

Abstract
Members of the multiple antibiotic resistance regulator (MarR) family often regulate gene activity by responding to a specific ligand. In the absence of ligand, most MarR proteins function as repressors, while ligand binding causes attenuated DNA binding and therefore increased gene expression. Previously, we have shown that urate is a ligand for MftR (major facilitator transport regulator), which is encoded by the soil bacterium Burkholderia thailandensis. We show here that both mftR and the divergently oriented gene mftP encoding a major facilitator transport protein are upregulated in the presence of urate. MftR binds two cognate sites in the mftR-mftP intergenic region with equivalent affinity and sensitivity to urate. Mutagenesis of four conserved residues previously reported to be involved in urate binding to Deinococcus radiodurans HucR and Rhizobium radiobacter PecS significantly reduced protein stability and DNA binding affinity but not ligand binding. These data suggest that residues equivalent to those implicated in ligand binding to HucR and PecS serve structural roles and that MftR relies on distinct residues for ligand binding. MftR exhibits a two-step melting transition suggesting independent unfolding of the dimerization and DNA-binding regions; urate binding or mutations in the predicted ligand-binding sites result in one-step unfolding transitions. We suggest that MftR binds the ligand in a cleft between the DNA-binding lobes and the dimer interface but that the mechanism of ligand-mediated attenuation of DNA binding differs from that proposed for other urate-responsive MarR homologues. Since DNA binding by MftR is attenuated at 37 °C, our data also suggest that MftR responds to both ligand and a thermal upshift by attenuated DNA binding and upregulation of the genes under its control.
Multiple antibiotic resistance regulator (MarR) proteins are important transcriptional regulators. More than 12,000 genes have been identified, which encode MarR family transcription factors, most of them in eubacteria.1 Many proteins of this family bind small molecule ligands such as household disinfectants, antibiotics, or organic solvents, and some are modified by reactive oxygen species (for a review, see refs (2−5)). For many MarR homologues, the natural ligand is unknown, which poses a challenge for understanding their mechanism of action.6,7 Most proteins of this family bind intergenic regions separating their own gene and a gene under their control, thereby repressing expression of both. In the presence of a small molecule ligand or specific cysteine oxidation, DNA binding is attenuated, which relieves repression.1,8−13 As sensors of changing environments, MarR proteins often regulate expression of genes involved in stress responses, virulence, and multidrug resistance.12−17
Gene regulation that depends on environmental cues is, for example, elicited when a bacterium infects a host. Part of the early response to a bacterial infection is for the host to produce reactive oxygen species (ROS) in defense against the invading bacterial pathogen. The primary sources of ROS generation are xanthine oxidase and NADPH oxidase. Xanthine oxidase converts hypoxanthine to xanthine and xanthine to urate, and it transfers electrons from these substrates to molecular oxygen to produce ROS.18−20 The urate that is produced during the generation of ROS is a potent antioxidant that may attenuate the adverse effects of ROS on the host.21,22
An important corollary of the simultaneous production of both ROS and urate is that the invading bacterium may detect and respond to both. Several bacterial transcription factors, including MarR homologues, have been characterized that respond directly to such host-derived ROS.11,13,23 That urate may function as an effector of gene activity has also been reported, and a subset of MarR family proteins have been identified that bind urate as their ligand. Members of this MarR subfamily, the urate responsive transcriptional regulators (UrtR), contain an N-terminal extension, which is not present in the prototypical MarR from Escherichia coli and other homologues.24 UrtRs also have four conserved residues, which have been shown to be important for urate binding and the attendant attenuation of DNA binding.24 Previously characterized UrtR proteins include PecS from Rhizobium radiobacter (Agrobacterium tumefaciens), which causes crown gall disease, and the soil bacterium Streptomyces coelicolor.(25,26) In both species, the pecS gene is divergently oriented from pecM, which encodes an efflux pump that belongs to the Drug Metabolite Transporter superfamily. In presence of the ligand urate, DNA binding by PecS is attenuated and genes encoding PecS and PecM are upregulated.25,26 These PecS proteins are homologous to the previously characterized PecS from the phytopathogenic bacterium Erwinia chrysanthemi (Dickeya dadantii), where it regulates the expression of several virulence genes, including genes encoding enzymes involved in the biosynthesis of the secondary metabolite indigoidine as well as PecM, the efflux pump through which the antioxidant indigoidine is extruded.16,27,28 At the time of infection, this global regulator regulates multiple genes involved in virulence and disease progression.16,29
Recently, we reported a distinct urate-responsive MarR homologue, major facilitator transport regulator (MftR), which is encoded by Burkholderia thailandensis.(30) The gene encoding MftR is not divergently oriented from a pecM gene but instead a gene that encodes the efflux pump MFTP (major facilitator transport protein). MFTP is an MFS (Major Facilitator Superfamily) homologue, and it has similarity to EmrD, a drug efflux pump from E. coli. MFS efflux pumps are abundant.31 They can export a wide variety of cytotoxic molecules, which contributes to multidrug resistance.32 MftR binding to the mftR-mftP intergenic region revealed two cognate sites, each consisting of 9 bp imperfect inverted repeats. Among the intermediates of purine catabolism, urate efficiently attenuated DNA binding. However, unlike previously characterized homologues, xanthine and hypoxanthine also antagonized DNA binding, suggesting relaxed ligand specificity.25,30,33
Here, we report site-directed mutagenesis of MftR, which reveals a differential mode of ligand binding compared to that of previously characterized UrtR homologues. MftR binds two cognate sites in the mftR-mftP intergenic region with comparable affinity and sensitivity to urate, and in the presence of exogenous urate, divergently oriented genes mftR and mftP are upregulated. MftR exhibits a two-step melting transition and binds DNA with lower affinity at 37 °C. We propose that DNA binding by MftR is attenuated upon host infection by both ligand binding and thermal destabilization.
Experimental Procedures
Sequence Alignment and Phylogenetic Analysis
MarR homologues were aligned using the MUSCLE sequence alignment server.34 Amino acid residues were shaded using BOXSHADE v3.21. Secondary structure elements were predicted based on the structure of D. radiodurans HucR.35 In MEGA4, the neighbor-joining method with 500 bootstrap replicates was used to generate the phylogenetic tree.36 The tree was drawn to scale, and positions containing gaps were removed. The evolutionary distances are in the unit of number of amino acid substitutions per site.
Generation of Mutant MftR and Protein Purification
MftR was cloned and purified as described previously.30 In brief, genomic DNA was extracted from B. thailandensis E264 and used as the template to amplify mftR (BTH_I2391). The PCR product was digested with NdeI and EcoRI and cloned into pET28b for expression of protein with an N-terminal His6-tag. Recombinant plasmid was used to create mutants. To create the W11F, D56S, R63S, and R89N substitutions in MftR, an overhanging primers technique was used for whole plasmid amplification (for primer sequences, see Supporting Information, Table S1).37 Parental recombinant plasmid was digested using DpnI, and mutant plasmid was gel purified using a gel and PCR clean up kit (Promega). The plasmid was transformed into E. coli TOP10 (Invitrogen), and the mutated plasmid was verified by sequencing. Mutant proteins were expressed as described for wild-type MftR: plasmids were transformed into E. coli BL21(DE3)pLysS. A single colony was used to grow an overnight culture in LB with 50 μg/mL kanamycin at 37 °C; the culture was then diluted 1:100 and protein expression induced by the addition of 1 mM isopropyl-1-thio-β-d-galactopyranoside (IPTG) at OD600 of 0.4–0.5. After 1 h, cells were chilled on ice, pelleted, and stored at −80 °C.
