

Abstract
Thrombus formation is of paramount importance in the pathophysiology of acute ischemic stroke. Current antithrombotics used to treat or prevent cerebral ischemia are only moderately effective or bear an increased risk of severe bleeding. von Willebrand factor (VWF) has long been known to be a key player in thrombus formation at sites of vascular damage. While the association between VWF and coronary heart disease has been well studied, knowledge about the role of VWF in stroke is much more limited. However, in recent years, an increasing amount of clinical and preclinical evidence has revealed the critical involvement of VWF in stroke development. This review summarizes the latest insights into the pathophysiologic role of VWF-related processes in ischemic brain injury under experimental conditions and in humans. Potential clinical merits of novel inhibitors of VWF-mediated platelet adhesion and activation as powerful and safe tools to combat thromboembolic disorders including ischemic stroke are discussed.
Keywords: stroke, von Willebrand factor, platelets, glycoprotein Ib
Introduction
Ischemic stroke is a devastating disease that represents the primary reason for sustained disability and the second leading cause of death worldwide.1 Eighty percent of strokes are caused by arterial occlusion of cerebral arteries whereas the remaining 20% are caused by intracerebral hemorrhages. Currently, the only established therapeutic option for acute stroke is rapid thrombolysis using the clot-breaking agent tissue plasminogen activator (t-PA) in order to achieve recanalization of occluded cerebral vessels. However, due to the increased risk of bleeding associated with late t-PA administration, intravenous t-PA is recommended only within the limited therapeutic time window of 4.5 hours post-stroke and thus is available to less than 10% of patients.2 A recent trial to extend the therapeutic window up to 9 hours by use of recombinant desmoteplase, a novel plasminogen activator, failed3 as did a trial using the defibrinogenating agent ancrod.4
In terms of secondary stroke prevention the situation is very similar. Antiplatelet agents such as acetylsalicylic acid (ASA), dipyridamole, and the platelet P2Y12 receptor inhibitor clopidogrel show only limited efficacy and substantially increase the risk of fatal bleeding. This holds also true for anticoagulants, particularly warfarin, and even the introduction of novel substance classes such as direct thrombin inhibitors (e.g. dabigatran) or FXa blockers (e.g. rivaroxaban, apixaban) could not overcome the threat of hemorrhage. These limitations emphasize the need for a better understanding of the pathophysiologic mechanisms of thrombus formation in acute ischemic stroke5, in order to successfully improve treatment.
VWF: role in hemostasis, thrombosis and inflammation
In 1926, Finnish physician Erik von Willebrand reported a new type of inherited bleeding disorder that was distinct from hemophilia A.6 Thirty years later, the plasma protein that is central to the disease was identified and was named von Willebrand factor (VWF). Now, more than half a century later, much of the structure and function of VWF has been elucidated and its role in maintaining the delicate balance between bleeding and thrombosis has become an intriguing subject. VWF is a large, multimeric glycoprotein. Along with serving as a protective carrier molecule for clotting factor VIII, its main function is mediating initial platelet adhesion at sites of vascular injury. Indeed, whereas this is a prerequisite for normal hemostasis, adhesion of platelets is also the first step in thrombosis and an important mediator of inflammation.7
The critical role of VWF in normal hemostasis is exemplified by von Willebrand disease (VWD). VWD is the most common inherited bleeding disorder in humans, caused by quantitative or qualitative defects in VWF.8 Bleeding symptoms range from mild (type 1) to severe (type 3) and include mucosal hemorrhages, such as epistaxis, menorrhagia, and bleeding from the gums and gastrointestinal tract.
VWF is exclusively synthesized in endothelial cells and megakaryocytes and circulates as multimers of varying size (up to 20,000 kDa). The multi-domain structure of the monomeric VWF building blocks (Figure 1) is fundamental to the function of VWF (Figure 2). Rapid binding of VWF (A3 domain) to exposed fibrillar collagen type I and III immobilizes VWF at sites of vascular damage. This and/or high shear blood leads to a conformational changes, exposing the binding site for platelet glycoprotein (GP)Ibα in the VWF A1 domain. The reversible nature of the GPIbα-VWF A1 interactions allows deceleration and rolling of platelets, which, especially under high shear forces, is necessary for the definitive arrest. Firm adhesion of platelets at the site of vascular injury is further supported by engagement of the platelet collagen receptors (GPVI and integrin α2β1) and leads to platelet activation. Upon platelet activation, soluble platelet agonists such as ADP, ATP and thromboxane A2 are released and platelet integrins like GPIIb/IIIa shift to a high-affinity state. Subsequent platelet aggregation is promoted by binding of activated platelet GPIIb/IIIa to its primary ligand fibrinogen and to the Arg-Gly-Asp (RGD) sequence found in the C1 domain of VWF. Additional incoming platelets are recruited to the growing thrombus, primarily via engagement of their GPIbα receptors (Figure 2). Hence, the GPIb complex is essential for both initial platelet adhesion to sites of vascular injury and recruitment of new platelets to the growing thrombus.
Figure 1. Schematic representation of VWF.

