

Abstract
Inflammation is closely intertwined with pathogenesis of Parkinson's disease (PD). Increasing evidence suggests that inhibition of glia-mediated inflammation might represent a promising therapeutic target for PD. Glia maturation factor (GMF), an inflammatory protein, predominantly localized in astrocytes is previously isolated, sequenced and cloned in our laboratory. In the present investigation, we demonstrate that GMF-deficiency in astrocytes upregulates the antioxidant status and limit the extent of lipid peroxidation and production of reactive oxygen species (ROS) along with diminished nuclear factor-κB-mediated inflammatory responses in 1-methyl-4-phenylpyridinium (MPP+)-induced toxicity. Primary astrocytes obtained from wild-type (Wt) and GMF-deficient (GMF-KO) mice were treated with 5, 10, and 20 μM MPP+ for 24, 48, and 72 h in vitro. Our results show decreased release of ROS and increased level of glutathione in astrocytes obtained from GMF-KO mice when compared to astrocytes derived from Wt mice following MPP+ treatment. Additionally, we found decreased activity of NF-κB, and reduced levels of proinflammatory tumor necrosis factor- α, interleukin-1β (IL-1β), IL-17, IL-33, and chemokine (C–C motif) ligand 2 (CCL2) in GMF-KO astrocytes when compared to Wt astrocytes. Our overall results suggest that GMF-KO astrocytes are significantly resistant to MPP+ toxicity when compared to Wt astrocytes.
Keywords: Glia maturation factor, Astrocytes, MPP+, Cytotoxicity, NF-κB, Cytokines
Introduction
Parkinson's disease (PD) is a neurodegenerative movement disorder caused by selective decay of dopaminergic neurons in the nigrostriatal pathway. Although, the cause of PD is not clearly understood, exposure to certain environmental neurotoxic pollutants such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) are known to be associated with the pathogenesis of PD in human and in animal models of PD. Several studies have suggested involvement of glia activation in the pathogenesis of PD along with other components of neurodegenerative processes, such as oxidative stress, inflammation, and apoptosis. Human postmortem studies have also suggested that altered glial functions and oxidative damage to lipids, proteins, and DNA in the PD brains (Dexter et al. 1994).
Astrocytes are abundant neuron-supporting glial cells that equipped with powerful arsenal of antioxidative molecules and neurotrophic factors. Astrocytes most likely improve dopaminergic neuronal survival by uptake of toxic molecules, such as glutamate and α-synuclein (Hazell et al. 1997; Lee et al. 2010; Miyazaki et al. 2013). Numerous studies have demonstrated that astrocytes play an important role in neurodegenerative disorders and exert a protective function against oxidative stress because of their effects on the metabolism of the antioxidant glutathione (GSH) and the defense against reactive oxygen species (ROS) (Wilson, 1997). Indeed, astrocyte-derived antioxidants reduce oxidative stress level in the surrounding neurons (Makar et al. 1994; Chen et al. 2009). Recently, it has been reported that astrocytes expressed metallothionein, protecting dopaminergic neurons through their quinone-quenching or free radical-scavenging properties (Miyazaki et al., 2011). Thus, increased numbers of healthy astrocytes, which possess antioxidative potential, are potentially viable therapeutic approach to prevent the degenerative process of PD. The neurotoxin 1-methyl-4-phenylpyridinium (MPP+), a high-affinity inhibitor of mitochondrial complex I, is a metabolite of MPTP oxidation formed by monoamine oxidase-B in astrocytes and it is a common neurotoxin used to investigate the alteration of astrocytic functions relevant to PD.
Glia maturation factor (GMF), a highly conserved brain protein, was previously isolated, sequenced and cloned in our laboratory (Lim et al. 1990; Kaplan et al. 1991; Zaheer et al. 1993). It has been previously documented that GMF is an intracellular regulator of stress-activated signal transduction and activates p38 MAP kinase and transcription factor NF-κB in astrocytes (Lim and Zaheer 1996; Zaheer and Lim 1996; Lim et al. 2000; Zaheer et al. 2001). We have also shown that the overexpression of GMF in astrocytes leads to the destruction of primary oligodendrocytes, the myelin-forming cells in the CNS, by interactions between astrocytes, microglia, and oligodendrocytes (Zaheer et al., 2007a). In addition, GMF has an apparently high rate of oxidase activity causing the formation of ROS, which can initiate lipid peroxidation and damage cell membranes (Kaimori et al. 2003; Zaheer et al. 2004). Recently, we have demonstrated involvement of proinflammatory GMF in the pathogenesis of several neurodegenerative diseases (Zaheer et al. 2007b; 2008; 2013). However, the role of GMF in MPP+-induced toxicity in astrocytes was not investigated. Ever since, deficiency of GMF modulates oxidative stress and inflammation in number of pathological conditions (Zaheer et al. 2007b; 2008; 2013), and is also crucial for the survival of dopaminergic neurons in the pathogenesis of PD (unpublished results). Therefore, in the present investigation, we report that GMF-deficient astrocytes are more resistant to MPP+-induced toxicity than astrocytes from Wt mice and this resistance may be associated with limitation of oxidative threat and down regulation of NF-κB-mediated inflammation.
Materials and Methods
Reagents
Enzyme-linked immunosorbent assay (ELISA) kits for tumor necrosis factor- alpha (TNF-α), chemokine (C–C motif) ligand 2 (CCL2), interleukin-17 (IL-17) and IL-33 and IL-1 β were purchased from R&D Systems (Minneapolis, MN). Assay kits for ROS and NF-κB were purchased from Abcam (Cambridge, MA). Assay kits for GSH and thiobarbituric acid reactive substance (TBARS) were from Cayman Chemical Company (Ann Arbor, MI). Antibody for iNOS was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). MPP+ iodide was purchased from Sigma-Aldrich (St. Louis, MO). Pregnant C57BL/6 mice were from Charles River (Wilmington, MA). GMF-deficient (GMF-KO) mice were maintained by backcross breeding to C57BL/6 for 10–12 generations. These mice were bred and maintained in the animal colony at The University of Iowa according to the guidelines of Institutional Animal Care and Use Committee (IACUC).