Cell pellets were thawed on ice for 1 h and cells resuspended in chilled wash buffer containing 50 mM sodium phosphate buffer (pH 7.0) and 300 mM NaCl. Before preparing lysate by centrifugation at 10 000g for 60 min, lysozyme (1.0 mg/mL), 10× DNase I buffer, and 2 μL of DNase I were added to each 5 mL cell suspension and incubated for 1 h. The supernatant was collected and mixed with HIS-Select Nickel Affinity beads previously washed with 10 volumes of chilled double distilled water and two times with wash buffer. After 1 h of incubation with beads at 4 °C, the mixture was directly transferred to a gravity flow column, and protein was eluted by increasing the concentrations of imidazole from 10 mM to 150 mM. Peak fractions, which contained pure protein, were pooled. Proteins were concentrated, and buffer was exchanged to wash buffer with 10% glycerol by using an Amicon centrifugal filter device (Millipore). The purity of proteins (WT and mutants) was verified by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and staining the gel with Coomassie brilliant-blue. Concentration was calculated using the BCA protein assay kit (Pierce).
To determine oligomeric states, proteins were cross-linked in a total volume of 10 μL with 0.5% (v/v) glutaraldehyde on ice for 30 min. An equal volume of Laemmli sample buffer was added to terminate the reaction, and the cross-linked proteins were subjected to SDS–PAGE.
Circular Dichroism Spectroscopy
A Jasco J-815 circular dichroism spectrophotometer (Jasco, Inc.) was used to measure far UV circular dichroism spectrum. To measure ellipticity, 0.2 mg/mL MftR and its variants were in CD buffer (50 mM sodium phosphate buffer (pH 7.0), 100 mM NaCl, and 2.5% glycerol), and a quartz cuvette with 0.1 cm path length was used. All protein samples were equilibrated at room temperature for 20 min (except the W11F mutant, which was kept on ice for equilibration). Measurements were conducted in triplicate with 1 nm steps. Predicted secondary structure was calculated using the K2D program from DichroWeb.38−40 The goodness of fit was determined from the NRMSD value, which was in the range of 0.094 to 0.110.
For melting profiles, samples were diluted to 0.8 mg/mL in CD buffer. Samples were scanned from 225 to 219 nm over the temperature range of 20–65 °C with 1 °C increments. Each sample was also reverse scanned (for W11F, the temperature range was 18–65 °C). Thermal equilibration time after each temperature step was 15 s. To verify the native state, samples were also scanned at 240–200 nm at 25 °C (W11F was scanned at 18 °C). The thermal denaturation curve for each wavelength was fitted using the four-parameter sigmoidal equation of Sigma Plot 9.
Fluorescence Spectroscopy and Fluorescence Quenching
A PTI QuantaMaster4/2006SE spectrofluorometer was used to measure the fluorescence spectra from 310 to 440 nm with excitation at 295 nm at 25 °C using a 0.3 cm path length cuvette. For the measurement, WT and mutants were resuspended in FL buffer (40 mM Tris-HCl (pH 8.0), 0.2 mM EDTA, 0.1% (w/v) Brij58, 100 mM NaCl, and 10 mM MgCl2) to a final concentration of 0.03 mg/mL. Reactions were incubated for 2 min before measuring the fluorescence. To measure the effect of urate, urate was dissolved in 0.4 M NaOH and serially diluted with 0.4 M NaOH. To correct for the inner filter effect and for the normalization of data, the absorbance of FL buffer, free ligand in FL buffer, and reaction mixture were measured. Correction of observed fluorescence and fluorescence quenching calculation and fitting to Hill equation were carried out as described previously.41
In Vivo Determination of mRNA Levels Using qRT-PCR
The B. thailandensis culture was grown overnight at 37 °C in LB media. The overnight culture was diluted 1:100 with LB media containing 10 mM urate. Urate was dissolved in 0.4 N NaOH and sterilized by passing through a 0.2 μM nylon syringe filter. The control culture was grown in LB to which an equal volume of 0.4 N NaOH was added. Both cultures were grown for 6 h before cells were collected using centrifugation. Cells were immediately suspended in chilled DEPC treated water and then collected by centrifugation. Total RNA was isolated using the hot phenol method with slight modifications.42 Instead of using phenol and chloroform in two steps, acid saturated phenol/chloroform (Ambion) was used twice. RNA quality was measured using Nanodrop 2000c (Thermo Scientific). cDNA generated using AMV reverse transcriptase (New England BioLabs) mixed with RNA, 25 mM dNTP, and 25 mM MgCl2 and kept for 1 h at 42 °C was used in quantitative PCR (qPCR). qPCR was carried out using an Applied BioSystems 7500 real time PCR system (Life Technologies). Primers used are shown in Supporting Information, Table S1. As a control, gapdh was used. SYBR Green I (Sigma) was used for detection. qRT-PCR generated data was analyzed using the comparative CT method (2–ΔΔCT).43
Thermal Stability Assay
Fluorescent dye SYPRO Orange (Invitrogen) (5×) was added to thermal stability buffer (200 μM Tris (pH 8.0) and 200 mM NaCl) with MftR (6 μM). DNA containing either of the two palindromes in the mftRO and mftpO intergenic region (36 bp) was prepared by annealing complementary oligonucleotides by heating to 95 °C followed by slow cooling to room temperature in TE′ (10 mM Tris, pH 8.0, 0.1 mM EDTA) with 100 mM NaCl. Oligonucleotides were purified using 12% polyacrylamide denaturing gels. DNA was added to 1:1 stoichiometry with protein. To measure the effect of ligand, urate (dissolved in 0.4 M NaOH) was added to protein in a ratio of 1:1 and at ∼16-fold excess. Mutant proteins (W11F, D56S, R63S, and R89N) were analyzed in the same way as wild-type MftR. Control samples excluded protein. An Applied BioSystems 7500 real time PCR system was used with increasing temperature from 5 °C to 94 °C in 1 degree increments, and fluorescence emission was corrected using the control sample without protein. The four-parameter sigmoidal equation of Sigma Plot 9 was used to fit the sigmoidal part of the melting curve. Data represent the mean of three replicates.
DNA Binding Assays
DNA binding was determined using electrophoretic mobility shift assays (EMSA). The 130 bp intergenic region (mftO) between mftR (BTH_I2391) and mftP (BTH_I2392) was amplified as previously described.30 DNA was radiolabeled using γ-32P-ATP and T4 polynucleotide kinase. Protein and DNA were mixed in binding buffer (0.5 M Tris-HCl pH 8.0, 250 mM NaCl, 0.1 M EDTA, 0.1 mM dithiothreitol, 0.05% Brij58, 10 μg/mL BSA, and 5% glycerol) and incubated at room temperature for 30 min. The high concentration of Tris was used to prevent pH changes upon subsequent addition of urate dissolved in 0.4 M NaOH. This mixture was loaded on a running 10% polyacrylamide gel (39:1 acrylamide/bis(acrylamide)), previously prerun for 30 min in 0.5 X Tris borate EDTA (TBE) at room temperature. After electrophoresis, gels were dried and exposed to phosphorimaging screens. Data were visualized using a storm 840 phosphorimager (GE Healthcare) and quantified with ImageQuant 5.1. Fractional complex formation was analyzed using KaleidaGraph 4.0 (Synergy Software) by fitting to f = fmax·[X]nH/(Kd + [X]nH) (where nH is the Hill coefficient, Kd is the apparent equilibrium dissociation constant reflecting half-maximal saturation of the DNA (not the microscopic dissociation constant), and [X] is the protein concentration). For DNA with a single site, this equation simplifies to a single-site binding isotherm (nH = 1). For the W11F mutant, EMSA were performed at 4 °C, and the effect of temperature on DNA binding by WT MftR was assessed by EMSA performed at 37 °C (incubation of binding reaction as well as electrophoresis). EMSA with HucR was performed as described by Perera et al.33
To determine the effect of urate, increasing concentrations of urate were added to the reaction mixtures. Since urate was dissolved in 0.4 M NaOH, equal volumes of 0.4 M NaOH were added to every reaction. After 30 min of incubation, samples were electrophoresed and data retrieved as described above. Fractional complex formation was fitted to f = A + Be–kL (where f is fraction saturation, k is decay constant, L is the ligand concentration, A is the saturation plateau, and B represents the decay amplitude). IC50 was calculated as the ligand concentration at which 50% of complex formation is inhibited. While the first-order decay equation does not consider the molecular events associated with MftR binding to DNA containing two DNA sites, it is suitable for comparing the ligand sensitivity of MftR variants. Densitometric data were derived from three independent experiments.
