (10 μM) disappears gradually with continued dorsomorphin superfusion. (B) I-V plots of NMDA-induced currents recorded with pipettes containing A769662 in the presence (n = 7) and absence of dorsomorphin. Note that dorsomorphin significantly altered the voltage dependence of currents evoked by NMDA. The A769662 control plot is the same as that shown in Fig. 2A2. The dorsomorphin I-V plot was constructed at least 30 min after starting superfusion. Asterisks: *, P < 0.05; **, P < 0.01.
3.3 Western immunoblot
The effects of A769662 and dorsomorphin on Thr-172 phosphorylation of AMPK (P-AMPK) were verified using Western immunoblot analysis. Brain slices were incubated for 60 minutes with varied concentrations of A769662 (0-100 μM), and some were pretreated for 45 minutes with dorsomorphin (30 uM) to approximate the doses and timing used in our electrophysiology experiments. Immunoblots in Fig. 4A show that A769662 produced a concentration-dependent increase in P-AMPK intensity. In contrast, total AMPK intensity was not changed by A769662 (n = 3; P > 0.05, one-way RM ANOVA). Figure 4B shows effects of A769662 on P-AMPK that have been normalized to levels of total AMPK. A769662 produced a concentration-dependent increase in P-AMPK (n = 3; P < 0.01, F = 13.84, one-way RM ANOVA). These data are also shown in Fig. 4C in which P-AMPK is expressed as percentage of the zero A769662 control. At the 30 μM concentration, A769662 increased P-AMPK to 438 ± 80% of control. Fig. 4C also shows the effect of pretreating slices with 30 μM dorsomorphin (n = 4). Dorsomorphin caused a significant decrease in the ability of A769662 to increase P-AMPK levels (P < 0.01, F4,12 = 8.99). These data support the conclusion that dorsomorphin causes a significant reduction in A769662-induced activation of AMPK.
Fig. 4.
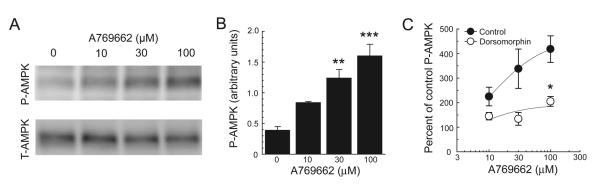
Western blot analysis showing antagonism of A769662-induced increased in P-AMPK by dorsomorphin. (A) A769662 increases the expression of phosphorylated AMPK alpha subunit (P-AMPK). (B) A769662 causes a concentration-dependent increase in P-AMPK (n = 3). (C) Dorsomorphin (30 μM, n = 4) antagonizes the A769662-induced increase (n = 3) in P-AMPK. Data are expressed as percentages of control P-AMPK in zero A769662. Asterisks: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
3.4 NMDA-induced conductance increase is mediated by K-ATP current
To test for involvement of K+ in the NMDA-dependent conductance increase, we investigated the ability of the sulfonylurea agent tolbutamide and other K+ channel blockers to modify NMDA currents in STN neurons recorded with pipettes that contained the AMPK activator A769662 (5 μM). As shown in the current trace in Fig. 5A1, NMDA (10 μM) evoked a small inward current followed by an outward current soon after starting whole-cell recording with A769662 in the pipette. In the presence of tolbutamide (100 μM), however, outward current evoked by NMDA was replaced by a larger inward current. On average, NMDA evoked 39 ± 13 pA of outward current at −70 mV in control (n = 10). In the presence of tolbutamide, however, NMDA caused an inward current of 68 ± 9 pA in those same neurons (P < 0.01, paired t test). Addition of Ba2+ (300 μM) to tolbutamide further increased the amplitude of this inward current to 104 ± 19 pA (n = 10; P < 0.05, paired t test). Tolbutamide-sensitive NMDA current was estimated to reverse at −95 ± 3 mV (n = 17), which is close the expected reversal potential for K+.
Fig. 5.

Augmentation of NMDA-induced currents by AMPK is sulfonylurea-sensitive. NMDA-induced outward currents were recorded with pipettes that contained A697662 (5 μM). (A1) Current trace shows that outward current evoked by NMDA (at −70 mV) was blocked by tolbutamide (100 μM). Note that the addition of Ba2+ (300 μM) increased the amplitude of NMDA-evoked inward current. (A2) Summarized I-V plots show that tolbutamide significantly altered the voltage dependence of NMDA-evoked currents in the presence of A769662, and the addition of Ba2+ caused a further shift in voltage dependence (n = 8). Also note that tolbutamide changed the slope conductance of NMDA-evoked currents from positive to negative, and this was further accentuated by Ba2+. (B1) NMDA-induced inward currents in tolbutamide were not further increased by apamin (1 μM). (B2) I-V plots show that the voltage dependence of NMDA currents in tolbutamide (100 μM) was not altered by the addition of apamin (n = 6). (C1) NMDA currents recorded in tolbutamide and penitrem A (10 μM). (C2) Summarized I-V plots show that the region of negative slope conductance of NMDA-gated currents in tolbutamide was not significantly altered by the addition of penitrem A (n = 12). Symbols near data points indicate significant differences (* and #, P < 0.05; ** and ##, P < 0.01; * and **, control vs tolbutamide; # and ##, tolbutamide vs tolbutamide plus Ba2+).
Voltage-dependent effects of tolbutamide with and without Ba2+ on NMDA-gated currents are shown Fig.5A2. Tolbutamide significantly altered the voltage dependence of NMDA currents (n = 10; P < 0.0001, F4,152 = 27.41). NMDA-induced current was associated with a positive slope conductance of 3.36 ± 0.63 nS with A769662 in pipettes, but this conductance decreased to 0.35 ± 0.35 nS (n = 10) in the presence of tolbutamide in those same neurons (P < 0.01, paired t test). Addition of Ba2+ further altered the voltage dependence of NMDA currents (n = 10; P < 0.0001, F4,152 = 9.47). NMDA-evoked currents in tolbutamide plus Ba2+ had a negative slope conductance (1.94 ± 0.41 nS) that was significantly larger than that recorded in tolbutamide alone (P < 0.01, paired t test).