Primary Cultures of Mouse astrocyte
Primary cultures of astrocytes were prepared essentially as described previously by us (Zaheer et al. 2001) and by others (Assouline et al. 1983; Delgado and Ganea 2003). In brief, mixed glial culture were established from newborn mouse brain by trypsin dissociation and cultured for 8–10 days as previously described. Astrocytes were purified by differential attachment as follows. The mixed glial cells were subcultured by trypsinization and reseeding. Twenty minutes after plating, when approximately two thirds of the cells (mainly astrocytes) have attached, the medium was changed to remove the floating unattached cells. The adhered cells were permitted to grow to a confluent monolayer. Cells were subcultured at least twice to achieve homogeneous monolayers of astrocytes. Astrocytes were grown in DMEM/F12 (Life Technologies, Grand Island, NY) containing 5 % fetal bovine serum. Astrocyte cultures were >98 % positive for glial fibrillary acidic, a specific marker for astrocytes.
MPP+ Treatments
For MPP+ treatments, primary cultures of mouse astrocytes obtained from Wt and GMF-KO mice were seeded into 24 well plates or cell culture flasks (25 cm2) at 1×106 cells /ml in complete medium for overnight at 37 °C. Then, the cells were incubated with various concentrations of MPP+ (5, 10, and 20 μM) for 24, 48, and 72 h in serum free medium. After the termination of incubation, cells were collected for the assays of TBARS, GSH, ROS, and the culture supernatant were collected for estimation of proinflammatory cytokines and chemokine. Protein was assayed in culture supernatant by BCA method.
Cytotoxicity Assay
Viability of astrocytes was quantitated by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay (Hansen et al. 1989). In order to investigate the influence of MPP+ on cell viability, we treated GMF-KO astrocytes and Wt astrocytes with MPP+ at different concentrations (5–100 μM). Ten microliters of MTT solution (5 mg/ml) were added to each well and incubated for 4 h at 37 °C. The blue formazine formed in viable cells was dissolved by adding an equal volume of lysis buffer made of 20 % SDS dissolved in a 1:1 mixture of water and N,N-dimethyl-formamide, pH 4.7. The optical density was measured with a plate reader at 595 nm. Cell death was assessed in 50 μl cell supernatant by using a commercially available kit (CytoTox 96 nonradioactive cytotoxicity assay kit, Promega, WI, USA) according to the manufacturer's instructions. The amount of lactate dehydrogenase (LDH) release into the culture medium upon cell lysis was measured by the conversion of a tetrazolium salt into red formazan product. The absorbance, proportional to the lysed cells, was measured at 490 nm. The amount of LDH released by the cells in the presence of 1 % Triton X-100 was considered maximal absorbance.
Lipid Peroxidation, and Glutathione and Nitric Oxide Assays
TBARS, as a marker of lipid peroxidation, and GSH and nitric oxide (NO) were detected according to the kit manufacturer's instructions. The TBARS results were expressed as nanomoles TBARS formed per hour per milligram protein. GSH was calculated in terms micromoles GSH formed per milligram protein using a molar extinction coefficient of 13.6×103 M−1 cm−1. The NO results were expressed as micromoles NO release per milligram protein.
Measurement of ROS Generation
ROS formation was detected by a commercially available kit according to the manufacturer's instruction using a compound, DCFDA. In brief, after cell sonication in a lysis buffer (0.1 % SDS, Tris-HCl, pH 7.4) at 4 °C, samples were centrifuged at 6,000g for 20 min at 4 °C. Supernatants were collected and subjected to fluorescence analysis at 485 nm excitation and 535 nm emissions using a microplate reader.
NF-κB Assay
NF-κB activity was determined by mouse phospho-Rel A/NF-κB p65 immunoassay kit (Abcam, Cambridge, MA) according to the manufacturer's instruction. Briefly, cells were seeded in 96-well plate and incubated with 20 μM concentration of MPP for 24, 48, and 72 h at 37 °C. After preincubation in blocking buffer, cells were incubated overnight with primary antibody at 4 °C followed by appropriate secondary antibody. Cells were then exposed to substrate solution for 20–40 min and read the plate using a fluorescence plate reader.
Immunoblotting
Western blot was performed to detect iNOS in the cell fraction as described previously (Zaheer and Lim, 1996). After exposure to MPP+, cells were lysed in RIPA cell lysis buffer (50 mM Tris-HCl pH7.4, 150 mM NaCl, 1 mM EDTA, 1 % NP-40, 0.5 % deoxycholate) containing protease (Roche Diagnostics, Indianapolis, IN) and phosphatase inhibitor cocktails (Cell Signaling Technology, Danvers, MA). Samples containing equal amounts of protein (35 μg) were separated in 10 % SDS-polyacrylamide gels. Proteins were transferred onto PVDF membrane and incubated overnight at 4 °C with primary rabbit polyclonal antibody against NOS2 (Santa Cruz, CA; 1:200 dilutions) followed by secondary antibody conjugated to horseradish peroxidase (HRP). Proteins recognized by the antibody were visualized by enhanced chemiluminescence Femto kit (Thermo Scientific, Rockford, IL). Blots were stripped and reprobed for β-actin (Sigma) as a loading control. Bands intensity was measured by densitometry and quantified using NIH-Image J software.
Cytokines and Chemokine Assay by ELISA
TNF-α, IL-1β, IL-17, IL-33, and CCL2 protein concentrations in the culture supernatants were estimated by ELISA as specified in the manufacturer's protocol. The lower detection limits of these ELISA are in the range of 8–12 pg/ml.
Statistical Analysis
Results are expressed as mean±SEM. Statistical analysis of the data was done by independent student t test. The p value less than 0.05 were considered statistically significant.
Results
GMF Deficiency Protects Astrocytes from MPP+-Induced Toxicity
The primary cultures of astrocytes from GMF-KO mice and GMF-containing Wt mice were incubated with variable concentrations (0–100 μM) of MPP+ and the cell viability was measured by MTT assays. MPP+ at 24 (Fig. 1a) and 48 h (Fig. 1b) caused a concentration-dependent decrease of viable astrocytes derived from Wt mice. In contrast, cell viability was higher in GMF-KO astrocytes when compared with Wt astrocytes following MPP+ treatment. The amount of LDH release into the culture medium upon cell lysis was measured by the conversion of a tetrazolium salt into red formazan product as described in Experimental Procedures. Results in Fig. 1c, d demonstrate significantly higher amounts of LDH released from Wt astrocytes following MPP+ treatment for 24 and 48 h when compared to GMF-KO astrocytes. Our results clearly indicate that GMF deficiency protects astrocytes from MPP+-induced cytotoxicity.
Fig. 1.

