

Abstract
Age-related changes in endoplasmic reticulum (ER) are associated with stress of this cell organelle. Unfolded protein response (UPR) is a normal physiological reaction of a cell in order to prevent accumulation of unfolded and misfolded proteins in the ER and improve the normal ER function. However, in pathologic conditions such as atherosclerosis, obesity, and diabetes, ER function becomes impaired, leading to the development of ER stress. In chronic ER stress, defective posttranslational protein folding results in deposits of aberrantly folded proteins in the ER and the induction of cell apoptosis mediated by UPR sensors C/EBPα-homologous protein (CHOP) and inositol requiring protein-1 (IRE1). Since ER stress and ER-induced cell death play a nonredundant role in the pathogenesis of atherosclerosis and diabetic macrovascular complications, pharmaceutical targeting of ER stress components and pathways may be beneficial in the treatment and prevention of cardiovascular pathology.
1. Introduction
The endoplasmic reticulum (ER) is a complex cytoplasmic membrane structure presented in eukaryotic cells. ER is involved in protein folding, lipid synthesis, and regulation of the intracellular calcium balance [1]. Secretory and membrane proteins, which are synthesized in ER, undergo proper folding in the ER lumen. ER chaperones such as glucose-regulated protein 78 kDa (GRP78 or BiP) and GRP94, oxidoreductases, and high calcium concentrations are essential for proper protein folding and assembling [2]. GRP78 is a Ca2+-dependent chaperone that is responsible for the folding of hydrophobic protein regions [3]. Protein disulfide isomerase (PDI) is involved in the formation of disulfide bonds whereas ER thiol oxidase (ERO1) initiates disulfide transfer to oxidized proteins [4]. Aging-related changes in ER are associated with stress of this cell organelle [5]. The oxidative protein folding is associated with advanced production of reactive oxygen species (ROS) that may lead to extensive oxidative stress and cell apoptosis [6]. Indeed, the ER is vulnerable to various stressors capable of disturbing the redox homeostasis in the ER lumen.
2. Unfolded Protein Response
Incompletely folded or misfolded proteins are subjected to ER-associated degradation (ERAD) that occurs in cytoplasm. ER-mediated protein synthesis and folding are strictly regulated. Impairments in ER folding capacity may result in the accumulation of unfolded proteins and induce ER stress. In the ER, three proteins are able to sense increase in misfolded polypeptides and initiate the unfolded protein response (UPR). The UPR sensors include activating transcription factor-6 (ATF6), inositol requiring protein-1 (IRE1), and protein kinase RNA-like ER kinase (PERK). All three proteins have domains exposed to the ER lumen and are capable of binding GRP78 [7]. In normal conditions, GRP78 is bound to the molecules of UPR sensors. In ER stress, GRP78 dissociates from the UPR sensors that leads in turn to induction of UPR (Figure 1).
Figure 1.
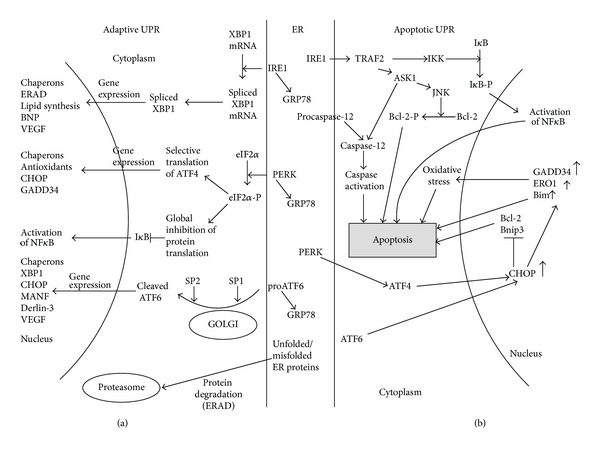
The adaptive and proapoptotic UPR pathways. (a) Adaptive UPR mechanism. In nonstressed conditions, the ER chaperone GRP78 binds to all three ER stress sensors such as PERK, IRE1, and ATF6. In ER stress, GRP78 dissociates from the ER sensors, and this leads to their activation. eIF2α is phosphorylated by PERK and dephosphorylated by GADD34. Phosphorylated eIF2α blocks global protein translation but remains selective translation of several proteins including transcriptional factor ATF4. ATF4 then initiates expression of UPR-related genes. Upon activation, ATF6 translocates from the ER to the Golgi complex where it is cleaved by proteases S1P and S2P. Cleaved ATF6 acts as a transcriptional factor activating expression of several UPR- and non-UPR genes including XBP1. Activated IRE1 specifically splices XBP1 mRNA. Spliced XBP1 shows transcription factor activity to induce UPR- and non-UPR genes. Proteasome plays an important role in degradation of unfolded and misfolded proteins. Thus, production of proteasome components is also stimulated to increase utilization of misfolded proteins through the mechanism of ERAD. (b) Proapoptotic UPR mechanism. The apoptotic pathway is induced in chronic and prolonged ER stress. CHOP plays a key role in mediating ER stress-induced apoptosis. CHOP expression is stimulated by ATF4- and ATF6. CHOP represses expression of antiapoptotic proteins Bcl-2 and Bnip3 and activates translocation of proapoptotic protein Bim to the ER membrane. IRE1α forms a complex with the adaptor protein TRAF2, which consequently activates ASK1 and JNK. Activation of JNK induces apoptosis cell through phosphorylation of several Bcl-2 family members. The IRE1α/TRAF2 complex also binds to IκB kinase, and this results in activation of transcription factor NF-κB. Prolonged ER stress activates caspase 12 that in turn activates caspase-9/3 thereby leading to the mitochondria-independent apoptotic pathway.
The UPR applies several mechanisms to minimize ER stress. One of those mechanisms involves the activation of chaperone synthesis in order to improve and intensify the intraluminal protein folding. Another mechanism includes protein translation arrest in order to prevent further protein load into the ER [26]. The ER folding capacity may be also improved indirectly, through stimulating ER biogenesis [6]. In a case of chronic long-term or acute ER stress, when the adaptive UPR is unable to stop the stress, apoptotic pathways are activated in the stressed cell [27]. ER sensor proteins such as IRE1 and PERK are involved in both the adaptive and the proapoptotic UPR pathways.
2.1. IRE1
Among ER stress sensors, IRE1 is the most evolutionarily preserved. In normal conditions, IRE1 and GRP78 interact with each other, and this prevents IRE1 activation [6]. In ER stress, GRP78 becomes released from the complex with IRE1. IRE1 is then activated by self-phosphorylation. The active IRE1 is able to specifically splice mRNA encoding X-box binding protein (XBP)1 thereby inducing translation of functionally active XBP1 [1]. XBP1 induces transcription of chaperones and other UPR-related proteins and enhances the degradation of misfolded proteins [28]. By degrading mRNAs other than XBP1, IRE1 contributes to reducing protein load to the ER [29].
However, in long-lasting ER stress, IRE1 can be involved in activation of proinflammatory pathways and apoptosis. IRE1 forms a complex with the adaptor protein tumor necrosis factor (TNF) receptor-associated factor (TRAF)2 [30] that in turn recruits mitogen-activated protein kinase, apoptosis signal-regulating kinase (ASK) [31], and caspase 12 [32]. The complex activates IκB kinase followed by IκB kinase-mediated suppression of the inhibitor of κB protein and induction of the nuclear factor (NF)-κB. Since NF-κB controls expression of many proinflammatory genes, IRE1 is therefore suggested to provide a link between the ER stress and inflammation [33].
2.2. PERK
This ER stress sensor molecule belongs to the family of serine threonine kinase and has a high degree of homology with ERE1. Both IRE1 and PERK share similar mechanisms of activation involving dissociation of GRP78 from the luminal binding domain and self-phosphorylation upon stress conditions. After activation, PERK downregulates eukaryotic translation initiation factor 2α (eIF2α) that is needed for cap recognition and therefore is essential for further induction of cap-dependent transcription. eIF2α inactivation results in marked decrease of protein load to the ER [6]. Interestingly, phosphorylated eIF2α is responsible for translation of several mRNAs including mRNA for transcriptional factor ATF4. This factor is responsible for the induction of the negative feedback regulatory loop since it activates expression of GADD34, a regulatory subunit of the phosphatase that dephosphorylates eIF2α and restores cap-dependent translation [34]. Indeed, ATF4 regulates protein translation during ER stress.
ATF4 stimulates expression of C/EBPα-homologous protein (CHOP, or GADD153). CHOP expression can be also induced by ATF6 and XBP1, but the PERK-eIF2α-dependent pathway is prevalent [35]. CHOP is a transcription factor that induces apoptosis through several mechanisms including upregulation of ERO1α, which then mediates Ca2+-dependent apoptotic pathway, and downregulation of antiapoptotic factors Bcl-2 and Bnip3 [36, 37].
ERO1α is involved in reoxidation of PDI yielding production of hydrogen peroxide, a byproduct of disulfide bond formation [38]. Therefore, ER stress-induced upregulation of ERO1α may lead to ROS overproduction and advanced oxidative stress that in turn contribute to cell apoptosis [5]. ERO1α activation stimulates inositol-1,4,5-trisphosphate receptor-1 (IP3R1), a ER-associated Ca2+ channel [39] that triggers depletion of the intraluminal calcium reservoir. Increase in cytoplasmic Ca2+ promotes stimulation of calcium/calmodulin-dependent protein kinase II, which plays a key role in induction of several proapoptotic pathways including activation of the death receptor FAS and mitochondrial release of apoptogens [40]. CHOP is directly involved in induction of expression and translocation to the ER membrane of the proapoptotic protein Bim [41].
2.3. ATF6
Upon initiation of ER stress, ATF6 is cleaved by two (site 1 and site 2) proteases associated with the Golgi complex. After cleavage, the cytosolic N-domain of ATF6 translocates into the nucleus where it triggers expression of many UPR-related genes including GRP78 and XBP1 [26]. ATF6 activates expression of Derlin-3 that enhances the ERAD activity [42]. Before degradation by the proteasome, most of misfolded proteins are ubiquitinated and extracted by the cytosolic ATPase p97 [43, 44].
