

Abstract
Background
Systemic mastocytosis (SM) is a heterogenous, clonal mast cell (MC) proliferation, rarely associated with clonal hematologic non-mast cell lineage disease (SM-AHNMD). KITD816V is regarded as driver-mutation in SM-AHNMD.
Methods
DNA isolated from peripheral blood (PB) of an SM-CMML patient was investigated with targeted next generation sequencing. Variants were verified by Sanger sequencing and further characterized in the SM part of the bone marrow trephine (BMT), normal tissue, and FACS sorted PB cell subpopulations.
Findings
Low coverage deep-sequencing (mean 10x) on a GS 454 Junior revealed two as yet unreported SNVs (CBFA2T3 and CLTCL1), both germ-line mutations. High coverage (mean 1674x) targeted re-sequencing on an Ion Proton revealed 177 variants in coding regions. Excluding SNPs, the final list comprised 11 variants. Among these, TET2 (p.Thr1027fs, p.Cys1263Ser) and RUNX1 (p.Asn109Ser) were identified in in the peripheral blood and the SM part of BMT, but not in normal tissue. Furthermore, Sanger sequencing of PB cells revealed similar signal intensities for both TET2 mutations in FACS sorted CD34+ precursor cells and CD16+ granulocytes comparable to signals in the SM part of BMT. In contrast, RUNX1 exhibited a double intensity in CD34+ cells compared to the SM part of BMT and a homozygous variant signal in granulocytes. Both TET2 and RUNX1 mutations were not detectable in B- and T-cells.
Conclusion
We present a heterozygous triple-mutation pattern (KIT, TET2, RUNX1) in mast cells (SM disease part) with additional LOH of RUNX1 in granulocytes (CMML disease part). These identified mutations allow a more detailed insight into a multistep pathogenesis which suggests a common tumor progenitor in SM-CMML.
Keywords: Chronic myelomonocytic leukemia, Systemic mastocytosis, SM-CMML, Next Generation Sequencing, c-KIT mutation, TET-2 mutation, RUNX1 mutation
Background
Systemic mastocytosis (SM) rarely occurs in combination with so called “associated clonal haematological non-mast cell lineage disease” (AHNMD) [1-5]. In most instances, the AHNMD component is of myeloid origin, including acute myeloid leukemia (AML), chronic myelomonocytic leukemia (CMML) or primary myelofibrosis (PMF) [1,6-8]. Recently, KITD816V mutations were identified in both disease components of SM-AHNMD with the highest frequency in SM-CMML [9], suggesting a common precursor.
Methods and findings
We report on a 73 year old male patient who was initially diagnosed with CMML. Untreated for three years, extended investigations for disease progression showed a normal karyotype and neither a JAK-2 mutation nor a BCR-ABL fusion gene. Due to abnormal BM mast cell infiltrates the patient met the WHO 08 [1] criteria for SM-CMML with proven KITD816V mutation in different cell subpopulations as detailed in (Figure 1A-D). The patient died of disease under treatment with azacitidin (vidaza®) four months after this diagnosis. Autopsy findings showed additionally to the bone marrow involvement widespread disease in different extramedullary haematopoietic and visceral organs. There was no transformation into acute leukemia.
Figure 1.