Primary cultures of astrocytes derived from Wt and GMF-KO mice were incubated with various doses of MPP+ (0–100 μM) for 24 and 48 h. MPP+-induced cytotoxicity was measured by MTT reduction and LDH release assays. a MTT assay show higher cell viability in GMF-KO astrocytes when compared to Wt astrocytes, and b Wt astrocytes released more LDH when compared to GMF-KO astrocytes following MPP+ treatments. *p<0.05 (n=4), compared with the Wt astrocytes and GMF-KO astrocytes treated with MPP+ by student t test analysis
GMF Deficiency Protects Astrocytes from MPP+-Induced Oxidative Damage
The primary cultures of astrocytes derived from GMF-KO mice and Wt mice were incubated with 5, 10, and 20 μM MPP+ for 24, 48, and 72 h. TBARS level following MPP+ treatment in the absence of GMF (GMF-KO astrocytes) or presence of GMF (Wt astrocytes) was measured in the culture supernatant to demonstrate the extent of GMF-dependent oxidative damage to lipids. The results show significantly (p<0.05) decreased level of TBARS content (2.68±0.46; 3.81±0.22; and5.27±1.32 nmol TBARS formed/mg protein) in GMF-KO astrocytes as compared to control Wt astrocytes (6.57±0.49; 9.43±0.28; and 8.61±1.79 nmol TBARS formed/mg protein) at 24 h with 5, 10 and 20 μM MPP+ treatments (Fig. 2a). We found almost similar effect at 48 and 72 h of MPP+ treatments. After 24, 48, and 72 h of MPP+ treatment at 5 μM concentration, we found consistent increase of TBARS level in Wt astrocytes (6.57±0.49; 8.86±1.94; and 11.51±1.26 nmol TBARS formed/mg protein), however; GMF-KO astrocytes showed significant and marked reduction of TBARS level (2.68±0.46; 4.06±0.98; and 4.49± 0.71 nmol TBARS formed/mg protein) as compared to control Wt astrocytes. Thus, this culture system allowed us to examine specific effects of GMF-KO in MPP+-induced oxidative damage to lipids in astrocytes.
Fig. 2.

Primary cultures of astrocytes derived from Wt and GMF-KO mice were incubated with MPP+ (5, 10, 20 μM) for 24, 48, and 72 h. Levels TBARS (a), GSH (b), and ROS (c) were assayed in the cell lysate. TBARS and GSH (n=4); ROS (n=5); Values are means±SEM. *p<0.05, compared with the Wt astrocytes and GMF-KO astrocytes treated with MPP+
Decrease in cellular GSH level increases the sensitivity of astrocytes to toxic insults and induces changes in mitochondrial function. The GSH content following MPP+ treatment in the GMF-KO astrocytes and Wt astrocytes was estimated to demonstrate the role of GMF in MPP+-induced free radicals imbalance in astrocytes. A significant increase (p<0.05) in GSH content (19.50±1.50; 14.12±0.66; and 12.86± 1.69 μmol GSH/mg protein) was detected in GMF-KO astrocytes when compared to Wt astrocytes (9.84±0.29; 7.29±0.53; and 5.97±0.80 μmol GSH/mg protein) following MPP+ treatment for 24 h in a dose-dependent (5, 10, and 20 μM) fashion (Fig. 2b). Similarly, we found significant (p<0.05) increase in GSH content in the GMF-KO astrocytes as compared to Wt astrocytes following MPP+ treatment at 48 and 72 h in a dose-dependent manner.
To determine whether deficiency of GMF suppress the MPP+-induced ROS production in astrocytes, we measured total ROS production in Wt astrocytes and GMF-KO astrocytes following MPP+ treatment at different time points. The formation of ROS within the cells was determined by monitoring a conversion of DCFH2-DA to DCF. ROS level was significantly (p<0.05) decreased in GMF-KO astrocytes (51.47±8.33; 50.49±4.82; and 152.94±20.53 DCF) when compared to Wt astrocytes (214.10±29.91; 222.05±25.01; and 222.54±19.64 DCF) at 5, 10, and 20 μM respectively, at 24 h after MPP+ treatment. Similarly, we found significant (p<0.05) decrease in ROS in the GMF-KO astrocytes as compared to Wt astrocytes following MPP+ treatment at 48 and 72 h (Fig. 2c).
GMF-Dependent Downregulation of NF-κB Activity in Astrocytes Following MPP+ Treatment
It is known that NF-κB regulates the expression of iNOS and several proinflammatory cytokines and chemokines. ELISA results (Fig. 3) show significant decrease (p<0.05) in NF-κB activity in GMF-KO astrocytes (117.69±21.31; 236.32±31.42; and 204.91±21.05 phosphorylation of RelA/NF-kB p65) when compared to Wt astrocytes (242.83±25.23; 422.23±29.69; and 513.76±50.56 phosphorylation of RelA/NF-kB p65) following 20 μM MPP+ treatment for 24, 48, and 72 h. Since we observed that astrocytes are more vulnerable to MPP+-induced toxicity with 20 μM, we have chosen a single dose of MPP+ for our experiments reported in Fig. 3.
Fig. 3.
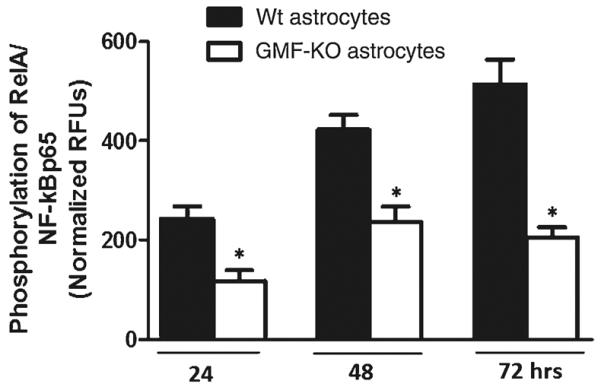
Reduction in NF-κB activity in GMF-KO astrocytes compared to Wt primary mouse astrocytes. Astrocytes were seeded and treated with 20 μM MPP+ for 24, 48, and 72 h and levels of NF-κB activity were measured by ELISA according to manufacturer's protocol. A significant time-dependent suppression of NF-κB activation following MPP+ treatment was seen in GMF-KO astrocytes as compared with Wt astrocytes. Values are means±SEM (n=6). *p<0.05, compared with the Wt astrocytes and GMF-KO astrocytes treated with MPP+
Western blot analysis was performed to determine the expression of iNOS in GMF-KO astrocytes and Wt astrocytes following MPP+ treatment (20 μM). Expression of iNOS was significantly (p<0.05) decreased in GMF-KO astrocytes when compared to Wt astrocytes following MPP+ toxicity (Fig. 4). Total NO level was significantly decreased in GMF-KO astrocytes when compared to Wt astrocytes following MPP+ toxicity in dose and time-dependent fashion (Fig. 5). Thus, these results demonstrate that GMF-deficient astrocytes are resistant to MPP+-induced oxidative damage due to decrease lipid peroxidation, ROS, iNOS, and NO production as well as by upregulation of glutathione content.
