

Abstract
Inwardly rectifying potassium (Kir) channels are gated by the membrane phospholipid phosphatidylinositol-4,5-bisphosphate (PIP2). Among them, Kir3 channel gating requires additional molecules, such as the βγ subunits of G proteins or intracellular sodium. Using an interactive computational-experimental approach, we show that sodium sensitivity of Kir channels involves the side-chains of an aspartate and a histidine located across from each other in a critical loop in the cytosolic domain, as well as the backbone carbonyls of two additional residues and a water molecule. The location of the coordination site in the vicinity of a conserved arginine shown to affect channel-PIP2 interactions suggests that sodium triggers a structural switch that frees the critical arginine. Mutations of the aspartate and the histidine that affect sodium sensitivity also enhance the channel’s sensitivity to PIP2. Furthermore, based on the molecular characteristics of the coordination site, we identify and confirm experimentally a novel sodium-sensitive phenotype in Kir5.1.
Kir channels are important in setting the resting membrane potential and modulating membrane excitability1–3. A common feature of Kir channels and many other ion channels that has emerged in recent years is that they all require the membrane phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2, 1) for activation4,5. Residues in both the C- and N- termini affect PIP2 sensitivity and activation of Kir channels.
Among these, the atrial KACh channels are heterotetrameric proteins that consist of Kir3.1 (GIRK1) and Kir3.4 (GIRK4) subunits. Heterologous coexpression of these two subunits in Xenopus oocytes or mammalian cell lines produce active heteromeric channels6–8. Single point mutations at a critical position located in the pore helix of either channel subunit result in homotetrameric highly active channels, Kir3.4_S143T or Kir3.4* and Kir3.1_F137S or Kir3.1* 10,11, while wild-type homotetramers typically show little or no activity.
Unlike most Kir channels, Kir3 channels are activated by PIP2 only in the presence of an additional gating molecule such as the βγ subunits of G proteins and/or intracellular sodium that enhance channel-PIP2 interactions12. Activation by internal sodium has been shown for several Kir3 channels including the heteromers Kir3.1/Kir3.213–15 and Kir3.1/Kir3.416 and the homomers Kir3.214 and Kir3.4*17. Kir3.1/Kir3.4 activation during Na+ accumulation has been suggested to be involved in the direct arrhythmogenic effects of cardiac glycosides, drugs widely used in heart failure or for improvement of the inotropic state of the heart16.
Studies aimed at identifying regions of Kir3 channels that mediate their activation by sodium have converged to a region located in the C-terminus proximal to the plasma membrane. In Kir3.4*, this region was found to include residues 214–25217. In Kir3.2, a similar region included the first 45 residues in the C-terminus that are downstream of the second transmembrane (TM2) helix15. Both studies identified a corresponding aspartate (position 223 in Kir3.4* and in position 226 in Kir3.2) as a prerequisite for sodium-dependent activation of Kir3 channels. Following a comparison of the dose-response relationships for sodium-dependent activation in Kir3.1/Kir3.2 heteromers and Kir3.2 homomers, it was deduced that Kir3.1 that possessed an asparagine at the equivalent position (position 217) was insensitive to sodium. Fitting of the dose-response curves with the Hill equation showed that while for the Kir3.2 homomers the Hill coefficient was about 4, for the Kir3.1/Kir3.2 or the Kir3.1/Kir3.4 heteromers it was about 215,16. This suggested that each of the four subunits within the homomeric Kir3.2 channel bind a sodium ion, while in the heteromeric Kir3.1/Kir3.2 or Kir3.1/Kir3.4 channels, only two of the four subunits are sodium sensitive.
In Kir3.4*, a highly conserved arginine (R225) in the loop that contains the critical aspartate (D223) affects the sensitivity of the channel to PIP25,17. It was suggested that D223 may normally act to inhibit electrostatic interactions between PIP2 and this critical arginine residue. Sodium binding would attenuate this inhibition, allowing the arginine residue to enhance the channel’s sensitivity to PIP217.
Kir2.1 has an asparagine at the position equivalent to 217 in Kir3.1 and 223 in Kir3.4, whose mutation to histidine, N216H, was implicated18 in ventricular arrhythmias associated with Andersen’s syndrome (LQT7). Although Kir2.1 is Na+ insensitive, the N216D mutant exhibits a Na+ sensitive phenotype17.
Fig. 1a shows sequences of the loop that contain the aspartate residue (highlighted) that is required for Na+ sensitivity. Both the Na+-sensitive Kir3.2 and Kir3.4 channels not only possess the Asp residue but also a histidine residue located across from the critical aspartate in a loop that contains several residues that affected PIP2 sensitivity (e.g. Fig. 1b - R225, I229 in Kir3.4).
Figure 1.
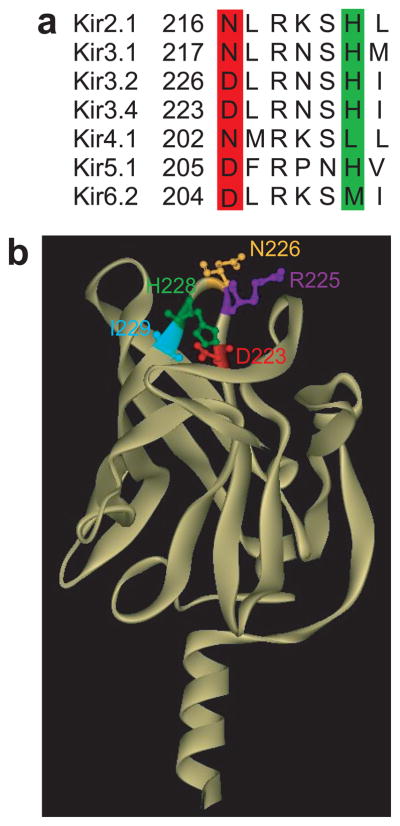
Positions of key residues in a loop of Kir channels where Na+ is coordinated. (a) Alignment of the loop segment of Kir2.1, Kir3.1, Kir3.2, and Kir3.4, Kir4.1, Kir5.1 and Kir6.2, which includes the residues found in this study to coordinate the Na+ ion (Asp in the red highlighted position and His in the green highlighted position). (b) Cytosolic domain of the homology model of Kir3.4 based on the crystallographic structure of the cytosolic domain of Kir3.1 showing along with the three residues of focus in the present study (D223 -in red, H228 -in green, R225 -in purple) the neighboring asparagine N226 (yellow), and isoleucine I229 (cyan) residues.