Results
Genomic Locus and Secondary Structure of MftR
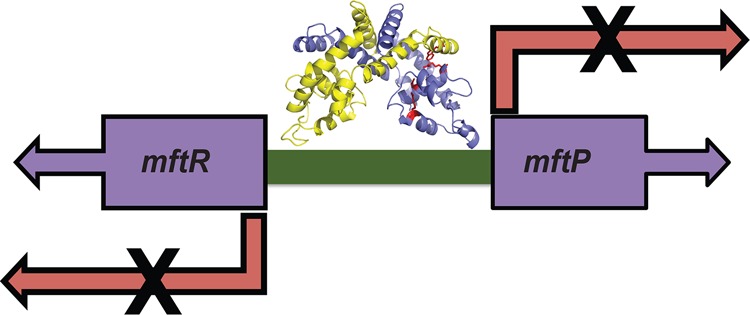
The B. thailandensis gene BTH_I2391 encodes a predicted MarR homologue that is divergently oriented from gene BTH_I2392 annotated as a major facilitator transport protein (MFTP) (Figure 1). mftP is separated from mftR by an intergenic region of 114 bp. The mftR-mftP locus is conserved among Burkholderia species, for example, in B. mallei and B. pseudomallei, where the intergenic region, mftO, also shares two similar palindromic sequences. B. mallei and B. pseudomallei cause glanders and melioidosis, respectively. Being highly infectious and causing infections that are difficult to treat, they are considered as category B priority pathogens.44
Figure 1.

B. thailandensismftR-mftP intergenic region. Genes are represented by arrows. The sequence of the intergenic region, mftO, is shown with two imperfect palindromes shown in bold face. mftrO represents a DNA construct with the palindrome near mftR, and mftpO is the region upstream of mftP.
As noted above, alignment of UrtR homologues previously revealed the conservation of four residues involved in urate binding (shown with arrows in Figure 2A). The N-terminal extension, α1, is a signature of urate-responsive MarR homologues, and it is absent from canonical MarR homologues such as E. coli MarR and MTH313.7,35,45 In the Deinococcus-radiodurans-encoded UrtR homologue HucR, this extension was shown to adopt an α-helix that braces the helices that form the dimer interface.35 UrtR homologues conserve sequence in helices α3 and α5; α3 contains residues involved in ligand binding by HucR and PecS, and α5 is the DNA recognition helix. Conservation of the recognition helices is also reflected in the conservation of cognate DNA sites among UrtR homologues.24
Figure 2.

Sequence alignment of MarR homologues. (A) Alignment generated using MUSCLE. Residues involved in urate binding or in communicating its binding to the recognition helix are shown using arrows. The alignment includes MTH313 (Methanobacterium thermoautotrophicum), D. radiodurans HucR, S. coelicolor PecS, D. dadantii PecS, Pectobacterium atrosepticum PecS, R. radiobacter PecS, MftR from B. thailandensis, B. mallei (BMA_0906), and B. pseudomallei (BURPS_1958). Secondary structure elements are based on the structure of HucR. (B) Phylogenetic tree of selected MarR homologues based on their amino acid sequence. Tree includes MarR homologues from panel A and B. oklahomensis and Rhizobium mesoamericanum PecS. The evolutionary distances are in units of the number of amino acid substitutions per position, and the scale bar represents an evolutionary distance of 0.1.
A phylogenetic tree was created to analyze the evolutionary relationship between MftR from Burkholderia spp. and other MarR homologues, particularly PecS homologues, which also belong to the UrtR family (Figure 2B). Phylogenetic analysis revealed that MftR homologues are clustered together and that urate responsive MarR homologues (D. dadantii PecS, S. coelicolor PecS, and D. radiodurans HucR) are more closely related, while E. coli MarR and MTH313 are distantly related. That MftR homologues from Burkholderia cluster together suggests common ancestry.
Among MarR homologues for which structures are known, HucR has ∼39% identity and ∼51% similarity with MftR and was used as the template to generate a model of MftR (Figure 3). MftR contains the N-terminal extension that forms an α-helix in HucR. Four residues found to bind urate in HucR (W11, D56, R63, and R89) and communicating ligand binding to the DNA recognition helices are predicted to occupy the same position in the model of MftR (shown in red in Figure 3). MftR was purified to apparent homogeneity (Supporting Information, Figure S1); far UV circular dichroism spectroscopy showed that the secondary structure composition of MftR is about 57% α-helix and 8% β-sheet (Supporting Information, Figure S2A). This is comparable to the HucR secondary structure composition of 55% α-helix and 5% β-sheet.35
Figure 3.
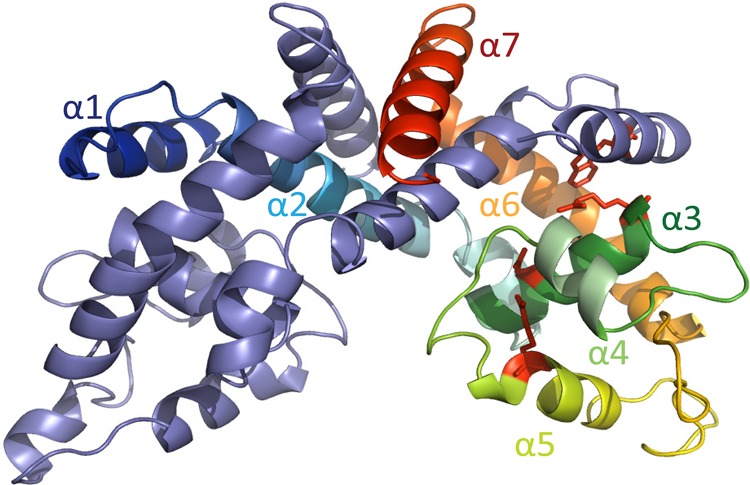
Predicted model of MftR. MftR model based on the structure of HucR (2fbk), created using SwissModel in automated mode. One monomer is colored blue to red (amino-terminus to carboxy-terminus; helices are shown as α1 to α7) and the other is in purple. Conserved residues, which are predicted to bind urate, are in red stick representation.
MftR Binding to mftO Regulates Expression of Divergently Oriented mftR-mftP Genes
MftR was previously shown to bind two sites in the mftR-mftP intergenic region (mftO; Figure 1) with modest negative cooperativity, and the ligand urate was shown to attenuate DNA binding.30 Such binding mode predicts repression of mftR and mftP gene expression in the absence of ligand. Therefore, we determined mRNA levels of mftR and mftP in vivo in the presence of urate. Growing cells at 37 °C resulted in measurable levels of transcript (Figure 4A). However, growth with 10 mM urate resulted in elevated expression of mftR (5.1 ± 0.7-fold) and mftP (13.7 ± 3.5-fold) (Figure 4). This suggests that MftR binding to mftO represses the transcription of mftR and mftP in vivo and that accumulation of urate leads to increased gene activity.
Figure 4.

In vivo transcript level of mftR and mftP. (A) PCR product (mftP) obtained from cDNA generated with increasing concentration of total RNA extracted from cells not exposed to exogenous urate. Lane M is the 100 bp marker, and lanes 1 to 5 show the PCR product obtained with increasing concentrations of RNA (25–100 ng/μL). (B) PCR product (mftP) with RNA extracted from cells grown in the presence of 10 mM urate. Lane M is the 100 bp marker and lanes 1 to 5 show the PCR product obtained with increasing concentrations of RNA (25–100 ng/μL). (C) Relative abundance of mftR and mftP transcript levels after the addition of 10 mM urate. Relative abundance of transcript level was calculated with the comparative CT method, with the reference control gene gapdh. Error bars represent the standard deviation of three experiments.