Fig. 4.
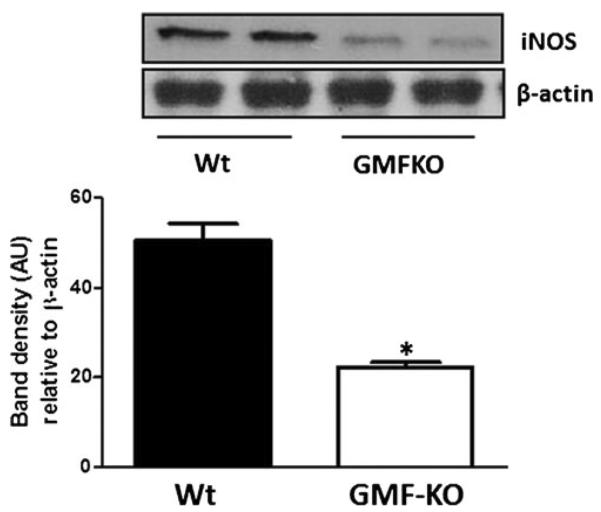
Downregulation of MPP+-dependent proinflammatory iNOS expression in GMF-KO astrocytes. Astrocytes derived from Wt and GMF-KO mice were incubated with MPP+ (20 μM) for 72 h. Cell lysates (35 μg protein per lane) were subjected to SDS-polyacrylamide gel electrophoresis followed by electroblotting. The blots were probed with anti-iNOS antibody and anti-β-actin antibody. Actin served as internal marker showing equal sample loading. Bands intensity was measured by densitometry and quantified using NIH-Image J software. Expression of iNOS was significantly decreased in GMF-KO astrocytes when compared to Wt astrocytes at 20 μM concentration of MPP+ after 72 h. Values are means±SEM (n=3). *p<0.05, compared with the Wt astrocytes and GMF-KO astrocytes treated with MPP+, AU arbitrary units
Fig. 5.
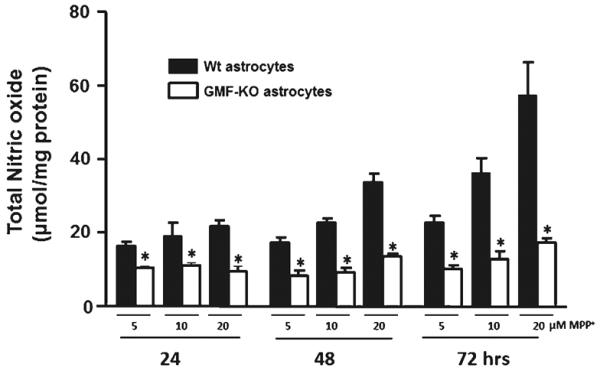
Nitric oxide (NO) levels were significantly decreased in GMF-KO astrocytes following MPP+ treatment in a dose- (5, 10, and 20 μM) and time-dependent (24, 48, and 72 h) manner. Astrocytes were incubated with MPP+ for 24, 48, and 72 h and then the culture media were collected for NO assay using commercial kit. Values are expressed as means±SEM (n=5). *p<0.05, compared with the Wt astrocytes and GMF-KO astrocytes treated with MPP
GMF-Deficient Astrocytes Release Reduced Level of Cytokines and Chemokine Following MPP+ Treatment
TNF-α, IL-1β, IL-17, IL-33, and chemokine CCL2 are reported be over-expressed under neurotoxin insults. Results in Fig. 6 show significantly (p<0.05) decreased release of TNF-α (116.86±14.05; 121.08±12.62; and 119.89±27.45 pg/ml), IL-1β (87.11±5.98; 128.27±13.48; and 131.84±6.56 pg/ml), IL-17 (93.18±15.75; 97.19±13.48; and 117.67±9.77 pg/ml), IL-33 (57.06±11.36; 49.86±12.25; and 62.33±7.02 pg/ml), and CCL2 (54.42±7.70; 87.78±8.95; and 80.87±12.06 pg/ml) at 5, 10, and 20 μM, respectively, in GMF-KO astrocytes when compared to the release from Wt astrocytes TNF-α (177.57±11.51; 193.48±33.20; and 217.89±29.19), IL-1 β (141.06±26.74; 229.79±34.48; and 271.40±11.53), IL-17 (179.83±26.77; 192.54±23.72; and 257.16±37.25), IL-33 (97.53±13.77; 136.55±24.89; and 228.70±30.03), and CCL2 (131.76±16.27; 154.78±16.26; and 153.02±15.73) pg/ml with concentration 5, 10, and 20 μM MPP+, respectively, following MPP+ toxicity at 24 h (Fig. 6). Similarly, we found the significant (p<0.05) decrease in proinflammatory cytokines and chemokine in the GMF-KO astrocytes as compared to Wt astrocytes following MPP+ toxicity at 48 and 72 h in a dose-dependent manner (Fig. 6). These results indicate that MPP+-induced release of proinflammatory cytokines and chemokine were significantly diminished in GMF-KO astrocytes.
Fig. 6.