Because we showed previously that NMDA-evoked K-ATP current was Ca2+-dependent (Shen and Johnson, 2010), we considered the possibility that the Ba2+-sensitive outward current that remained in the presence of tolbutamide might be mediated by Ca2+-activated K+ (gKCa) channels. However, the current trace in Fig. 5B1 shows that NMDA-induced inward currents recorded in tolbutamide were not increased further by the small conductance gKCa blocker apamin. On average, NMDA (10 μM) evoked 63 ± 6 pA of inward current in the presence of tolbutamide (100 μM), and this inward current was not changed (72 ± 3 pA) by the addition of apamin (1 μM) in those same neurons (n = 6; P > 0.05, paired t test). I-V plots in Fig. 5B2 show that the addition of apamin to tolbutamide did not significantly alter the voltage dependence of NMDA currents recorded in the presence of A769662 (n = 6; P = 0.289, F4,88 = 1.27). We also tested the effect of penitrem A (10 μM), a blocker of large conductance gKCa channels, on NMDA-induced currents (Fig. 5C1). I-V plots in Fig. 5C2 show that the addition of penitrem A to tolbutamide did not further alter the voltage dependence of NMDA currents (n = 12; P > 0.05, F4,184 = 0.95). These data indicate that the augmentation by AMPK of the NMDA-evoked conductance increase is mediated by K-ATP channels and not by small or big conductance Ca2+-activated K+ channels. Because adding Ba2+ to tolbutamide more effectively blocked outward current than did tolbutamide alone, it is possible that NMDA evokes another type of K+ current that remains unidentified. However, it is also possible that Ba2+-sensitive current in tolbutamide is due to use of a submaximally effective concentration of tolbutamide.
3.5 Augmentation of NMDA-evoked K-ATP conductance by AMPK is Ca2+-, calmodulinand CaMKKβ-dependent
We proceeded to define the possible roles of Ca2+ and Ca2+-dependent pathways on AMPK augmentation of NMDA-evoked K-ATP current. As seen in the current trace in Fig. 6A1, superfusing the slice with low Ca2+ (0.2 mM) aCSF blocked the outward current evoked by NMDA (10 μM). On average, NMDA evoked an inward current of 37 ± 14 pA at −70 mV with 5 μM A769662 in the pipette when the slice was perfused with low Ca2+ aCSF, which was significantly different from the 58 ± 30 pA of outward current evoked by NMDA in normal aCSF (n = 6; P < 0.05, paired t test). Low Ca2+ also significantly modified the NMDA I-V relationship, as shown in Fig. 6A2 (P < 0.0001; F4,88 = 7.78). NMDA-evoked currents in normal aCSF had a positive slope conductance of 5.1 ± 2.1 nS, which was significantly different from the 0.1 ± 0.2 nS as recorded in low Ca2+ in the same neurons (n = 6; P < 0.05, paired t test). These data suggest that Ca2+ influx participates in the AMPK effect on NMDA-evoked K-ATP current.
Fig. 6.

Augmentation of NMDA-evoked K-ATP conductance by AMPK is dependent upon Ca2+, calmodulin, and CaMKKβ. All recordings were made with pipettes that contained A769662 (5 μM). (A1) Current traces showing that low Ca2+ aCSF (0.2 mM) blocks NMDA-induced outward current. (A2) Summarized I-V plots for NMDA currents in control and low Ca2+ aCSF. (B1) Current trace shows that NMDA failed to evoke outward current (at −70 mV) when pipettes contained BAPTA (10 mM) in place of EGTA. (B2) Summarized I-V plots show that the presence of BAPTA significantly altered the voltage-dependence of currents evoked by NMDA (n = 6) compared to the A769662 control. (C1) Current trace shows that bath application of the calmodulin inhibitor CGS9343B (40 μM) blocked NMDA-evoked outward current (at –70 mV). (C2) Summarized I-V plots show that CGS9343B (40 μM) significantly altered the voltage-dependence of currents evoked by NMDA (n = 7) compared to A769662 control. (D1) Current trace shows that STO609 (10 μM), an inhibitor of CaMKKβ, blocked NMDA-evoked outward current (at −70 mV). Slices were superfused with STO609 at least 30 min prior to beginning whole-cell recording. (D2) Summarized I-V plots showing that STO609 significantly altered the voltage dependence of NMDA current (n = 7) compared to the A769662 control. STO609 was superfused at least 30 min prior to construction of the I-V plot. I-V plots for A769662 control in B2 and D2 are historical controls and are the same as that shown in Fig. 2A2. The data shown in A2 and C2 are paired data in which data for the A769662 control were obtained from the same neurons treated with CGS9343B or low Ca2+ aCSF. Asterisks: *, P < 0.05; **, P < 0.01.
STN neurons were also recorded with pipettes that contained the Ca2+ chelator BAPTA (10 mM), which has much faster Ca2+-buffering kinetics than the EGTA that we use in normal internal solutions. Pipettes also contained the AMPK activator A769662 (5 μM). As shown in the current trace in Fig. 6B1, repeated applications of NMDA (10 μM) consistently evoked inward currents. On average, NMDA (10 μM) evoked 22 ± 6 pA of inward current at −70 mV during the initial application, which increased slightly but significantly to 27 ± 5 pA 30 min after starting whole cell recording (n = 7; P < 0.05, paired t test). Figure 6B2 shows the I-V plot for NMDA currents recorded after at least 30 min of intracellular BAPTA dialysis. Exposure to BAPTA plus A769662 caused a significant shift in the I-V curve for NMDA currents compared to the control IV plot for A769662 (P < 0.0001, F4,120 = 22.03). The negative slope conductance of NMDA current measured between −70 and −100 mV in BAPTA (0.2 ± 0.2 nS, n = 7) was also significantly different from the positive slope conductance of 3.9 ± 0.8 nS (n = 5) that was recorded in A769662 control cells (P < 0.01, t test). These results show that intracellular BAPTA markedly reduced the AMPK-dependent NMDA-activated K-ATP conductance increase, which supports the hypothesis that Ca2+ mediates the AMPK-induced augmentation of K-ATP current.
Having established Ca2+ dependence, we proceeded to test for involvement of downstream Ca2+-dependent targets. To test for involvement of calmodulin, we attempted to block AMPK-dependent potentiation of NMDA-evoked K-ATP currents with the calmodulin inhibitor CGS9343B (40 μM). As seen in the current trace in Fig. 6C1, NMDA (10 μM) evoked inward currents that were followed by outward currents with A769662 in the pipette. However, superfusing the slice with CGS9343B for 10 min potentiated the inward current and reduced the outward current that were evoked by NMDA. On average, NMDA evoked an inward current of 33 ± 3 pA at −70 mV in the presence of CGS9343B, which was significantly different from the 34 ± 9 pA of outward current evoked by NMDA under control conditions in the same neurons (n = 6; P < 0.01, paired t test). CGS9343B also significantly modified the NMDA I-V relationship, as shown in Fig. 6C2 (P < 0.0001, F4,88 = 8.57). Under the control condition, NMDA evoked currents with a positive slope conductance of 2.8 ± 0.6 nS. However, in the presence of CGS9343B, NMDA currents had a slope conductance of 0.1 ± 0.2 nS in the same neurons (n = 6; P < 0.01, paired t test). These data suggest that calmodulin participates in the AMPK effect on NMDA-evoked K-ATP current.