Here we show using a computational-experimental interactive approach how Na+ was coordinated in Kir channels, and how it affected the interactions between the aspartate and the highly conserved arginine located two residues away. According to the model, the sodium coordination site in Kir channels involved the side-chains of the aspartate and the histidine as well as the backbone carbonyls of two additional residues and a water molecule. Guided by these results, we identified Kir5.1 to be sodium dependent as well, and showed how its sodium sensitivity could be removed via mutagenesis of residues in the corresponding loop. Kir5.1 expression in renal tubular epithelia involved in Na+ reabsorption and in brainstem neurons that act as chemoreceptors, may provide important clues towards the physiological importance of the Na+ sensitivity of K+ channels.
Results
Coordination of Na+ in Kir3.1_N217D
Recently, the crystallographic structure of the cytosolic domain of Kir3.1 was determined19,20. Based on this structure20, we examined the interactions between N217 and an arginine at position 219. Minimization of the structure showed that there was no interaction between these two positions (Supplementary Fig. 1a online). However, when position 217 was mutated to an aspartate, hydrogen bonding was observed between positions 217 and 219 (Fig. 2a): the side-chain carbonyl OD1 of the aspartate interacts with the side-chain hydrogen HH12 of the guanidinium group of the arginine. Experimentally, Kir3.1*_N217D exhibited very small whole-cell basal currents (Supp. Fig. 1c). Furthermore, the N217D mutant has been shown15 to confer sodium sensitivity to a chimera of Kir3.1 that has the transmembrane domain of Kir3.2. While this chimera with an asparagine at position 217 exhibited currents that were similar in magnitude to those of the Kir3.1/Kir3.2 heteromeric channel21, the N217D mutant of the chimera exhibited significantly reduced currents. Reduction of current was also observed when Kir3.1_N217D was co-expressed with Kir3.2 as compared to the currents exhibited by Kir3.1/Kir3.215. These results suggest that introduction of Na+ dependence to Kir3.1 can significantly reduce basal currents. We therefore examined the effect of Na+ on the interaction between D217 and R219 in Kir3.1. A Na+ ion and a water molecule are practically equivalent in an electron density map22. We therefore solvated the cytosolic domain of the channel, and based on the experimental result that mutation of the aspartate to asparagine in several Kir channels affects sodium sensitivity, we replaced water molecules that were located within 5Å from D217 with Na+ ions, one at a time.
Figure 2.

H-bonding pattern between position 217 and residues in its vicinity in (a) Kir3.1_N217D after minimization. The model was based on the crystallographic structure of Kir3.1 (PDB accession number 1N9P19). (b) H-bonding pattern between the Na+ ion and the residues that surround it in its coordination site in Kir3.1_N217D. (c) Minimal distance between positions 223 and 225 as obtained from MD simulations of Kir3.4, Kir3.4_D223N, and Kir3.4_H228M. (d) RMSD of the backbone residues of the cytosolic domain relative to the starting minimized structure that corresponds to the MD simulations of Kir3.4, Kir3.4_D223N, and Kir3.4_H228M. The calculations were performed using Simulaid: http://atlas.physbio.mssm.edu/~mezei/simulaid/simulaid.html). Figures a, and b were drawn using PyMol48.
There were a total of seven water molecules within this radius, and while in most cases the Na+ ions moved away from the cytosolic domain, in two cases following minimization the Na+ ions moved to the position displayed in Fig. 2b. One of these cases is depicted in Supplementary Fig. 1b online and shows a 4.6Å displacement of the Na+ that occurred during minimization. The final position obtained in the minimization suggests that Na+ is coordinated in Kir3.1 by the side-chain carbonyl OD2 of D217 and by the backbone carbonyls of M223 and L264. It is also within interaction distance from the H222 (Fig. 2b). In addition, a water molecule was found to be within hydrogen bonding distance from the sodium ion (Supplementary Fig. 1d online). This result is in agreement with existing structures of sodium binding proteins that have demonstrated that sodium is ligated by oxygen atoms provided by water as well as main-chain carbonyl, carboxyl or hydroxyl groups with an average coordination number of five to six23. No interaction is observed between D217 and R219, suggesting that Na+ breaks the hydrogen bond between these two residues, freeing R219 to affect sensitivity to PIP2.
Coordination site of Na+: experimental validation using the Kir3.4* channel
As the N217D mutation in Kir3.1* results in a significant decrease in whole-cell currents (Supplementary Fig. 1c online), we proceeded to validate the coordination site of Na+ using the Kir3.4* model system that exhibits significantly higher basal currents. Kir3.4* that possesses naturally an aspartate at the equivalent position exhibits strong Na+ sensitivity, while its D223N mutant lacks Na+ sensitivity17. As noted above, minimization of Kir3.1_N217D has suggested that the side-chains of D217 and H222 are involved in the coordination of the Na+.
Although Kir6.2 has the critical aspartate at the equivalent position (Fig. 1a), it lacks Na+ sensitivity. Kir6.2 possesses a methionine at the equivalent position to the H222 whose side-chain coordinates the Na+ in the Kir3.1 model (Fig. 2b). We have therefore examined the effect of the H228M mutation at the equivalent position in Kir3.4 on the interactions between D223 and R225. Molecular dynamics (MD) simulations of Kir3.4 and Kir3.4_D223N (Fig. 2c) using a homology model based on the structure of the cytosolic domain of Kir3.119 (see Methods), showed that there was loss of hydrogen bonding between position 223 and R225 following the D223N mutation. In contrast, following a conformational change, hydrogen bonding was formed in Kir3.4, and persisted during the rest of the simulation. The average minimal distance between positions 223 and 225 after the conformational change was 1.8±0.2Å. On the other hand, in Kir3.4_D223N hydrogen bonding did not form. The average minimal distance was 7.1±0.4Å after an initial conformational change. The simulated structures equilibrated within 0.6ns (Fig. 2d). In order to further characterize the conformational transitions that occur during the simulations, we applied clustering analysis. Applying the clustering algorithm ART-2′ (24–26) implemented in CHARMM(27) and using a clustering radius of 1.75Å, we classified the structures obtained in the simulations into clusters according to the distances between the atoms in the side-chains of the residues at positions 223 and 225 that could form hydrogen bonding (see Supplementary. Fig. 2a online). Supplementary Table 1 online includes the details of the clusters formed. The dominant clusters obtained from the simulation of Kir3.4 and Kir3.4_D223N are represented in Supplementary Figs. 2b,c online by the backbone atoms of the critical loop and the side-chain atoms of positions 223 and 225 in the cluster center conformation.