Reduced expressions of inflammatory cytokines/chemokine in primary cultures of GMF-KO astrocytes compared to Wt astrocytes following MPP+ treatment. Astrocytes were incubated with MPP+ for 24, 48, and 72 h at 5, 10, and 20 μM MPP+. After the incubation period was over the culture media were collected for the assay of proinflammatory cytokines and chemokine by ELISA. Levels of TNF-α, IL-1β, IL-17, IL-33, and CCL2 were significantly decreased in the GMF-KO astrocytes when compared to the Wt astrocytes in dose- (5, 10, and 20 μM) and time-dependent manner (24, 48, and 72 h) following MPP+ treatment. Values are means±SEM (n=5). *p<0.05, compared with the Wt astrocytes and GMF-KO astrocytes treated with MPP+
Discussion
Enhanced expression of GMF in the brain has been documented in many conditions associated with inflammation and neurodegeneration (Zaheer et al., 2007a; Zaheer et al., 2007b; Thangavel et al., 2012). Numerous studies have shown that inflammation is common to a variety of neurodegenerative diseases, including Parkinson's disease. Chronic inflammation usually involves activation of glial cells resulting in the production of inflammatory mediators such as cytokines/chemokines, ROS and RNS. In this regard, our novel approach to down regulate GMF expression in CNS to stop the deleterious inflammation associated with neurodegeneration is novel because such an approach may be beneficial in Parkinson's disease. We have recently demonstrated, using GMF-deficient (GMF-KO) mice that an absence of endogenous GMF delayed the onset and drastically reduced loss of dopaminergic neurons in substantia nigra and striatum of mice following MPTP administration (unpublished results). In this study, we report that GMF-KO astrocytes showed limited oxidative damage and secretion of inflammatory mediators following MPP+ treatment, demonstrating neuroinflammatory function of GMF in MPP+ toxicity. Our present results show decreased release of ROS and increased level of GSH in GMF-KO astrocytes when compared to Wt astrocytes following MPP+ treatment. We also found decreased activity of NF-κB, and expression of reduced levels of proinflammatory TNF-α, IL-1β, IL-17, IL-33, and CCL2 in GMF-KO astrocytes compared to Wt astrocytes following MPP+ treatment. Our results suggests that GMF-KO astrocytes are more resistant to PD relevant MPP+ toxicity compared to Wt astrocytes by reducing oxidative loads along with diminished levels of NF-κB and proinflammatory molecules. Our previous studies have demonstrated that enhanced expression of GMF in glia aggravates inflammation (Zaheer et al. 2002; 2008).
Inflammation mediated by the activation of astrocytes plays a crucial role in the neurodegeneration disorders such as PD. Activated astrocytes release a numerous proinflammatory cytokines, including TNF-α and chemokines such as CCL2, as well as ROS and NO (Regan et al. 2001; Matos et al. 2008). These factors may contribute to the neuroinflammatory process, resulting in dopaminergic neuronal cell death in PD. In the present study, we investigated whether deficiency of GMF confers modulatory effects on MPP+-induced toxicity in the mouse astrocytes in vitro. We demonstrate that GMF-KO astrocytes showed decreased release of a variety of proinflammatory mediators such as TNF-α and IL-1β through the suppression of NF-κB and its responsive gene, which suggests that deficiency of GMF could effectively ameliorate MPP+-induced toxicity. Previous studies have indicated that GMF deficiency contended the oxidative loads and limits the inflammatory response via the downregulation of NF-κB and its responsive genes. Our present study with LDH and MTT assays indicate that GMF-KO protects mouse astrocytes from MPP+-induced toxicity.
Oxidative stress has been proposed as one major factor leading to cell death. ROS, NO production, and lipid peroxidation may initiate neurotoxic cascade of events. Thus, the excess of free radicals may lead to peroxidative impairment of membrane lipids and consequently disrupt neuronal functions and cause cellular death. GSH is the major antioxidant present in high amounts in astrocytes as compared with neuronal cells and free radicals. It eliminates H2O2 and organic peroxides by GPx. A reduction in the level of GSH may impair H2O2 clearance and promote formation of OH•, the most toxic molecule to the brain, leading to more oxidant load and further oxidative damage (Dringen 2000; Khan et al. 2013). In the present study, MPP+ treatment with different concentrations at three different time points to astrocytes caused an overproduction of free radicals which, in turn, caused oxidative damages to membrane lipid and protein levels, and ultimately led to a decrease in GSH content. Our results suggest that GMF could protect astrocytes from oxidative stress generated by MPP+ which is consistent with previous reports where MPP+-induced toxicity leads to oxidative damage (Sundar Boyalla et al. 2011; Xiao et al. 2011).
Glial activation is considered a rapid cellular response to inflammation (McNaught and Jenner 1999; Koprich et al. 2008). Activation of astrocytes induces cytotoxic mediators such as NO and inflammatory cytokines, which may contribute to the disease progression including PD. Regulation of NF-κB is known to be redox-sensitive which depends on the availability of ROS. Several in vivo studies have reported the activation of NF-kB in response to MPP+ toxicity (Liu et al. 2010; Song et al. 2012). The present findings are in agreement with our earlier studies where overexpression of GMF led to NF-kB activation and small interfering RNA-mediated GMF knockdown almost completely blocked the GMF-dependent NF-kB activation (Lim et al. 2000; Zaheer et al. 2007a). NO production resulting from induced NOS2 gene expression and subsequent iNOS enzyme activation is a primary contributor to the inflammatory response. High expression of inflammatory cytokines are implicated in PD, with the observation of TNF-α, IL-1β, IL-17, and IL-33 elevation in MPP+-induced toxicity. These cytokines may have deleterious effects on the dopaminergic neurons in PD. Cytokines also directly bind to their receptors on the cell surfaces on dopaminergic neurons to initiate the process of neurodegeneration. Once activated, these cytokine receptors could trigger intracellular death related signaling pathways. In turn, this sequence of events results in an increased proinflammatory cytokine production, enhancement of leukocyte infiltration, and upregulation of adhesion molecules that contribute to both necrotic and apoptotic cell death (del Zoppo et al. 2000; Tansey and Goldberg 2010). Increased expression of proinflammatory cytokines and chemokines occurs in the CNS diseases such as Alzheimer's disease, Multiple Sclerosis, and PD. CCL2 facilitates the entry of inflammatory cells that are important for the CNS tissue damage and demyelination in the neurodegenerative diseases. Our present study showed that MPP+-induced CCL2 release from astrocytes, which could attract glial cells and inflammatory infiltrates at the site of inflammation in PD brain. Since the release of CCL2 is reduced in the GMF-KO mice, GMF-KO strategy may reduce the neuroinflammatory processes by decreased accumulation of inflammatory infiltrates. Recently, IL-17 secreting Th17 cells have been shown to infiltrate the mouse brain following neurodegeneration (Reynolds et al. 2010; Browne et al. 2013). Although IL-17 plays an important role in disease pathogenesis in autoimmune diseases (Zhang et al. 2013), Alzheimer's disease (Nie et al. 2013; Zhang et al. 2013), PD and post stroke neurodegeneration (Swardfager et al. 2013), its role in PD is not yet clear. Astrocytes, microglia, and neurons express receptor for IL-17, activation of which is known to activate NF-kB and GSK3 (Zepp et al. 2011). Moreover, IL-17 is known to induce activation of microglia (Murphy et al. 2010), and neuronal injury (Wang et al. 2009). Recently, Reynolds et al. have reported the upregulation of IL-17 concentration in the mouse brain following MPTP treatments (Reynolds et al. 2010). In agreement with this study, we also found upregulated release of IL-17 in mouse primary astrocytes and deficiency of GMF decreases its release significantly.