3. Role of ER Stress in Atherosclerosis
Prolonged ER stress observed in atherosclerotic lesions is an important contributor to proatherogenic progression [45]. ER stress was found in all major cell type in atherosclerosis including macrophages, vascular smooth muscle cells (VSMCs), and endothelial cells (ECs).
3.1. ER Stress in Macrophages
In normal macrophages, low density lipoprotein (LDL) cholesterol particles are loaded from late endosomes to the ER. In the ER, cholesterol is esterified and accumulates to form inert lipid droplets [46]. In atherosclerotic macrophages, ER-mediated cholesterol reesterification is markedly reduced or failed resulting in heavy intracellular deposits of nonesterified cholesterol in foam cells [47]. Electron microscopy observations revealed that ER in atherosclerotic macrophages undergoes a remarkable change (Figure 2). In foam cells, intraluminal ER oxidoreductases oxidize cholesterol to 7-ketocholesterol (7-KC) and other oxysterols. Oxysterols are highly cytotoxic and may induce cell death through ROS-mediated oxidative damage and other mechanisms [48].
Figure 2.
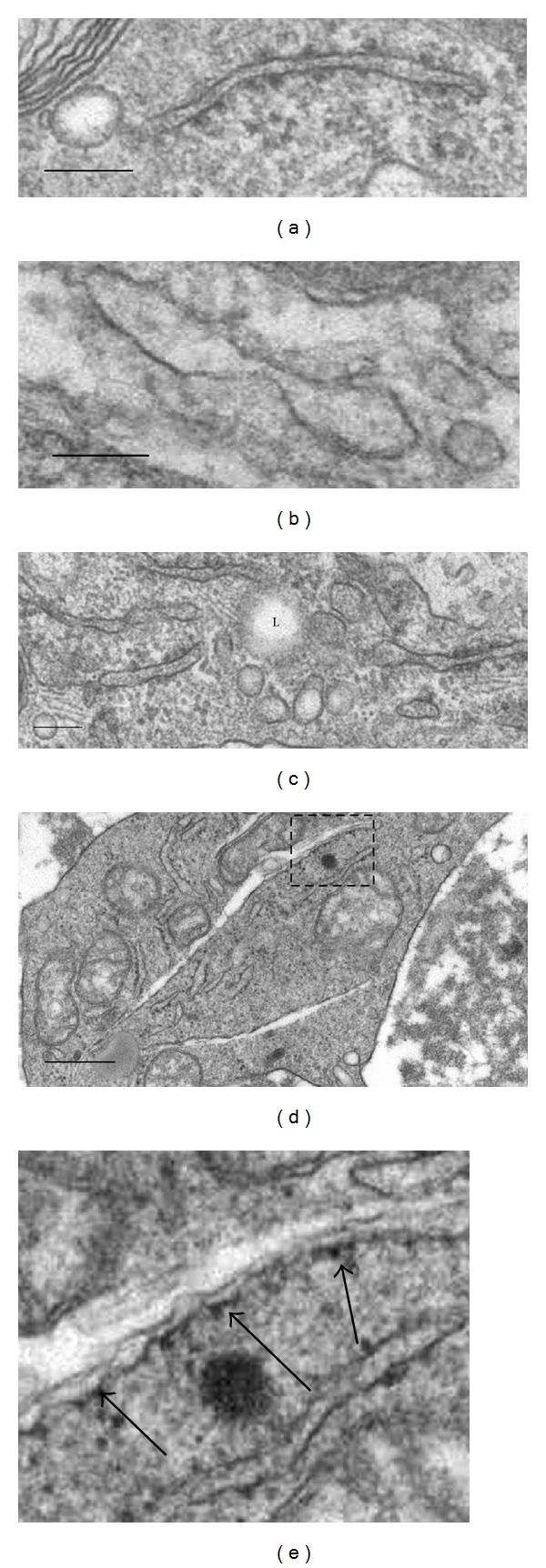
Structural alterations of cisterns of granular endoplasmic reticulum (ER) in macrophages residing in human atherosclerotic lesions (identified by means of electron microscopy) (a–e). In contrast to intact ER cistern appearance (a), some ER cisterns display a notable expansion of the intracisternal space (b) and demonstrate focal disappearance of ribosomes from the internal membranes of cisterns (a, b). In some macrophages, the expansion of the intracisternal space is accompanied by degenerative alterations of ER cistern (d, e). (e) is a detail of (d). The arrows in (e) show ribosomes which are still present on the internal surface of a degenerating ER cistern. In (c), L: lipid droplet. Bars = 100 nm (a–c), 500 nm (d).
Prolonged ER stress contributes to apoptosis of lesional macrophages. Apoptosis associated with robust expression of CHOP was shown in human lesions [45] and atherosclerotic plaques of apolipoprotein (apo)E-deficient mice [49]. Inactivating Chop in apoE-deficient mice leads to decreased rates of macrophage apoptosis and plaque necrosis [50, 51]. CHOP contributes to ER stress-induced macrophage death by inducing Fas activation, depletion of ER-associated calcium stores, and release of apoptogens from mitochondria [52].
In early plaques, apoptotic cells are quickly phagocytized by macrophages [53]. This process is driven by anti-inflammatory cytokines such as transforming growth factor- (TGF-) β and interleukin- (IL-) 10 [54]. In advanced plaques, macrophages cannot efficiently clear dying cells that become necrotic [55]. This results in the formation of the inflammatory necrotic core [56].
In some circumstances, ER stress alone is not strong enough to induce apoptosis in macrophages. Additional stimuli such as the activation of pattern recognition receptors (PRRs) are required to initiate cell death [56]. PPRs include various Toll-like receptors (TLRs) and scavenger receptors. In plaque macrophages, PPRs may be activated by oxidized lipids, and this leads to the induction of apoptosis via CD36-TLR2 pathway and is accompanied with NADPH oxidase-mediated oxidative stress [57]. NADPH oxidase contains a subunit Nox2 whose inhibition minimizes ER stress-induced macrophage death [58]. These findings suggest a central role of this enzyme as a link between the oxidative stress and ER stress in promoting macrophage apoptosis. In addition, upregulation of NADPH oxidase further aggravates apoptotic process through stimulating PERK-CHOP-dependent mechanism.
At low doses, ER stress inducers such as modified (oxidized and acetylated) LDL, 7-KC, and peroxynitrite donor SIN are able to stimulate macrophage PRRs and cause NADPH oxidase-mediated ROS production [56, 59]. In a “two-hit” hypothesis, ER stress in plaque macrophages should be induced by a low-dose ER stressor such as PRR ligands that in turn triggers macrophage apoptosis [57, 59].
Lipoprotein(a) [Lp(a)], an LDL-like lipoprotein, and oxidized phospholipids are established to represent strong risk factors for human atherosclerosis [59]. To initiate apoptosis in ER-stressed macrophages, both atherogenic lipid inducers utilize similar mechanisms involving the activation of CD36-TLR2 signaling and oxidative stress [57].
Lp(a) is a major carrier of oxidized phospholipids in human blood [60]. According to the “two-hit” hypothesis, Lp(a) could therefore mediate apoptosis in human plaque macrophages.
3.2. ER Stress in Endothelial Cells
In EC, various ER stress inducers were shown to initiate UPR. For example, shear stress activates IRE1-dependent UPR [61, 62] whereas oxidized phospholipids and homocysteine induce both IRE1- and CHOP-mediated pathways [63–65]. In dynamic models of shear stress, a variety of UPR-related molecules including ATF6, GRP78, IRE1, and XBP1 were upregulated in ECs [61, 62, 66, 67]. XBP1 is always overexpressed in advanced plaques, a finding that may reflect a proatherogenic role of long-term XBP1 upregulation whereas limited stimulation of this ER stress effector may be protective against ER stress [61].
ER stress induced by modified (oxidized and glycated) LDL results in the development of oxidative stress and oxidation-mediated inhibition of sarcoplasmic/endoplasmic reticulum Ca2+-dependent ATPase (SERCA), a calcium pomp resided in the ER [8]. AMP kinase (AMPK) α2 suppresses SERCA oxidation, and inhibition of this kinase in LDL receptor- (Ldlr-) deficient mice leads to advanced ER stress and atherogenesis [9]. Thus, alterations in calcium homeostasis caused by oxidative stress play a crucial role in ER stress-mediated endothelial dysfunction in atherosclerotic vessels. ER stress-induced apoptosis diminishes the barrier function of the vascular endothelium and induces procoagulant phenotypic changes in ECs that may be directly responsible for increased risk of thrombosis and other late atherosclerotic complications [68].
3.3. ER Stress in VSMCs
A stable plaque phenotype may be critically disturbed by apoptosis in VSMCs that alters the formation of the protective fibrous cap [69]. In VSMCs, CHOP-mediated apoptotic mechanism may be induced by numerous ER stressors such as 7-KC, homocysteine, glucosamine, free cholesterol, and others [70–74]. CHOP-dependent apoptosis is accompanied with enhanced formation and release of ROS, and N-acetylcysteine, an anti-oxidant, may therefore protect cultured VSMCs against apoptotic death [73].
Elevated plasma levels of homocysteine are considered to increase atherosclerotic risk in humans and animal models [71, 75]. Hyperhomocysteinemia is believed to induce ER stress through alterations of calcium balance [76] and upregulation of sterol response element binding protein-2 (SREBP-2) that increases lipid deposits in VSMCs [77, 78]. Glucosamine that accumulates in vascular cells in diabetes may have a primary responsibility for ER stress induction in VSMCs of diabetic patients associated with GRP78 upregulation [74]. However, the mechanisms of ER stress-mediated apoptosis in VSMCs are significantly less studied compared to those of macrophages and ECs.
4. ER Stress and Obesity
The human body is able to accumulate extra fat in the adipose tissue to survive in starvation. Normally, fat is deposited in adipocytes. However, regular intake of fat-rich diet and alterations in lipid metabolism may lead to the phenomenon of ectopic fat storage, when fat accumulates not only in adipocytes but also in nonadipocyte cells. In obesity, higher free fatty acids levels may enhance lipid accumulation in macrophages and promote formation of foam cells [79].