Finally, we tested the hypothesis that potentiation of NMDA-induced K-ATP current required Ca2+-calmodulin-dependent protein kinase kinase beta (CaMKKβ), which is a known Ca2+-dependent activator of AMPK (Salminen et al., 2011). The current trace in Fig. 6D1 shows that bath application of the CaMKKβ inhibitor STO609 (10 μM) completely blocked NMDA-induced outward currents, despite the presence of A769662 in pipettes. In these experiments slices were superfused with STO609 at least 30 min prior to beginning whole-cell recordings. On average, NMDA (10 μM) evoked 22 ± 7 pA of inward current at −70 mV in the initial application in the presence of STO 609, and this inward current increased to 51 ± 9 pA after 30 min of STO609 superfusion (n = 7; P < 0.05, paired t test). Figure 6D2 shows that the I-V relationship for NMDA currents recorded after 30 min with STO609 was significantly different from the A769662 control I-V plot (P < 0.0001, F4,120 = 12.93). This figure also shows that the positive slope conductance of NMDA currents in the A769662 control group was significantly different from the conductance of 0.3 ± 0.3 nS (n = 7) recorded in the presence of STO609 (P < 0.01, t test).These data suggest that AMPK augmentation of NMDA-induced K-ATP currents is Ca2+ dependent and is likely mediated by calmodulin and phosphorylation of AMPK by CaMKKβ.
3.6 Potentiation by AMPK activation of NMDA-dependent K-ATP conductance increase requires NO and cGMP
Not only has AMPK been shown to activate nitric oxide synthase (NOS) (Chen et al., 1999), but NO has also been shown to cause further AMPK activation (Lira et al., 2007; Murphy et al., 2009). Because we had shown previously that NMDA-evoked K-ATP current is dependent upon NO and cGMP (Shen and Johnson, 2010), we proceeded to test whether or not the potentiation of K-ATP current by AMPK was also dependent upon these second messengers. All recordings were done using pipettes that contained A769662 (5 μM). Current traces in Fig. 7A1 show that NMDA-induced outward current is blocked by superfusing the slice with the NOS inhibitor L-NAME (200 μM). On average, NMDA (10 μM) evoked 10 ± 14 pA of inward current at −70 mV in the presence of L-NAME, which was significantly different than the 36 ± 17 pA of outward current that was evoked by NMDA in the absence of L-NAME in the same neurons (n = 6; P < 0.05, paired t test). L-NAME also significantly modified the NMDA I-V relationship, as shown in Fig. 7A2 (n = 6; P < 0.0001, F4,88 = 11.81). In the absence of L-NAME, NMDA current was associated with a positive slope conductance of 2.8± 0.5 nS. However, this conductance was reduced to 0.7 ± 0.5 nS in the presence of L-NAME in the same neurons (P < 0.05, paired t test). These results suggest that NO is required for augmentation of NMDA-induced K-ATP current by AMPK.
Fig. 7.

Augmentation of the NMDA-activated K-ATP conductance by AMPK requires NO and cGMP. All recordings were made with pipettes that contained A769662 (5 μM). (A1) Current trace shows that bath application of the NOS inhibitor L-NAME (200 μM) blocked NMDA-evoked outward current (at –70 mV). Note that only inward current was evoked by NMDA in the presence of L-NAME. (A2) Summarized I-V plots show that L-NAME (200 μM) significantly altered the voltage-dependence of currents evoked by NMDA (n = 6). (B1) Current trace shows that the guanylyl cyclase inhibitor ODQ (20 μM) blocked NMDA-evoked outward current (at –70 mV) in an STN neuron. Note that NMDA evoked an inward current in the presence of ODQ, whereas NMDA evoked outward current after ODQ was washed from the slice. (B2) Summarized I-V plots show that ODQ (20 μM) significantly altered the voltage-dependence of currents evoked by NMDA and changed the slope conductance of NMDA-evoked currents from positive to negative (n = 7). Asterisks: *, P < 0.05; **, P < 0.01.
We tested for involvement of cGMP-dependent pathways by examining the ability of the guanylyl cyclase inhibitor ODQ to block AMPK-dependent potentiation of NMDA-induced K-ATP currents. Current traces in Fig. 7B1 show that superfusing the slice with ODQ for 15 min blocked the outward current evoked by NMDA (10 μM). On average, NMDA evoked an inward current of 84 ± 17 pA at −70 mV in the presence of ODQ, which was significantly different from the 78 ± 34 pA of outward current evoked by NMDA without ODQ (n = 7; P < 0.05, paired t test). I-V plots in Fig. 7B2 show that ODQ also significantly modified the NMDA I-V relationship (n = 7; P < 0.0001, F4,104 = 10.83). Also, NMDA-evoked currents had a negative slope conductance of 0.7 ± 0.5 nS in the presence of ODQ, and this was significantly different from the positive slope conductance of 4.7 ± 1.6 nS recorded before ODQ in the same neurons (n =7; P < 0.05, paired t test). These results suggest that augmentation of NMDA-induced K-ATP current requires participation of a cGMP-dependent pathway.
3.7 AMPK activation fails to augment diazoxide currents
We considered the possibility that AMPK might augment K-ATP currents by increasing the surface expression of K-ATP channels (Sukhodub et al., 2007; Lim et al., 2009). Therefore, we examined the effect of an AMPK activator on outward currents evoked by bath application of the K-ATP channel opener diazoxide (200 μM). Current traces in Fig. 8A1 show that diazoxide evoked reproducible outward currents (at −70 mV) when recorded with pipettes that contained A769662 (5 μM). Slices were superfused with diazoxide for 10 min at intervals of 20 min. This figure also shows that tolbutamide (100 μM) rapidly aborted the diazoxide current. Data in Fig. 8A2 show that diazoxide outward current recorded immediately after starting whole-cell recording (0 min) averaged 23 ± 4 pA, which was not significantly different from currents recorded 20 and 40 min after beginning recordings (P > 0.05, one-way ANOVA). Moreover, current evoked by diazoxide at 0 min with A769662 was not significantly different from the 26 ± 5 pA of outward current evoked using pipettes without A769662 (P > 0.05, Mann-Whitney rank sum test). We next tested whether or not repeated applications of NMDA might enhance the effect of an AMPK activator on K-ATP current. The current traces in Fig. 8B1 show that repeated applications of diazoxide evoked reproducible outward currents despite repeatedly evoking NMDA currents with pipettes that contained A769662. In these experiments diazoxide was superfused every 30-35 min, and NMDA (10 μM) was superfused between diazoxide applications. The summary data in Fig. 7B2 show that amplitudes of diazoxide-induced currents were not significantly different despite repeated applications of NMDA in the presence of A769662 (P > 0.05, one-way ANOVA). These data suggest that augmentation of NMDA-induced K-ATP currents by AMPK is not due to increased surface expression of K-ATP channels.
Fig. 8.