The intergenic region mftO contains two imperfect palindromes (Figure 1). To assess if differential gene expression might be due to differential MftR binding to these sites, DNA constructs containing either of the two identified palindromes were designed and named mftrO and mftpO (Figure 1) and used in electrophoretic mobility shift assays. MftR formed a stable complex with both mftrO and mftpO (Figure 5A and data not shown) as evidenced by an apparent dissociation constant (Kd) of 0.7 ± 0.1 nM and 0.6 ± 0.1 nM, respectively (Figure 5B). With increasing concentration of urate, the binding of MftR to mftrO and mftpO was attenuated (with an IC50 of 3.7 ± 0.3 mM and 2.2 ± 0.0 mM) (Figure 5C–D and data not shown). Evidently, MftR binds comparably to mftrO and mftpO and with equivalent sensitivity to ligand. The observed differential gene regulation in vivo is therefore not likely to derive from differences in MftR binding to its cognate sites.
Figure 5.

MftR binds both palindromes in its operator DNA, and the complexes are sensitive to urate. (A) EMSA showing mftpO (3.0 nM) titrated with increasing concentrations of MftR (0.1–200 nM; lanes 2–15); reaction in lane 1 contains DNA only. Complex and free DNA are identified at the right as C and F, respectively. (B) Fractional complex formation plotted as a function of MftR concentration. Binding isotherm with mftrO (○; solid line) and mftpO (+; dashed line). (C) Effect of urate on the binding of MftR to mftpO. Lane 1 contains DNA only. Reaction in lane 2 contains no ligand. The MftR-mftpO complex was titrated with increasing concentrations of urate (3–18 mM; lanes 3–11). (D) Normalized complex fraction as a function of urate concentration. MftR-mftrO complex (○; solid line) and MftR-mftpO complex (+; dashed line) titrated with increasing concentrations of urate. Error bars represent the standard deviation of three independent repeats.
Ligand Binding by MftR Impacts Two Parts of the Protein
In contrast to HucR and PecS proteins that respond primarily to the ligand urate by attenuated DNA binding, DNA binding by B. thailandensis MftR is also appreciably reduced by other intermediates in the purine degradation pathway, specifically xanthine and hypoxanthine, indicating relaxed ligand specificity.25,30,33 To determine the effect of the four residues previously implicated in urate binding to HucR and PecS, the equivalent residues in MftR were therefore mutated to generate W11F, D56S, R63S, and R89N MftR variants. For both HucR and PecS, substitution of residues corresponding to MftR residues W11, D56, and R63 generally ablates the response to urate, while mutating the equivalent of R89 in the DNA recognition helix severely compromises DNA binding.25,33
All MftR protein variants were purified to apparent homogeneity (Supporting Information, Figure S1A). All structures of MarR proteins, including that of HucR, reveal highly intertwined monomers,7,35 suggesting that any mutations that preclude dimerization would also exhibit significantly altered (or abolished) secondary structure. Glutaraldehyde cross-linking revealed equivalent formation of dimeric species for all protein variants, suggesting that mutations did not interfere with dimerization (Supporting Information, Figure S1B). The far UV CD spectra of MftR variants indicated similar secondary structure composition when compared to those of MftR and HucR (Supporting Information, Figure S2), indicating that mutations did not significantly affect the overall protein fold.
Since these substitutions have the potential to affect protein stability, the thermal stability of WT and mutants was determined using differential scanning fluorometry; SYPRO Orange was used as a fluorescent reporter of protein unfolding as a function of temperature. WT MftR exhibited a two-step melting transition, which is unusual for MarR homologues (Figure 6A). Domain 1 (D1) has a significantly lower melting temperature (Tm = 49.4 °C) than domain 2 (D2; Tm = 57.8 °C) (Table 1 and Figure 6A), perhaps corresponding to independent unfolding transitions for the DNA binding lobes and the dimerization region. By comparison, D1 has a melting temperature similar to that of HucR (51.1 °C) and S. coelicolor PecS (47.3 °C), and D2 has a melting temperature similar to that of R. radiobacter PecS (61.3 °C) and S. coelicolor TamR (59.9 °C).10,25,26,46 D56S (Tm = 50.5 °C) and R89N (Tm = 53.0 °C) substitutions destabilized MftR less than R63S (Tm = 43.0 °C), while the W11F mutant was severely destabilized (Tm = 24.7 °C; Table 1 and Figure 6B–C). All mutants showed one-step melting transitions.
Figure 6.

Melting temperature of MftR and mutant proteins determined by differential scanning fluorometry. Thermal denaturation is represented by the fluorescence intensity resulting from the binding of SYPRO Orange to denatured protein as a function of temperature. (A) Thermal denaturation profile of WT MftR. (B) Thermal denaturation profile of D56S, R63S, and R89N. (C) Melting profile of W11F; a higher initial fluorescence may reflect the presence of already denatured protein. (D) Melting profile of WT with the addition of 100 μM urate. (E) Denaturation profile of D56S, R63S, and R89N with urate. (F) Denaturation profile of W11F with urate. (G) Thermal denaturation profile of MftR with 6 μM mftpO. (H) Denaturation profile of mutant D56S, R63S, and R89N with mftpO. (I) Symbols used in panels A–H for MftR and mutants.
Table 1. Thermal Stability of MftR and Mutants.
| 6 μM urate | 100 μM urate | mftrO | mftpO | ||
|---|---|---|---|---|---|
| MftR D1 | 49.4 ± 0.3 | 49.2 ± 0.2 | 61.3 ± 1.0 | 53.0 ± 0.2 | 52.7 ± 0.2 |
| MftR D2 | 57.9 ± 0.1 | 58.0 ± 0.1 | 60.3 ± 0.1 | 60.9 ± 0.3 | |
| W11F | 24.7 ± 0.1 | 27.0 ± 0.1 | 28.8 ± 0.7 | a | a |
| D56S | 50.5 ± 0.2 | 52.3 ± 0.3 | 51.3 ± 0.1 | 48.4 ± 0.1 | 48.0 ± 0.1 |
| R63S | 43.0 ± 0.1 | 48.7 ± 0.4 | 64.1 ± 3.9 | 42.8 ± 0.2 | 43.0 ± 0.2 |
| R89N | 53.0 ± 0.2 | 55.9 ± 0.2 | 63.0 ± 0.6 | 54.0 ± 0.1 | 55.6 ± 0.9 |
Did not yield quantifiable data.
All MftR mutants were destabilized compared to WT MftR. However, the magnitude of the fluorescence was variable, with the D56S mutant protein in particular showing very low levels of fluorescence. We therefore repeated the determination of thermal stability using CD spectroscopy. These experiments verified that all MftR mutations resulted in reduced thermal stability, and they showed that the calculated Tm values are comparable to those measured using SYPRO Orange (Supporting Information, Figure S2 and Table S2; all values obtained by CD spectroscopy are ∼2 °C higher due to the different compositions of the CD buffer).
These experiments also revealed that thermal melting is irreversible, likely due to protein aggregation, as evidenced by the formation of a white precipitate upon denaturation. This precludes a thermodynamic analysis of the unfolding transition, and only the Tm is reported. Even though the unfolding was irreversible due to aggregation, information about relative stability may still be obtained. Irreversible thermal denaturation is also a characteristic of HucR.46 For the R63S and D56S mutants that lead to lower SYPRO Orange fluorescence upon denaturation, we surmise that these protein variants aggregated during (as opposed to after) the formation of the fully denatured state, thus resulting in lower fluorescence yields. Such reduced fluorescence is commonly seen following protein denaturation, as such aggregates exclude the bound dye.47 The observed reduction in dye binding to the R63S and D56S variants clearly shows that the thermally induced transitions are inherently different compared to those of wild-type MftR. We also note that the denaturation of MftR domain D1, which leads to a significant increase in SYPRO Orange fluorescence (Figure 6A), was associated with only a modest change in ellipticity (Supporting Information, Figure S2A). Taken together, these experiments show that all substitutions destabilize either one or both MftR domains D1 and D2 and/or result in accelerated protein aggregation.
Combining protein and urate at a stoichiometric ratio of 1:1 did not significantly change protein stability (using 6 μM urate and protein; Table 1), whereas a higher concentration of urate (100 μM) resulted in increased thermal stability, suggesting ligand binding to both WT and mutant proteins. A one-step unfolding transition was observed for WT, with a Tm of 61.3 °C, suggesting predominant stabilization of domain D1 (Figure 6D). Whereas only a marginal increase in the Tm of D56S was observed on urate binding, W11F had a Tm of 28.8 °C, an increase of ∼4 °C compared to that of the protein alone, while R63S and R89N were significantly stabilized (Tm of 64.1 and 62.9 °C, respectively; Figure 6E).