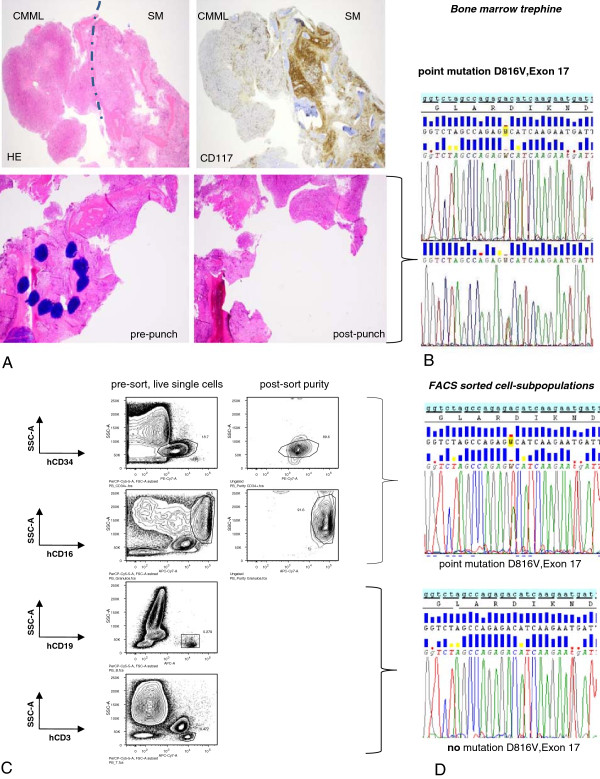
Assessment of the KITD816V mutation on the bone marrow trephine. Aupper line: HE stain of the bone marrow trephine showing both, CMML and SM part in distinct areas in this patient (left). A CD117 immunostain (right) highlights the mast cells in the SM compartment, however cells within the CMML compartment are negative. Lower line: HE control before and after punching out the SM area (containing around 90% of MC in the CD117 staining) submitted to PCR and Sanger Sequencing. B Forward and reverse sequences indicating the KITD816V mutation of microdissected SM area of the bone marrow trephine. C FACS sorted cells as indicated with a purity of around 90% for CD34+ stem cells and granulocytes. D Shows the sequencing results of the PCR amplification product for KITD816V, and forward strands for isolated cell-subpopulations respectively.
We applied a cancer gene panel containing 427 genes, custom-designed to include all exons of these genes on a solid capture array by NimbleGen (Roche). Captured targets (Additional file 1: Table S1) were deep-sequenced on a GS 454-Junior (Roche) revealing 1033 gene variants with a mean coverage of 10-fold. The analysis with the GS Reference Mapper software v.2.5 (Roche; default settings and hg19 as reference genome) identified, next to the successfully verified KITD816V mutation, sixty-four (6%) variants in the coding region of 42 genes (Additional file 2: Table S2). Of the 64 variants, 61 (95%) were single nucleotide polymorphisms. The remaining were two as yet unreported SNVs (CBFA2T3 and CLTCL1) and one 1-bp deletion (BLM; COSM252959). All three mutations were confirmed by Sanger sequencing (Genetic Analyzer 3130xl (Applied Biosystems); [10]) using DNA from PB, the SM part of BMT and normal tissue, meeting the criteria for germ-line mutations (representative sequences in Figure 2A). Since, however, the assumed 1-bp deletion of BLM at AA515del1 – Asn515fs*16 could be identified in a non-tumoral human embryonic kidney cell line (HEK293T), we interpreted this aberration as a PCR/sequencing artefact associated with a mononucleotide repeat at this site. This finding was additionally confirmed by sub-cloning of the PCR-product in a bacterial expression vector and sequencing with different primer sets (Additional file 3: Figure S1). Interestingly, previous reports describe exactly the same deletion in gastrointestinal cancer and in T-ALL cell lines [11,12].
Figure 2.
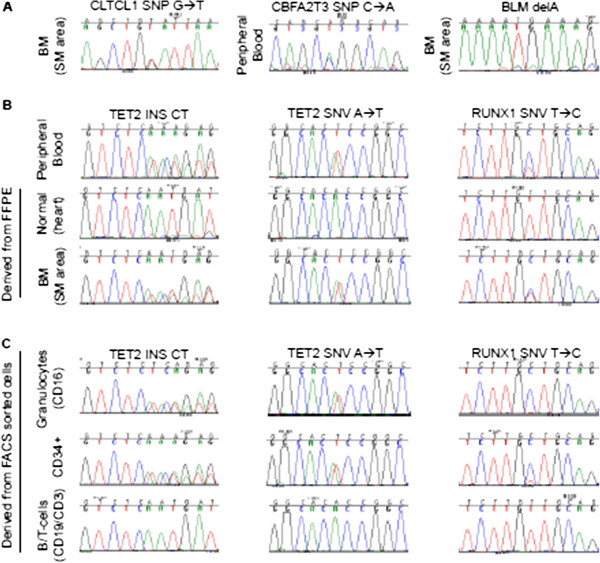
Verification of identified variants by Sanger sequencing. A: Representative sequences of CBFA2T3, CLTCL1, and BLM using DNA derived from the SM part of BMT, PB, and the SM part of BMT, respectively. B: Sequencing of TET2 and RUNX1 using DNA derived from PB, the SM part of BMT, and normal tissue. C: Sequencing of TET2 and RUNX1 using DNA derived from FACS sorted PB cell populations.