AMPK activator A769662 does not potentiate currents evoked by diazoxide. (A1) Current traces show that currents evoked by bath applications of diazoxide (200 μM) do not increase over time when recorded with pipettes that contained A769662 (5 μM). This figure also shows that diazoxide currents are completely blocked by tolbutamide (100 μM). (A2) Summary histogram showing that A769662 does not change the amplitude of diazoxide current. Diazoxide currents were evoked every 20 min after beginning whole-cell recordings. The bar labeled “control” represents diazoxide currents recorded without A769662 in pipettes. (B1) Current traces showing that repeated applications of NMDA (10 μM) do not alter diazoxide-induced currents when recorded with pipettes that contained A769662. (B2) Summary histogram showing lack of effect of repeated NMDA applications on diazoxide currents. NMDA and diazoxide were applied to the bath every 30-35 min. The bar labeled “control” represents currents evoked before NMDA application using pipettes with A769662. Numbers above columns indicate number of cells.
3.8 Functional consequences of AMPK-dependent K-ATP channel activation by NMDA
To evaluate changes in firing pattern, we examined the influences of AMPK, NMDA and tolbutamide on the depolarization-induced plateau potential that underlies one type of burst firing in STN neurons. Depolarizing pulses of current (200 pA, 100 ms duration) were used to evoke plateau potentials. Plateau potential duration was measured as the time from depolarization onset to return to baseline. A small holding current was injected to establish an initial membrane potential of −70 mV prior to drug application. As shown in Fig. 9A, a brief depolarizing current pulse evoked a burst of spikes that rides on top of a plateau potential. NMDA (10 μM) prolonged the plateau potential from a control duration of 337 ± 68 ms to 1494 ± 448 ms (n = 7; P < 0.05, paired t test) when recorded with control internal solutions. In contrast, Fig. 9B shows that NMDA caused a significant shortening of the plateau potential (from 705 ± 204 ms to 196 ± 30 ms) when recording with pipettes that contained 5 μM A769662 (n = 7; P < 0.05, t test). However, superfusing the slice with either 100 μM tolbutamide (Fig. 9C) or 30 μM dorsomorphin (Fig. 9D) again permitted NMDA to lengthen plateau potentials. On average, NMDA prolonged the plateau potential by 393 ± 147% in tolbutamide (n = 7) and by 329 ± 26% in dorsomorphin (n = 6), both of which were significantly different from the 54 ± 9% reduction in plateau duration caused by NMDA in A769662 alone. These results suggest that AMPK-dependent activation of K-ATP channels causes a significant antagonism of the excitatory effect of NMDA receptor stimulation on plateau potential duration.
Fig. 9.

A769662 shortens plateau potentials by activating K-ATP. Current-clamp whole-cell recordings of plateau potentials obtained from STN neurons without (control) and with A769662 in pipettes. Although NMDA lengthened the plateau potential under control conditions (A), NMDA shortened the plateau potential when recording with a pipette that contained A769662 (B). Both tolbutamide (C) and dorsomorphin (D) blocked the effect of A769662, thus allowing NMDA to lengthen the plateau potential. Each plateau potential was evoked by a brief depolarizing current pulse (200 pA; 100 ms duration) as illustrated under the voltage trace in “D”. A holding current was applied to establish an initial membrane potential of −70 mV. Wash indicates recording after NMDA is washed from the slice. Data were obtained from four different neurons.
We next investigated the influence of AMPK activation on NMDA-induced burst firing. Using pipettes that contained normal internal solution, superfusing the slice with NMDA (10 μM) evoked burst firing, as seen in Fig. 10A. Superfusing slices with diazoxide (100 μM) caused reversible suppressed burst firing in all neurons tested (n = 8). Tolbutamide (100 μM) tended to facilitate NMDA-evoked burst firing as evidenced by an increase in number of spikes per burst (79 ± 50 in control versus 131 ± 37 during tolbutamide; P = 0.11) and by an increase in the burst duration (1516 ± 344 in control versus 2979 ± 333 during tolbutamide; n = 3; P < 0.05, t tests). In addition, Fig. 10A shows that NMDA-induced burst firing could also be suppressed by superfusing 0.5 mM AICAR, which is an AMP mimic that is known to activate AMPK (n = 3).
Fig. 10.
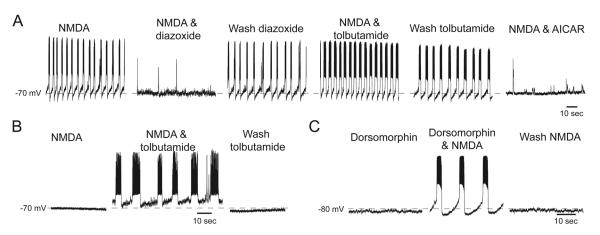
AMPK activation suppresses NMDA-evoked burst firing. (A) Current-clamp recordings showing NMDA-induced K-ATP channel-dependent burst firings in a STN neuron recorded with control internal solutions. Burst firing is inhibited by diazoxide (100 μM) and facilitated by tolbutamide (100 μM). The AMPK activator AICAR (0.5 mM) also inhibits NMDA-induced burst firing. (B) NMDA (10 μM) failed to evoke burst firing when recording with a pipette that contained A769662 (5 μM). However, bursting was evoked during superfusion with either tolbutamide (B) or dorsomorphin (C) despite recording with A769662 in the pipette.
We also studied NMDA-induced burst firing when recording with pipettes that contained A769662 (Figs 10B & 10C). Although NMDA-induced burst firing was initially suppressed in the presence of A769662, bursting was induced by NMDA when K-ATP channels were blocked with tolbutamide (100 μM), as seen in Fig. 10B (n = 4). Figure 10C shows that superfusing the slice with dorsomorphin (30 μM) also facilitated the ability of NMDA to evoke bursting in the presence of A769662 (n = 10). These results support the conclusion that AMPK exerts an inhibitory influence on STN neuronal excitability by facilitation of NMDA-dependent K-ATP channel activity.
4. DISCUSSION
In the present study, we demonstrated that pharmacological activation of AMPK augments K-ATP current that is evoked by NMDA receptor stimulation in STN neurons. This augmentation is Ca2+-dependent and requires participation of calmodulin and the upstream kinase CaMKKβ. Our studies also present evidence for involvement of NO and cGMP-dependent pathways in the augmentation of K-ATP channel function by AMPK. In addition, others have shown that AMPK can phosphorylate NOS and promote NO synthesis (Chen et al., 1999), and NO can further accelerate the phosphorylation of AMPK (Lira et al., 2007). Moreover, AMPK has been shown to interact directly with K-ATP channel subunits to increase K-ATP channel function (Yoshida et al., 2012; Du et al., 2014). Thus, our results add to a growing body of literature showing that AMPK activation facilitates K-ATP channel function. A summary schematic of proposed pathways involved in the actions of AMPK on K-ATP function is presented in Fig. 11.
Fig. 11.