Changes in thermal stability of MftR and mutant proteins were equivalent when measuring Tm for stoichiometric ratios of protein and DNA representing either cognate site (mftrO and mftpO) (Figure 6G–H, Table 1). WT MftR in complex with either mftrO or mftpO still exhibited a two-step unfolding transition, and both D1 (Tm = 52.7 °C) and D2 (Tm = 60.9 °C) were modestly stabilized as compared to WT MftR alone. W11F when mixed with mftRO and mftpO showed aggregation. In contrast, D56S was modestly destabilized on DNA binding (Tm ∼48 °C), while the Tm for R63S and R89N was not significantly altered.
Mutations Reduce Affinity for mftO
To determine whether the mutations affect DNA binding, EMSAs were performed with MftO, which contains both cognate sites. When experiments were performed at room temperature, no DNA binding was observed for W11F, likely due to its thermal instability. EMSAs with this mutant were therefore performed at 4 °C. All mutants except R89N bound DNA, forming two clearly distinguishable complexes (C1 and C2; Supporting Information, Figure S3). The failure of R89N to bind DNA is consistent with the effect of the equivalent mutation in HucR and PecS (for Kd values measured for HucR and PecS variants harboring the equivalent substitutions, see Supporting Information, Table S3).25,33 All other mutations significantly reduced DNA binding affinity, with W11F yielding a Kd that is 4 times higher, D56S a Kd that is approximately 140 times higher, and R63S a Kd approximately 10-fold higher than that of WT MftR (Table 2 and Supporting Information, Figure S3). The observation that two complexes are formed with MftO that contains two MftR sites (yet would be long enough for nonspecific binding of additional proteins) suggests retention of specificity. That all mutant proteins exhibit a comparable modest negative cooperativity is also consistent with a common mode of binding.
Table 2. Binding Affinity and Inhibition Constant of MftR and Variants.
| Kd (nM) | nH | IC50 (mM) | |
|---|---|---|---|
| MftR | 1.6 ± 0.1 | 0.8 ± 0.1 | 3.0 ± 0.1 |
| W11F | 6.3 ± 0.5 | 0.6 ± 0.2 | 5.9 ± 0.8 |
| D56S | 220.0 ± 1.8 | 0.7 ± 0.1 | 3.1 ± 0.1 |
| R63S | 15.1 ± 1.3 | 0.9 ± 0.1 | 8.0 ± 0.2 |
Considering the gradual increase in fluorescence, reflecting the thermal instability of WT MftR above 30 °C, DNA binding was examined at 37 °C. At this temperature, which would correspond to the body temperature of a mammalian host, a 4-fold higher Kd (6.9 ± 1.9 nM) was observed compared to that at room temperature (Supporting Information, Figure S3F). To evaluate if such behavior is specific to MftR, we also performed this experiment with HucR. No difference was observed in Kd as a function of temperature (1.0 ± 0.2 nM at room temperature and 1.2 ± 0.2 nM at 37 °C; Supporting Information, Figure S4).
Taken together, these data show that the effect on DNA binding of MftR mutations is different from that of the equivalent mutations in HucR or PecS. For HucR, only substitution of Arg in the DNA recognition helix has a significant effect on DNA binding, causing the mutant protein to bind DNA nonspecifically and with low affinity, while the other three substitutions have little effect on DNA binding affinity (Supporting Information, Table S3).33 Similarly, substitution of Arg in the recognition helix of PecS abolishes DNA binding.25 While substitution of MftR residue R63 resulted in an ∼10-fold decrease in DNA binding affinity, only a marginal (∼2-fold) decrease in binding affinity is observed when the equivalent mutation is made in PecS. Trp in α1 of PecS is important for folding, as evidenced by its substitution causing protein aggregation, but substitution of Asp results in only a modest decrease in DNA-binding affinity, in contrast to the ∼140-fold decrease observed for MftR-D56S. In addition, our data indicate that DNA binding is reduced at the physiological temperature associated with host infection; in contrast, binding of HucR to its cognate site is not affected by an increase in temperature to 37 °C.
For both HucR and PecS, mutagenesis of individual residues in the ligand-binding pocket (corresponding to MftR substitutions W11F, D56S, and R63S) largely abolishes the response to urate.25,33 The effect of urate on DNA binding by the equivalent MftR mutants was therefore measured. With increasing concentration of urate, the binding of each mutant to mftO was attenuated (Supporting Information, Figure S5). IC50 was equivalent for WT and D56S (Table 3) with W11F and R63S exhibiting an IC50 only 2 and 2.5 times higher than that of WT MftR (Table 3). The ability of urate to attenuate DNA binding by mutant proteins is consistent with the observed increase in thermal stability on ligand binding. Evidently, the association of ligand with MftR is different from that observed for HucR and PecS and is consistent with the previously reported ability of related ligands to disrupt DNA binding by MftR.30
Table 3. Fluorescence Quenching of MftR and Mutants with the Addition of Urate.
| K (μM) | nH | |
|---|---|---|
| MftR | 6.1 ± 2.1 | 0.8 ± 0.1 |
| W11F | 2.4 ± 2.0 | 0.7 ± 0.2 |
| D56S | 8.4 ± 2.5 | 0.8 ± 0.2 |
| R63S | 6.6 ± 0.5 | 2.9 ± 1.3 |
| R89N | 4.8 ± 2.0 | 1.0 ± 0.2 |
The binding of urate to MftR variants was further investigated by measurement of intrinsic tryptophan fluorescence. The fluorescence intensities of WT and mutants were variable in the region of 310 to 440 nm (Figure 7A). For W11F, the fluorescence was almost negligible, which suggests that it is the primary source of fluorescence in WT MftR, which has an additional tryptophan (W98). W98 is predicted to be located in the loop near the DNA recognition helix; by comparison, HucR has an additional tryptophan in α3 that is quenched by a nearby Tyr.35 Presumably, the fluorescence of W98 is likewise quenched.33 The other mutants also exhibited lower fluorescence intensity at 325 nm compared to that of WT. Mutant W11F and D56S displayed maximum fluorescence at 327 nm, while R63S and R89N had fluorescence maxima at 329 nm. These data indicate a change in the environment of tryptophan in the mutant proteins.
Figure 7.

Fluorescence of MftR and mutant proteins and response to urate. (A) Fluorescence profiles of MftR mutants. Mutation of W11 to F (solid black dashed line) causes a significant loss in the intrinsic fluorescence. WT MftR (●), D56S (×), R63S (solid gray line), and R89N (◊). (B) Fluorescence quenching upon urate addition. WT MftR profile is shown as a solid black line and open square with ×. W11F (+) profile with dashed line, and D56S (⧫) with gray lines.
Titration of protein variants with increasing concentration of urate resulted in a concentration-dependent fluorescence quenching at 325 nm (Figure 7B and Table 3). None of the individual mutations significantly altered the affinity for urate, which is consistent with the ability of urate to attenuate DNA complex formation. The ∼6 μM affinity for urate also rationalizes the modest effect of 6 μM urate in modulating protein stability, as only ∼50% of MftR would have urate bound at this concentration of ligand. It may also indicate that occupancy of both ligand-binding sites is required for changes in protein stability to be manifest.
Discussion
Differential Upregulation of mftR and mftP in the Presence of Urate
When B. thailandensis cultures were grown in the presence of urate, mftR and mftP genes were upregulated ∼5- and 14-fold, respectively (Figure 4). The significant attenuation of MftR binding to the mftR-mftP intergenic region in vitro on binding to urate supports the conclusion that MftR controls the expression of these genes in vivo (Supporting Information, Figure S5). Considering that MftR has equivalent affinity for each of the identified palindromes and that urate is equally potent as an antagonist of DNA binding to each site (Figure 5), we infer that the differential upregulation of mftR and mftP is not due to preferential association of MftR with either cognate site. Instead, different promoter strengths of the divergent promoters may be responsible for the observed differences in gene expression. We also cannot rule out the possibility that other transcription factors contribute to the observed regulation in vivo.