VWF is a multimeric protein composed of dimeric building blocks. The VWF domains, the binding sites of major binding partners and the cleavage site for ADAMTS13 are indicated. Adapted from De Meyer et al.8
Figure 2. Schematic representation of VWF-mediated platelet adhesion and aggregation.
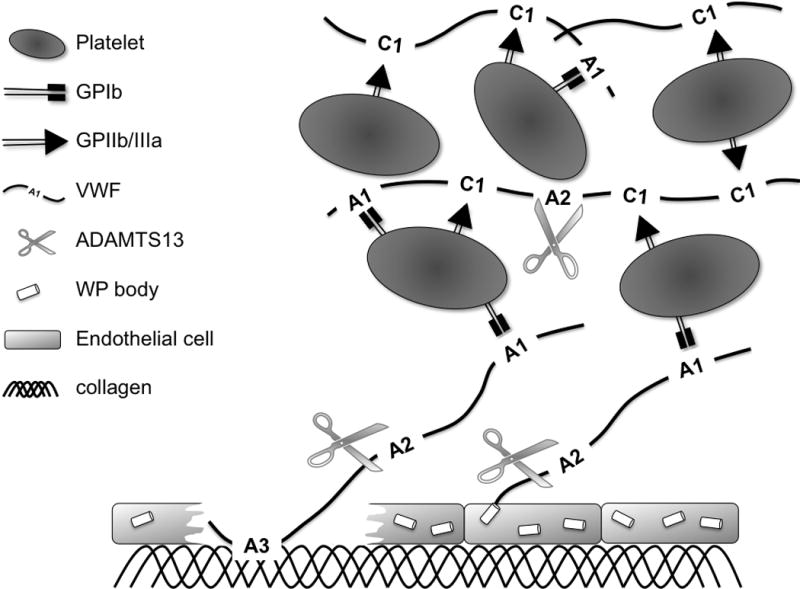
VWF that is immobilized on collagen with its A3 domain or released from endothelial Weibel-Palade (WP) bodies recruits platelets by binding with its A1 domain to platelet GPIbα. Together with fibrinogen (not shown), VWF further cross-links platelets via binding of its C1 domain to platelet GPIIb/IIIa.
Besides its role in thrombus formation, VWF has also been shown to support inflammatory processes. In vitro experiments demonstrated that VWF promotes leukocyte adhesion by acting as a ligand for the leukocyte receptors P-selectin glycoprotein ligand 1 and β2 integrin.9 VWF-bound platelets also were shown to support leukocyte tethering and rolling under high shear stress10. More recently, Petri et al. showed that VWF promotes the extravasation of leukocytes from blood vessels in a strictly platelet and GPIbα dependent way.11
ADAMTS13: a biological regulator of VWF activity
VWF activity is correlated with multimer size, with ultra-large (UL) multimers spontaneously binding to platelets. UL-VWF is released from endothelial and platelet storage granules upon stimulation with various thrombogenic or inflammatory secretagogues, but also during hypoxia.12, 13 To prevent spontaneous thrombosis, ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type 1 repeats, 13) cleaves the Y1605-M1606 bond in the VWF A2 domain (Figure 1 and 2), thereby digesting UL-VWF into smaller, less reactive molecules. As seen in thrombotic thrombocytopenic purpura (TTP), ADAMTS13 deficiency is associated with thrombotic occlusion of microvessels from multiple organs, including the brain.14–16 Experimental studies in mice have demonstrated the antithrombotic and anti-inflammatory properties of ADAMTS13.17, 18
VWF and stroke risk
Because of the pivotal role of VWF in platelet adhesion and thrombus formation, it seems logical to anticipate a correlation between high plasma levels of VWF and development of cardiovascular disease. While this relationship has been well studied for coronary heart disease19, much less is known about the association between VWF levels and stroke. When comparing stroke patients with healthy controls, several studies found an association between high VWF levels and stroke,20–28 and even different etiologic subtypes of ischemic stroke.29 However, increased VWF levels are a well-known marker of endothelial activation and/or dysfunction and a conclusive interpretation of many of these case-control studies on the causative or consequential nature of high VWF levels in stroke patients has been obscured by the post-stroke time point of VWF measurement. By also measuring the VWF propeptide, a recent investigation of two independent case-control studies (COCOS and ATTAC) elegantly indicated that endothelial cell activation and subsequent Weibel-Palade body secretion is indeed an important mechanism underlying the association between total VWF levels and the occurrence of a first ischemic stroke.30 Several prospective studies, such as the recent longitudinal analysis embedded in the Rotterdam study31, clearly identified high plasma levels of VWF as a strong predictor of stroke.32–37 Importantly, correlation between VWF levels and stroke-related mortality persisted after adjustment for common risk factors such as age, stroke severity, and atrial fibrillation indicating that VWF is an independent predictor.38 On a genetic level, VWF polymorphisms have been identified that significantly raise the risk of ischemic stroke.39–41 Not all of these are associated with higher VWF levels, suggesting that other mechanisms, such as increased VWF activity, could contribute to the risk of stroke as well.