In obese people, adipocytes are particularly vulnerable to ER stress and apoptosis due to abnormal fat deposits and upregulated lipid metabolism [80]. Macrophages resided in the adipose tissue phagocytize both the extra fat droplets and apoptotic adipocytes releasing high amounts of ROS by mitochondria. Excessive ROS production drives further progression of cellular stress and increases secretion of adipokines in adipocytes [81]. Adipokines promote preferential differentiation of macrophages towards the proinflammatory M1 phenotype [82].
Adiposity is associated with enhanced M1 macrophage-dependent production of multiple proinflammatory mediators such as IL-1β, IL-6, TNF-α, and CXCL10. M1 macrophages inhibit adipocyte hypertrophy and adipogenesis [83] and support low-grade inflammation in the adipose and nonadipose tissues including vessels [84]. In lesional macrophages, adiposity promotes ER stress by activation of the macrophage fatty acid-binding protein-4, also known as adipocyte fatty acid binding protein aP2 that mediates transfer of saturated fatty acids (SFAs) [19]. Increase in SFA levels leads to the induction of apoptosis in macrophages. ApoE-deficient mice lacking aP2 have reduced atherosclerotic lesions, in which expression of XBP1 and PERK is downregulated and macrophage apoptosis is decreased [85]. Inactivation of aP2 protects macrophages from palmitate-induced ER stress and apoptosis [19]. In aP2-deficient macrophages, expression of transcription factor LXRα is activated, and this factor stimulates transcription of stearoyl-CoA-desaturase 1, an enzyme converting SFAs to monounsaturated fatty acids, which are significantly less potent of inducing ER stress [86]. Indeed, activation of LXRα in aP2-deficient macrophages prevents ER stress while overexpression of aP2 in macrophages and adipocytes, in contrast, supports ER stress induction and atherogenesis.
This protective effect is mediated by increased expression of transcription factor LXRα in aP2-deficient macrophages. This factor activates expression of stearoyl-CoA-desaturase 1, converting SFAs to monounsaturated fatty acids that are significantly less capable of inducing ER stress [86].
5. ER Stress and Diabetes
5.1. Insulin Resistance-Induced ER Stress in Macrophages
In diabetic subjects with atherosclerosis, the proatherogenic role of ER stress and CHOP-mediated macrophage apoptosis is significantly enhanced that results in the development of advanced plaques with the especially large necrotic core [87, 88]. Macrophages were shown to have functional insulin receptors, and insulin resistance (IR) is a potent inducer of chronic ER stress in macrophages [89]. High insulin doses suppress insulin signaling in macrophages [88]. Under diabetic conditions, insulin signal transduction in macrophages is also inhibited by diacylglycerol-dependent activation of protein kinase C [90].
Expression of the scavenger receptor SRA is markedly upregulated in IR macrophages. Indeed, according to the “two-hit” hypothesis, these macrophages should be particularly sensitive to PRR-driven ER stress and apoptosis [91]. Experiments with cultured IR macrophages loaded with lipoprotein-derived free cholesterol do show markedly increased apoptosis that suggest a key role of SRA-induced mechanism of ER stress in mediating death of IR macrophages [92]. In these macrophages, MEK/ERK/cAMP-responsive element-binding protein (CREBP) signaling and calcium homeostasis are disturbed [89]. Alterations in intracellular calcium balance involve depletion of ER calcium stores and reduced SERCA activity. Antidiabetic agent exenatide rescues IR macrophages from apoptosis by activation of the macrophage glucagon-like peptide 1 (GLP-1) receptor followed by restoring MEK/ERK signaling and inhibition of Ca2+-dependent apoptosis [89].
In IR and ER-stressed macrophages, activity of the serine/threonine-specific protein kinase Akt is lowered and Akt- and NF-κB-dependent pathways responsible for cell survival are suppressed [92]. Insufficient Akt activity in IR macrophages is associated with preferential localization of transcription factor FoxO1 in the nucleus [93]. Normally, Akt-dependent phosphorylation of FoxO1 in response to insulin signaling initiates translocation of this factor to cytoplasm where it is inactivated by proteolytic degradation. Macrophages deficient for FoxO1, 3, and 4 are resistant to ER stress-driven apoptosis [93]. The preferential nuclear location of FoxO1 correlates with enhanced expression of IκBε, an inhibitor of NF-κB, which in turn increases apoptosis of IR macrophages [94].
5.2. Glucosamine-Induced ER Stress
Diabetic hyperglycemia significantly increases cardiovascular risk inducing vascular dysfunction through inhibitory effects on proliferation of vascular cells and enhancement of their apoptosis [95–98]. Several pathologic mechanisms link diabetic hyperglycemia and atherosclerosis. Activation of the aldose reductase pathway alters redox homeostasis and promotes oxidative stress-mediated damage of vascular cells [99]. High glucose induces overactivity of protein kinase C that leads to reduced endothelial vasodilation [100] and increased ROS production [101]. Nonenzymatic glycation is markedly increased in diabetic patients, and this results in uncontrolled production of advanced glycation end-products (AGEs) [102] whose accumulation in blood plasma is related to enhanced modification of lipoproteins thereby increasing their atherogenicity [103]. Receptor for AGE (RAGE) is expressed in macrophages, ECs, and VSMCs [104], and AGE-RAGE interaction induces signaling pathways associated with increased ROS production and activation of inflammatory response in vascular cells [105].
In the hexosamine pathway, glutamine: fructose-6 phosphate amidotransferase (GFAT) catalyzes conversion of glucose to glucosamine-6 phosphate (G-6P) [106]. Diabetic hyperglycemia activates the hexosamine pathway that leads to the production of elevated G-6P levels in vascular cells [107, 108]. UDP-N-acetylglucosamine (UDP-GlcNAc), an end-product of the hexosamine pathway, is involved in both O- and N-linked protein glycation. N-glycosylation is an important stage of posttranslational modifications of newly synthesized proteins performed in the ER lumen [109]. Inhibition of N-glycosylation by tunicamycin (UDP-GlcNAc antagonist) activates the UPR [110].
GFAT is a rate-limiting enzyme in the hexosamine pathway. Overactivity of this enzyme in diabetic conditions promotes ER stress via stimulation of expression of UPR-related genes and contributes to downstream events including lipid accumulation and activation of proinflammatory and apoptotic pathways [111]. In contrast, GFAT inhibition attenuates ER stress [74]. Cultured human aortic VSMCs and macrophages treated with glucosamine develop apoptosis [74, 112, 113]. Therefore, suppression of GFAT could have a therapeutic potential in prevention of glucosamine-induced ER stress and apoptosis.
Glycogen synthase kinase (GSK)-3 whose expression is activated in glucosamine-induced ER stress may represent another potential target for antiatherogenic therapy [114]. GSK-3β activation in the aorta apoE-deficient hyperglycemic hyperhomocysteinemic mice fed on high-fat diet correlates with advanced atherosclerosis [115]. GSK-3α and β are two enzyme isoforms implicated in a variety of signaling pathways [116]. Upon the UPR induction, the inactive enzyme phosphorylated at Ser(21/9) is rapidly degraded in lysosomes that yields increase in GSK-3 activity [117]. Inhibition of GSK-3 displays both atheroprotective and anti-ER stress effects in cell cultures [118, 119] and hyperglycemic murine models [120].
6. Therapeutic Targeting of UPR Components and Its Clinical Potential
Targeting of proteins in ER stress and ER stress-induced apoptosis may be of high therapeutic value for treatment of human diseases in which ER stress plays a substantial role (Table 1). Promoters of GRP78 and GRP94 genes share significant sequence homology that explains the high concordance in coordinated expression of both ER chaperones [121]. Activation of ER chaperones plays an important role in adaptive UPR since it improves protein folding and prevents ER stress-induced apoptosis. Overexpression or stimulation of GRP78/94 had beneficial effect on ER-stressed cardiomyocytes [122–124] and showed cardioprotective properties in experiments in vivo [11–13].
Table 1.
Therapeutics targeting molecular components of ER stress and ER stress-induced apoptosis.
| Drug | Mechanism | Potential indication | Reference |
|---|---|---|---|
| 5-Aminoimidazole-4-carboxyamide-1-β-D-ribofuranoside (AICAR) | Reduction of ER stress by AMPK activation | Ischemic heart disease, heart failure, cardiac hypertrophy, atherosclerosis | [8–10] |
|
| |||
| BiP inducer X | Induction of GRP78 | Heart failure, stroke | [11, 12] |
|
| |||
| Curcumin | Induction of GRP94 | Heart failure, atherosclerosis, thrombosis, diabetes, diabetic cardiomyopathy, inflammation, dyslipidemia | [13] |
|
| |||
| CS-866 | Reduction of ER stress by pressure-overload | Heart failure, cardiac hypertrophy | [14] |
|
| |||
| EN460 | ERO1α inhibitor | Prevention/reduction of ER stress-induced oxidative stress | [15, 16] |
|
| |||
| Benzodiazepinones | ASK1 inhibitor | Atherosclerosis, cerebrovascular ischemia | [16] |
|
| |||
| QM295 | ERO1α inhibitor | Prevention/reduction of ER stress-induced oxidative stress | [15] |
|
| |||
| Isoproterenol | Proteasome activation and assembly | Heart failure, atherosclerosis | [17] |
|
| |||
| Pioglitazone | Reduction of ER stress | Heart failure, atherosclerosis | [18] |
|
| |||
| Phenylbutyrate | Chemical chaperone | Heart failure, atherosclerosis, pulmonary hypertension | [19–21] |
|
| |||
| Pravastatin | Reduction of ER stress by pressure-overload | Heart failure, cardiac hypertrophy | [22] |
|
| |||
| Salubrinal | Prevention of eIF2a dephosphorylation | Heart failure, cardiac hypertrophy | [23] |
|
| |||
| SB203580 | CHOP phosphorylation | Heart failure, cardiac hypertrophy, atherosclerosis | [24] |
|
| |||
| SP600125 | Prevention of CHOP induction by stretch | Heart failure, cardiac hypertrophy, atherosclerosis | [24] |
|
| |||
| Sunitinib | IRE1 activation | Heart failure, atherosclerosis | [25] |
|
| |||
| Tauroursodeoxycholic acid (TUDCA) | Chemical chaperone | Heart failure, atherosclerosis | [19] |
Chemical chaperones such as phenylbutyrate and tauroursodeoxycholic acid (TUDCA) are able to stabilize proteins in their native conformation thereby mimicking properties of native ER chaperones [125]. Murine macrophages and adipocytes treated with chemical chaperones showed resistance to ER stress [19]. At present, phenylbutyric acid (PBA) in its sodium salt form is approved for therapy of urea cycle disorders [126] and is in process of clinical testing for treatment of some genetic disorders related to protein misfolding [127, 128]. PBA was shown to reduce ER stress and normalize glucose levels in diabetic mice [129]. Taking into account clinical approval of PBA for therapy of several diseases, it would be interesting to check whether PBA is efficient for treatment of cardiovascular pathology.