MD simulation also showed that the H228M mutation affected the interaction between D223 and R225. By creating a hydrophobic environment in the center of the loop, the methionine kept the two charged amino acids at a distance that exceeded hydrogen bonding distance most of the time. This can be seen in Supplementary Figs. 2e,f online that depict the orientations of the side-chains of position 223, 225 and 228 in Kir3.4 and in Kir3.4_H228M. For Kir3.4_H228M, the average minimal distance was 4.4±0.6Å following a relaxation of the structure from its initial conformation in the simulation (Fig. 2c). The dominant cluster obtained from the simulation of Kir3.4_H228M is represented in Supplementary Fig. 2d online by the backbone atoms of the critical loop and the side- chains of D223 and R225 in the cluster center conformation. This average minimal distance is between the values obtained for Kir3.4 and Kir3.4_D223N. Thus, hydrogen bonding is formed between D223 and R225 in the H228M mutant only rarely.
In order to further substantiate these results, we have repeated the simulations in the presence of water (see Supplementary Fig. 3c online) using the primary hydration shell (PHS) approach(28) as described in the Methods part. The results obtained from these simulations were in agreement with those obtained in the absence of solvent, as can be seen in Supplementary. Figs. 3a,b online. Specifically, the average minimal distances between positions 223 and 225 as obtained in the presence of the primary hydration shell were 1.7±0.1Å, 6.5±0.8Å, and 4.2±0.2Å for Kir3.4, Kir3.4_D223N and Kir3.4_H228M, respectively. This suggests that the interaction between positions 223 and 228 is formed independent of the water that surrounds the cytosolic domain.
Examination of the whole-cell currents of the D223N and the H228M mutants (Fig. 3d) shows that both of them exhibited basal currents similar to each other, which were approximately 50% higher than the basal currents exhibited by the control Kir3.4* channel. Figs. 3a–c show representative traces obtained from macropatches of oocytes expressing Kir3.4* and its D223N or H228M mutants. While Kir3.4* exhibited high sensitivity to sodium following a short application (1–3min) of PIP2 (Fig. 3a), sodium did not enhance the activation of the D223N mutant (Fig. 3b) and had only a very limited effect in the case of the H228M (Fig. 3c). Comparison of the Na+ dose-response curves for the H228M with that of Kir3.4* showed that for all concentrations, the Na+ stimulation of channel activity is significantly lower than that of the wild-type (Supplementary Fig. 4a online). This implies that the efficacy of the H228M mutant is lower than that of the wild-type. Yet, with a Hill coefficient of 4, and comparable EC50 values (Kir3.4*: 33mM and Kir3.4*_H228M: 26mM), it appears that the potency is not affected significantly by the H228M mutation. Furthermore, while PIP2 alone did not lead to significant activation of Kir3.4*, following the D223N and the H228M mutations that the simulations suggest disrupt the hydrogen bonding between R225 and D223, PIP2 alone was sufficient to activate the channels. Supplementary Fig. 4b online depicts the PIP2 dose-response curves for Kir3.4* and for the two mutants. Comparison of the curves shows that the dose-response curves for both mutants are left-shifted with respect to the wild-type, showing that the PIP2 sensitivity of the channel is indeed enhanced by these mutations. While the EC50 for Kir3.4* is 14.6μM, for the D223N and H228M mutations, it is 1.5μM and 1.7μM, respectively, which is about 10 fold lower than for Kir3.4*.
Figure 3.

Experimental evidence for Kir3.4* residues involved in Na+ sensitivity (a)–(c) Representative traces of inside-out macropatch recordings of Xenopus oocytes. Na+ (30mM) and PIP2 (2.5μM) were applied as indicated by the bars in the control solution (ND96K+EGTA) (a) Kir3.4* (b) Kir3.4*_D223N (c) Kir3.4*_H228M. (d) Whole-cell basal currents of Kir3.4*, Kir3.4*_D223N, and Kir3.4*_H228M recorded in Xenopus oocytes at −80mV. (e)–(f) Representative traces of inside-out macropatch recordings of Xenopus oocytes. Na+ (30mM) and PIP2 (2.5μM) were applied as indicated by the bars in the control solution (ND96K+EGTA). Both Kir3.4*_N226K (e) and Kir3.4*_I229L (f) show strong sensitivity to Na+ similar to Kir3.4*.
Kir3.4* residues that do not affect Na+ sensitivity
In order to further validate our results concerning the coordination site of the sodium in Kir channels, we examined two positions in the loop segment that are occupied by different residues in Kir3.4 and Kir2.1, but whose side-chains do not interact with the sodium ion in the Kir3.1 model (Fig. 2b). MD simulations suggest that both in the N226K and the I229L mutants in Kir3.4, positions 223 and 225 interact, although not as strongly as they do in Kir3.4 itself. After relaxation of the initial conformation in the simulation, the average minimal distance between positions 223 and 225 is 3.0±0.3Å in the N226K mutant and 2.3±0.4Å in the I229L mutant (Supplementary Fig. 5 online).
The whole-cell currents of the N226K and the I229L mutants exhibited similar basal currents to those exhibited by the D223N and the H228M mutants, of approximately 50% higher than those exhibited by the control Kir3.4* channel (data not shown). This effect is likely to be mediated via an increase in PIP2 sensitivity, somehow caused by the mutations, as shown explicitly for the I229L mutant (Supplementary Fig. 4b online). Yet, the representative traces obtained from macropatches of oocytes expressing the N226K and I229L mutant (Figs. 3e–f) show that unlike the D223N and the H228M mutants (Figs. 3b–c), both mutants, similarly to the control Kir3.4* (Fig. 3a), exhibit high sensitivity to sodium following a short application of PIP2. While in the absence of sodium, these mutants do not exhibit significant currents following a short application of PIP2, longer exposures to PIP2 could stimulate currents independently of sodium as was previously observed for the I229L mutant17. Yet, even under these conditions, the mutants exhibit sodium sensitivity (data not shown). These results are consistent with the notion that sodium is required to interfere with the interaction between the aspartate (D223 in Kir3.4) and the arginine (R225 in Kir3.4): in Kir3.4 and in both the N226K and the I229L mutants these residues interact, while in the D223N mutant no interaction is observed between these positions and in the H228M mutant interaction between these positions is observed only rarely. The summary in Fig. 4 depicts a strong correlation between the sodium sensitivity and the interactions between the aspartate and the arginine in the Kir3.4* mutations tested.