To achieve a higher resolution we performed a high coverage targeted re-sequencing with identical blood derived DNA using the Ion AmpliSeq™ Comprehensive Cancer Panel comprising all exons of 409 known cancer genes (Life Technologies). Forty ng DNA was used as input and the target regions were 13-fold PCR amplified. After ligation of Torrent specific adapters, the amplicons were subjected to clonal expansion on spheres using the Ion One Touch 2 system (Life Technologies). Subsequently, the amplicons were dispersed on a PI chip and sequenced on the Ion Proton platform [13]. Mean coverage was 1674-fold whereas more than 93% of the amplicons were covered at least 100-fold and more than 81% at least 500-fold allowing sensitive variant detection. Alignment (hg19), local re-alignment (2-fold), and probabilistic variant detection (90% probability and 10-fold coverage) were performed using CLC Genomics Workbench version 5.5. Additional filtering resulted in variants with following characteristics: i) non-synonymous variants and INDELs resulting in frame-shifts, ii) phred-score > = 20 and variant coverage > = 50x, and iii) forward/reverse read ratio > = 0.25. These filter criteria resulted in identifying 177 variants (Additional file 4: Table S3). From these, 144 were present in the 1000 Genome database and dbSNP and were therefore excluded in the following analysis. Under additional more stringent filtering criteria for 1-bp INDELs in homopolymeric regions (CLC default settings for 454/Ion) a final list of 11 variants emerged (Additional file 5: Table S4) including KITD816V. Interestingly, variants of two genes previously described in SM-AHNMD [14]TET2 (p.Thr1027fs, p.Cys1263Ser) and RUNX1 (p.Asn109Ser) were detected. Although detectable by the 454-Junior, their coverage was too low to be called by the GS Reference Mapper. Of note, 44 variants of the 64 variants identified by the 454-Junior/GS Reference Mapper platform lay in target regions of the CCP (Additional file 2: Table S2). From these, 35 were verified by the Ion Torrent/CLC platform, one was differently aligned, and eight were not detected.
Thus, the variants in TET2 and RUNX1 represented most probably true positive calls potentially involved in SM-CMML pathogenesis. Although, they have not yet been reported in the Cosmic database, the mutations highly likely influence the protein function: i) the frame-shift mutation in TET2 occurring in front of the two conserved regions of TET2 (1104–1478, 1845–2002) and the SNV lying within the first region [15], ii) the mutation in RUNX1 located in the RUNT domain which is responsible for proper DNA binding and heterodimerization [16].
The 2-bp insertion (p.Thr1027fs) and the SNV (p.Cys1263Ser) in TET2, and the SNV (p.Asn109Ser) in RUNX1 were confirmed in PB and the SM part of BMT (Figure 2B). Very low signals of the variants were detected in normal tissue, most probably due to infiltrating mast cells. Whereas no signals were found in B- and T-lymphocytes, similar signal intensities for both TET2 mutations were seen in CD34+ precursor cells and CD16+ granulocytes comparable to the signals in the SM part of BMT (Figure 2C). In contrast, RUNX1 exhibited a double intensity of the variant in CD34+ cells compared to the SM part of BMT and a homozygous variant signal in granulocytes.