IL-33 has been shown to be increased in the tissues of various inflammatory and autoimmune diseases (Li et al. 2012; Pei et al. 2014). Recently, we have shown an increase of IL-33 release by recombinant GMF from mouse astrocytes and that IL-33 induces neuronal degeneration through the release of proinflammatory molecules such as NO and TNF-α (Kempuraj et al. 2013). Our present study indicates that MPP+ also increased the release of IL-33 as well as NO from the astrocytes, may play an important role in MPP+-induced neuronal degeneration and that this effect was reduced in GMF-KO astrocytes indicating the GMF deficiency may lead to neuroprotection. Additionally, MPP+ increased the release of TNF-α and IL-1β, the cytokines that are already known to induce neurodegeneration (Marx et al. 2001). Thus, GMF deficiency is associated with concomitant reduction of the associated oxidative loads, NF-kB downregulation, reduced iNOS expression, and cytokine levels after MPP+ toxicity. The inhibition of NF-kB translocation, iNOS expression, and cytokine level show that GMF has an inflammatory activity and deficiency of GMF could ameliorate the MPP+-induced toxicity. It could also be useful in slowing neuronal death and therefore, halting progression of the disease.
It has been already established that GMF is an important regulator of CNS inflammation (Lim et al. 2000; Zaheer et al. 2008; 2011) and inhibition of endogenous GMF by targeting GMF will provide an avenue for effective and selective strategy to slow deleterious oxidative and inflammatory processes in PD.
Using GMF-KO astrocytes, we determined how the lack of GMF affects the oxidative status following MPP+ insult. By studying side-by-side signal transudations in the total absence of endogenous GMF and in the presence of GMF, a stark contrast was obtained that facilitated to delineate the function of GMF inside the cell. These results suggest that deficiency of GMF might delay the onset and slow the progression of MPP+-induced toxicity in astrocytes by resistant to oxidative threats and downregulation of NF-kB mediated inflammatory responses. Further in-depth mechanistic study is needed to clarify the protective effect against MPP+-induced oxidative damage in animal models of PD.
Acknowledgments
This work was supported by the Department of Veterans Affairs Merit Review award (to AZ) and by the National Institute of Neurological Disorders and Stroke grants NS073670 (to AZ).
Abbreviations
- CCL2
(C–C motif) ligand 2
- DA
Dopamine
- ELISA
Enzyme-linked immunosorbent assay
- GMF
Glia maturation factor
- GMF-KO
GMF knockout
- GSH
Glutathione
- HRP
Horseradish peroxidase
- IL-1
Interleukin-1
- iNOS
Inducible nitric oxide synthase
- LDH
Lactate dehydrogenase
- MPP
1-Methyl-4-phenyl-pyridine
- MPTP
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MTT
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NF-kB
Nuclear transcription factor-κB
- PD
Parkinson's disease
- ROS
Reactive oxygen species
- SDS
Sodium dodecyl sulfate
- TBARS
Thiobarbituric acid reactive substance
- TNF-α
Tumor necrosis factor-alpha
- Wt
Wild-type
References
- Assouline JG, Bosch EP, Lim R. Purification of rat Schwann cells from cultures of peripheral nerve: an immunoselective method using surfaces coated with anti-immunoglobulin antibodies. Brain Res. 1983;277:389–392. doi: 10.1016/0006-8993(83)90953-8. [DOI] [PubMed] [Google Scholar]
- Browne TC, McQuillan K, McManus RM, O'Reilly JA, Mills KH, Lynch MA. IFN-gamma Production by amyloid beta-specific Th1 cells promotes microglial activation and increases plaque burden in a mouse model of Alzheimer's disease. J Immunol. 2013;190:2241–2251. doi: 10.4049/jimmunol.1200947. [DOI] [PubMed] [Google Scholar]
- Chen PC, Vargas MR, Pani AK, Smeyne RJ, Johnson DA, Kan YW, Johnson JA. Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson's disease: critical role for the astrocyte. Proc Natl Acad Sci U S A. 2009;106:2933–2938. doi: 10.1073/pnas.0813361106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Zoppo G, Ginis I, Hallenbeck JM, Iadecola C, Wang X, Feuerstein GZ. Inflammation and stroke: putative role for cytokines, adhesion molecules and iNOS in brain response to ischemia. Brain Pathol. 2000;10:95–112. doi: 10.1111/j.1750-3639.2000.tb00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado M, Ganea D. Vasoactive intestinal peptide prevents activated microglia-induced neurodegeneration under inflammatory conditions: potential therapeutic role in brain trauma. FASEB J. 2003;17:1922–1924. doi: 10.1096/fj.02-1029fje. [DOI] [PubMed] [Google Scholar]
- Dexter DT, Holley AE, Flitter WD, Slater TF, Wells FR, Daniel SE, Lees AJ, Jenner P, Marsden CD. Increased levels of lipid hydroperoxides in the parkinsonian substantia nigra: an HPLC and ESR study. Mov Disord. 1994;9:92–97. doi: 10.1002/mds.870090115. [DOI] [PubMed] [Google Scholar]
- Dringen R. Metabolism and functions of glutathione in brain. Prog Neurobiol. 2000;62:649–671. doi: 10.1016/s0301-0082(99)00060-x. [DOI] [PubMed] [Google Scholar]
- Hansen MB, Nielsen SE, Berg K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell killing. J Immunol Methods. 1989;119:203–210. doi: 10.1016/0022-1759(89)90397-9. [DOI] [PubMed] [Google Scholar]
- Hazell AS, Itzhak Y, Liu H, Norenberg MD. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) decreases glutamate uptake in cultured astrocytes. J Neurochem. 1997;68:2216–2219. doi: 10.1046/j.1471-4159.1997.68052216.x. [DOI] [PubMed] [Google Scholar]