Figure 4.

Summary data of the effect of perfusion of Na+ (30mM) on Kir3.4* currents obtained from inside-out patches of Xenopus oocytes after a short application of PIP2 (2.5μM) as a function of the minimal distance between positions 223 and 225 as obtained from MD simulations.
Coordination site of Na+: experimental validation using the Kir3.1/Kir3.2 channel
As noted above, both the homomeric Kir3.2 channel, and the heteromeric Kir3.1/Kir3.2 are sodium sensitive14,15 (see Supplementary Figs. 6b,c online). Sodium sensitivity of this channel has been linked to the aspartate in Kir3.2 (Supplementary Fig. 6d online), which is located in the equivalent position to position 217 in Kir3.1 (Fig. 1a). We proceeded to examine the effect of mutation of the histidine to a methionine on Kir3.2 on the sodium sensitivity of the channel in the same conditions as those used previously to show the effect of the D226N on sodium sensitivity15. Similarly to the effect of the corresponding mutation in Kir3.4*, the result in Kir3.2_H231M was a significant reduction in sodium sensitivity (Supplementary Figs. 6b,e online), further supporting the importance of the histidine to the sodium sensitivity characteristics of Kir channels. As with Kir3.4*, the Kir3.2 Asp to Asn and the His to Met mutations yielded currents significantly larger than control (Supplementary Fig. 6a online).
Validation of the coordination site of Na+ using the Kir2.1 channel
Kir2.1 has an asparagine at the position equivalent to D223 in Kir3.4 (Fig. 1a), and has been shown to be Na+ insensitive17. On the other hand, its N216D mutant exhibited Na+ sensitivity, albeit weaker than in Kir3.4*. Kir2.1 possesses a histidine at the position that corresponds to position 228 in Kir3.4*. We examined whether the corresponding mutation in Kir2.1, H221M, would reverse the effect of the N216D mutation by monitoring the effect of intracellular sodium on the rundown observed when a macropatch was detached. As can be seen in Figs. 5a,b, application of intracellular sodium significantly slowed down the rundown of Kir2.1_N216D currents, consistent with the interpretation that the presence of sodium enhanced the sensitivity of the channel to PIP2. This effect on the slowing of the rundown, however, was eliminated by the introduction of the additional H221M mutation as can be seen in Fig. 5c, implying that the H221M mutation indeed reversed the increased sensitivity to sodium introduced by the N216D mutation in agreement with our model (Fig. 2b), and with the results obtained for Kir3.4* (Fig. 3a–d) and Kir3.2 (Supplementary Fig. 6 online). Examination of these Kir2.1 mutants using HA-tagged constructs29 suggested that these mutations did not affect membrane expression (data not shown).
Figure 5.

Experimental evidence for Kir2.1 and Kir5.1 residues involved in Na+ sensitivity (a)–(c) Representative traces of inside-out patches of Xenopus oocytes, showing Kir2.1 current rundown. Na+ (30mM) was perfused in the control solution (ND96K+EGTA). The rundown of Kir2.1_N216D is slowed down following application of sodium. This effect is reversed for the Kir2.1_N216DH221M mutant. (d)–(f) Representative traces of inside-out macropatch recordings of currents from Kir4.1 as a homomer or heteromer with Kir5.1expressed in Xenopus oocytes. Na+ (30mM) was applied as indicated by the bars in the control solution (ND96K+EGTA) The rundown of Kir4.1/Kir5.1 is slowed down following application of sodium. (d) Kir4.1 (e) Kir4.1/Kir5.1 (f) Kir4.1/Kir5.1_D205N
Kir5.1 is Na+ sensitive
Examination of the sequences of all other Kir channels shows that Kir5.1 is the only other member of a Kir subfamily besides Kir3 members that possesses both the Asp and His residues involved in Na+ sensitivity (Fig. 1a). We proceeded to examine the sodium sensitivity characteristics of Kir5.1. Since Kir5.1 is not functional on its own we have co-expressed it with Kir4.1. Kir4.1 itself does not possess the aspartate or the histidine, and was not sodium sensitive (Fig. 5d) as could be seen when intracellular sodium was perfused during the rundown observed after the macropatch was detached. On the other hand, as can be seen in Fig. 5e, application of intracellular sodium significantly slowed the rundown of Kir4.1/Kir5.1 currents, confirming the sodium sensitivity of Kir5.1. To further investigate the source of the sodium sensitivity in Kir5.1, we examined the effect of mutation D205N on the sodium sensitivity of the channel. As can be seen in Fig. 5f, the D205N mutation removed the sodium sensitivity of the channel. Interestingly, the H210M did not affect Na+ sensitivity (Supplementary Fig. 7a online). Comparison of the four residues included in the loop between D205 and H210 with the equivalent residues in other Kir channels, revealed that three of these residues are unique to Kir5.1: F206, P208 and N209. We thus exchanged the loop segment (DFRPNH) with the corresponding residues in the sodium insensitive Kir6.2 (DLRKSM). The resulting channel, which still included D205 but in which the histidine was mutated to a methionine, was sodium insensitive as can be seen in Supplementary Fig. 7b online. This result suggests that the constituents of the residues in the loop between the aspartate and the histidine play an important role in affecting the sodium sensitivity of this channel.
Discussion
Kir3 channels are activated by PIP2 only in the presence of gating molecules, such as the βγ subunits of G proteins and/or intracellular sodium. In this paper we suggest that sodium is coordinated by residues located in a critical loop in the cytosolic domain. This loop includes an arginine that has been found to be critical to channel-PIP2 interactions (R225 in Kir3.4*).