Inasmuch the SNPs and seven variants identified (Additional file 5: Table S4) are implicated in the disease development needs to be confirmed. Importantly however, we present an in-depth analysis of the sub-clonal relation of SM-CMML which reveals essential concomitant key mutations in KIT, TET2, and RUNX1 in the emergence of different disease compartments. Since the mutant allelic fraction in the blood of KIT (0.44), TET2 (0.45, 0.44), and RUNX1 (0.53) assessed by NGS (Proton platform) were in a similar range (Additional file 5: Table S4), we assume that these mutations belong to the same CD34+ sub-clonal cell population. This is supported by the fact that the electropherograms of the Sanger sequencing exhibited the same intensities for KIT and TET2 mutations in blood and the SM-area as well as in the sorted CD34+ and granulocytic cell fraction (Figure 2). Additionally, RUNX1 mutant signals assessed by Sanger sequencing were highest in the granulocytic cell fraction and balanced with wild-type in the SM-area (purity 90%) which implies that an additional LOH of RUNX1 occured in a CD34+ mast cells precursor. This is as well reflected by the fact that the mutant allelic fraction of RUNX1 (0.53) in the blood assessed by NGS (Proton platform) was increased by 0.09 in comparison to the other three SNVs (mean mutant allelic fraction KIT, 2x TET2: 0.44). Since the KIT (CD117) protein is expressed in the SM, but not in the CMML compartment (Figure 1) ascribes the KITD816V mutation a pivotal role in the disease initiation, particularly of the SM fraction. Whereas TET2 and RUNX1 were necessary sequential mutations for progression to overt SM, homozygous loss of wild-type RUNX1 seemingly was the driver mutation enabling a clonal expansion of the CMML lineage as suggested by our disease model (Figure 3). This further underlines the concept of common tumour progenitor cells leading to a spectrum of the same disease rather than two different ones as the term SM-AHNMD evokes. The presence of RUNX1 and TET2 mutations in mature granulocytes and in MC of the BMT in our study is quite novel, since so far only one recent study showed concomitant mutations of KIT and TET2 in mast cells and granulocytes in SM-AHNMD [14]. The increased number of driving-mutations could explain the rapid clinical progression in our patient and is in line with the previous study, advocating additional molecular aberrations a prognostic role [14].
Figure 3.
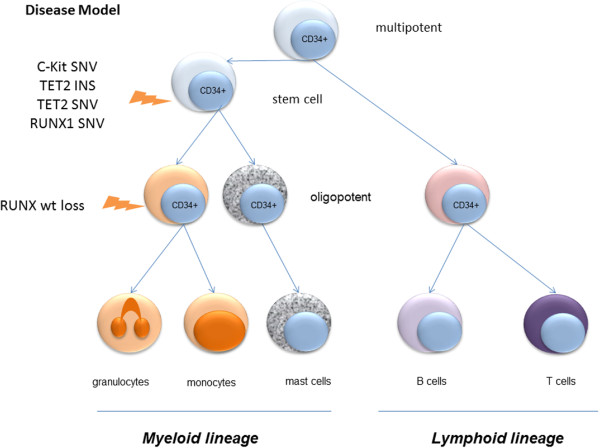
Hypothetical disease model of SM-CMML pathogenesis.
In summary, the current SM-CMML derived from the same mutated tumour progenitor cells (KIT SNV, TET2 frame-shift, TET2 SNV, RUNX1 SNV) which acquired additional mutations (i.e. RUNX1 wild-type loss) in different sub-clones as hypothesised in our model to finally manifest in the pathological state of SM-CMML.
Consent
The study was approved by the official authorities of the ethical committee of the County of Zurich (StV2-2007) and a written consent was obtained by the patient.
Abbreviations
AHNMD: Associated clonal hematologic non-mast cell lineage disease; BMT: Bone marrow trephine; CMML: Chronic Myelomonocytic Leukemia; PB: Peripheral blood; SM: Systemic Mastocytosis; SNP: Single nucleotide polymorphism; SNV: Single nucleotide variation.
Competing interests
The authors declare that they have no competing interest.
Authors’ contribution
MR is responsible for the NGS and the related verification analysis and wrote the manuscript. AB performed NGS and Sanger for NGS verification. QZ performed data analysis. RM performed FACS analysis. TR and DRZ did the microdissection and C-KIT analysis of all samples and contributed to the manuscript. JSG did the molecular BCR-ABL analysis and wrote the manuscript. MT did the initial diagnosis of SM-CMMLs, designed the study and wrote the manuscript. MGM, HM and PJW wrote and contributed to the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Target regions of the NimbleGen capture array and the AmpliSeq Comprehensive Cancer panel.