A major down-regulator of VWF activity is ADAMTS13. Accordingly, low levels of ADAMTS13 come along with an increased risk of cardiovascular disease, including ischemic stroke.25, 28 Analogous to VWF, single nucleotide polymorphisms in ADAMTS13 were found to be associated with the incidence of ischemic stroke in a Swedish population.42
Together, these proof-of-principle studies established that plasma levels of VWF and/or ADAMTS13 are associated with the risk of stroke in the general population. However, determination of VWF and/or ADAMTS13 levels on a regular basis to judge the risk of thromboembolic disease in individual patients cannot be recommended until larger prospective trials and standardized test systems are available.
Experimental studies: role of VWF in acute stroke
The importance of VWF as a risk factor for stroke occurrence and mortality in humans recently also stimulated experimental studies in models of acute stroke. Using a mouse model of transient middle cerebral artery occlusion (tMCAO), we showed that mice that are deficient in VWF due to VWF gene abrogation (VWF−/−)43 are protected from brain ischemia/reperfusion injury.44–46 Infarct sizes in VWF−/− were ~60% of the infarct volumes in wild-type controls one day after tMCAO, which was accompanied by reduced fibrin accumulation in the infarcted brain hemisphere. Accordingly, neurological scores assessing motor function and coordination were significantly better in VWF−/− mice compared to controls. Importantly, genetic disruption of VWF did not increase the risk of intracerebral bleeding in the context of ischemic stroke.45 Reconstitution of plasma VWF by hydrodynamic gene transfer fully restored the susceptibility of VWF−/− mice to cerebral ischemia underlining the causative role of VWF in this setting.44, 45 This is in line with the well-established antithrombotic effects of VWF deficiency in several experimental arterial and venous thrombosis models.43, 47–50 Further illustrating the critical role of VWF in ischemia/reperfusion injury are the findings that ADAMTS13−/− mice are more susceptible to focal cerebral ischemia.46, 51 These mice developed significantly larger infarctions, with an increased accumulation of immune cells and thrombi in the ischemic brain tissue, resulting in more severe neurological deficits.51 On the other hand, intravenous administration of recombinant ADAMTS13 into wild-type mice immediately before reperfusion significantly reduced infarct volume.46
By reconstituting VWF−/− mice with different VWF mutants, we recently showed that binding of VWF to both collagen and GPIbα, but not to GPIIb/IIIa, are mandatory steps in stroke development.44 The involvement of collagen and GPIbα-mediated platelet adhesion in stroke is corroborated by the findings that blocking platelet collagen receptor GPVI or GPIbα also confers a protective effect in the mouse tMCAO model.52 Blockade of GPIIb/IIIa did not affect stroke size and led to an increased incidence of intracerebral hemorrhage, whereas blocking of GPIbα or GPVI did not increase the frequency of intracerebral bleeding.52 Finally, mice in which downstream signaling of GPIb via phospholipase D1 is abrogated and mice in which the extracellular part of GPIb is replaced by human interleukin-4 receptor (GPIbα/IL4Rα)53 are also protected against focal cerebral ischemia without causing excessive bleeding (54 and SFDM and DDW, unpublished observations, 2010). These observations further underline that blockage of the GPIbα-VWF axis or collagen-platelet axis might be a safe approach in ischemic stroke.
Inhibitors of VWF: a promising class of antithrombotics on the brink of reaching the clinic
From the above, it is clear that pharmacological interference in VWF-mediated platelet adhesion and thrombus formation could have clinical benefit as a promising strategy in stroke treatment. Although no such VWF-blockers have yet achieved regulatory approval for marketing, there are promising preclinical and clinical studies that demonstrate the antithrombotic potential of agents that inhibit VWF function by blocking the VWF-collagen or VWF-GPIbα interaction (Figure 3). In this section, we will discuss candidate molecules that could prove useful in stroke therapy based on the encouraging results they have demonstrated in the inhibition of VWF-mediated thrombosis. These inhibitors include monoclonal antibodies against VWF (82D6A3, AJvW2 and its humanized form AJW200) or GPIbα (6B4 and its humanized form h6B4), the nanobody® ALX-0081, the aptamer ARC1779, and the recombinant GPIbα fragment GPG-290 (Table 1).55–57 A detailed overview of the key features of each of these inhibitors is given in Table S1 (please see http://stroke.ahajournals.org).
Figure 3. Schematic representation of mode-of-action of various VWF inhibitors.