Our model, which is based on the crystallographic structure of the cytosolic domain of Kir3.1, suggests that sodium is coordinated by the side-chains of N217D and H222, and by the backbone carbonyls of M223 and L264. MD simulations of Kir3.4 suggest that in Kir3.4, D223 (equivalent to position 217 in Kir3.1) and R225 interact in the absence of sodium, while in the D223N mutant R225 does not interact with position 223, and in the H228M mutant (equivalent to position 222 in Kir3.1) interactions between these positions occur rarely. In view of possible inaccuracies in the standard force fields for alkali ions, the accuracy of computational studies involving sodium ions could be questioned30. We thus proceeded to confirm the results experimentally. Our data show that in agreement with the notion that sodium ions play a role of a switch that breaks the hydrogen bonding between the aspartate and the arginine, an Asn in place of the Asp does not exhibit sodium sensitivity and a Met in place of the His exhibits significantly reduced or no sodium sensitivity in the Kir3.4*, Kir3.2, Kir2.1, and Kir5.1 channels.
The side-chain of histidine has been previously shown to be associated with sodium binding sites in small dipeptides. In general, sodium is known to bind peptides primarily through their oxygen atom22,31, which in many cases includes the backbone carbonyls of the amino acids involved. However, for the GlyHis and the HisGly dipeptides, sodium also binds the imidazole side-chain of a histidine that serves as an additional chelation site32. This further corroborates our finding that the binding site of the sodium in Kir channels involves the side-chain of the histidine in addition to that of the aspartate, and the backbone carbonyls of two additional residues. Furthermore, it is possible that similar binding sites may also exist in other ion channels, where intracellular sodium may act as a second messenger to regulate function33. For example, intracelluar sodium activates the sodium activated potassium channels (IK(Na))34,35, and downregulates the activity of amiloride-sensitive epithelial Na+ channels36.
In addition to the four residues suggested to coordinate the sodium ion, a water molecule was found to be within hydrogen bonding distance from the sodium ion. This result is in agreement with existing structures of sodium binding proteins that have suggested an average coordination number of five to six that includes the participation of water molecules as well23,37.
Fig. 4 summarizes the data obtained for the Kir3.4* channel and its mutants. As shown in the figure, our results suggest the existence of a strong correlation between sodium sensitivity and the strength of the interaction between an aspartate and a critical arginine for channel activation by PIP2. While it is not clear whether this arginine affects channel activation directly or allosterically, it is likely that the availability of this arginine modulates the efficacy and/or affinity of PIP238,39. Accordingly, the less the aspartate and the arginine interact, the less sensitivity to sodium by the channel would be observed. This suggests that in order to enable activation of Kir3 channels, the critical arginine should be free to affect the channel’s sensitivity to PIP2. This can be achieved in several ways. First, intracellular sodium can interact with the aspartate and prevent it from interacting with the arginine, in essence pulling it away from the arginine. Alternatively, our data suggest two additional mechanisms that can free the critical arginine in a manner that does not require sodium for PIP2-dependent activation of the channel: (a) by exchange of the negatively charged aspartate by another residue that does not compete with PIP2 for the positively charged arginine located two positions away; an appropriate exchange was the polar asparagine, which both the Kir3.1 and Kir2.1 channels possess and are sodium insensitive naturally; (b) by introduction of a hydrophobic residue in the vicinity of the aspartate and the arginine prevented them from interacting with each other; an appropriate exchange was the introduction of the hydrophobic methionine instead of the polar histidine across from the aspartate.
These results suggest a sodium binding motif in which an aspartate and a histidine are located in a loop across from each other. In particular, our results suggest that in Kir2 and Kir3 channels, DLR(K/N)SH is a sodium binding motif.
Another Kir channel that possesses an aspartate and a histidine is Kir5.1 that our data identifies to be sodium sensitive as well. The loop residues in between the aspartate and the histidine in this case are quite different from those in other Kir channels. As a result, while the corresponding D205N mutation eliminated sodium sensitivity, the H210M did not. Mutation of the loop residues in between the aspartate and the histidine to those that exist in the sodium-insensitive channel Kir6.2 in addition to the H210M mutation resulted in loss of sodium sensitivity. This suggests that the loop residues between the aspartate and the histidine are crucial to sodium sensitivity. Altogether, the common residues in the critical loop in the cytosolic domain of Kir5.1, Kir3 channels and Kir2.1_N216D (Fig. 1a) suggest that the sodium binding motif in Kir channels is DXRXXH.
Examination of the critical loop in the various crystallographic structures of the cytosolic domains of Kir channels (Kir2.120, Kir3.119,20,40, and Kir3.241) shows variability in the structure of the loop. Moreover, in the structure of the cytosolic domain of sodium-sensitive Kir3.2 this loop is absent, further indicating a likely flexibility of the loop. It is thus possible that in addition to affecting the interaction between the aspartate and the arginine, the sodium ions may also determine the conformation of this critical loop.
The physiological role of Na+-sensitive potassium channels is not clear. Our discovery that Kir5.1 confers Na+ sensitivity to other channels it heteromerizes with opens interesting possibilities for the physiological role of Na+ sensitivity of K+ channels. Kir5.1 is expressed in kidney tubules42, and brainstem neurons43 among other parts of the body. A negative membrane potential of kidney tubular cells contributes significantly towards Na+ reabsorption from the tubular lumen into the cell. Na+ sensitive K+ conductances along the Na+ reabsorbing length of the nephron would ensure that the membrane potential is maintained at negative levels near the equilibrium potential for potassium. Interestingly, Kir5.1 also enhances the CO2/pH sensitivities of channels it heteromerizes with and has been suggested to contribute to the central CO2 chemosensitivity of brainstem cardio-respiratory nuclei43. An additional possible role for Na+ sensitivity in the physiology of these neurons remains to be explored.