TUDCA was shown to display antiapoptotic and anti-ER stress properties for many types of cells and many diseases including atherosclerosis. TUDCA was able to block ER stress and slow lesion progression in Ldlr-deficient mice [9] and efficiently prevent apoptosis and ER stress induced by oxidized LDL in murine macrophages transgenic for human APOE4, a genetic risk variant for Alzheimer disease and atherosclerosis [130]. The antiapoptotic function of TUDCA can be released through restoring calcium homeostasis and SERCA activity [131] and downregulation of proapoptotic protein Bad [132].
Salubrinal specifically inhibits eIF2α phosphatases [23] and therefore supports blocking protein synthesis mediated by phosphorylated eIF2α [133]. Salubrinal is able to stop ER stress-induced apoptosis by inhibiting synthesis of members of proapoptotic signaling such as CHOP and caspase-12 in cardiac myocytes [134] and upregulating GRP78 in neurons [23]. However, in pancreatic β-cells, salubrinal induced activation of ATF4-CHOP mechanism that resulted in severe ER stress and apoptosis [133]. Thus, various cell types differently respond to salubrinal, and this limits its utility as a broad spectrum antiapoptotic drug [135].
CHOP is crucial in inducing ER stress-mediated apoptosis and hence development of CHOP inhibitors would be beneficial in prevention of atherosclerosis and treatment of heart failure and cardiac hypertrophy [50]. To date, no pharmacological agents specific for CHOP are available but there are drugs able to target molecular components of CHOP-mediated signaling. For example, SB203580, an inhibitor of p38 mitogen-activated protein kinase disrupts CHOP phosphorylation [136]. Mitogen-activated protein kinase blockers indirectly inhibit CHOP-dependent signaling in ER stress-induced apoptosis. JNK inhibitor SP600125 showed ability to suppress mechanical stretch-induced activation of CHOP [24].
Inhibition of ERO1α results in disruption of ER stress induced by oxidative stress and CHOP. Furthermore, several selective ERO1α inhibitors were developed. The inhibitor EN460 inactivates ERO1α by blocking its reoxidation [15]. Inhibitors EN460 and QM295 are able to launch the adaptive UPR signaling that prevents ER stress [15, 16]. Advanced ROS production induced by ERO1α overactivation may be efficiently suppressed by the antioxidant N-acetylcysteine [137] and by the treatment with curcumin and masoprocol that protect PDI from oxidative inactivation [138].
Restoring proteasome function, which is inhibited in ER stress [139], by protein kinase A activators such as isoproterenol or forskolin helps to attenuate ER stress-induced apoptosis [17]. TNF-α is significantly upregulated in ER stress, and inhibition of this cytokine by pravastatin [22] or TNF-α-specific antibody [24] results in significant protection of cardiomyocytes and other cells against apoptotic death. Hyperactivity of ASK1, a downstream target of IRE1-mediated signaling, contributes to the development of cardiac hypertrophy and heart failure, and inhibition of ASK1 by benzodiazepinones may be helpful for therapy of these cardiopathies [140, 141].
AMPK regulates switching from anabolic pathways (fatty acid synthesis, protein synthesis, etc.) to catabolism (fatty acid oxidation, glucose transport, etc.) thereby elevating energy levels in the cell [142]. The RNAse activity of IRE1 is probably required to activate AMPK that leads to the induction of the proper UPR and increases cell survival [143]. AMPK activation has the cardioprotective effect through reducing cardiac ER stress [10, 144]. Inactivation of AMPK is associated with severe ER stress and atherosclerosis that can be reduced by ER stress suppressors such as tempol or TUDCA [8, 9]. In contrast, multiple AMPK agonists such as 5-aminoimidazole-4-carboxyamide-1-β-D-ribofuranoside (AICAR), atorvastatin, A-769662, and PT1 reduce cardiovascular disease by blocking ER stress [10, 145]. Currently, AMPK activators are implicated in the treatment of obesity and metabolic syndrome. However, these drugs may be very helpful in antiatherogenic and cardioprotective therapy [146].
7. Conclusion: Limitations and Challenges in Anti-ER Stress Therapy
The UPR can be targeted by two ways including the activation of components of the adaptive mechanism of UPR and inhibition of those involved in the proapoptotic pathways of UPR. However, several questions should be answered to increase our understanding of mechanisms by which UPR targeting may help in therapy of cardiovascular disease. For example, one ER stressor (ATF6) has a cardioprotective role [147, 148] while two others (IRE1α and PERK) are involved in both the adaptive and proapoptotic UPR pathways. To date, the mechanisms controlling the switch from cell survival to death are not fully understood. Indeed, we do not know precisely when to activate or inhibit ER stress sensor proteins for treatment.
A variety of chemical inhibitors of protein kinases including receptor tyrosine kinase inhibitors are available. Some of those including sunitinib can directly activate IRE1 that results in XBP1 splicing and decreased ER stress [25]. Sunitinib malate is approved for use in treating renal cell carcinoma and gastrointestinal stromal tumor. However, in patients with a previous history of hypertension and coronary heart disease, sunitinib increases risk for cardiovascular events [149]. Thus, kinase inhibitors especially those that have a broad target spectrum should be carefully evaluated to prevent acute side effects.
In preclinical studies, chemical chaperones showed promising results in the improvement of ER folding capacity [125]. However, there are some limitations that seriously affect the therapeutic efficiency of these agents. Typically, high doses of these small molecule drugs are required to reach the desired effect. In addition, the UPR components are broadly expressed and their inhibition/activation in one tissue or organ may negatively influence the function of another tissue or organ. Targeting cell-specific ER components such as cAMP-responsive element-binding transcription factor H (CREBH) may be a promising strategy. The implementation of nanotherapeutic targeting approaches would be helpful for resolving these problems and providing new advances in efficient prevention of ER stress and treatment of ER stress-related diseases.
Using nanoparticles loaded with a therapeutic agent and coated with a monoclonal antibody against a tissue-specific antigen is a promising strategy for targeted delivery of a drug at high local concentrations. However, the development of nanotherapeutic tools for targeting cardiovascular ER stress-induced apoptosis is still in its infancy. Delie et al. [150] constructed polymeric nanoparticles capable of recognizing the COOH-terminal ER retention domain of GRP78, which is markedly overexpressed in prostate and ovarian cancer. The nanoparticles were able to deliver a cytotoxic agent, paclitaxel, to GRP-78-positive cancer cells. Niu et al. [151] reported a cardioprotective effect of nanoparticles loaded with cerium oxide (CeO2), a ROS scavenger, in transgenic mice with cardiac-specific expression of monocyte chemoattractant protein- (MCP-) 1 that causes ischemic cardiomyopathy associated with the activation of ER stress. In heart failure, MCP-1 is involved in cardiomyocyte death through ROS-induced ER stress and apoptosis mediated by MCP-1-induced protein (MCPIP), a proapoptotic transcription factor [152]. CeO2 nanoparticles injected intravenously inhibited progressive left ventricular dysfunction and dilatation in MCP mice by reducing oxidative stress and ER stress associated with suppression of expression of key ER-stress-related proteins [151].
The development of therapeutic nanoparticles capable of prolonged circulation in the bloodstream may provide an effective alternative method for treating ER stress in atherosclerosis and other cardiovascular diseases. For example, liposomal encapsulation of a drug and further liposomal pegylation significantly increase drug stability and residence time in blood as well as decreasing its cardiotoxicity [153]. In a rat ischemia/reperfusion model of cardiac injury, Takahama et al. [154] showed significantly advanced cardioprotective properties for prolonged adenosine encapsulated in pegylated liposomes compared to free adenosine. Knowledge regarding the mechanisms of the UPR and ER-stress-related diseases has rapidly accumulated in recent years, but many questions remain unanswered. Investigations of the mechanisms and pharmacological actions of ER stress are important in providing new mechanistic insights and developing novel targets for ER stress-related diseases. We believe that a more deep understanding of ER stress will open promising avenues for the development of clinically useful drugs.
Acknowledgments
The authors wish to thank the Russian Ministry of Education and Science, the Russian Scientific Foundation (Grant 14-15-00112), and the School of Medical Sciences, University of New South Wales, Sydney, for supporting their work.
Abbreviations
- AGE:
Advanced glycation end-product
- AICAR:
5-Aminoimidazole-4-carboxyamide-1-β-D-ribofuranoside
- AMPK:
AMP kinase
- Apo:
Apolipoprotein
- ASK1:
Apoptosis signal-regulating kinase-1
- ATF:
Activating transcription factor
- Bcl-2:
B cell lymphoma-2
- Bim:
BH3-only protein
- CHOP:
C/EBPα-homologous protein
- CREBH:
cAMP-responsive element-binding transcription factor H
- CREBP:
cAMP-responsive element-binding protein
- eIF2α:
Eukaryotic translation initiation factor 2α
- EC:
Endothelial cell
- ER:
Endoplasmic reticulum
- ERAD:
ER-associated degradation
- ERO1:
ER oxidase 1
- GFAT:
Glutamine:fructose-6 phosphate amidotransferase
- GLP-1:
Glucagon-like peptide 1
- G6P:
Glucosamine-6 phosphate
- GRP:
Glucose-regulated protein
- GSK-3:
Glycogen synthase kinase-3
- IL:
Interleukin
- IP3R1:
Inositol-1,4,5-trisphosphate receptor-1
- IRE1:
Inositol requiring protein-1
- 7-KC:
Ketocholesterol
- LDL:
Low density lipoprotein
- Ldlr:
LDL receptor
- Lp(a):
Lipoprotein(a)
- MCP-1:
Monocyte chemoattractant protein-1
- MCPIP:
MCP-1-induced protein
- PARM-1:
Prostate androgen-regulated mucin-like protein 1
- PBA:
Phenylbutyrate
- PERK:
Protein kinase RNA-like ER kinase
- PDI:
Protein disulfide isomerase
- PRR:
Pattern recognition receptor
- RAGE:
AGE receptor
- ROS:
Reactive oxygen species
- SERCA:
Sarcoplasmic/endoplasmic reticulum Ca2+-dependent ATPase
- SFA:
Saturated fatty acid
- SRA:
Scavenger receptor
- SREBP-2:
Sterol response element binding protein-2
- TLR:
Toll-like receptor
- TNF:
Tumor necrosis factor
- TRAF2:
TNF receptor-associated factor 2
- TUDCA:
Tauroursodeoxycholic acid
- UDP-GlcNAc:
UDP-N-acetylglucosamine
- UPR:
Unfolded protein response
- VSMC:
Vascular smooth muscle cell
- XBP1:
X-box binding protein 1.