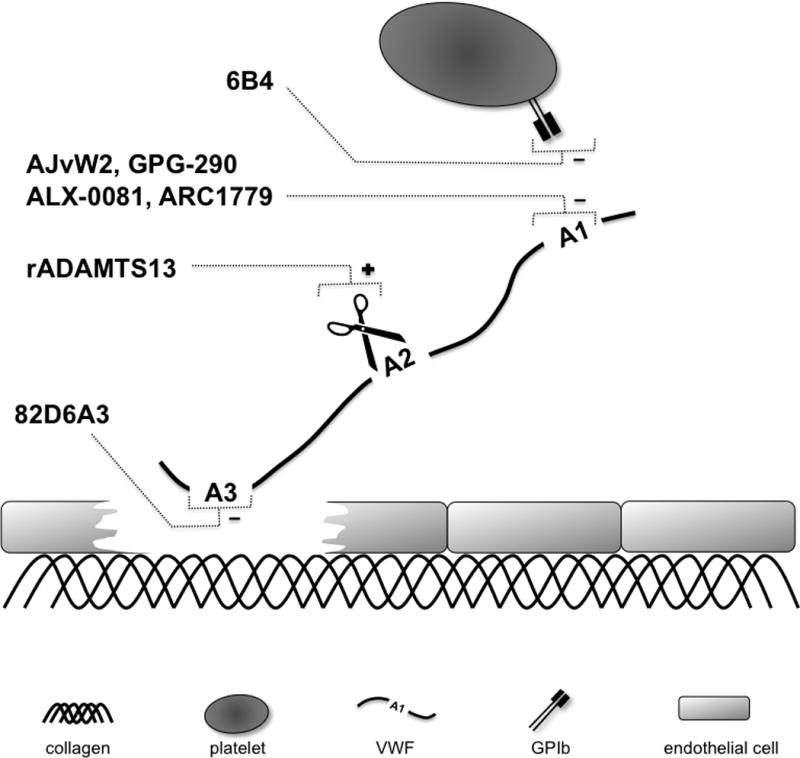
VWF-mediated platelet adhesion can be blocked by inhibiting binding of VWF to either collagen or GPIbα, or by cleaving VWF by ADAMTS13.
Table 1. Inhibitors of VWF-mediated platelet adhesion.
A detailed description of each of these inhibitors is given in Table S1 (please see http://stroke.ahajournals.org).
| Compound | Description |
|---|---|
| 82D6A3 | Monoclonal antibody against VWF A3 domain that inhibits binding of VWF to collagen |
| 6B4 h6B4 |
Fab-fragment of a monoclonal antibody against platelet GPIbα that inhibits binding of VWF to GPIb |
| AJvW2 AJW200 |
Monoclonal antibody against VWF A1 domainα that inhibits binding of VWF to GPIbα |
| GPG-290 | Chimeric recombinant protein containing gain-of-function GPIbα fragment that binds to A1 domain thereby inhibiting binding of VWF to GPIbα |
| ARC1779 | Aptamer against VWF A1 domain that inhibits binding of VWF to GPIbα |
| ALX-0081 ALX-0681 |
Nanobody® against VWF A1 domain that inhibits binding of VWF to GPIbα |
| rADAMTS13 | Recombinant protein that cleaves VWF multimers by cleaving the Y1605-M1606 bond in the VWF A2 domain |
While most attention has been focused on the development of VWF-GPIbα inhibitors, the anti-VWF antibody 82D6A3 is different in that it inhibits the binding of VWF to collagen. Preclinical studies in baboons have demonstrated that 82D6A3 has a strong antithrombotic efficacy.58 This observation indicates that, despite the existence of other binding partners for VWF in the extracellular matrix, VWF binding to fibrillar collagen has an important role in mediating thrombosis. A humanized version of 82D6A3 has been constructed for further preclinical and clinical testing.59
AJvW2, AJW200, 6B4 and h6B4 are monoclonal antibodies designed to disrupt the VWF-GPIbα interaction by binding to the VWF A1 domain and GPIbα respectively. Both antibodies have been tested extensively in preclinical thrombosis models.60–67 The clinical efficacy and tolerance of AJW200 has been demonstrated in human volunteers without bleeding complications.68
GPG-290 is a homodimeric recombinant fragment of human GPIbα composed of the first 290 amino acids and conjugated to human IgG1Fc to form a homodimer. GPG-290 contains two gain-of-function mutations (G233V/M239V) resulting in its enhanced affinity for the VWF A1 domain (and possibly other GPIb ligands). Its antithrombotic activity was demonstrated in preclinical murine and canine models of arterial and venous thrombosis.48, 69, 70
The aptamer ARC1779 represents an interesting new class of inhibitors. Aptamers are nucleic acid macromolecules that tightly bind to a specific molecular target.71 In solution, the chain of nucleotides forms intra-molecular interactions that fold the molecule into a complex three-dimensional shape that enhances affinity for its target molecule. A potential advantage of this class of molecules is that they can be inactivated by a complementary aptamer antidote if necessary. ARC1779 is a 40-nucleotide DNA/RNA aptamer conjugated to a 20 kDa PEG to enhance its pharmacokinetic properties. ARC1779 binds to the VWF A1 domain, and was reported to effectively inhibit thrombus formation in several preclinical settings.72, 73 The aptamer was well tolerated in healthy volunteers in a Phase I trial. Importantly, in a Phase II trial, ARC1779 effectively increased platelet counts in critically ill TTP patients by blocking spontaneous VWF-mediated platelet aggregation74–76 and even prevented desmopressin-induced thrombocytopenia in patients with VWD type 2B.77 High affinity aptamers to murine VWF have recently been developed, which will allow the investigation of anti-VWF aptamers in murine preclinical models of thrombosis, including stroke.78 Interestingly, a recent trial showed that ARC1779 was able to reduce cerebral emboli signals in patients undergoing carotid endarterectomy.79 Nanobodies are antibody-derived therapeutic proteins that contain the structural and functional properties of naturally occurring heavy-chain antibodies. The cameloid bivalent Nanobody® ALX-0081 specifically targets exposed GPIbα binding sites in VWF. By blocking GPIbα binding to VWF, ALX-0081 showed strong antithrombotic potency in preclinical baboon studies and in blood obtained from patients undergoing percutaneous coronary intervention (PCI).80, 81 In a Phase I clinical study, this nanobody was found to be well tolerated and safe. ALX-0081 (and ALX-0681, a subcutaneous formulation of ALX-0081) entered a Phase II study in patients undergoing PCI and recently also a Phase II study in TTP patients.