Variants in the coding region identified by 454 GS Junior and overlap with the variants identified by the AmpliSeq Comprehensive Cancer panel and Ion Proton sequencing.
BLM Sanger sequencing. The index patient, the cell line HEK293T, and the sub-cloned PCR product of BLM were Sanger sequenced with BLM primers. The same sub-cloned BLM PCR product was as well sequenced with M13 primers located in the cloning vector.
Variants detected by the AmpliSeq Comprehensive Cancer panel and Ion Proton sequencing.
Filtered somatic variants potentially involved in SM-CMML pathogenesis detected by the AmpliSeq Comprehensive Cancer panel and Ion Proton sequencing.
Contributor Information
Markus Rechsteiner, Email: Markus.Rechsteiner@usz.ch.
Rouven Müller, Email: Rouven.Mueller@usz.ch.
Tanja Reineke, Email: tanja.reineke@googlemail.com.
Jeroen Goede, Email: Jeroen.Goede@usz.ch.
Annette Bohnert, Email: Annette.Bohnert@usz.ch.
Qing Zhong, Email: Qing.Zhong@usz.ch.
Markus G Manz, Email: Markus.Manz@usz.ch.
Holger Moch, Email: Holger.Moch@usz.ch.
Peter J Wild, Email: Peter.Wild@usz.ch.
Dieter R Zimmermann, Email: Dieter.Zimmermann@usz.ch.
Marianne Tinguely, Email: Marianne.Tinguelykovarik@uzh.ch.
Acknowledgment
This work has been funded by the Novartis Foundation for Medicine and Biology (MT: No 04C58). We thank Dr. M. Umbricht, Sonja Brun-Schmid, Norbert Wey and the laboratory of in situ techniques, Institute of Surgical Pathology for their excellent support.
References
- Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editor. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC; 2008. [Google Scholar]
- Wang SA, Hutchinson L, Tang G, Chen SS, Miron PM, Huh YO, Jones DM, Bueso-Ramos C, Verstovsek S, Medeiros LJ, Miranda RN. Systemic mastocytosis with associated clonal hematological non-mast cell lineage disease: clinical significance and comparison of chomosomal abnormalities in SM and AHNMD components. Am J Hematol. 2013;88(3):219–224. doi: 10.1002/ajh.23380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschandl P, Mullauer L, Kittler H. Systemic mastocytosis associated with chronic myelomonocytic leukemia and xanthogranuloma. Dermatol Pract Concept. 2012;2(3):203a203. doi: 10.5826/dpc.0203a03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horny HP, Sotlar K, Sperr WR, Valent P. Systemic mastocytosis with associated clonal haematological non-mast cell lineage diseases: a histopathological challenge. J Clin Pathol. 2004;57(6):604–608. doi: 10.1136/jcp.2003.014860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullarkat VA, Bueso-Ramos C, Lai R, Kroft S, Wilson CS, Pullarkat ST, Bu X, Thein M, Lee M, Brynes RK. Systemic mastocytosis with associated clonal hematological non-mast-cell lineage disease: analysis of clinicopathologic features and activating c-kit mutations. Am J Hematol. 2003;73(1):12–17. doi: 10.1002/ajh.10322. [DOI] [PubMed] [Google Scholar]
- Stoecker MM, Wang E. Systemic mastocytosis with associated clonal hematologic nonmast cell lineage disease: a clinicopathologic review. Arch Pathol Lab Med. 2012;136(7):832–838. doi: 10.5858/arpa.2011-0325-RS. [DOI] [PubMed] [Google Scholar]