METHODS
Molecular modeling via molecular dynamics
MD simulations were performed using CHARMM27 version 26. The crystallographic structure of the cytosolic domain of Kir3.1 (PDB accession number 1N9P19) was used as the initial structure in the simulations of Kir3.1, and as the basis for the homology model of the cytosolic domain of Kir3.4. The identity between the cytosolic domains of Kir3.1 and Kir3.4 is 63.3%. 20.5% of the remaining residues are similar and only 16.2% are completely different. The high sequence identity of these channels justifies using the cytosolic domain of Kir3.1 for constructing a homology model of Kir3.4. Within the sequence of the critical loop examined (Fig. 1a), the sequence of the channels differs in two residues only. Following a minimization stage using the Steepest Descent and the adopted-basis Newton Raphson algorithms, a 1.8nsec MD simulation at room temperature was carried out for Kir3.4 and its mutants with a time step of 0.001psec and using the leap frog Verlet integrator with Langevin dynamics27. The force field for the energy calculation was the CHARMM force field27. The non-bonded cut-off distance CUTNB that was used in combination with the FSWITCH option in CHARMM was 13Å. The environment was modeled by a distance-dependent dielectric. As the cut-off distance could affect the MD results44, the model obtained based on the MD simulations was confirmed experimentally.
Simulations using the primary hydration shell method
The primary hydration shell (PHS) method28 is a method that provides both an efficient representation of solvation effects and a flexible nonspherical restraining potential. In order to replace the bulk representation of the solvent, a restraining force that balances the instantaneous pressure inside the primary solvent shell is applied.
The MD simulations were performed using CHARMM, version 2627.
The cytosolic domain was surrounded by a 30Å sphere of water molecules whose origin was located at its center of mass. Water molecules located within 2.6Å from the cytosolic domain were deleted and the combined system was minimized. After removing all waters located 5Å from the cytosolic domain, the system was further minimized. The structure was then equilibrated and the simulation was run at room temperature for 2nsec with a time step of 0.001psec, a restraining force of 0.95Kcal/moleÅ, and using the leap frog Verlet integrator with Langevin dynamics employed in the presence of the miscellaneous mean field potential (MMFP) in CHARMM keeping the center of mass of channel fixed and allowing the application of the PHS model.
Expression of Recombinant Channels in Xenopus Oocytes
Point mutations on the background of the control Kir3.4_S143T (Kir3.4*) or Kir3.1_F137S (Kir3.1*)10,11,45 were generated using the Quickchange site-directed mutagenesis kit (Stratagene, LaJolla, CA). cRNAs were transcribed in vitro using the “Message Machine” kit (Ambion, Austin, TX). cRNA concentration of Kir3.1* or Kir3.4* or Kir2.1 was estimated from two successive dilutions, which were electrophoresed in parallel on formaldehyde gels and compared to known concentrations of an RNA marker (GIBCO, Gaithersburg, MD). Oocytes were isolated and microinjected as previously described46. Expression of channel proteins in Xenopus oocytes was accomplished by injection of the desired amount of cRNA. Oocytes were injected with cRNA, 2 ng of channel. All oocytes were maintained at 17°C. Two-electrode voltage clamp recordings were performed 2 days following injection. The use of Xenopus laevis frogs was approved by the institutional IACUC committee.
Two-Electrode Voltage-Clamp Recording and Analysis
Whole-cell currents were measured by conventional two-microelectrode voltage clamp with a GeneClamp 500 amplifier (Axon Instruments, Union City, CA), as previously reported46. A high-potassium (HK) solution was used to superfuse oocytes (in mM: 96 KCl, 1 NaCl, 1 MgCl2, 5 KOH/HEPES [pH 7.4]). Basal currents represent the difference of inward currents obtained (at −80 mV) in the presence of 3 mM BaCl2 in HK solution from those in the absence of Ba2+. Each experiment shown was performed on 3–5 oocytes of the same batch. A minimum of two batches of oocytes were tested for each normalized recording shown. Recordings from different batches of oocytes were normalized by the mean of whole-cell basal currents from oocytes expressing the control channel Kir3.1* or Kir3.4*. Statistics (i.e., mean and standard error of the mean) of each construct were calculated from all of the normalized data recorded from different batches of oocytes.
Macropatch Recording and Analysis
Macropatch channel activity was recorded on oocytes under the inside-out mode of standard patch-clamp methods47, as previously described17. The bath and pipette solutions were composed of 96 mM KCl, 1 mM MgCl2, 5 mM EGTA and 10 mM HEPES, pH 7.4. For the Kir3.1/Kir3.2 channel and its mutants 2mM of MgCl2 were used. PtdIns(4,5)P2 was from Boehringer Mannheim. PtdIns(4,5)P2 was sonicated in cool water or on ice for 3 min before application, and was used at a final concentration of 2.5 μM. MgATP was from Sigma-Aldrich, and was used at a final concentration of 5mM. Na+ was added to the bath solution at a concentration of 30mM. Currents were recorded at a holding membrane potential of −100 mV. Recordings were made using the Axon 200A patch-clamp amplifier. Data were sampled at 5 kHz, filtered at 2 kHz and stored on a PC-compatible computer. Analysis was carried out using Clampfit 9.
Supplementary Material
Acknowledgments
We thank E. Findeis, T. Borges, V. Petrou and H. Vaananen for oocyte preparation. This work was supported by a NIH grant (HL-59949) to D.E.L. D.E.L is an Established Investigator of the American Heart Association.
Footnotes
AUTHOR CONTRIBUTIONS
ARD designed and performed the molecular modeling studies, the mutagenesis, and the TEVC experiments, and wrote the manuscript. JLS (Kir3.4*, Kir2.1), QZ (Kir4.1/Kir5.1) and RR (Kir3.1/Kir3.2, Kir4.1) performed the inside-out macropatch recordings. ARM carried out the PIP2 dose-response curve and ZZ performed the Na+ dose-response curve. DEL initiated and supervised the work, he edited the manuscript produced by ARD and revised the final form of the manuscript.