Apart from blocking binding of VWF to either collagen or GPIbα, another way of reducing VWF activity is decreasing its size by ADAMTS13. Recombinant ADAMTS13 is being developed as a new therapeutic agent and, as mentioned earlier, was protective in a preclinical mouse model of ischemic stroke.46 Recombinant human ADAMTS13 has received orphan designation for the treatment of TTP (EU/3/08/588).
Potential use of VWF antagonism in stroke therapy
Since VWF antagonists are starting to enter the first clinical trials, it is interesting to speculate on the potential role of these new drugs in stroke therapy. Inhibitors of the VWF-GPIbα or VWF-collagen interaction have no direct thrombolytic properties, so their use is unlikely to result in dissolution of an already existing thrombus in the acute setting of ischemic stroke. However, when used in combination with t-PA, VWF antagonists could prevent ongoing microvascular thrombus formation during the reperfusion phase after successful or spontaneous thrombolysis82, 83, reducing the occurrence of re-thrombosis and/or secondary stroke progression. Whether dual therapy of VWF inhibitors and t-PA would allow the use of lower and thereby safer doses of t-PA remains to be established. The use of t-PA-dependent experimental thromboembolic stroke models will have to shed more light on the possible beneficial effects of combining thrombolytic therapy with VWF inhibition. Former attempts to prevent the apposition of further clots into an already existing thrombus during the early stage of ischemic stroke by applying heparins failed.84 While heparins could counteract deterioration of stroke symptoms in some studies, the net clinical benefit was outweighed by an increased risked of severe hemorrhages. Consequently, full-intensity parenteral anticoagulation with heparins is no longer recommended in the acute phase of cerebral ischemia.85 In view of the anticipated safer benefit-over-risk ratio in terms of bleeding, it will be interesting to see whether VWF antagonists are more successful than heparins during the acute phase of ischemic stroke. Nevertheless, close CT or MRI monitoring of potential hemorrhagic transformation should accompany potential future applications of VWF inhibitors in stroke patients. Interestingly, recombinant ADAMTS13 promoted thrombus dissolution in injured mouse mesenteric arterioles17, calling for further studies of the thrombolytic potential of ADAMTS13 in acute ischemic stroke.
With regard to stroke prevention, VWF inhibition could become useful for short-term therapy during procedures associated with increased risk of cerebral thromboemoblism, such as angiography, carotid endarterectomy, or heart surgery under conditions of extracorporeal circulation. Promising results were recently obtained with ARC1779, which significantly reduced cerebral embolization in patients undergoing carotid endarterectomy.79 In the absence of oral formulations, the potential of VWF blockers to prevent stroke or systemic embolism in chronic conditions, e.g. in patients with atrial fibrillation, cannot yet be judged.
Conclusion
There is now compelling evidence on preclinical and clinical levels that interactions between the cerebral blood vessels and platelets employing VWF, GPIbα and collagen are instrumental in ischemic brain disease. Early clinical testing of lead compounds targeting VWF-mediated platelet adhesion in healthy individuals or selected disease states points towards a favorable bleeding risk profile and higher efficacy as compared to established antithrombotic drugs. Based on the encouraging results in rodent stroke models, larger translational studies in non-human primates and proof-of-principle clinical trials are now warranted to further judge the risk-to-benefit ratio of interfering with the early steps of platelet activation. If these trials are as promising as the current experimental data, an exciting decade exploring VWF-targeted stroke strategies lies ahead. Back in 1926, Dr. von Willebrand may not have fully grasped what great implications his seminal report6 on a bleeding disease would have on our understanding and treatment of thrombotic disorders. More than ninety years later, stroke may very well become one of them.
Supplementary Material
Acknowledgments
We thank Lesley Cowan for helping with the preparation of the manuscript. We thank Dr. Robert Schaub and Dr. Karen Vanhoorelbeke for critical reading and helpful advice.
Funding Sources
S.F.D.M. is a post-doctoral fellow of the Research Foundation Flanders (Fonds voor Wetenschappelijk Onderzoek Vlaanderen). G.S. and C.K. are supported by the Deutsche Forschungsgemeinschaft, Bonn, Germany, SFB 688, projects A13 and B1.