- Kaimori JY, Takenaka M, Nakajima H, Hamano T, Horio M, Sugaya T, Ito T, Hori M, Okubo K, Imai E. Induction of glia maturation factor-beta in proximal tubular cells leads to vulnerability to oxidative injury through the p38 pathway and changes in antioxidant enzyme activities. J Biol Chem. 2003;278:33519–33527. doi: 10.1074/jbc.M301552200. [DOI] [PubMed] [Google Scholar]
- Kaplan R, Zaheer A, Jaye M, Lim R. Molecular cloning and expression of biologically active human glia maturation factor-beta. J Neurochem. 1991;57:483–490. doi: 10.1111/j.1471-4159.1991.tb03777.x. [DOI] [PubMed] [Google Scholar]
- Kempuraj D, Khan MM, Thangavel R, Xiong Z, Yang E, Zaheer A. Glia maturation factor induces interleukin-33 release from astrocytes: implications for neurodegenerative diseases. J Neuroimmune Pharmacol. 2013;8:643–650. doi: 10.1007/s11481-013-9439-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MM, Kempuraj D, Thangavel R, Zaheer A. Protection of MPTP-induced neuroinflammation and neurodegeneration by Pycnogenol. Neurochem Int. 2013;62:379–388. doi: 10.1016/j.neuint.2013.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koprich JB, Reske-Nielsen C, Mithal P, Isacson O. Neuroinflammation mediated by IL-1beta increases susceptibility of dopamine neurons to degeneration in an animal model of Parkinson's disease. J Neuroinflammation. 2008;5:8. doi: 10.1186/1742-2094-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Suk JE, Patrick C, Bae EJ, Cho JH, Rho S, Hwang D, Masliah E, Lee SJ. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem. 2010;285:9262–9272. doi: 10.1074/jbc.M109.081125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Li Y, Liu X, Gao X, Wang Y. IL-33 blockade suppresses the development of experimental autoimmune encephalomyelitis in C57BL/6 mice. J Neuroimmunol. 2012;247:25–31. doi: 10.1016/j.jneuroim.2012.03.016. [DOI] [PubMed] [Google Scholar]
- Lim R, Zaheer A. In vitro enhancement of p38 mitogen-activated protein kinase activity by phosphorylated glia maturation factor. J Biol Chem. 1996;271:22953–22956. doi: 10.1074/jbc.271.38.22953. [DOI] [PubMed] [Google Scholar]
- Lim R, Zaheer A, Lane WS. Complete amino acid sequence of bovine glia maturation factor beta. Proc Natl Acad Sci U S A. 1990;87:5233–5237. doi: 10.1073/pnas.87.14.5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim R, Zaheer A, Yorek MA, Darby CJ, Oberley LW. Activation of nuclear factor-kappaB in C6 rat glioma cells after transfection with glia maturation factor. J Neurochem. 2000;74:596–602. doi: 10.1046/j.1471-4159.2000.740596.x. [DOI] [PubMed] [Google Scholar]
- Liu L, Xu H, Jiang H, Wang J, Song N, Xie J. Ghrelin prevents 1-methyl-4-phenylpyridinium ion-induced cytotoxicity through anti-oxidation and NF-kappaB modulation in MES23.5 cells. Exp Neurol. 2010;222:25–29. doi: 10.1016/j.expneurol.2009.11.009. [DOI] [PubMed] [Google Scholar]
- Makar TK, Nedergaard M, Preuss A, Gelbard AS, Perumal AS, Cooper AJ. Vitamin E, ascorbate, glutathione, glutathione disulfide, and enzymes of glutathione metabolism in cultures of chick astrocytes and neurons: evidence that astrocytes play an important role in antioxidative processes in the brain. J Neurochem. 1994;62:45–53. doi: 10.1046/j.1471-4159.1994.62010045.x. [DOI] [PubMed] [Google Scholar]
- Marx CE, Jarskog LF, Lauder JM, Lieberman JA, Gilmore JH. Cytokine effects on cortical neuron MAP-2 immunoreactivity: implications for schizophrenia. Biol Psychiatry. 2001;50:743–749. doi: 10.1016/s0006-3223(01)01209-4. [DOI] [PubMed] [Google Scholar]
- Matos M, Augusto E, Oliveira CR, Agostinho P. Amyloid-beta peptide decreases glutamate uptake in cultured astrocytes: involvement of oxidative stress and mitogen-activated protein kinase cascades. Neuroscience. 2008;156:898–910. doi: 10.1016/j.neuroscience.2008.08.022. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Jenner P. Altered glial function causes neuronal death and increases neuronal susceptibility to 1-methyl-4-phenylpyridinium-and 6-hydroxydopamine-induced toxicity in astrocytic/ventral mesencephalic co-cultures. J Neurochem. 1999;73:2469–2476. doi: 10.1046/j.1471-4159.1999.0732469.x. [DOI] [PubMed] [Google Scholar]
- Miyazaki I, Asanuma M, Kikkawa Y, Takeshima M, Murakami S, Miyoshi K, Sogawa N, Kita T. Astrocyte-derived metallothionein protects dopaminergic neurons from dopamine quinone toxicity. Glia. 2011;59:435–451. doi: 10.1002/glia.21112. [DOI] [PubMed] [Google Scholar]
- Miyazaki I, Asanuma M, Murakami S, Takeshima M, Torigoe N, Kitamura Y, Miyoshi K. Targeting 5-HT1A receptors in astrocytes to protect dopaminergic neurons in parkinsonian models. Neurobiol Dis. 2013;59:244–256. doi: 10.1016/j.nbd.2013.08.003. [DOI] [PubMed] [Google Scholar]
- Murphy AC, Lalor SJ, Lynch MA, Mills KH. Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain Behav Immun. 2010;24:641–651. doi: 10.1016/j.bbi.2010.01.014. [DOI] [PubMed] [Google Scholar]
- Nie K, Zhang Y, Gan R, Wang L, Zhao J, Huang Z, Tang H, Wang L. Polymorphisms in immune/inflammatory cytokine genes are related to Parkinson's disease with cognitive impairment in the Han Chinese population. Neurosci Lett. 2013;541:111–115. doi: 10.1016/j.neulet.2013.02.024. [DOI] [PubMed] [Google Scholar]
- Pei C, Barbour M, Fairlie-Clarke KJ, Allan D, Mu R, Jiang HR. Emerging role of interleukin-33 in autoimmune diseases. Immunology. 2014;141:9–17. doi: 10.1111/imm.12174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan RF, Wang Y, Ma X, Chong A, Guo Y. Activation of extracellular signal-regulated kinases potentiates hemin toxicity in astrocyte cultures. J Neurochem. 2001;79:545–555. doi: 10.1046/j.1471-4159.2001.00590.x. [DOI] [PubMed] [Google Scholar]