Conflict of Interests
The authors report no conflict of interests.
References
- 1.Minamino T, Kitakaze M. ER stress in cardiovascular disease. Journal of Molecular and Cellular Cardiology. 2010;48(6):1105–1110. doi: 10.1016/j.yjmcc.2009.10.026. [DOI] [PubMed] [Google Scholar]
- 2.Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nature Reviews Drug Discovery. 2008;7(12):1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- 3.Gething M-J. Role and regulation of the ER chaperone BiP. Seminars in Cell and Developmental Biology. 1999;10(5):465–472. doi: 10.1006/scdb.1999.0318. [DOI] [PubMed] [Google Scholar]
- 4.Simmen T, Lynes EM, Gesson K, Thomas G. Oxidative protein folding in the endoplasmic reticulum: tight links to the mitochondria-associated membrane (MAM) Biochimica et Biophysica Acta: Biomembranes. 2010;1798(8):1465–1473. doi: 10.1016/j.bbamem.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Najdoo N. ER and aging—protein folding and the ER stress response. Ageing Research Reviews. 2009;8(3):150–159. doi: 10.1016/j.arr.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 6.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nature Reviews Molecular Cell Biology. 2007;8(7):519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 7.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nature Cell Biology. 2000;2(6):326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 8.Dong Y, Zhang M, Wang S, et al. Activation of AMP-activated protein kinase inhibits oxidized LDL-triggered endoplasmic reticulum stress in vivo. Diabetes. 2010;59(6):1386–1396. doi: 10.2337/db09-1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dong Y, Zhang M, Liang B, et al. Reduction of AMP-activated protein kinase α2 increases endoplasmic reticulum stress and atherosclerosis in vivo. Circulation. 2010;121(6):792–803. doi: 10.1161/CIRCULATIONAHA.109.900928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Terai K, Hiramoto Y, Masaki M, et al. AMP-activated protein kinase protects cardiomyocytes against hypoxic injury through attenuation of endoplasmic reticulum stress. Molecular and Cellular Biology. 2005;25(21):9554–9575. doi: 10.1128/MCB.25.21.9554-9575.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kudo T, Kanemoto S, Hara H, et al. A molecular chaperone inducer protects neurons from ER stress. Cell Death and Differentiation. 2008;15(2):364–375. doi: 10.1038/sj.cdd.4402276. [DOI] [PubMed] [Google Scholar]
- 12.Oida Y, Hamanaka J, Hyakkoku K, et al. Post-treatment of a BiP inducer prevents cell death after middle cerebral artery occlusion in mice. Neuroscience Letters. 2010;484(1):43–46. doi: 10.1016/j.neulet.2010.08.015. [DOI] [PubMed] [Google Scholar]
- 13.Aggarwal BB, Harikumar KB. Potential therapeutic effects of curcumin, the anti-inflammatory agent, against neurodegenerative, cardiovascular, pulmonary, metabolic, autoimmune and neoplastic diseases. International Journal of Biochemistry and Cell Biology. 2009;41(1):40–59. doi: 10.1016/j.biocel.2008.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okada K, Minamino T, Tsukamoto Y, et al. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: Possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation. 2004;110(6):705–712. doi: 10.1161/01.CIR.0000137836.95625.D4. [DOI] [PubMed] [Google Scholar]
- 15.Blais JD, Chin K, Zito E, et al. A small molecule inhibitor of Endoplasmic Reticulum Oxidation 1 (ERO1) with selectively reversible thiol reactivity. Journal of Biological Chemistry. 2010;285(27):20993–21003. doi: 10.1074/jbc.M110.126599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim I, Shu CW, Xu W, et al. Chemical biology investigation of cell death pathways activated by endoplasmic reticulum stress reveals cytoprotective modulators of ASK1. The Journal of Biological Chemistry. 2009;284(3):1593–1603. doi: 10.1074/jbc.M807308200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Asai M, Tsukamoto O, Minamino T, et al. PKA rapidly enhances proteasome assembly and activity in in vivo canine hearts. Journal of Molecular and Cellular Cardiology. 2009;46(4):452–462. doi: 10.1016/j.yjmcc.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 18.Yoshiuchi K, Kaneto H, Matsuoka T, et al. Pioglitazone reduces ER stress in the liver: direct monitoring of in vivo ER stress using ER stress-activated indicator transgenic mice. Endocrine Journal. 2009;56(9):1103–1111. doi: 10.1507/endocrj.k09e-140. [DOI] [PubMed] [Google Scholar]
- 19.Erbay E, Babaev VR, Mayers JR, et al. Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nature Medicine. 2009;15(12):1383–1391. doi: 10.1038/nm.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daosukho C, Chen Y, Noel T, et al. Phenylbutyrate, a histone deacetylase inhibitor, protects against Adriamycin-induced cardiac injury. Free Radical Biology and Medicine. 2007;42(12):1818–1825. doi: 10.1016/j.freeradbiomed.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lenin R, Maria MS, Agrawal M, Balasubramanyam J, Mohan V, Balasubramanyam M. Amelioration of glucolipotoxicity-induced endoplasmic reticulum stress by a chemical chaperone in human THP-1 monocytes. Experimental Diabetes Research. 2012;2012:10 pages. doi: 10.1155/2012/356487.356487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao H, Liao Y, Minamino T, et al. Inhibition of cardiac remodeling by pravastatin is associated with amelioration of endoplasmic reticulum stress. Hypertension Research. 2008;31(10):1977–1987. doi: 10.1291/hypres.31.1977. [DOI] [PubMed] [Google Scholar]
- 23.Boyce M, Py BF, Ryazanov AG, et al. A pharmacoproteomic approach implicates eukaryotic elongation factor 2 kinase in ER stress-induced cell death. Cell Death and Differentiation. 2008;15(3):589–599. doi: 10.1038/sj.cdd.4402296. [DOI] [PubMed] [Google Scholar]
- 24.Cheng WP, Wang BW, Shyu KG. Regulation of GADD153 induced by mechanical stress in cardiomyocytes. European Journal of Clinical Investigation. 2009;39(11):960–971. doi: 10.1111/j.1365-2362.2009.02193.x. [DOI] [PubMed] [Google Scholar]
- 25.Korennykh AV, Egea PF, Korostelev AA, et al. The unfolded protein response signals through high-order assembly of Ire1. Nature. 2009;457(7230):687–693. doi: 10.1038/nature07661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rutkowski DT, Kaufman RJ. A trip to the ER: coping with stress. Trends in Cell Biology. 2004;14(1):20–28. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 27.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nature Cell Biology. 2011;13(3):184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glimcher LH. XBP1: the last two decades. Annals of the Rheumatic Diseases. 2010;69(supplement 1):i67–i71. doi: 10.1136/ard.2009.119388. [DOI] [PubMed] [Google Scholar]
- 29.Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313(5783):104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- 30.Urano F, Wang X, Bertolotti A, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287(5453):664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 31.Nishitoh H, Matsuzawa A, Tobiume K, et al. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes and Development. 2002;16(11):1345–1355. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakagawa T, Zhu H, Morishima N, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β . Nature. 2000;403(6765):98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 33.Kaneko M, Niinuma Y, Nomura Y. Activation signal of nuclear factor-κB in response to endoplasmic reticulum stress is transduced via IRE1 and tumor necrosis factor receptor-associated factor 2. Biological and Pharmaceutical Bulletin. 2003;26(7):931–935. doi: 10.1248/bpb.26.931. [DOI] [PubMed] [Google Scholar]
- 34.Ma Y, Hendershot LM. Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. The Journal of Biological Chemistry. 2003;278(37):34864–34873. doi: 10.1074/jbc.M301107200. [DOI] [PubMed] [Google Scholar]
- 35.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death & Differentiation. 2004;11(4):381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- 36.Zinszner H, Kuroda M, Wang X, et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes and Development. 1998;12(7):982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bc12 and perturbing the cellular redox state. Molecular and Cellular Biology. 2001;21(4):1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gross E, Sevier CS, Heldman N, et al. Generating disulfides enzymatically: reaction products and electron acceptors of the endoplasmic reticulum thiol oxidase Ero1p. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(2):299–304. doi: 10.1073/pnas.0506448103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li G, Mongillo M, Chin K, et al. Role of ERO1-α-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. Journal of Cell Biology. 2009;186(6):783–792. doi: 10.1083/jcb.200904060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Timmins JM, Ozcan L, Seimon TA, et al. Calcium/calmodulin-dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. Journal of Clinical Investigation. 2009;119(10):2925–2941. doi: 10.1172/JCI38857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Puthalakath H, O'Reilly LA, Gunn P, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129(7):1337–1349. doi: 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- 42.Belmont PJ, Chen WJ, San Pedro MN, et al. Roles for endoplasmic reticulum-associated degradation and the novel endoplasmic reticulum stress response gene derlin-3 in the ischemic heart. Circulation Research. 2010;106(2):307–316. doi: 10.1161/CIRCRESAHA.109.203901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsukamoto O, Minamino T, Kitakaze M. Functional alterations of cardiac proteasomes under physiological and pathological conditions. Cardiovascular Research. 2010;85(2):339–346. doi: 10.1093/cvr/cvp282. [DOI] [PubMed] [Google Scholar]