This work was supported by National Heart, Lung, and Blood Institute of the National Institutes of Health grant R01 HL041002 (to D.D.W.)
Footnotes
Disclosures
None
References
- 1.Lopez AD, Mathers CD, Ezzati M, Jamison DT, Murray CJ. Global and regional burden of disease and risk factors, 2001: systematic analysis of population health data. Lancet. 2006;367:1747–1757. doi: 10.1016/S0140-6736(06)68770-9. [DOI] [PubMed] [Google Scholar]
- 2.Hacke W, Kaste M, Bluhmki E, Brozman M, Davalos A, Guidetti D, et al. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med. 2008;359:1317–1329. doi: 10.1056/NEJMoa0804656. [DOI] [PubMed] [Google Scholar]
- 3.Hacke W, Furlan AJ, Al-Rawi Y, Davalos A, Fiebach JB, Gruber F, et al. Intravenous desmoteplase in patients with acute ischaemic stroke selected by MRI perfusion–diffusion weighted imaging or perfusion CT (DIAS-2): a prospective, randomised, double-blind, placebo-controlled study. Lancet Neurol. 2009;8:141–150. doi: 10.1016/S1474-4422(08)70267-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levy DE, del Zoppo GJ, Demaerschalk BM, Demchuk AM, Diener H-C, Howard G, et al. Ancrod in acute ischemic stroke: results of 500 subjects beginning treatment within 6 hours of stroke onset in the ancrod stroke program. Stroke. 2009;40:3796–3803. doi: 10.1161/STROKEAHA.109.565119. [DOI] [PubMed] [Google Scholar]
- 5.Stoll G, Kleinschnitz C, Nieswandt B. Molecular mechanisms of thrombus formation in ischemic stroke: novel insights and targets for treatment. Blood. 2008;112:3555–3562. doi: 10.1182/blood-2008-04-144758. [DOI] [PubMed] [Google Scholar]
- 6.von Willebrand EA. Hereditar pseudohemofili. Fin Laekaresaellsk Hand. 1926;68:87–112. [Google Scholar]
- 7.Wagner DD, Frenette PS. The vessel wall and its interactions. Blood. 2008;111:5271–5281. doi: 10.1182/blood-2008-01-078204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Meyer SF, Deckmyn H, Vanhoorelbeke K. von Willebrand factor to the rescue. Blood. 2009;113:5049–5057. doi: 10.1182/blood-2008-10-165621. [DOI] [PubMed] [Google Scholar]
- 9.Pendu R, Terraube V, Christophe OD, Gahmberg CG, de Groot PG, Lenting PJ, et al. P-selectin glycoprotein ligand 1 and beta2-integrins cooperate in the adhesion of leukocytes to von Willebrand factor. Blood. 2006;108:3746–3752. doi: 10.1182/blood-2006-03-010322. [DOI] [PubMed] [Google Scholar]
- 10.Bernardo A, Ball C, Nolasco L, Choi H, Moake JL, Dong JF. Platelets adhered to endothelial cell-bound ultra-large von Willebrand factor strings support leukocyte tethering and rolling under high shear stress. J Thromb Haemost. 2005;3:562–570. doi: 10.1111/j.1538-7836.2005.01122.x. [DOI] [PubMed] [Google Scholar]
- 11.Petri B, Broermann A, Li H, Khandoga AG, Zarbock A, Krombach F, et al. von Willebrand factor promotes leukocyte extravasation. Blood. 2010;116:4712–4719. doi: 10.1182/blood-2010-03-276311. [DOI] [PubMed] [Google Scholar]
- 12.Pinsky DJ, Naka Y, Liao H, Oz MC, Wagner DD, Mayadas TN, et al. Hypoxia-induced exocytosis of endothelial cell Weibel-Palade bodies. A mechanism for rapid neutrophil recruitment after cardiac preservation. J Clin Invest. 1996;97:493–500. doi: 10.1172/JCI118440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krishnan S, Siegel J, Pullen G, Hevelow M, Dampier C, Stuart M. Increased von Willebrand factor antigen and high molecular weight multimers in sickle cell disease associated with nocturnal hypoxemia. Thromb Res. 2008;122:455–458. doi: 10.1016/j.thromres.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou Z, Nguyen TC, Guchhait P, Dong JF. Von Willebrand factor, ADAMTS-13, and thrombotic thrombocytopenic purpura. Semin Thromb Hemost. 2010;36:71–81. doi: 10.1055/s-0030-1248726. [DOI] [PubMed] [Google Scholar]
- 15.Lindblom A, Thorsen S, Hillarp A, Björk P. Minor stroke as singular manifestation of hereditary thrombotic thrombocytopenic purpura in a young man. Int Angiol. 2009;28:336–339. [PubMed] [Google Scholar]