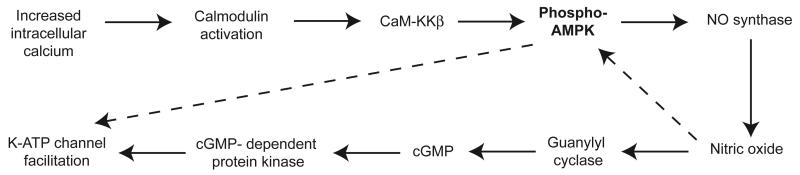
Schematic illustrating proposed pathways by which AMPK augments K-ATP channel function. Arrows with solid lines indicate pathways that are supported by our experimental results, whereas arrows with dashed lines indicate pathways that are supported by the literature.
4.1 Role of AMPK as an energy sensor
Activated by a rising AMP/ATP ratio, AMPK acts as a master regulator that strives to maintain cellular energy homeostasis (Hardie et al., 2003). When activated by phosphorylation, AMPK up-regulates those processes that increase ATP, such as increasing glucose uptake, glycolysis, fatty acid oxidation, and mitochondrial biogenesis. At the same time, AMPK switches off pathways that consume ATP, such as protein and glycogen synthesis (Russo et al., 2013). Exercise of skeletal muscle activates AMPK, as does energy-compromising conditions such as hypoglycemia, hypoxia, and ischemia (Hardie et al., 2003). AMPK also increases the translocation of the glucose transporter GLUT4 to plasma membranes and thus facilitates the actions of insulin to control blood glucose. Because hypoglycemia and hypoxia activate AMPK (Amato and Man, 2011), it may be important to note that living brain slice preparations are very sensitive to fluctuations in glucose level and oxygen tension. Although our brain slices were superfused with a hyperglycemic (11 mM glucose) solution that is saturated with oxygen, levels of glucose and oxygen drop off markedly toward the center of the slice, which can alter neuronal physiology (An et al., 2008; Jiang and Haddad, 1991). Depending upon the location of the recorded neuron in the slice, it is reasonable to expect that local alterations in glucose level and oxygen tension could potentially influence basal AMPK activity and K-ATP channel function in brain slices.
4.2 AMPK and K-ATP channel function
Although we found that AMPK activation markedly potentiated K-ATP currents evoked by NMDA, our results also showed that AMPK activation did not augment K-ATP currents evoked by diazoxide. Furthermore, diazoxide-induced current was not augmented after serial exposures to NMDA in the presence of an AMPK activator, which argues against the hypothesis that augmentation of diazoxide current requires concomitant NMDA receptor activation. Because AMPK activation failed to increase diazoxide-induced currents, this suggests that AMPK does not increase the surface expression of K-ATP channels in STN neurons. This conclusion contrasts with the work of others who found that AMPK activation increased the surface expression of K-ATP channel subunits in pancreatic β-cells (Smith et al., 2006; Lim et al., 2009) and cardiac myocytes (Sukhodub et al., 2007; Yoshida et al., 2012). Our results also do not agree with studies reporting that AMPK activation inhibited K-ATP channel activity as a mechanism for inducing insulin release in pancreatic β-cells (Wang et al., 2005; Langelueddecke et al., 2012; Düfer et al., 2010). In the present study, we suggest that AMPK activation augments NMDA-induced K-ATP currents by potentiating the actions of NO- and cGMP-coupled pathways, which are well-known to potentiate K-ATP function (Han et al., 2001; Shen and Johnson, 2010; Lin et al., 2004; Chai et al., 2011). In contrast, the action of diazoxide is unchanged during AMPK activation because the opening of K-ATP channels by diazoxide is not dependent upon these second messenger pathways.
4.3 AMPK actions in other central neurons
Although the role of AMPK as an energy sensor is well documented, the possibility that AMPK is also an important regulator of neuronal activity is just beginning to be explored. AMPK is widely expressed in the brain, although expression of subunit subtypes varies amongst specific types of neuron and during different stages of development (Turnley et al., 1999). Kuramoto et al (2007) showed that the AMPK activator metformin reduced the rundown in GABAB receptor-mediated outward currents produced by repeated applications of baclofen in cultured hippocampus neurons. Moreover, these investigators demonstrated that AMPK subunits and GABAB receptors were colocalized when expressed in HEK cells. Ikematsu et al (2011) showed that activation of AMPK by A769662 phosphorylates the voltage-dependent delayed rectifier K+ channel Kv2.1. This action of AMPK caused a hyperpolarizing shift in the voltage dependence of Kv2.1 that reduced the excitatory responses of hippocampal neurons to depolarizing current pulses. AMPK has also been shown to phosphorylate TREK K+ channels and BK gKCa channels and reduce their conductances in carotid body neurons (Kréneisz et al., 2009; Ross et al., 2011). AMPK activators have also been shown to inhibit the expression of long-term potentiation in hippocampal neurons (Potter et al., 2010). A well-documented central action of AMPK has been identified in the hypothalamus that regulates appetite. Thus, AMPK activation has been shown to trigger Ca2+ influx and cFOS expression in neuropeptide Y neurons in the arcuate nucleus and induce feeding behavior in rats (Kohno et al., 2011; Dickson and Luckman, 1997). Moreover, the orexigenic hormone ghrelin stimulates appetite by promoting AMPK phosphorylation in arcuate neurons (Kirsz and Zieba, 2011). Although knowledge of AMPK actions in neurons is limited, our data and the results of others suggest that AMPK activation dampens neuronal excitability, which would be consistent with its role to promote the conservation of metabolic energy.
4.4 Functional considerations
Our studies demonstrate that AMPK activation can regulate the firing pattern of STN neurons. By activating K-ATP current, we have shown that AMPK activating agents shorten the duration of plateau potentials and suppress NMDA-induced burst firing. Thus, activation of K-ATP channels by NMDA receptor stimulation would tend to dampen the excitatory influence of glutamate, and our data suggest that AMPK facilitates this inhibitory influence of K-ATP channels. Because burst firing in STN neurons is associated with symptoms of Parkinson’s disease (Wichmann and Dostrovsky, 2011), suppression of bursting may be a potential strategy for improving symptoms of this disease. Our results suggest that AMPK activation in the STN might be explored as a possible treatment strategy for controlling symptoms of Parkinson’s disease. However, it is important to note that the hyperpolarizing influence of K-ATP currents in dopamine neurons would be expected to reduce dopamine release and worsen symptoms of Parkinson’s disease. Therefore, augmentation of K-ATP channel function by AMPK would have to be limited to STN and perhaps other basal ganglia outflow neurons in order to expect benefit in Parkinson’s disease.