Domain Organization of MftR
The structures of MarR proteins reveal a highly conserved fold in which three helices from each monomer (corresponding to HucR helices α2, α6, and α7) form a tightly intertwined dimer interface, with the central helices α3, α4, and α5 forming the DNA binding domain (Figures 2–3).6,7,15,35,45 The sequence conservation and secondary structure composition of MftR is consistent with conservation of this overall fold (Supporting Information, Figure 2A). The two-step unfolding transition observed for MftR therefore most likely reflects independent unfolding transitions for these two regions of the protein. A three-state unfolding of the MftR dimer may arise from native dimer N2 undergoing a transition to a partly unfolded dimeric intermediate I2, followed by the formation of unfolded monomers U (N2 ↔ I2 ↔ 2U). Alternatively, dissociation of monomers may precede unfolding (N2 ↔ 2I ↔ 2U).
The MftR residues W11, D56, R63, and R89 are predicted to occupy equivalent positions in HucR and MftR. In the structure of HucR, residues corresponding to R89 (α5) and D56 (α3) form a salt bridge that participates in anchoring the DNA recognition helix α5 (dark green in Figure 3) to α3 (yellow; Figure 3). The failure of R89N to yield a detectable complex with DNA in EMSA is consistent with this interpretation and with the observation that the equivalent substitution in HucR and PecS likewise results in severely compromised DNA binding.25,33 D56S also binds DNA with significantly reduced affinity, suggesting that the interaction of D56 with R89 is important for proper disposition of the DNA recognition helix. The observed modest destabilization of D56S on DNA binding is consistent with its lower DNA binding affinity, which may imply the need for conformational changes that disrupt stabilizing contacts in order to position DNA recognition helices for optimal DNA interaction.
On the basis of these considerations, the 51–53 °C melting temperature of the D56S and R89N mutants is predicted to reflect disruption of a salt bridge and therefore a destabilization of the DNA binding lobe, leading to the inference that domain D1 (Tm ∼ 49 °C) in WT MftR corresponds to the dimer interface, while D2 (Tm ∼ 58 °C) represents the DNA binding region. This inference is supported by the observation that denaturation of D1 is associated with only a modest change in ellipticity (Supporting Information, Figure S2A) but a significant increase in binding of SYPRO Orange (Figure 6A); if the dimer interface is “loosened” with an increase in temperature to yield a partly unfolded dimeric species, or if MftR monomers dissociate, overall helical content may not be significantly affected, whereas hydrophobic patches may be exposed to which the dye can bind. R63 is predicted to reside at the end of α3 (dark green; Figure 3), near W11 from α1 of the second monomer, which braces the long helices α6 that connect the dimerization and DNA-binding regions of the protein (Figure 3). The location of these residues at the juncture of these two protein regions would rationalize the more severe destabilization observed when these residues are substituted to generate R63S (Tm ∼43 °C) and W11F (Tm ∼25 °C), with melting temperatures reflecting destabilization of both MftR regions D1 and D2. Taken together, our data suggest a three-state unfolding of MftR in which the first transition corresponds to an unfolding event involving the dimer interface that either leads to a partially folded dimeric intermediate or to complete disentanglement of monomers. These possibilities cannot be distinguished based on these experiments. The second transition would then correspond to unfolding of the transient intermediate, an event that includes unfolding of the DNA-binding lobes.
Thermal unfolding of MftR begins above ∼30 °C, as evidenced by a gradual increase in fluorescence of the SYPRO Orange reporter of protein unfolding. Consistent with this observation, MftR binds DNA with ∼4-fold reduced affinity at 37 °C. RovA, a MarR-type regulator from Yersinia pseudotuberculosis that participates in the establishment of infection, was recently reported to have a thermosensing loop in the dimerization domain.48 Upon host entry, the thermal upshift results in a structural rearrangement in the RovA dimer, which leads to attenuated DNA binding and regulation of virulence-associated processes.48 In RovA, the residues responsible for reduced DNA binding at 37 °C reside in the loop between the two C-terminal helices that constitute the dimerization domain (corresponding to HucR helices α6 and α7). MftR also binds DNA less efficiently at 37 °C compared to that at room temperature. The inference that the less stable domain D1 corresponds to the dimerization region suggests that MftR likewise responds to a thermal upshift by conformational changes in the dimerization domain that result in attenuated DNA binding. We also note that the ∼4-fold reduction in DNA-binding affinity observed when RovA binds its cognate sites at 37 °C is comparable to the observed increase in Kd for MftR binding from 1.6 to 6.9 nM. Communication between DNA-binding lobes and the dimer interface is also reflected in the modest stabilization of both domains D1 and D2 on DNA binding (Table 1).
When Burkholderia species invade plants or mammals, an oxidative burst is encountered during which urate may be produced. Urate production is therefore a signal for successful host colonization. In addition, infection of mammalian species would be associated with a thermal upshift. Our data suggest that DNA binding by MftR is attenuated both by exposure to the physiological temperature associated with infection of mammalian hosts and by urate, resulting in upregulation of mftR and mftP. The observation that mftP and mftR transcripts are readily detectable when cells are grown in the absence of urate at 37 °C (Figure 4A) is consistent with the inference that repression may be more efficient at lower temperatures.
Ligand Binding to MftR
Structures of MarR homologues in complex with ligand reveal a shared ligand-binding pocket in a deep crevice between the dimerization domain and the DNA-binding lobes.1,7,15 This crevice corresponds to the urate-binding pocket identified in HucR and PecS.25,33 That MftR binds urate with modest negative cooperativity is consistent with the existence of two sites; such negative cooperativity of urate binding was also observed for HucR and PecS.25,33,41
MftR conserves the N-terminal helix and the four residues previously shown to be involved in urate binding and attenuation of DNA binding by HucR and PecS.25,33 The proposed mode of urate interaction with HucR involves the Trp of α1 and Arg of α3 interacting with urate by a hydrogen bond and a salt bridge, respectively. At the bottom of the binding pocket, Arg of the recognition helix (α5) forms a salt bridge with Asp of α3; the binding of urate would cause a charge repulsion of Asp in α3 that would in turn displace the DNA recognition helix α5, resulting in attenuated DNA binding.33 For MftR, however, it was already reported that hypoxanthine and xanthine also inhibit DNA binding, albeit less efficiently than urate.30 This contrasts with the observation that these ligands have little or no effect on DNA binding by HucR and PecS.25,33 Since hypoxanthine is uncharged, this suggests that binding of ligand to MftR induces a conformational change to attenuate DNA binding without a strict requirement for charge repulsion; consistent with this inference, D56 can be substituted without significantly affecting urate-mediated attenuation of DNA binding. The ability of the uncharged xanthine to attenuate DNA binding by MftR also suggests that a salt bridge to R63 in α3 is not critical for ligand binding and is consistent with the observation that this residue can be substituted without loss of urate binding. R89 in the recognition helix is not predicted to interact directly with the ligand; this prediction is borne out by the observation that the R89N mutant binds urate with an affinity comparable to that of WT MftR.