References
- 1.Tamargo J, Caballero R, Gómez R, Valenzuela C, Delpon E. Pharmacology of cardiac potassium channels. Cardio Res. 2004;62:9– 33. doi: 10.1016/j.cardiores.2003.12.026. [DOI] [PubMed] [Google Scholar]
- 2.Isomoto S, Kondo C, Kurachi Y. Inwardly rectifting potassium channels: their molecular heterogeneity and function. Jap J Physiol. 1997;47:11–39. doi: 10.2170/jjphysiol.47.11. [DOI] [PubMed] [Google Scholar]
- 3.Nerbonne JM, Nichols CG, Schwarz TL, Escande D. Genetic Manipulation of Cardiac K+ Channel Function in Mice What Have We Learned, and Where Do We Go From Here? Circ Res. 2001;89:944–956. doi: 10.1161/hh2301.100349. [DOI] [PubMed] [Google Scholar]
- 4.Hilgemann DW, Feng S, Nasuhoglu C. The complex and intriguing lives of PIP2 with ion channels and transporters. Sci STKE. 2001;RE19 doi: 10.1126/stke.2001.111.re19. [DOI] [PubMed] [Google Scholar]
- 5.Lopes CMB, Zhang H, Rohacs T, Yang J, Logothetis DE. Alterations in Conserved Interactions between PIP2 and Kir Channels Underlie Channelopathies. Neuron. 2002;34:933–944. doi: 10.1016/s0896-6273(02)00725-0. [DOI] [PubMed] [Google Scholar]
- 6.Krapivinsky G, Gordon EA, Wickman K, Velimirovic B, Krapivinsky L, Clapham DE. The G-protein-gated atrial K+ channel IKACh is a heteromultimer of two inwardly rectifying K+-channel proteins. Nature. 1995;374:135–141. doi: 10.1038/374135a0. [DOI] [PubMed] [Google Scholar]
- 7.Chan KW, Langan MN, Sui JL, Kozak JA, Pabon A, Ladias JA, Logothetis DE. A recombinant inwardly rectifying potassium channel coupled to GTP-binding proteins. J Gen Physiol. 1996;107:381–397. doi: 10.1085/jgp.107.3.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hedin KE, Lim NF, Clapham DE. Cloning of a Xenopus laevis inwardly rectifying K+ channel subunit that permits GIRK1 expression of IKACh currents in oocytes. Neuron. 1996;16:423–429. doi: 10.1016/s0896-6273(00)80060-4. [DOI] [PubMed] [Google Scholar]
- 9.Kofuji P, Davidson N, Lester HA. Evidence that neuronal G-protein-gated inwardly rectifying K+ channels are activated by G beta gamma subunits and function as heteromultimers. Proc Natl Acad Sci USA. 1995;92:6542–6546. doi: 10.1073/pnas.92.14.6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan KW, Sui JL, Vivaudou M, Logothetis DE. Control of channel activity through a unique amino acid residue of a G protein-gated inwardly rectifying K+ channel subunit. Proc Natl Acad Sci USA. 1996b;93:14193–14198. doi: 10.1073/pnas.93.24.14193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vivaudou M, Chan KW, Sui JL, Jan LY, Reuveny E, Logothetis DE. Probing the G-protein regulation of GIRK1 and GIRK4, the two subunits of the KACh channel, using functional homomeric mutants. J Biol Chem. 1997;272:31553–31560. doi: 10.1074/jbc.272.50.31553. [DOI] [PubMed] [Google Scholar]
- 12.Sui JL, Petit-Jacques J, Logothetis DE. Activation of the atrial KACh channel by the βγ subunits of G proteins or intracellular Na+ ions depends on the presence of phosphatidylinositol phosphates. Proc Natl Acad Sci USA. 1998;95:1307–1312. doi: 10.1073/pnas.95.3.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lesage F, Guillemare E, Fink M, Duprat F, Heurteaux C, Fosset M, Romey G, Barhanin J, Lazdunski M. Molecular properties of neuronal G-protein-activated inwardly rectifying K+ channels. J Biol Chem. 1995;270:28660–7. doi: 10.1074/jbc.270.48.28660. [DOI] [PubMed] [Google Scholar]
- 14.Ho IH, Murrell-Lagnado RD. Molecular determinants for sodium-dependent activation of G protein-gated K+ channels. J Biol Chem. 1999;274:8639–48. doi: 10.1074/jbc.274.13.8639. [DOI] [PubMed] [Google Scholar]
- 15.Ho IH, Murrell-Lagnado RD. Molecular mechanism for sodium-dependent activation of G protein-gated K+ channels. J Physiol. 1999b;520:645–51. doi: 10.1111/j.1469-7793.1999.00645.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sui JL, Chan KW, Logothetis DE. Na+ activation of the muscarinic K+ channel by a G-protein-independent mechanism. J Gen Physiol. 1996;108:381–391. doi: 10.1085/jgp.108.5.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang H, He C, Yan X, Mirshahi T, Logothetis DE. Activation of inwardly rectifying K+ channels by distinct PtdIns(4,5)P2 interactions. Nat Cell Biol. 1999;1:183–188. doi: 10.1038/11103. [DOI] [PubMed] [Google Scholar]
- 18.Tristani-Firouzi M, et al. Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome) J Clin Invest. 2002;110:381–388. doi: 10.1172/JCI15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nishida M, MacKinnon R. Structural Basis of Inward Rectification: Cytoplasmic Pore of the G Protein-Gated Inward Rectifier GIRK1 at 1.8 A Resolution. Cell. 2002;111:957–965. doi: 10.1016/s0092-8674(02)01227-8. [DOI] [PubMed] [Google Scholar]
- 20.Pegan S, Arrabit C, Zhou W, Kwiatkowski W, Collins A, Slesinger PA, Choe S. Cytoplasmic domain structures of Kir2.1 and Kir3.1 show sites for modulating gating and rectification. Nat Neurosci. 2005;8:279–287. doi: 10.1038/nn1411. [DOI] [PubMed] [Google Scholar]
- 21.Stevens EB, Woodward R, Ho IH, Murrell-Lagnado R. Identification of regions that regulate the expression and activity of G protein-gated inward rectifier K+ channels in Xenopus oocytes. J Physiol. 1997;503:547–62. doi: 10.1111/j.1469-7793.1997.547bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Di Cera E, Guinto ER, Vindigni A, Dang QD, Ayala YM, Wuyi M, Tulinsky A. The Na+ binding site of thrombin. J Biol Chem. 1995;270:22089–22092. doi: 10.1074/jbc.270.38.22089. [DOI] [PubMed] [Google Scholar]