- 16.Sevy A, Doche E, Squarcioni C, Poullin P, Serratrice J, Nicoli F, et al. Stroke in a young patient treated by alteplase heralding an acquired thrombotic thrombocytopenic purpura. J Clin Apheresis. 2011;26:152–155. doi: 10.1002/jca.20276. [DOI] [PubMed] [Google Scholar]
- 17.Chauhan AK, Motto DG, Lamb CB, Bergmeier W, Dockal M, Plaimauer B, et al. Systemic antithrombotic effects of ADAMTS13. J Exp Med. 2006;203:767–776. doi: 10.1084/jem.20051732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chauhan AK, Kisucka J, Brill A, Walsh MT, Scheiflinger F, Wagner DD. ADAMTS13: a new link between thrombosis and inflammation. J Exp Med. 2008;205:2065–2074. doi: 10.1084/jem.20080130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vischer UM. von Willebrand factor, endothelial dysfunction, and cardiovascular disease. J Thromb Haemost. 2006;4:1186–1193. doi: 10.1111/j.1538-7836.2006.01949.x. [DOI] [PubMed] [Google Scholar]
- 20.Licata G, Tuttolomondo A, Di Raimondo D, Corrao S, Di Sciacca R, Pinto A. Immuno-inflammatory activation in acute cardio-embolic strokes in comparison with other subtypes of ischaemic stroke. Thromb Haemost. 2009;101:929–937. [PubMed] [Google Scholar]
- 21.Kozuka K, Kohriyama T, Nomura E, Ikeda J, Kajikawa H, Nakamura S. Endothelial markers and adhesion molecules in acute ischemic stroke–sequential change and differences in stroke subtype. Atherosclerosis. 2002;161:161–168. doi: 10.1016/s0021-9150(01)00635-9. [DOI] [PubMed] [Google Scholar]
- 22.Bath PM, Blann A, Smith N, Butterworth RJ. Von Willebrand factor, P-selectin and fibrinogen levels in patients with acute ischaemic and haemorrhagic stroke, and their relationship with stroke sub-type and functional outcome. Platelets. 1998;9:155–159. doi: 10.1080/09537109876618. [DOI] [PubMed] [Google Scholar]
- 23.Stott DJ, Spilg E, Campbell AM, Rumley A, Mansoor MA, Lowe GD. Haemostasis in ischaemic stroke and vascular dementia. Blood Coagul Fibrinolysis. 2001;12:651–657. doi: 10.1097/00001721-200112000-00006. [DOI] [PubMed] [Google Scholar]
- 24.Lip GYH, Blann AD, Farooqi IS, Zarifis J, Sagar G, Beevers DG. Sequential alterations in haemorheology, endothelial dysfunction, platelet activation and thrombogenesis in relation to prognosis following acute stroke: The West Birmingham Stroke Project. Blood Coagul Fibrinolysis. 2002;13:339–347. doi: 10.1097/00001721-200206000-00010. [DOI] [PubMed] [Google Scholar]
- 25.Bongers TN, de Bruijne ELE, Dippel DWJ, de Jong AJ, Deckers JW, Poldermans D, et al. Lower levels of ADAMTS13 are associated with cardiovascular disease in young patients. Atherosclerosis. 2009;207:250–254. doi: 10.1016/j.atherosclerosis.2009.04.013. [DOI] [PubMed] [Google Scholar]
- 26.Catto AJ, Carter AM, Barrett JH, Bamford J, Rice PJ, Grant PJ. von Willebrand factor and factor VIII: C in acute cerebrovascular disease. Relationship to stroke subtype and mortality. Thromb Haemost. 1997;77:1104–1108. [PubMed] [Google Scholar]
- 27.Qizilbash N, Duffy S, Prentice CR, Boothby M, Warlow C. Von Willebrand factor and risk of ischemic stroke. Neurology. 1997;49:1552–1556. doi: 10.1212/wnl.49.6.1552. [DOI] [PubMed] [Google Scholar]
- 28.Bongers TN, de Maat MPM, van Goor M-LPJ, Bhagwanbali V, van Vliet HHDM, Gómez García EB, et al. High von Willebrand factor levels increase the risk of first ischemic stroke: influence of ADAMTS13, inflammation, and genetic variability. Stroke. 2006;37:2672–2677. doi: 10.1161/01.STR.0000244767.39962.f7. [DOI] [PubMed] [Google Scholar]
- 29.Hanson E, Jood K, Karlsson S, Nilsson S, Blomstrand C, Jern C. Plasma levels of von Willebrand factor in the etiologic subtypes of ischemic stroke. J Thromb Haemost. 2011;9:275–281. doi: 10.1111/j.1538-7836.2010.04134.x. [DOI] [PubMed] [Google Scholar]
- 30.van Schie MC, De Maat MPM, Dippel DWJ, de Groot PG, Lenting PJ, Leebeek FWG, et al. von Willebrand factor propeptide and the occurrence of a first ischemic stroke. J Thromb Haemost. 2010;8:1424–1426. doi: 10.1111/j.1538-7836.2010.03863.x. [DOI] [PubMed] [Google Scholar]