Urate-bound MftR showed a one-step melting curve with a Tm of ∼61 °C, suggesting predominant stabilization of domain D1 (Tm ∼49 °C), inferred to correspond to an unfolding event involving the dimerization region or dissociation of MftR monomers. Such stabilizing interactions might derive from direct contacts to residues in helices α1, α2, or α6 that are predicted to line the binding pocket based on the HucR structure; in addition to W11 from α1, candidate residues include a His from α6 and a Gln from α2. By comparison, binding of the anionic phenolic ligand protocatechuate to PcaV involves direct contacts to His and Arg from helix one (corresponding to HucR helix α2).1 Notably, urate binding to the R63S and R89N mutants completely reversed the destabilization imposed by these substitutions and resulted in a Tm comparable to that observed for WT MftR in complex with urate. This suggests that urate binds between the DNA binding lobe and dimer interface, resulting in stabilization of both the DNA-binding lobe and the dimer interface. In contrast, urate binding only modestly stabilized W11F (Tm ∼29 °C); evidently, stabilizing contacts to the ligand were insufficient to overcome the destabilization imposed by the W11F substitution. Urate binding also did not stabilize D56S, suggesting that both stabilizing contacts to the DNA binding lobe (perhaps residues in α3) and the dimerization region were compromised by this mutation. Taken together, our data suggest the binding of urate in a cleft that bridges the dimerization and DNA-binding regions likely by contacts to residues in helices α1, α2, and α6 from the dimer interface and α3 from the DNA-binding HTH motif. Urate binding to MftR is predicted to lead to structural rearrangements, which attenuate DNA binding; the significantly altered unfolding transitions observed on ligand binding are consistent with this premise.
Phylogenetic analyses show that UrtR proteins cluster together, separate from other MarR homologues (Figure 2 and ref (24)). We have also shown that sequence conservation of UrtR DNA recognition helices correlates with conservation of cognate sites in gene promoters.24 However, UrtR proteins appear to have diverged with regard to ligand specificity and mode of ligand binding, despite apparently featuring a shared ligand-binding pocket in the cleft that bridges the DNA-binding lobes and the dimerization region. D. radiodurans HucR, which regulates the expression of a uricase gene, and PecS, which controls the expression of a gene encoding the efflux pump PecM, are specific for urate, with little or no effect of other intermediates in purine metabolism.25,26,33,46 MftR features relaxed ligand specificity, but urate remains the most efficient ligand as measured by its ability to attenuate DNA binding.30 In contrast, S. coelicolor encodes another UrtR homologue, TamR, which is responsible for regulating the activity of genes encoding proteins involved in maintaining flux through the citric acid cycle: DNA binding by TamR is attenuated by trans-aconitate and closely related compounds but not by urate.10
Four conserved residues are characteristic of UrtR homologues: amino acids corresponding to MftR residues R89 and D56 appear to be important for positioning the DNA recognition helices properly, as reflected in attenuated DNA binding on their substitution. In addition, the negative charge of Asp is necessary for conformational changes associated with attenuated DNA binding in HucR and PecS on binding the negatively charged urate;25,33 in MftR, such charge repulsion is not required.30 Tryptophan in α1 may be conserved among UrtR homologues primarily for structural reasons, as reflected in the significant thermal instability of MftR-W11F and in the observed aggregation of the equivalent PecS mutant.25 Similarly, the residue corresponding to R63 in MftR may be structurally important, as evidenced by the thermal instability imposed on its substitution. In HucR and PecS, however, this residue is also important for conferring specificity for the negatively charged urate. Taken together, we propose that the four residues that are characteristic of UrtR proteins are conserved primarily for structural reasons. Ligand specificity is conferred by select residues lining the identified ligand-binding pocket; in HucR and PecS, these residues include amino acids corresponding to W11, D56, and R63, while the relaxed ligand specificity of MftR requires the interaction of bound ligand with distinct residues.
In conclusion, our data suggest that MftR shares with other urate-responsive MarR homologues a ligand-binding pocket that bridges the DNA-binding lobes and the dimerization region. Residues seen to be strictly conserved among UrtR proteins may play mainly structural roles, although they may also participate in conferring specificity for the negatively charged ligand urate. In contrast, the relaxed ligand specificity of MftR is consistent with other residues lining the ligand-binding pocket participating in direct contacts to the ligand. The two-step thermal unfolding transition of MftR is unusual; we propose that the thermal upshift associated with infection of a mammalian host leads to structural rearrangements in the dimer interface that manifest in attenuated DNA binding. MftR may therefore respond to both the ligand and an increase in ambient temperature by attenuated DNA binding and upregulation of the gene encoding the MFTP efflux pump.
Acknowledgments
We thank Professor Evgueni E. Nesterov for the use of his spectrofluorometer and spectrophotometer and Professor Claudia Husseneder for the use of the Nanodrop. We thank Dinesh K. Deochand for purified HucR protein and Chinmay V. Tikhe for assistance with the Nanodrop.
Glossary
Abbreviations
- EMSA
electrophoretic mobility shift assay
- MarR
multiple antibiotic resistance regulator
- MftR
major facilitator transport regulator
- MFTP
major facilitator transport protein
- MFS
major facilitator superfamily
- ROS
reactive oxygen species
- UrtR
urate-responsive transcriptional regulator
Supporting Information Available
Primer sequences, melting temperatures for all MftR variants as determined by CD spectroscopy, apparent dissociation constants for HucR and PecS mutants binding to their respective cognate sites, SDS–PAGE gels of purified MftR variants and dimers produced after glutaraldehyde cross-linking, CD spectra and melting curves for all MftR variants, EMSAs of MftR variants binding to MftO, HucR binding to its cognate site at room temperature and 37 °C, and effect of urate on DNA binding by MftR variants. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by National Institutes of Health grant 1R15GM107825 (to AG).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Davis J. R.; Brown B. L.; Page R.; Sello J. K. (2013) Study of PcaV from Streptomyces coelicolor yields new insights into ligand-responsive MarR family transcription factors. Nucleic Acids Res. 41, 3888–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson S. P.; Grove A. (2006) Ligand-responsive transcriptional regulation by members of the MarR family of winged helix proteins. Curr. Issues Mol. Biol. 8, 51–62. [PubMed] [Google Scholar]
- Ellison D. W.; Miller V. L. (2006) Regulation of virulence by members of the MarR/SlyA family. Curr. Opin. Microbiol. 9, 153–159. [DOI] [PubMed] [Google Scholar]
- Perera I. C.; Grove A. (2010) Molecular mechanisms of ligand-mediated attenuation of DNA binding by MarR family transcriptional regulators. J. Mol. Cell Biol. 2, 243–254. [DOI] [PubMed] [Google Scholar]
- Grove A. (2013) MarR family transcription factors. Curr. Biol. 23, R142–143. [DOI] [PubMed] [Google Scholar]
- Holley T. A.; Stevenson C. E.; Bibb M. J.; Lawson D. M. (2013) High resolution crystal structure of Sco5413, a widespread actinomycete MarR family transcriptional regulator of unknown function. Proteins 81, 176–182. [DOI] [PubMed] [Google Scholar]
- Saridakis V.; Shahinas D.; Xu X.; Christendat D. (2008) Structural insight on the mechanism of regulation of the MarR family of proteins: high-resolution crystal structure of a transcriptional repressor from Methanobacterium thermoautotrophicum. J. Mol. Biol. 377, 655–667. [DOI] [PubMed] [Google Scholar]
- Cohen S. P.; Levy S. B.; Foulds J.; Rosner J. L. (1993) Salicylate induction of antibiotic resistance in Escherichia coli: activation of the mar operon and a mar-independent pathway. J. Bacteriol. 175, 7856–7862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J. R.; Sello J. K. (2010) Regulation of genes in Streptomyces bacteria required for catabolism of lignin-derived aromatic compounds. Appl. Microbiol. Biotechnol. 86, 921–929. [DOI] [PubMed] [Google Scholar]