- 44.Su H, Wang X. The ubiquitin-proteasome system in cardiac proteinopathy: a quality control perspective. Cardiovascular Research. 2010;85(2):253–262. doi: 10.1093/cvr/cvp287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Myoishi M, Hao H, Minamino T, et al. Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome. Circulation. 2007;116(11):1226–1233. doi: 10.1161/CIRCULATIONAHA.106.682054. [DOI] [PubMed] [Google Scholar]
- 46.Brown MS, Goldstein JL. Lipoprotein metabolism in the macrophage: implications for cholesterol deposition in atherosclerosis. Annual Review of Biochemistry. 1983;52:223–261. doi: 10.1146/annurev.bi.52.070183.001255. [DOI] [PubMed] [Google Scholar]
- 47.Maxfield FR, Tabas I. Role of cholesterol and lipid organization in disease. Nature. 2005;438(7068):612–621. doi: 10.1038/nature04399. [DOI] [PubMed] [Google Scholar]
- 48.Feng B, Yaol PM, Li Y, et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nature Cell Biology. 2003;5(9):781–792. doi: 10.1038/ncb1035. [DOI] [PubMed] [Google Scholar]
- 49.Zhou J, Lhoták Š, Hilditch BA, Austin RC. Activation of the unfolded protein response occurs at all stages of atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation. 2005;111(14):1814–1821. doi: 10.1161/01.CIR.0000160864.31351.C1. [DOI] [PubMed] [Google Scholar]
- 50.Thorp E, Li G, Seimon TA, Kuriakose G, Ron D, Tabas I. Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of Apoe−/− and Ldlr−/− mice lacking CHOP. Cell Metabolism. 2009;9(5):474–481. doi: 10.1016/j.cmet.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsukano H, Gotoh T, Endo M, et al. The endoplasmic reticulum stress-C/EBP homologous protein pathway-mediated apoptosis in macrophages contributes to the instability of atherosclerotic plaques. Arteriosclerosis, Thrombosis, and Vascular Biology. 2010;30(10):1925–1932. doi: 10.1161/ATVBAHA.110.206094. [DOI] [PubMed] [Google Scholar]
- 52.Ozcan L, Tabas I. Pivotal role of calcium/calmodulin-dependent protein kinase II in ER stress-induced apoptosis. Cell Cycle. 2010;9(2):223–224. doi: 10.4161/cc.9.2.10596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schrijvers DM, De Meyer GRY, Herman AG, Martinet W. Phagocytosis in atherosclerosis: molecular mechanisms and implications for plaque progression and stability. Cardiovascular Research. 2007;73(3):470–480. doi: 10.1016/j.cardiores.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 54.Henson PM, Bratton DL, Fadok VA. Apoptotic cell removal. Current Biology. 2001;11(19):R795–R805. doi: 10.1016/s0960-9822(01)00474-2. [DOI] [PubMed] [Google Scholar]
- 55.Tabas I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25(11):2255–2264. doi: 10.1161/01.ATV.0000184783.04864.9f. [DOI] [PubMed] [Google Scholar]
- 56.Devries-Seimon T, Li Y, Yoa PM, et al. Cholesterol-induced macrophage apoptosis requires ER stress pathways and engagement of the type A scavenger receptor. The Journal of Cell Biology. 2005;171(1):61–73. doi: 10.1083/jcb.200502078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Seimon TA, Nadolski MJ, Liao X, et al. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metabolism. 2010;12(5):467–482. doi: 10.1016/j.cmet.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li G, Scull C, Ozcan L, Tabas I. NADPH oxidase links endoplasmic reticulum stress, oxidative stress, and PKR activation to induce apoptosis. Journal of Cell Biology. 2010;191(6):1113–1125. doi: 10.1083/jcb.201006121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Seimon TA, Obstfeld A, Moore KJ, Golenbock DT, Tabas I. Combinatorial pattern recognition receptor signaling alters the balance of life and death in macrophages. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(52):19794–19799. doi: 10.1073/pnas.0609671104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Erqou S, Kaptoge S, Perry PL, et al. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. Journal of the American Medical Association. 2009;302(4):412–423. doi: 10.1001/jama.2009.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zeng L, Zampetaki A, Margariti A, et al. Sustained activation of XBP1 splicing leads to endothelial apoptosis and atherosclerosis development in response to disturbed flow. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(20):8326–8331. doi: 10.1073/pnas.0903197106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Civelek M, Manduchi E, Riley RJ, Stoeckert CJ, Jr., Davies PF. Chronic endoplasmic reticulum stress activates unfolded protein response in arterial endothelium in regions of susceptibility to atherosclerosis. Circulation Research. 2009;105(5):453–461. doi: 10.1161/CIRCRESAHA.109.203711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gargalovic PS, Gharavi NM, Clark MJ, et al. The unfolded protein response is an important regulator of inflammatory genes in endothelial cells. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26(11):2490–2496. doi: 10.1161/01.ATV.0000242903.41158.a1. [DOI] [PubMed] [Google Scholar]
- 64.Gora S, Maouche S, Atout R, et al. Phospholipolyzed LDL induces an inflammatory response in endothelial cells through endoplasmic reticulum stress signaling. FASEB Journal. 2010;24(9):3284–3297. doi: 10.1096/fj.09-146852. [DOI] [PubMed] [Google Scholar]
- 65.Outinen PA, Sood SK, Pfeifer SI, et al. Homocysteine-induced endoplasmic reticulum stress and growth arrest leads to specific changes in gene expression in human vascular endothelial cells. Blood. 1999;94(3):959–967. [PubMed] [Google Scholar]
- 66.Feaver RE, Hastings NE, Pryor A, Blackman BR. GRP78 upregulation by atheroprone shear stress via p38-, α2β1-dependent mechanism in endothelial cells. Arteriosclerosis, Thrombosis, and Vascular Biology. 2008;28(8):1534–1541. doi: 10.1161/ATVBAHA.108.167999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chien S. Effects of disturbed flow on endothelial cells. Annals of Biomedical Engineering. 2008;36(4):554–562. doi: 10.1007/s10439-007-9426-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bombeli T, Schwartz BR, Harlan JM. Endothelial cells undergoing apoptosis become proadhesive for nonactivated platelets. Blood. 1999;93(11):3831–3838. [PubMed] [Google Scholar]
- 69.Clarke MC, Figg N, Maguire JJ, et al. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nature Medicine. 2006;12(9):1075–1080. doi: 10.1038/nm1459. [DOI] [PubMed] [Google Scholar]
- 70.Cheng WP, Hung HF, Wang BW, Shyu KG. The molecular regulation of GADD153 in apoptosis of cultured vascular smooth muscle cells by cyclic mechanical stretch. Cardiovascular Research. 2008;77(3):551–559. doi: 10.1093/cvr/cvm057. [DOI] [PubMed] [Google Scholar]
- 71.Zhou J, Werstuck GH, Lhoták Š, et al. Association of multiple cellular stress pathways with accelerated atherosclerosis in hyperhomocysteinemic apolipoprotein E-deficient mice. Circulation. 2004;110(2):207–213. doi: 10.1161/01.CIR.0000134487.51510.97. [DOI] [PubMed] [Google Scholar]
- 72.Kedi X, Ming Y, Yongping W, Yi Y, Xiaoxiang Z. Free cholesterol overloading induced smooth muscle cells death and activated both ER- and mitochondrial-dependent death pathway. Atherosclerosis. 2009;207(1):123–130. doi: 10.1016/j.atherosclerosis.2009.04.019. [DOI] [PubMed] [Google Scholar]
- 73.Pedruzzi E, Guichard C, Ollivier V, et al. NAD(P)H oxidase Nox-4 mediates 7-ketocholesterol-induced endoplasmic reticulum stress and apoptosis in human aortic smooth muscle cells. Molecular and Cellular Biology. 2004;24(24):10703–10717. doi: 10.1128/MCB.24.24.10703-10717.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Werstuck GH, Khan MI, Femia G, et al. Glucosamine-induced endoplasmic reticulum dysfunction is associated with accelerated atherosclerosis in a hyperglycemic mouse model. Diabetes. 2006;55(1):93–101. [PubMed] [Google Scholar]
- 75.Zhou J, Austin RC. Contributions of hyperhomocysteinemia to atherosclerosis: Causal relationship and potential mechanisms. BioFactors. 2009;35(2):120–129. doi: 10.1002/biof.17. [DOI] [PubMed] [Google Scholar]
- 76.Dickhout JG, Sood SK, Austin RC. Role of endoplasmic reticulum calcium disequilibria in the mechanism of homocysteine-induced ER stress. Antioxidants and Redox Signaling. 2007;9(11):1863–1873. doi: 10.1089/ars.2007.1780. [DOI] [PubMed] [Google Scholar]
- 77.Colgan SM, Tang D, Werstuck GH, Austin RC. Role of ERO1-α-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. International Journal of Biochemistry and Cell Biology. 2007;39(10):1843–1851. doi: 10.1016/j.biocel.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 78.Werstuck GH, Lentz SR, Dayal S, et al. Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. Journal of Clinical Investigation. 2001;107(10):1263–1273. doi: 10.1172/JCI11596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cnop M, Foufelle F, Velloso LA. Endoplasmic reticulum stress, obesity and diabetes. Trends in Molecular Medicine. 2012;18(1):59–68. doi: 10.1016/j.molmed.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 80.Keuper M, Blüher M, Schön MR, et al. An inflammatory micro-environment promotes human adipocyte apoptosis. Molecular and Cellular Endocrinology. 2011;339(1-2):105–113. doi: 10.1016/j.mce.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 81.Cheng X, Folco EJ, Shimizu K, Libby P. Adiponectin induces pro-inflammatory programs in human macrophages and CD4+ T cells. Journal of Biological Chemistry. 2012;287(44):36896–36904. doi: 10.1074/jbc.M112.409516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Oh J, Riek AE, Weng S, et al. Endoplasmic reticulum stress controls M2 macrophage differentiation and foam cell formation. The Journal of Biological Chemistry. 2012;287(15):11629–11641. doi: 10.1074/jbc.M111.338673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lumeng CN, DeYoung SM, Bodzin JL, Saltiel AR. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes. 2007;56(1):16–23. doi: 10.2337/db06-1076. [DOI] [PubMed] [Google Scholar]