5. CONCLUSIONS
Our studies suggest that AMPK activation augments K-ATP current that is evoked by NMDA receptor stimulation in STN neurons. Facilitation of K-ATP current requires Ca2+-dependent activation of the upstream kinase CaMKKβ, and also requires participation of NO- and cGMP-dependent pathways. Because diazoxide-induced currents were not affected, our data suggest that augmentation of K-ATP current by AMPK is not mediated by increased surface expression of K-ATP channel subunits. By increasing the inhibitory influence of K-ATP channels, we suggest that AMPK activation tempers the excitatory influence of NMDA receptor stimulation. Selective activation of AMPK in the STN might be explored in the future as a possible therapeutic strategy for the treatment of Parkinson’s disease symptoms.
AMPK activation augmented K-ATP currents evoked by NMDA in STN neurons
Potentiation of K-ATP current was dependent upon Ca2+, NO, and cGMP
AMPK actions were blocked by dorsomorphin and STO-609
AMPK activators did not potentiate diazoxide currents
AMPK dampens the excitatory effect of NMDA via potentiating K-ATP current
Acknowledgments
This work was supported by NIH grant NS038715 and by the Portland Veterans Affairs Parkinson’s Disease Research, Education, and Clinical Center.
Abbreviations
- aCSF
artificial cerebrospinal fluid
- AMPK
5′-Adenosine monophosphate-activated protein kinase
- CaMKKβ
Calmodulin-dependent protein kinase kinase beta
- gKCa
Calcium-activated potassium conductance
- NMDA
N-methyl-D-aspartate
- NO
nitric oxide
- NOS
nitric oxide synthase
- STN
Subthalamic nucleus
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Amato S, Man H-Y. Bioenergy sensing in the brain. The role of AMP-activated protein kinase in neuronal metabolism, development and neurological diseases. Cell Cycle. 2011;10:3452–3460. doi: 10.4161/cc.10.20.17953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An JH, Su Y, Radman T, Bikson M. Effects of glucose and glutamine concentration in the formulation of the artificial cerebrospinal fluid (ACSF) Brain Res. 2008;1218:77–96. doi: 10.1016/j.brainres.2008.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft FM. Adenosine 5′-triphosphate-sensitive potassium channels. Ann Rev Neurosci. 1988;11:97–118. doi: 10.1146/annurev.ne.11.030188.000525. [DOI] [PubMed] [Google Scholar]
- Beall C, Hamilton DL, Gallagher J, Logie L, Wright K, Soutar MP, Dadak S, Ashford FB, Haythorne E, Du Q, Jovanovic A, McCrimmon RJ, Ashford MLJ. Mouse hypothalamic GT1-7 cells demonstrate AMPK-dependent intrinsic glucose-sensing behaviour. Diabetologia. 2012;55:2432–2444. doi: 10.1007/s00125-012-2617-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti F, Lanotte M, Colloca L, Ducati A, Zibetti M, Lopiano L. Electrophysiological properties of thalamic, subthalamic and nigral neurons during the anti-parkinsonian placebo response. J Physiol (Lond) 2009;587:3869–3883. doi: 10.1113/jphysiol.2009.169425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman H, Wichmann T, DeLong MR. Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science. 1990;249:1436–1438. doi: 10.1126/science.2402638. [DOI] [PubMed] [Google Scholar]
- Beurrier C, Congar P, Bioulac B, Hammond C. Subthalamic nucleus neurons switch from single-spike activity to burst-firing mode. J Neurosci. 1999;19:599–609. doi: 10.1523/JNEUROSCI.19-02-00599.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan MD, Magill PJ, Terman D, Bolam JP, Wilson CJ. Move to the rhythm: oscillations in the subthalamic nucleus-external globus pallidus network. Trend Neurosci. 2002;25:525–531. doi: 10.1016/s0166-2236(02)02235-x. [DOI] [PubMed] [Google Scholar]
- Bevan MD, Wilson CJ. Mechanisms underlying spontaneous oscillation and rhythmic firing in rat subthalamic neurons. J Neurosci. 1999;19:7617–7628. doi: 10.1523/JNEUROSCI.19-17-07617.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Y, Zhang D-M, Lin Y-F. Activation of cGMP-dependent protein kinase stimulates cardiac ATP-sensitive potassium channels via a ROS/calmodulin/CaMKII signaling cascade. PLoS ONE. 2011;6(3):e18191. doi: 10.1371/journal.pone.0018191. doi: 10.1371/journal.pone.0018191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z-P, Mitchelhill KI, Michell BJ, Stapleton D, Rodriguez-Crespo I, Witters LA, Power DA, Ortiz de Montellano PR, Kemp BE. AMP-activated protein kinase phosphorylation of endothelial NO synthase. Fed Eur Biochem Soc Lett. 1999;443:285–289. doi: 10.1016/s0014-5793(98)01705-0. [DOI] [PubMed] [Google Scholar]
- Chu J-U, Jeong MJ, Song K-I, Lee H-C, Kim J, Kim Y-J, Choi IK, Suh J-KF, Youn I. Spontaneous synchronized burst firing of subthalamic nucleus neurons in rat brain slices measured on multi-electrode arrays. Neurosci Res. 2012;72:324–340. doi: 10.1016/j.neures.2012.01.004. [DOI] [PubMed] [Google Scholar]
- Dickson SL, Luckman SM. Induction of c-fos messenger ribonucleic acid in neuropeptide Y and growth hormone (GH)-releasing factor neurons in the rat arcuate nucleus following systemic injection of the GH secretagogue, GH-releasing peptide-6. Endocrinol. 1997;138:771–777. doi: 10.1210/endo.138.2.4907. [DOI] [PubMed] [Google Scholar]
- Do MTH, Bean BP. Subthreshold sodium currents and pacemaking of subthalamic neurons: modulation by slow inactivation. Neuron. 2003;39:109–120. doi: 10.1016/s0896-6273(03)00360-x. [DOI] [PubMed] [Google Scholar]
- Du R-H, Dai T, Cao W-J, Lu M, Ding J, Hu G. Kir6.2-containing ATP-sensitive K+ channel is required for cardioprotection of resveratrol in mice. Cardiovascular Diabetology. 2014;13:35. doi: 10.1186/1475-2840-13-35. doi: 10.1186/1475-2840-13-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Düfer M, Noack K, Krippeit-Drews P, Drews G. Activation of the AMP-activated protein kinase enhances glucose-stimulated insulin secretion in mouse β-cells. Islets. 2010;2:156–163. doi: 10.4161/isl.2.3.11412. [DOI] [PubMed] [Google Scholar]