- Huang H.; Grove A. (2013) The transcriptional regulator TamR from Streptomyces coelicolor controls a key step in central metabolism during oxidative stress. Mol. Microbiol. 87, 1151–1166. [DOI] [PubMed] [Google Scholar]
- Lee J. W.; Soonsanga S.; Helmann J. D. (2007) A complex thiolate switch regulates the Bacillus subtilis organic peroxide sensor OhrR. Proc. Natl. Acad. Sci. U.S.A. 104, 8743–8748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Z.; Lou H.; Zhu R.; Zhu J.; Zhang D.; Zhao B. S.; Zeng S.; Chen X.; Chan J.; He C.; Chen P. R. (2014) The multiple antibiotic resistance regulator MarR is a copper sensor in Escherichia coli. Nature Chem. Biol. 10, 21–28. [DOI] [PubMed] [Google Scholar]
- Fuangthong M.; Helmann J. D. (2002) The OhrR repressor senses organic hydroperoxides by reversible formation of a cysteine-sulfenic acid derivative. Proc. Natl. Acad. Sci. U.S.A. 99, 6690–6695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomovskaya O.; Lewis K.; Matin A. (1995) EmrR is a negative regulator of the Escherichia coli multidrug resistance pump EmrAB. J. Bacteriol. 177, 2328–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y. M.; Jeng W. Y.; Ko T. P.; Yeh Y. J.; Chen C. K.; Wang A. H. (2010) Structural study of TcaR and its complexes with multiple antibiotics from Staphylococcus epidermidis. Proc. Natl. Acad. Sci. U.S.A. 107, 8617–8622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hommais F.; Oger-Desfeux C.; Van Gijsegem F.; Castang S.; Ligori S.; Expert D.; Nasser W.; Reverchon S. (2008) PecS is a global regulator of the symptomatic phase in the phytopathogenic bacterium Erwinia chrysanthemi 3937. J. Bacteriol. 190, 7508–7522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchmeier N.; Bossie S.; Chen C. Y.; Fang F. C.; Guiney D. G.; Libby S. J. (1997) SlyA, a transcriptional regulator of Salmonella typhimurium, is required for resistance to oxidative stress and is expressed in the intracellular environment of macrophages. Infect. Immun. 65, 3725–3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Rio L. A.; Corpas F. J.; Sandalio L. M.; Palma J. M.; Gomez M.; Barroso J. B. (2002) Reactive oxygen species, antioxidant systems and nitric oxide in peroxisomes. J. Exp. Bot. 53, 1255–1272. [PubMed] [Google Scholar]
- van der Vliet A. (2008) NADPH oxidases in lung biology and pathology: host defense enzymes, and more. Free Radical Biol. Med. 44, 938–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin H. M.; Hancock J. T.; Salisbury V.; Harrison R. (2004) Role of xanthine oxidoreductase as an antimicrobial agent. Infect. Immun. 72, 4933–4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper D. C.; Spitsin S.; Kean R. B.; Champion J. M.; Dickson G. M.; Chaudhry I.; Koprowski H. (1998) Uric acid, a natural scavenger of peroxynitrite, in experimental allergic encephalomyelitis and multiple sclerosis. Proc. Natl. Acad. Sci. U.S.A. 95, 675–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foti M. C.; Amorati R. (2009) Non-phenolic radical-trapping antioxidants. J. Pharm. Pharmacol. 61, 1435–1448. [DOI] [PubMed] [Google Scholar]
- Chen P. R.; Nishida S.; Poor C. B.; Cheng A.; Bae T.; Kuechenmeister L.; Dunman P. M.; Missiakas D.; He C. (2009) A new oxidative sensing and regulation pathway mediated by the MgrA homologue SarZ in Staphylococcus aureus. Mol. Microbiol. 71, 198–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera I. C.; Grove A. (2011) MarR homologs with urate-binding signature. Protein Sci. 20, 621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera I. C.; Grove A. (2010) Urate is a ligand for the transcriptional regulator PecS. J. Mol. Biol. 402, 539–551. [DOI] [PubMed] [Google Scholar]
- Huang H.; Mackel B. J.; Grove A. (2013) Streptomyces coelicolor encodes a urate-responsive transcriptional regulator with homology to PecS from plant pathogens. J. Bacteriol. 195, 4954–4965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reverchon S.; Rouanet C.; Expert D.; Nasser W. (2002) Characterization of indigoidine biosynthetic genes in Erwinia chrysanthemi and role of this blue pigment in pathogenicity. J. Bacteriol. 184, 654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouanet C.; Nasser W. (2001) The PecM protein of the phytopathogenic bacterium Erwinia chrysanthemi, membrane topology and possible involvement in the efflux of the blue pigment indigoidine. J. Mol. Microbiol. Biotechnol. 3, 309–318. [PubMed] [Google Scholar]
- Mhedbi-Hajri N.; Malfatti P.; Pedron J.; Gaubert S.; Reverchon S.; Van Gijsegem F. (2011) PecS is an important player in the regulatory network governing the coordinated expression of virulence genes during the interaction between Dickeya dadantii 3937 and plants. Environ. Microbiol. 13, 2901–2914. [DOI] [PubMed] [Google Scholar]
- Grove A. (2010) Urate-responsive MarR homologs from Burkholderia. Mol. Biosyst. 6, 2133–2142. [DOI] [PubMed] [Google Scholar]
- Li X. Z.; Nikaido H. (2009) Efflux-mediated drug resistance in bacteria: an update. Drugs 69, 1555–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fluman N.; Bibi E. (2009) Bacterial multidrug transport through the lens of the major facilitator superfamily. Biochim. Biophys. Acta 1794, 738–747. [DOI] [PubMed] [Google Scholar]
- Perera I. C.; Lee Y. H.; Wilkinson S. P.; Grove A. (2009) Mechanism for attenuation of DNA binding by MarR family transcriptional regulators by small molecule ligands. J. Mol. Biol. 390, 1019–1029. [DOI] [PubMed] [Google Scholar]
- Edgar R. C. (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinf. 5, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordelon T.; Wilkinson S. P.; Grove A.; Newcomer M. E. (2006) The crystal structure of the transcriptional regulator HucR from Deinococcus radiodurans reveals a repressor preconfigured for DNA binding. J. Mol. Biol. 360, 168–177. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Nei M.; Dudley J.; Tamura K. (2008) MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Briefings Bioinf. 9, 299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L.; Baumann U.; Reymond J. L. (2004) An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 32, e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade M. A.; Chacon P.; Merelo J. J.; Moran F. (1993) Evaluation of secondary structure of proteins from UV circular dichroism spectra using an unsupervised learning neural network. Protein Eng. 6, 383–390. [DOI] [PubMed] [Google Scholar]
- Whitmore L.; Wallace B. A. (2004) DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 32, W668–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmore L.; Wallace B. A. (2008) Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers 89, 392–400. [DOI] [PubMed] [Google Scholar]
- Wilkinson S. P.; Grove A. (2005) Negative cooperativity of uric acid binding to the transcriptional regulator HucR from Deinococcus radiodurans. J. Mol. Biol. 350, 617–630. [DOI] [PubMed] [Google Scholar]
- Collart M. A., and Oliviero S. (2001) Preparation of yeast RNA. Curr. Protoc. Mol. Biol. Chapter 13, Unit13 12, DOI: 10.1002/0471142727.mb1312s23. [DOI] [PubMed] [Google Scholar]
- Schmittgen T. D.; Livak K. J. (2008) Analyzing real-time PCR data by the comparative C(T) method. Nature Protoc. 3, 1101–1108. [DOI] [PubMed] [Google Scholar]
- Galyov E. E.; Brett P. J.; DeShazer D. (2010) Molecular insights into Burkholderia pseudomallei and Burkholderia mallei pathogenesis. Annu. Rev. Microbiol. 64, 495–517. [DOI] [PubMed] [Google Scholar]
- Alekshun M. N.; Levy S. B.; Mealy T. R.; Seaton B. A.; Head J. F. (2001) The crystal structure of MarR, a regulator of multiple antibiotic resistance, at 2.3 A resolution. Nat. Struct. Biol. 8, 710–714. [DOI] [PubMed] [Google Scholar]
- Wilkinson S. P.; Grove A. (2004) HucR, a novel uric acid-responsive member of the MarR family of transcriptional regulators from Deinococcus radiodurans. J. Biol. Chem. 279, 51442–51450. [DOI] [PubMed] [Google Scholar]
- Biggar K. K.; Dawson N. J.; Storey K. B. (2012) Real-time protein unfolding: a method for determining the kinetics of native protein denaturation using a quantitative real-time thermocycler. BioTechniques 53, 231–238. [DOI] [PubMed] [Google Scholar]
- Quade N.; Mendonca C.; Herbst K.; Heroven A. K.; Ritter C.; Heinz D. W.; Dersch P. (2012) Structural basis for intrinsic thermosensing by the master virulence regulator RovA of Yersinia. J. Biol. Chem. 287, 35796–35803. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.