- 84.Zhang H, Cui J, Zhang C. Emerging role of adipokines as mediators in atherosclerosis. World Journal of Cardiology. 2010;2:370–376. doi: 10.4330/wjc.v2.i11.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Makowski L, Boord JB, Maeda K, et al. Lack of macrophage fatty-acid-binding protein aP2 protects mice deficient in apolipoprotein E against atherosclerosis. Nature Medicine. 2001;7(6):699–705. doi: 10.1038/89076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hellemans KH, Hannaert J, Denys B, et al. Susceptibility of pancreatic beta cells to fatty acids is regulated by LXR/PPARα-dependent stearoyl-coenzyme a desaturase. PLoS ONE. 2009;4(9) doi: 10.1371/journal.pone.0007266.e7266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tabas I, Tall A, Accili D. The impact of macrophage insulin resistance on advanced atherosclerotic plaque progression. Circulation Research. 2010;106(1):58–67. doi: 10.1161/CIRCRESAHA.109.208488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liang CP, Han S, Senokuchi T, Tall AR. The macrophage at the crossroads of insulin resistance and atherosclerosis. Circulation Research. 2007;100(11):1546–1555. doi: 10.1161/CIRCRESAHA.107.152165. [DOI] [PubMed] [Google Scholar]
- 89.Liang C-P, Han S, Li G, Tabas I, Tall AR. Impaired MEK signaling and SERCA expression promote ER stress and apoptosis in insulin-resistant macrophages and are reversed by exenatide treatment. Diabetes. 2012;61(10):2609–2620. doi: 10.2337/db11-1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bergman BC, Hunerdosse DM, Kerege A, Playdon MC, Perreault L. Localisation and composition of skeletal muscle diacylglycerol predicts insulin resistance in humans. Diabetologia. 2012;55(4):1140–1150. doi: 10.1007/s00125-011-2419-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liang C, Han S, Okamoto H, et al. Increased CD36 protein as a response to defective insulin signaling in macrophages. Journal of Clinical Investigation. 2004;113(5):764–773. doi: 10.1172/JCI19528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Han S, Liang C, DeVries-Seimon T, et al. Macrophage insulin receptor deficiency increases ER stress-induced apoptosis and necrotic core formation in advanced atherosclerotic lesions. Cell Metabolism. 2006;3(4):257–266. doi: 10.1016/j.cmet.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 93.Senokuchi T, Liang CP, Seimon TA, et al. Forkhead transcription factors (FoxOs) promote apoptosis of insulin-resistant macrophages during cholesterol-induced endoplasmic reticulum stress. Diabetes. 2008;57(11):2967–2976. doi: 10.2337/db08-0520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Beg AA, Baltimore D. An essential role for NF-κB in preventing TNF-α-induced cell death. Science. 1996;274(5288):782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 95.Lehto S, Rönnemaa T, Haffner SM, Pyörälä K, Kallio V, Laakso M. Dyslipidemia and hyperglycemia predict coronary heart disease events in middle-aged patients with NIDDM. Diabetes. 1997;46(8):1354–1359. doi: 10.2337/diab.46.8.1354. [DOI] [PubMed] [Google Scholar]
- 96.Haffner SM. The importance of hyperglycemia in the nonfasting state to the development of cardiovascular disease. Endocrine Reviews. 1998;19(5):583–592. doi: 10.1210/edrv.19.5.0343. [DOI] [PubMed] [Google Scholar]
- 97.Quagliaro L, Piconi L, Assaloni R, Martinelli L, Motz E, Ceriello A. Intermittent high glucose enhances apoptosis related to oxidative stress in human umbilical vein endothelial cells: the role of protein kinase C and NAD(P)H-oxidase activation. Diabetes. 2003;52(11):2795–2804. doi: 10.2337/diabetes.52.11.2795. [DOI] [PubMed] [Google Scholar]
- 98.Popov D, Simionescu M. Cellular mechanisms and signalling pathways activated by high glucose and AGE-albumin in the aortic endothelium. Archives of Physiology and Biochemistry. 2006;112(4-5):265–273. doi: 10.1080/13813450601094573. [DOI] [PubMed] [Google Scholar]
- 99.Gleissner CA, Sanders JM, Nadler J, Ley K. Upregulation of aldose reductase during foam cell formation as possible link among diabetes, hyperlipidemia, and atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2008;28(6):1137–1143. doi: 10.1161/ATVBAHA.107.158295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tesfamariam B, Brown ML, Cohen RA. Elevated glucose impairs endothelium-dependent relaxation by activating protein kinase C. Journal of Clinical Investigation. 1991;87(5):1643–1648. doi: 10.1172/JCI115179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Inoguchi T, Li P, Umeda F, et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000;49(11):1939–1945. doi: 10.2337/diabetes.49.11.1939. [DOI] [PubMed] [Google Scholar]
- 102.Wells-Knecht KJ, Zyzak DV, Litchfield JE, Thorpe SR, Baynes JW. Mechanism of autoxidative glycosylation: identification of glyoxal and arabinose as intermediates in the autoxidative modification of proteins by glucose. Biochemistry. 1995;34(11):3702–3709. doi: 10.1021/bi00011a027. [DOI] [PubMed] [Google Scholar]
- 103.Smit AJ, Hartog JW, Voors AA, van Veldhuisen DJ. Advanced glycation endproducts in chronic heart failure. Annals of the New York Academy of Sciences. 2008;1126:225–230. doi: 10.1196/annals.1433.038. [DOI] [PubMed] [Google Scholar]
- 104.Yan SF, Ramasamy R, Naka Y, Schmidt AM. Glycation, inflammation, and RAGE: a scaffold for the macrovascular complications of diabetes and beyond. Circulation Research. 2003;93(12):1159–1169. doi: 10.1161/01.RES.0000103862.26506.3D. [DOI] [PubMed] [Google Scholar]
- 105.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54(6):1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 106.Nerlich AG, Sauer U, Kolm-Litty V, Wagner E, Koch M, Schleicher ED. Expression of glutamine:fructose-6-phosphate amidotransferase in human tissues: evidence for high variability and distinct regulation in diabetes. Diabetes. 1998;47(2):170–178. doi: 10.2337/diab.47.2.170. [DOI] [PubMed] [Google Scholar]
- 107.Wu G, Haynes TE, Yan W, Meininger CJ. Presence of glutamine:fructose-6-phosphate amidotransferase for glucosamine-6-phosphate synthesis in endothelial cells: effects of hyperglycaemia and glutamine. Diabetologia. 2001;44(2):196–202. doi: 10.1007/s001250051599. [DOI] [PubMed] [Google Scholar]
- 108.Fülöp N, Mason MM, Dutta K, et al. Impact of type 2 diabetes and aging on cardiomyocyte function and O-linked N-acetylglucosamine levels in the heart. The American Journal of Physiology: Cell Physiology. 2007;292(4):C1370–C1378. doi: 10.1152/ajpcell.00422.2006. [DOI] [PubMed] [Google Scholar]
- 109.Helenius A. How N-linked oligosaccharides affect glycoprotein folding in the endoplasmic reticulum. Molecular Biology of the Cell. 1994;5(3):253–265. doi: 10.1091/mbc.5.3.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 111.Sage AT, Walter LA, Shi Y, et al. Hexosamine biosynthesis pathway flux promotes endoplasmic reticulum stress, lipid accumulation, and inflammatory gene expression in hepatic cells. American Journal of Physiology: Endocrinology and Metabolism. 2010;298(3):E499–E511. doi: 10.1152/ajpendo.00507.2009. [DOI] [PubMed] [Google Scholar]
- 112.Hossain GS, van Thienen JV, Werstuck GH, et al. TDAG51 is induced by homocysteine, promotes detachment-mediated programmed cell death, and contributes to the development of atherosclerosis in hyperhomocysteinemia. The Journal of Biological Chemistry. 2003;278(32):30317–30327. doi: 10.1074/jbc.M212897200. [DOI] [PubMed] [Google Scholar]
- 113.Khan MI, Pichna BA, Shi Y, Bowes AJ, Werstuck GH. Evidence supporting a role for endoplasmic reticulum stress in the development of atherosclerosis in a hyperglycaemic mouse model. Antioxidants and Redox Signaling. 2009;11(9):2289–2298. doi: 10.1089/ars.2009.2569. [DOI] [PubMed] [Google Scholar]
- 114.Robertson LA, Kim AJ, Werstuck GH. Mechanisms linking diabetes mellitus to the development of a role forendoplasmic reticulum stress synthase kinase-3. Canadian Journal of Physiology and Pharmacology. 2006;84:39–48. doi: 10.1139/Y05-142. [DOI] [PubMed] [Google Scholar]
- 115.McAlpine CS, Bowes AJ, Khan MI, Shi Y, Werstuck GH. Endoplasmic reticulum stress and glycogen synthase kinase-3β activation in apolipoprotein E–deficient mouse models of accelerated atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2012;32:82–91. doi: 10.1161/ATVBAHA.111.237941. [DOI] [PubMed] [Google Scholar]