- Reynolds AD, Stone DK, Hutter JA, Benner EJ, Mosley RL, Gendelman HE. Regulatory T cells attenuate Th17 cell-mediated nigrostriatal dopaminergic neurodegeneration in a model of Parkinson's disease. J Immunol. 2010;184:2261–2271. doi: 10.4049/jimmunol.0901852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JX, Shaw PC, Wong NS, Sze CW, Yao XS, Tang CW, Tong Y, Zhang YB. Chrysotoxine, a novel bibenzyl compound selectively antagonizes MPP(+), but not rotenone, neurotoxicity in dopaminergic SH-SY5Y cells. Neurosci Lett. 2012;521:76–81. doi: 10.1016/j.neulet.2012.05.063. [DOI] [PubMed] [Google Scholar]
- Sundar Boyalla S, Barbara Victor M, Roemgens A, Beyer C, Arnold S. Sex- and brain region-specific role of cytochrome c oxidase in 1-methyl-4-phenylpyridinium-mediated astrocyte vulnerability. J Neurosci Res. 2011;89:2068–2082. doi: 10.1002/jnr.22669. [DOI] [PubMed] [Google Scholar]
- Swardfager W, Winer DA, Herrmann N, Winer S, Lanctot KL. Interleukin-17 in post-stroke neurodegeneration. Neurosci Biobehav Rev. 2013;37:436–447. doi: 10.1016/j.neubiorev.2013.01.021. [DOI] [PubMed] [Google Scholar]
- Tansey MG, Goldberg MS. Neuroinflammation in Parkinson's disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis. 2010;37:510–518. doi: 10.1016/j.nbd.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangavel R, Stolmeier D, Yang X, Anantharam P, Zaheer A. Expression of glia maturation factor in neuropathological lesions of Alzheimer's disease. Neuropathol Appl Neurobiol. 2012;38:572–581. doi: 10.1111/j.1365-2990.2011.01232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DD, Zhao YF, Wang GY, Sun B, Kong QF, Zhao K, Zhang Y, Wang JH, Liu YM, Mu LL, Wang DS, Li HL. IL-17 potentiates neuronal injury induced by oxygen-glucose deprivation and affects neuronal IL-17 receptor expression. J Neuroimmunol. 2009;212:17–25. doi: 10.1016/j.jneuroim.2009.04.007. [DOI] [PubMed] [Google Scholar]
- Wilson JX. Antioxidant defense of the brain: a role for astrocytes. Can J Physiol Pharmacol. 1997;75:1149–1163. [PubMed] [Google Scholar]
- Xiao H, Lv F, Xu W, Zhang L, Jing P, Cao X. Deprenyl prevents MPP(+)-induced oxidative damage in PC12 cells by the upregulation of Nrf2-mediated NQO1 expression through the activation of PI3K/Akt and Erk. Toxicology. 2011;290:286–294. doi: 10.1016/j.tox.2011.10.007. [DOI] [PubMed] [Google Scholar]
- Zaheer A, Fink BD, Lim R. Expression of glia maturation factor beta mRNA and protein in rat organs and cells. J Neurochem. 1993;60:914–920. doi: 10.1111/j.1471-4159.1993.tb03237.x. [DOI] [PubMed] [Google Scholar]
- Zaheer A, Lim R. In vitro inhibition of MAP kinase (ERK1/ERK2) activity by phosphorylated glia maturation factor (GMF) Biochemistry. 1996;35:6283–6288. doi: 10.1021/bi960034c. [DOI] [PubMed] [Google Scholar]
- Zaheer A, Mathur SN, Lim R. Overexpression of glia maturation factor in astrocytes leads to immune activation of microglia through secretion of granulocyte-macrophage-colony stimulating factor. Biochem Biophys Res Commun. 2002;294:238–244. doi: 10.1016/S0006-291X(02)00467-9. [DOI] [PubMed] [Google Scholar]
- Zaheer A, Yang B, Cao X, Lim R. Decreased copper-zinc superoxide dismutase activity and increased resistance to oxidative stress in glia maturation factor-null astrocytes. Neurochem Res. 2004;29:1473–1480. doi: 10.1023/b:nere.0000029558.82943.00. [DOI] [PubMed] [Google Scholar]
- Zaheer A, Yorek MA, Lim R. Effects of glia maturation factor overexpression in primary astrocytes on MAP kinase activation, transcription factor activation, and neurotrophin secretion. Neurochem Res. 2001;26:1293–1299. doi: 10.1023/a:1014241300179. [DOI] [PubMed] [Google Scholar]
- Zaheer A, Zaheer S, Sahu SK, Knight S, Khosravi H, Mathur SN, Lim R. A novel role of glia maturation factor: induction of granulocyte-macrophage colony-stimulating factor and proinflammatory cytokines. J Neurochem. 2007a;101:364–376. doi: 10.1111/j.1471-4159.2006.04385.x. [DOI] [PubMed] [Google Scholar]
- Zaheer A, Zaheer S, Sahu SK, Yang B, Lim R. Reduced severity of experimental autoimmune encephalomyelitis in GMF-deficient mice. Neurochem Res. 2007b;32:39–47. doi: 10.1007/s11064-006-9220-x. [DOI] [PubMed] [Google Scholar]
- Zaheer A, Zaheer S, Thangavel R, Wu Y, Sahu SK, Yang B. Glia maturation factor modulates beta-amyloid-induced glial activation, inflammatory cytokine/chemokine production and neuronal damage. Brain Res. 2008;1208:192–203. doi: 10.1016/j.brainres.2008.02.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaheer S, Thangavel R, Wu Y, Khan MM, Kempuraj D, Zaheer A. Enhanced expression of glia maturation factor correlates with glial activation in the brain of triple transgenic Alzheimer's disease mice. Neurochem Res. 2013;38:218–225. doi: 10.1007/s11064-012-0913-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaheer S, Wu Y, Sahu SK, Zaheer A. Suppression of neuro inflammation in experimental autoimmune encephalomyelitis by glia maturation factor antibody. Brain Res. 2011;1373:230–239. doi: 10.1016/j.brainres.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zepp J, Wu L, Li X. IL-17 receptor signaling and T helper 17-mediated autoimmune demyelinating disease. Trends Immunol. 2011;32:232–239. doi: 10.1016/j.it.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ke KF, Liu Z, Qiu YH, Peng YP. Th17 cell-mediated neuroinflammation is involved in neurodegeneration of abeta1-42-induced Alzheimer's disease model rats. PLoS One. 2013;8:e75786. doi: 10.1371/journal.pone.0075786. [DOI] [PMC free article] [PubMed] [Google Scholar]