- Dunn-Meynell AA, Rawson NE, Levin BE. Distribution and phenotype of neurons containing the ATP-sensitive K+ channel in rat brain. Brain Res. 1998;814:41–54. doi: 10.1016/s0006-8993(98)00956-1. [DOI] [PubMed] [Google Scholar]
- Göransson O, McBride A, Hawley SA, Ross FA, Shpiro N, Foretz M, Viollet B, Hardie DG, Sakamoto K. Mechanism of action of A-769662, a valuable tool for activation of AMP-activated protein kinase. J Biol Chem. 2007;282:32549–32560. doi: 10.1074/jbc.M706536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallows KR. Emerging role of AMP-activated protein kinase in coupling membrane transport to cellular metabolism. Curr Opin Nephrol Hypertens. 2005;14:464–471. doi: 10.1097/01.mnh.0000174145.14798.64. [DOI] [PubMed] [Google Scholar]
- Han J, Kim N, Kim E, Ho W-K, Earm YE. Modulation of ATP-sensitive potassium channels by cGMP-dependent protein kinase in rabbit ventricular myocytes. J Biol Chem. 2001;276:22140–22147. doi: 10.1074/jbc.M010103200. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Scott JW, Pan DA, Hudson ER. Management of cellular energy by the AMP-activated protein kinase system. Fed Eur Biochem Soc Lett. 2003;546:113–120. doi: 10.1016/s0014-5793(03)00560-x. [DOI] [PubMed] [Google Scholar]
- Hollerman JR, Grace AA. Subthalamic nucleus cell firing in the 6-OHDA-treated rat: basal activity and response to haloperidol. Brain Res. 1992;590:291–299. doi: 10.1016/0006-8993(92)91108-q. [DOI] [PubMed] [Google Scholar]
- Ikematsu N, Dallas ML, Ross FA, Lewis RW, Rafferty JN, David JA, Suman R, Peers C, Hardie DG, Evans AM. Phosphorylation of the voltage-gated potassium channel Kv2.1 by AMP-activated protein kinase regulates membrane excitability. Proc Natl Acad Sci (USA) 2011;108:18132–18137. doi: 10.1073/pnas.1106201108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Haddad GG. Effect of anoxia on intracellular and extracellular potassium activity in hypoglossal neurons in vitro. J Neurophysiol. 1991;66:103–111. doi: 10.1152/jn.1991.66.1.103. [DOI] [PubMed] [Google Scholar]
- Kass JI, Mintz IM. Silent plateau potentials, rhythmic bursts, and pacemaker firing: Three patterns of activity that coexist in quadristable subthalamic neurons. Proc Natl Acad Sci (USA) 2006;103:183–188. doi: 10.1073/pnas.0506781102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsz K, Zieba DA. Ghrelin-mediated appetite regulation in the central nervous system. Peptides. 2011;32:2256–2264. doi: 10.1016/j.peptides.2011.04.010. [DOI] [PubMed] [Google Scholar]
- Kitai ST, Deniau JM. Cortical inputs to the subthalamus: Intracellular analysis. Brain Res. 1981;214:411–415. doi: 10.1016/0006-8993(81)91204-x. [DOI] [PubMed] [Google Scholar]
- Kohno D, Sone H, Tanaka S, Kurita H, Gantulga D, Yada T. AMP-activated protein kinase activates neuropeptide Y neurons in the hypothalamic arcuate nucleus to increase food intake in rats. Neurosci Lett. 2011;499:194–198. doi: 10.1016/j.neulet.2011.05.060. [DOI] [PubMed] [Google Scholar]
- Kréneisz O, Benoit JP, Bayliss DA, Mulkey DK. AMP-activated protein kinase inhibits TREK channels. J Physiol (Lond) 2009;587:5819–5830. doi: 10.1113/jphysiol.2009.180372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuramoto N, Wilkins ME, Fairfax BP, Revilla-Sanchez R, Terunuma M, Tamaki K, Iemata M, Warren N, Couve A, Calver A, Horvath Z, Freeman K, Carling D, Huang L, Gonzales C, Cooper E, Smart TG, Pangalos MN, Moss SJ. Phospho-dependent functional modulation of GABA-B receptors by the metabolic sensor AMP-dependent protein kinase. Neuron. 2007;53:233–247. doi: 10.1016/j.neuron.2006.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langelueddecke C, Jakab M, Ketterl N, Lehner L, Hufnagl C, Schmidt S, Geibel JP, Fuerst J, Ritter M. Effect of the AMP-kinase modulators AICAR, metformin and Compound C on insulin secretion of INS-1E rat insulinoma cells under standard cell culture conditions. Cellular Phyiol Biochem. 2012;29:75–86. doi: 10.1159/000337589. [DOI] [PubMed] [Google Scholar]
- Lim A, Park S-H, Sohn J-W, Jeon J-H, Park J-H, Song D-K, Lee S-H, Ho W-K. Glucose deprivation regulates K-ATP channel trafficking via AMP-activated protein kinase in pancreatic β-cells. Diabetes. 2009;58:2813–2819. doi: 10.2337/db09-0600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y-F, Raab-Graham K, Jan YN, Jan LY. NO stimulation of ATP-sensitive potassium channels: involvement of Ras/mitogen-activated protein kinase pathway and contribution to neuroprotection. Proc Natl Acad Sci (USA) 2004;101:7799–7804. doi: 10.1073/pnas.0402496101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lira VA, Soltow QA, Long JHD, Betters JL, Sellman JE, Criswell DS. Nitric oxide increases GLUT4 expression and regulates AMPK signaling in skeletal muscle. Am J Physiol Endocrinol Metab. 2007;293:E1062–E1068. doi: 10.1152/ajpendo.00045.2007. [DOI] [PubMed] [Google Scholar]
- Loucif KC, Wilson CL, Baig R, Lacey MG, Stanford IM. Functional interconnectivity between the globus pallidus and the subthalamic nucleus in the mouse brain slice. J Physiol (Lond) 2005;567:977–987. doi: 10.1113/jphysiol.2005.093807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltete D, Jodoin N, Karachi C, Houeto JL, Navarro S, Cornu P, Agid Y, Welter ML. Subthalamic stimulation and neuronal activity in the substantia nigra in Parkinson’s disease. J Neurophysiol. 2007;97:4017–4022. doi: 10.1152/jn.01104.2006. [DOI] [PubMed] [Google Scholar]
- Murphy BA, Fakira KA, Song Z, Beuve A, Routh VH. AMP-activated protein kinase and nitric oxide regulate the glucose sensitivity of ventromedial hypothalamic glucose-inhibited neurons. Am J Physiol Cell Physiol. 2009;297:C750–C758. doi: 10.1152/ajpcell.00127.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Z-G, Bouali-Benazzouz R, Gao D-M, Benabid A-L, Benazzouz A. Time-course of changes in firing rates and firing patterns of subthalamic nucleus neuronal activity after 6-OHDA-induced dopamine depletion in rats. Brain Res. 2001;899:142–147. doi: 10.1016/s0006-8993(01)02219-3. [DOI] [PubMed] [Google Scholar]