- 23.Glusker JP. Structural aspects of metal liganding to functional groups in proteins. Adv Protein Chem. 1991;42:1–76. doi: 10.1016/s0065-3233(08)60534-3. [DOI] [PubMed] [Google Scholar]
- 24.Karpen ME, Tobias DJ, Brooks CL., III Statistical clustering techniques for the analysis of long molecular dynamics trajectories: analysis of 2.2-ns trajectories of YPGDV. Biochemistry. 1993;32:412–420. doi: 10.1021/bi00053a005. [DOI] [PubMed] [Google Scholar]
- 25.Carpenter GA, Grossberg S. ART 2: self-organization of stable category recognition codes for analog input patterns. Appl Opt. 1987;26:4919–4930. doi: 10.1364/AO.26.004919. [DOI] [PubMed] [Google Scholar]
- 26.Pao Y-H. Adaptive Pattern Recognition and Neural Networks. Addison-Wesley; New-York: 1989. [Google Scholar]
- 27.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comp Chem. 1983;4:187–217. [Google Scholar]
- 28.Beglov D, Roux B. Dominant Solvation Effects from Primary Shell of Hydration: Approximation for Molecular Dynamics Simulations. Biopolymers. 1995;35:171–178. [Google Scholar]
- 29.Zerangue N, Schwappach B, Jan YN, Jan LY. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane KATP channels. Neuron. 1999;22:537–548. doi: 10.1016/s0896-6273(00)80708-4. [DOI] [PubMed] [Google Scholar]
- 30.Lamoureux G, Roux B. Absolute hydration free energy scale for alkali and halide ions established from simulations with a polarizable force field. J Phys Chem B. 2006;110:3308–3322. doi: 10.1021/jp056043p. [DOI] [PubMed] [Google Scholar]
- 31.Wang P, Wesdemiotis C, Kapota C, Ohanessian G. The Sodium Ion Affinities of Simple Di-, Tri-, and Tetrapeptides. J Am Soc Mass Spectrom. 2007;18:541–52. doi: 10.1016/j.jasms.2006.10.024. [DOI] [PubMed] [Google Scholar]
- 32.Kapota C, Ohanessian G. The low energy tautomers and conformers of the dipeptides HisGly and GlyHis and of their sodium ion complexes in the gas phase. Phys Chem Chem Phys. 2005;7:3744–3755. doi: 10.1039/b508092d. [DOI] [PubMed] [Google Scholar]
- 33.Yu XM. The Role of Intracellular Sodium in the Regulation of NMDA-Receptor- Mediated Channel Activity and Toxicity. Mol Neurobiol. 2006;33:63–80. doi: 10.1385/MN:33:1:063. [DOI] [PubMed] [Google Scholar]
- 34.Dryer SE. Na+-activated K+ channels: A new family of large-conductance ion channels. Trends Neurosci. 1994;17:155–160. doi: 10.1016/0166-2236(94)90093-0. [DOI] [PubMed] [Google Scholar]
- 35.Salkoff L, Butler A, Ferreira G, Santi C, Wei A. High-conductance potassium channels of the SLO family. Nat Rev Neurosci. 2006;7:921–931. doi: 10.1038/nrn1992. [DOI] [PubMed] [Google Scholar]
- 36.Komwatana P, Dinudom A, Young JA, Cook DI. Cytosolic Na+ controls and epithelial Na+ channel via the Go guanine nucleotide-binding regulatory protein. Proc Natl Acad Sci USA. 1996;93:8107–8111. doi: 10.1073/pnas.93.15.8107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harding MM. The architecture of metal coordination groups in proteins. Acta Crystallogr D Biol Crystallogr. 2004;60:849–859. doi: 10.1107/S0907444904004081. [DOI] [PubMed] [Google Scholar]
- 38.Logothetis DE, Jin T, Lupyan D, Rosenhouse-Dantsker A. Phosphoinositide- mediated gating of inwardly rectifying K+ channels. Pflugers Arch. 2007a;455:83–95. doi: 10.1007/s00424-007-0276-5. [DOI] [PubMed] [Google Scholar]
- 39.Logothetis DE, Lupyan D, Rosenhouse-Dantsker A. Diverse Kir modulators act in close proximity to residues implicated in phosphoinositide binding. J Physiol. 2007b;582:953–965. doi: 10.1113/jphysiol.2007.133157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nishida M, Cadene M, Chait BT, Mackinnon R. Crystal structure of a Kir3.1- prokaryotic Kir channel chimera. Embo J. 2007;26:4005–4015. doi: 10.1038/sj.emboj.7601828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Inanobe A, Matsuura T, Nakagawa A, Kurachi Y. Structural diversity in the cytoplasmic region of G protein-gated inward rectifier K+ channels. Channels. 2007;1:39–45. [PubMed] [Google Scholar]
- 42.Tucker SJ, Imbrici P, Salvatore L, D’Adamo MC, Pessia M. pH dependence of the inwardly rectifying potassium channel, Kir5.1, and localization in renal tubular epithelia. J Biol Chem. 2000;275:16404–7. doi: 10.1074/jbc.C000127200. [DOI] [PubMed] [Google Scholar]
- 43.Wu J, Xu H, Shen W, Jiang C. Expression and coexpression of CO2-sensitive Kir channels in brainstem neurons of rats. J Membr Biol. 2004;197:179–91. doi: 10.1007/s00232-004-0652-4. [DOI] [PubMed] [Google Scholar]
- 44.Schreiber H, Steinhauser O. Cutoff size does strongly influence molecular dynamics results on solvated polypeptides. Biochemistry. 1992;31:5856–60. doi: 10.1021/bi00140a022. [DOI] [PubMed] [Google Scholar]
- 45.Chan KW, Sui JL, Vivaudou M, Logothetis DE. Specific regions of heteromeric subunits involved in enhancement of G protein-gated K+ channel activity. J Biol Chem. 1997;272:6548–6555. doi: 10.1074/jbc.272.10.6548. [DOI] [PubMed] [Google Scholar]
- 46.He C, Yan X, Zhang H, Mirshahi T, Jin T, Huang A, Logothetis DE. Identification of critical residues controlling G protein-gated inwardly rectifying K+ channel activity through interactions with the beta gamma subunits of G proteins. J Biol Chem. 2002;277:6088–6096. doi: 10.1074/jbc.M104851200. [DOI] [PubMed] [Google Scholar]
- 47.Hilgemann DW. In: Single-Channel Recording. Sakmann B, Neher E, editors. Plenum; New York: 1995. pp. 307–327. [Google Scholar]
- 48.DeLano WL. The PyMOL molecular graphics system. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.