- Fritsche-Polanz R, Fritz M, Huber A, Sotlar K, Sperr WR, Mannhalter C, Fodinger M, Valent P. High frequency of concomitant mastocytosis in patients with acute myeloid leukemia exhibiting the transforming KIT mutation D816V. Mol Oncol. 2010;4(4):335–346. doi: 10.1016/j.molonc.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotlar K, Bache A, Stellmacher F, Bultmann B, Valent P, Horny HP. Systemic mastocytosis associated with chronic idiopathic myelofibrosis: a distinct subtype of systemic mastocytosis associated with a [corrected] clonal hematological non-mast [corrected] cell lineage disorder carrying the activating point mutations KITD816V and JAK2V617F. J Mol Diagn. 2008;10(1):58–66. doi: 10.2353/jmoldx.2008.070061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotlar K, Colak S, Bache A, Berezowska S, Krokowski M, Bultmann B, Valent P, Horny HP. Variable presence of KITD816V in clonal haematological non-mast cell lineage diseases associated with systemic mastocytosis (SM-AHNMD) J Pathol. 2010;220(5):586–595. doi: 10.1002/path.2677. [DOI] [PubMed] [Google Scholar]
- Rechsteiner M, von Teichman A, Ruschoff JH, Fankhauser N, Pestalozzi B, Schraml P, Weber A, Wild P, Zimmermann D, Moch H. KRAS, BRAF, and TP53 deep sequencing for colorectal carcinoma patient diagnostics. J Mol Diagn. 2013;15(3):299–311. doi: 10.1016/j.jmoldx.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Kim NG, Choi YR, Baek MJ, Kim YH, Kang H, Kim NK, Min JS, Kim H. Frameshift mutations at coding mononucleotide repeats of the hRAD50 gene in gastrointestinal carcinomas with microsatellite instability. Cancer Res. 2001;61(1):36–38. doi: 10.1186/bcr362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi S, Takeuchi N, Fermin AC, Taguchi H, Koeffler HP. Frameshift mutations in caspase-5 and other target genes in leukemia and lymphoma cell lines having microsatellite instability. Leuk Res. 2003;27(4):359–361. doi: 10.1016/S0145-2126(02)00215-1. [DOI] [PubMed] [Google Scholar]
- Boland JF, Chung CC, Roberson D, Mitchell J, Zhang X, Im KM, He J, Chanock SJ, Yeager M, Dean M. The new sequencer on the block: comparison of Life Technology's Proton sequencer to an Illumina HiSeq for whole-exome sequencing. Hum Genet. 2013;132(10):1153–1163. doi: 10.1007/s00439-013-1321-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaab J, Schnittger S, Sotlar K, Walz C, Fabarius A, Pfirrmann M, Kohlmann A, Grossmann V, Meggendorfer M, Horny HP, Valent P, Jawhar M, Teichmann M, Metzgeroth G, Erben P, Ernst T, Hochhaus A, Haferlach T, Hofmann WK, Cross NC, Reiter A. Comprehensive mutational profiling in advanced systemic mastocytosis. Blood. 2013;122(14):2460–2466. doi: 10.1182/blood-2013-04-496448. [DOI] [PubMed] [Google Scholar]
- Metzeler KH, Maharry K, Radmacher MD, Mrozek K, Margeson D, Becker H, Curfman J, Holland KB, Schwind S, Whitman SP, Wu YZ, Blum W, Powell BL, Carter TH, Wetzler M, Moore JO, Kolitz JE, Baer MR, Carroll AJ, Larson RA, Caligiuri MA, Marcucci G, Bloomfield CD. TET2 mutations improve the new European LeukemiaNet risk classification of acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2011;29(10):1373–1381. doi: 10.1200/JCO.2010.32.7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh CP, Wang CQ, Ng CE, Ito Y, Araki M, Tergaonkar V, Huang G, Osato M. RUNX1 meets MLL: epigenetic regulation of hematopoiesis by two leukemia genes. Leukemia. 2013;27(9):1793–1802. doi: 10.1038/leu.2013.200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Target regions of the NimbleGen capture array and the AmpliSeq Comprehensive Cancer panel.
Variants in the coding region identified by 454 GS Junior and overlap with the variants identified by the AmpliSeq Comprehensive Cancer panel and Ion Proton sequencing.
BLM Sanger sequencing. The index patient, the cell line HEK293T, and the sub-cloned PCR product of BLM were Sanger sequenced with BLM primers. The same sub-cloned BLM PCR product was as well sequenced with M13 primers located in the cloning vector.
Variants detected by the AmpliSeq Comprehensive Cancer panel and Ion Proton sequencing.
Filtered somatic variants potentially involved in SM-CMML pathogenesis detected by the AmpliSeq Comprehensive Cancer panel and Ion Proton sequencing.