- Oakhill JS, Chen Z-P, Scott JW, Steel R, Castelli LA, Ling N, Macaulay SL, Kemp BE. β-Subunit myristoylation is the gatekeeper for initiating metabolic stress sensing by AMP-activated protein kinase (AMPK) Proc Natl Acad Sci (USA) 2010;107:19237–19241. doi: 10.1073/pnas.1009705107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang T, Zhang Z-S, Gu M, Qiu B-Y, Yu L-F, Cao P-R, Shao W, Su M-B, Li J-Y, Nan F-J, Li J. Small molecule antagonizes autoinhibition and activates AMP-activated protein kinase in cells. J Biol Chem. 2008;283:16051–16060. doi: 10.1074/jbc.M710114200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent A, Hazrati L-N. Functional anatomy of the basal ganglia. II. The place of subthalamic nucleus and external pallidum in basal ganglia circuitry. Brain Res Rev. 1995;20:128–154. doi: 10.1016/0165-0173(94)00008-d. [DOI] [PubMed] [Google Scholar]
- Potter WB, O’Riordan KJ, Barnett D, Osting SMK, Wagoner M, Burger C, Roopra A. Metabolic regulation of neuronal plasticity by the energy sensor AMPK. PLoS ONE. 2010;5:e8996. doi: 10.1371/journal.pone.0008996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross FA, Rafferty JN, Dallas ML, Ogunbayo O, Ikematsu N, McClafferty H, Tian L, Widner H, Rowe ICM, Wyatt CN, Shipston MJ, Peers C, Hardie DG, Evans AM. Selective expression in carotid body type I cells of a single splice variant of the large conductance calcium- and voltage-activated potassium channel confers regulation by AMP-activated protein kinase. J Biol Chem. 2011;286:11929–11936. doi: 10.1074/jbc.M110.189779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouzaire-Dubois B, Scarnati E. Pharmacological study of the cortical-induced excitation of subthalamic nucleus neurons in the rat: Evidence for amino acids as putative neurotransmitters. Neurosci. 1987;21:429–440. doi: 10.1016/0306-4522(87)90133-3. [DOI] [PubMed] [Google Scholar]
- Russo GL, Russo M, Ungaro P. AMP-activated protein kinase: a target for old drugs against diabetes and cancer. Biochem Pharmacol. 2013;86:339–350. doi: 10.1016/j.bcp.2013.05.023. [DOI] [PubMed] [Google Scholar]
- Salminen A, Kaarniranta K, Haapasalo A, Soininen H, Hiltunen M. AMP-activated protein kinase: a potential player in Alzheimer’s disease. J Neurochem. 2011;118:460–474. doi: 10.1111/j.1471-4159.2011.07331.x. [DOI] [PubMed] [Google Scholar]
- Shah AK, Gupta A, Dey CS. AICAR induced AMPK activation potentiates neuronal insulin signaling and glucose uptake. Arch Biochem Biophys. 2011;509:142–146. doi: 10.1016/j.abb.2011.03.005. [DOI] [PubMed] [Google Scholar]
- Shen K-Z, Johnson SW. Presynaptic dopamine D-2 and muscarine M-3 receptors inhibit excitatory and inhibitory transmission to rat subthalamic neurones in vitro. J Physiol (Lond) 2000;525:331–341. doi: 10.1111/j.1469-7793.2000.00331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K-Z, Johnson SW. Ca2+ influx through NMDA-gated channels activates ATP-sensitive K+ currents through a nitric oxide-cGMP pathway in subthalamic neurons. J Neurosci. 2010;30:1882–1893. doi: 10.1523/JNEUROSCI.3200-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K-Z, Johnson SW. Group I mGluRs evoke K-ATP current by intracellular Ca2+ mobilization in rat subthalamus neurons. J Pharmacol Exp Ther. 2013;345:139–150. doi: 10.1124/jpet.112.201566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AJ, Partridge CJ, Asipu A, Mair LA, Hunter M, Sivaprasadarao A. Increased ATP-sensitive K+ channel expression during acute glucose deprivation. Biochem Biophys Res Comm. 2006;348:1123–1131. doi: 10.1016/j.bbrc.2006.07.170. [DOI] [PubMed] [Google Scholar]
- Sukhodub A, Jovanovic S, Du Q, Budas G, Clelland A, Shen M, Sakamoto K, Tian R, Jovanovic A. AMP-activated protein kinase mediates preconditioning in cardiomyocytes by regulating activity and trafficking of sarcolemmal ATP-sensitive K+ channels. J Cell Physiol. 2007;210:224–236. doi: 10.1002/jcp.20862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnley AM, Stapleton D, Mann RJ, Witters LA, Kemp BE, Bartlett PF. Cellular distribution and developmental expression of AMP-activated protein kinase isoforms in mouse central nervous system. J Neurochem. 1999;72:1707–1716. doi: 10.1046/j.1471-4159.1999.721707.x. [DOI] [PubMed] [Google Scholar]
- Vucicevic L, Misirkic M, Janjetovic K, Vilimanovich U, Sudar E, Isenovic E, Prica M, Harhaji-Trajkovic L, Kravic-Stevovic T, Bumbasirevic V, Trajkovic V. Compound C induces protective autophagy in cancer cells through AMPK inhibition-independent blockade of Akt/mTOR pathway. Autophagy. 2011;7:40–50. doi: 10.4161/auto.7.1.13883. [DOI] [PubMed] [Google Scholar]
- Wang C-Z, Wang Y, Di A, Magnuson MA, Ye H, Roe MW, Nelson DJ, Bell GI, Philipson LH. 5-Amino-imidazole carboxamide riboside acutely potentiates glucose-stimulated insulin secretion from mouse pancreatic islets by K-ATP channel-dependent and β-independent pathways. Biochem Biophys Res Comm. 2005;330:1073–1079. doi: 10.1016/j.bbrc.2005.03.093. [DOI] [PubMed] [Google Scholar]
- Wichmann T, Bergman H, DeLong MR. The primate subthalamic nucleus. III. Changes in motor behavior and neuronal activity in the internal pallidum induced by subthalamic inactivation in the MPTP model of parkinsonism. J Neurophysiol. 1994;72:521–530. doi: 10.1152/jn.1994.72.2.521. [DOI] [PubMed] [Google Scholar]
- Wichmann T, Dostrovsky JO. Pathological basal ganglia activity in movement disorders. Neurosci. 2011;198:232–244. doi: 10.1016/j.neuroscience.2011.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Bao L, Kefaloyianni E, Taskin E, Okorie U, Hong M, Dhar-Chowdhury P, Kaneko M, Coetzee WA. AMP-activated protein kinase connects cellular energy metabolism to KATP channel function. J Mol Cell Cardiol. 2012;52:410–418. doi: 10.1016/j.yjmcc.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z-T, Munhall A, Shen K-Z, Johnson SW. Calcium dependent subthreshold oscillations determine bursting activity induced by N-methyl-D-aspartate in rat subthalamic neurons in vitro. Eur J Neurosci. 2004;19:1296–1304. doi: 10.1111/j.1460-9568.2004.03240.x. [DOI] [PubMed] [Google Scholar]