- 116.Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. Journal of Cell Science. 2003;116(7):1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nijholt DAT, Nölle A, van Haastert ES, et al. Unfolded protein response activates glycogen synthase kinase-3 via selective lysosomal degradation. Neurobiology of Aging. 2013;34(7):1759–1771. doi: 10.1016/j.neurobiolaging.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 118.Werstuck GH, Kim AJ, Brenstrum T, Ohnmacht SA, Panna E, Capretta A. Examining the correlations between GSK-3 inhibitory properties and anti-convulsant efficacy of valproate and valproate-related compounds. Bioorganic and Medicinal Chemistry Letters. 2004;14(22):5465–5467. doi: 10.1016/j.bmcl.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 119.Shi Y, Gerritsma D, Bowes AJ, Capretta A, Werstuck GH. Induction of GRP78 by valproic acid is dependent upon histone deacetylase inhibition. Bioorganic & Medicinal Chemistry Letters. 2007;17(16):4491–4494. doi: 10.1016/j.bmcl.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 120.Bowes AJ, Khan MI, Shi Y, Robertson L, Werstuck GH. Valproate attenuates accelerated atherosclerosis in hyperglycemic ApoE-deficient mice: Evidence in support of a role for endoplasmic reticulum stress and glycogen synthase kinase-3 in lesion development and hepatic steatosis. The American Journal of Pathology. 2009;174(1):330–342. doi: 10.2353/ajpath.2009.080385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chang SC, Erwin AE, Lee AS. Glucose-regulated protein (GRP94 and GRP78) genes share common regulatory domains and are coordinately regulated by common trans-acting factors. Molecular and Cellular Biology. 1989;9:2153–2162. doi: 10.1128/mcb.9.5.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Vitadello M, Penzo D, Petronilli V, et al. Overexpression of the stress protein Grp94 reduces cardiomyocyte necrosis due to calcium overload and simulated ischemia. The FASEB Journal. 2003;17(8):923–925. doi: 10.1096/fj.02-0644fje. [DOI] [PubMed] [Google Scholar]
- 123.Reddy RK, Mao C, Baumeister P, Austin RC, Kaufman RJ, Lee AS. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors. Role of ATP binding site in suppression of caspase-7 activation. Journal of Biological Chemistry. 2003;278(23):20915–20924. doi: 10.1074/jbc.M212328200. [DOI] [PubMed] [Google Scholar]
- 124.Fu HY, Minamino T, Tsukamoto O, et al. Overexpression of endoplasmic reticulum-resident chaperone attenuates cardiomyocyte death induced by proteasome inhibition. Cardiovascular Research. 2008;79(4):600–610. doi: 10.1093/cvr/cvn128. [DOI] [PubMed] [Google Scholar]
- 125.Engin F, Hotamisligil GS. Restoring endoplasmic reticulum function by chemical chaperones: an emerging therapeutic approach for metabolic diseases. Diabetes, Obesity and Metabolism. 2010;12(2):108–115. doi: 10.1111/j.1463-1326.2010.01282.x. [DOI] [PubMed] [Google Scholar]
- 126.Leonard JV, Morris AAM. Urea cycle disorders. Seminars in Neonatology. 2002;7(1):27–35. doi: 10.1053/siny.2001.0085. [DOI] [PubMed] [Google Scholar]
- 127.Cudkowicz ME, Andres PL, Macdonald SA, et al. Phase 2 study of sodium phenylbutyrate in ALS. Amyotrophic Lateral Sclerosis. 2009;10(2):99–106. doi: 10.1080/17482960802320487. [DOI] [PubMed] [Google Scholar]
- 128.Lee B, Rhead W, Diaz GA, et al. Phase 2 comparison of a novel ammonia scavenging agent with sodium phenylbutyrate in patients with urea cycle disorders: safety, pharmacokinetics and ammonia control. Molecular Genetics and Metabolism. 2010;100(3):221–228. doi: 10.1016/j.ymgme.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Özcan U, Yilmaz E, Özcan L, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313(5790):1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Cash JG, Kuhel DG, Basford JE, et al. Apolipoprotein E4 impairs macrophage efferocytosis and potentiates apoptosis by accelerating endoplasmic reticulum stress. The Journal of Biological Chemistry. 2012;287(33):27876–27884. doi: 10.1074/jbc.M112.377549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Takada A, Miki T, Kuno A, et al. Role of ER stress in ventricular contractile dysfunction in type 2 diabetes. PLoS ONE. 2012;7(6) doi: 10.1371/journal.pone.0039893.e39893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Miller SDW, Greene CM, McLean C, et al. Tauroursodeoxycholic acid inhibits apoptosis induced by Z alpha-1 antitrypsin via inhibition of bad. Hepatology. 2007;46(2):496–503. doi: 10.1002/hep.21689. [DOI] [PubMed] [Google Scholar]
- 133.Fu HY, Okada K, Liao Y, et al. Ablation of C/EBP homologous protein attenuates endoplasmic reticulum-mediated apoptosis and cardiac dysfunction induced by pressure overload. Circulation. 2010;122(4):361–369. doi: 10.1161/CIRCULATIONAHA.109.917914. [DOI] [PubMed] [Google Scholar]
- 134.Liu C-L, Li X, Hu G-L, et al. Salubrinal protects against tunicamycin and hypoxia induced cardiomyocyte apoptosis via the PERK-eIF2α signaling pathway. Journal of Geriatric Cardiology. 2012;9(3):258–268. doi: 10.3724/SP.J.1263.2012.02292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Liu C, Li X, Hu G, et al. Salubrinal protects against tunicamycin and hypoxia induced cardiomyocyte apoptosis via the PERK-eIF2α signaling pathway. Journal of Geriatric Cardiology. 2012;9(3):258–268. doi: 10.3724/SP.J.1263.2012.02292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wang X, Ron D. Stress-induced phosphorylation and activation of the transcription factor CHOP (GADD153) by p38 MAP kinase. Science. 1996;272(5266):1347–1349. doi: 10.1126/science.272.5266.1347. [DOI] [PubMed] [Google Scholar]
- 137.Guo R, Ma H, Gao F, Zhong L, Ren J. Metallothionein alleviates oxidative stress-induced endoplasmic reticulum stress and myocardial dysfunction. Journal of Molecular and Cellular Cardiology. 2009;47(2):228–237. doi: 10.1016/j.yjmcc.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pal R, Cristan EA, Schnittker K, Narayan M. Rescue of ER oxidoreductase function through polyphenolic phytochemical intervention: Implications for subcellular traffic and neurodegenerative disorders. Biochemical and Biophysical Research Communications. 2010;392(4):567–571. doi: 10.1016/j.bbrc.2010.01.071. [DOI] [PubMed] [Google Scholar]
- 139.Tsukamoto O, Minamino T, Okada K, et al. Depression of proteasome activities during the progression of cardiac dysfunction in pressure-overloaded heart of mice. Biochemical and Biophysical Research Communications. 2006;340(4):1125–1133. doi: 10.1016/j.bbrc.2005.12.120. [DOI] [PubMed] [Google Scholar]
- 140.Hikoso S, Ikeda Y, Yamaguchi O, et al. Progression of heart failure was suppressed by inhibition of apoptosis signal-regulating kinase 1 via transcoronary gene transfer. Journal of the American College of Cardiology. 2007;50(5):453–462. doi: 10.1016/j.jacc.2007.03.053. [DOI] [PubMed] [Google Scholar]
- 141.Homma K, Katagiri K, Nishitoh H, Ichijo H. Targeting ASK1 in ER stress-related neurodegenerative diseases. Expert Opinion on Therapeutic Targets. 2009;13(6):653–664. doi: 10.1517/14728220902980249. [DOI] [PubMed] [Google Scholar]
- 142.Winder WW. Energy-sensing and signaling by AMP-activated protein kinase in skeletal muscle. Journal of Applied Physiology. 2001;91(3):1017–1028. doi: 10.1152/jappl.2001.91.3.1017. [DOI] [PubMed] [Google Scholar]
- 143.Meares GP, Hughes KJ, Naatz A, et al. IRE1-dependent activation of AMPK in response to nitric oxide. Molecular and Cellular Biology. 2011;31(21):4286–4297. doi: 10.1128/MCB.05668-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sasaki H, Asanuma H, Fujita M, et al. Metformin prevents progression of heart failure in dogs role of AMP-activated protein kinase. Circulation. 2009;119(19):2568–2577. doi: 10.1161/CIRCULATIONAHA.108.798561. [DOI] [PubMed] [Google Scholar]
- 145.Jia F, Wu C, Chen Z, Lu G. Atorvastatin inhibits homocysteine -induced endoplasmic reticulum stress through activation of AMP-activated protein kinase. Cardiovascular Therapeutics. 2012;30(6):317–325. doi: 10.1111/j.1755-5922.2011.00287.x. [DOI] [PubMed] [Google Scholar]
- 146.Kim M, Tian R. Targeting AMPK for cardiac protection: opportunities and challenges. Journal of Molecular and Cellular Cardiology. 2011;51(4):548–553. doi: 10.1016/j.yjmcc.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Martindale JJ, Fernandez R, Thuerauf D, et al. Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circulation Research. 2006;98(9):1186–1193. doi: 10.1161/01.RES.0000220643.65941.8d. [DOI] [PubMed] [Google Scholar]
- 148.Toko H, Takahashi H, Kayama Y, et al. ATF6 is important under both pathological and physiological states in the heart. Journal of Molecular and Cellular Cardiology. 2010;49(1):113–120. doi: 10.1016/j.yjmcc.2010.03.020. [DOI] [PubMed] [Google Scholar]
- 149.Gupta R, Maitland ML. Sunitinib, hypertension, and heart failure: a model for kinase inhibitor-mediated cardiotoxicity. Current Hypertension Reports. 2011;13(6):430–435. doi: 10.1007/s11906-011-0229-4. [DOI] [PubMed] [Google Scholar]
- 150.Delie F, Ribaux P, Petignat P, Cohen M. Anti-KDEL-coated nanoparticles: a promising tumor targeting approach for ovarian cancer? Biochimie. 2012;94(11):2391–2397. doi: 10.1016/j.biochi.2012.06.010. [DOI] [PubMed] [Google Scholar]
- 151.Niu J, Azfer A, Rogers LM, Wang X, Kolattukudy PE. Cardioprotective effects of cerium oxide nanoparticles in a transgenic murine model of cardiomyopathy. Cardiovascular Research. 2007;73(3):549–559. doi: 10.1016/j.cardiores.2006.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Younce CW, Wang K, Kolattukudy PE. Hyperglycaemia-induced cardiomyocyte death is mediated via MCP-1 production and induction of a novel zinc-finger protein MCPIP. Cardiovascular Research. 2010;87(4):665–674. doi: 10.1093/cvr/cvq102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hofheinz RD, Gnad-Vogt SU, Beyer U, Hochhaus A. Liposomal encapsulated anti-cancer drugs. Anti-Cancer Drugs. 2005;16(7):691–707. doi: 10.1097/01.cad.0000167902.53039.5a. [DOI] [PubMed] [Google Scholar]
- 154.Takahama H, Minamino T, Asanuma H, et al. Prolonged targeting of ischemic/reperfused myocardium by liposomal adenosine augments cardioprotection in rats. Journal of the American College of Cardiology. 2009;53(8):709–717. doi: 10.1016/j.jacc.2008.11.014. [DOI] [PubMed] [Google Scholar]