- 31.Wieberdink RG, van Schie MC, Koudstaal PJ, Hofman A, Witteman JCM, de Maat MPM, et al. High von Willebrand Factor Levels Increase the Risk of Stroke. The Rotterdam Study. Stroke. 2010;41:2151–2156. doi: 10.1161/STROKEAHA.110.586289. [DOI] [PubMed] [Google Scholar]
- 32.Roldán V, Marín F, García-Herola A, Lip GYH. Correlation of plasma von Willebrand factor levels, an index of endothelial damage/dysfunction, with two point-based stroke risk stratification scores in atrial fibrillation. Thromb Res. 2005;116:321–325. doi: 10.1016/j.thromres.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 33.Lip GYH, Lane D, Van Walraven C, Hart RG. Additive role of plasma von Willebrand factor levels to clinical factors for risk stratification of patients with atrial fibrillation. Stroke. 2006;37:2294–2300. doi: 10.1161/01.STR.0000236840.00467.84. [DOI] [PubMed] [Google Scholar]
- 34.Pinto A, Tuttolomondo A, Casuccio A, Di Raimondo D, Di Sciacca R, Arnao V, et al. Immuno-inflammatory predictors of stroke at follow-up in patients with chronic non-valvular atrial fibrillation (NVAF) Clin Sci. 2009;116:781–789. doi: 10.1042/CS20080372. [DOI] [PubMed] [Google Scholar]
- 35.Conway DSG, Pearce LA, Chin BSP, Hart RG, Lip GYH. Prognostic value of plasma von Willebrand factor and soluble P-selectin as indices of endothelial damage and platelet activation in 994 patients with nonvalvular atrial fibrillation. Circulation. 2003;107:3141–3145. doi: 10.1161/01.CIR.0000077912.12202.FC. [DOI] [PubMed] [Google Scholar]
- 36.Tzoulaki I, Murray GD, Lee AJ, Rumley A, Lowe GDO, Fowkes FGR. Relative value of inflammatory, hemostatic, and rheological factors for incident myocardial infarction and stroke: the Edinburgh Artery Study. Circulation. 2007;115:2119–2127. doi: 10.1161/CIRCULATIONAHA.106.635029. [DOI] [PubMed] [Google Scholar]
- 37.Folsom AR, Rosamond WD, Shahar E, Cooper LS, Aleksic N, Nieto FJ, et al. Prospective study of markers of hemostatic function with risk of ischemic stroke. The Atherosclerosis Risk in Communities (ARIC) Study Investigators. Circulation. 1999;100:736–742. doi: 10.1161/01.cir.100.7.736. [DOI] [PubMed] [Google Scholar]
- 38.Carter AM, Catto AJ, Mansfield MW, Bamford JM, Grant PJ. Predictive variables for mortality after acute ischemic stroke. Stroke. 2007;38:1873–1880. doi: 10.1161/STROKEAHA.106.474569. [DOI] [PubMed] [Google Scholar]
- 39.van Schie MC, de Maat MPM, Isaacs A, van Duijn CM, Deckers JW, Dippel DWJ, et al. Variation in the von Willebrand Factor gene is associated with VWF levels and with the risk of cardiovascular disease. Blood. 2011;117:1393–1399. doi: 10.1182/blood-2010-03-273961. [DOI] [PubMed] [Google Scholar]
- 40.Dai K, Gao W, Ruan C. The Sma I polymorphism in the von Willebrand factor gene associated with acute ischemic stroke. Thromb Res. 2001;104:389–395. doi: 10.1016/s0049-3848(01)00389-9. [DOI] [PubMed] [Google Scholar]
- 41.van Schie MC, van Loon JE, de Maat MP, Leebeek FW. Genetic determinants of von Willebrand Factor levels and activity in relation to the risk of cardiovascular disease. A review. J Thromb Haemost. 2011;9:899–908. doi: 10.1111/j.1538-7836.2011.04243.x. [DOI] [PubMed] [Google Scholar]
- 42.Hanson E, Jood K, Nilsson S, Blomstrand C, Jern C. Association between genetic variation at the ADAMTS13 locus and ischemic stroke. J Thromb Haemost. 2009;7:2147–2148. doi: 10.1111/j.1538-7836.2009.03617.x. [DOI] [PubMed] [Google Scholar]
- 43.Denis C, Methia N, Frenette PS, Rayburn H, Ullman-Culleré M, Hynes RO, et al. A mouse model of severe von Willebrand disease: defects in hemostasis and thrombosis. Proc Natl Acad Sci U S A. 1998;95:9524–9529. doi: 10.1073/pnas.95.16.9524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Meyer SF, Schwarz T, Deckmyn H, Denis CV, Nieswandt B, Stoll G, et al. Binding of von Willebrand Factor to Collagen and Glycoprotein Ibalpha, But Not to Glycoprotein IIb/IIIa, Contributes to Ischemic Stroke in Mice. Arterioscler Thromb Vasc Biol. 2010;30:1949–1951. doi: 10.1161/ATVBAHA.110.208918. [DOI] [PubMed] [Google Scholar]
- 45.Kleinschnitz C, De Meyer SF, Schwarz T, Austinat M, Vanhoorelbeke K, Nieswandt B, et al. Deficiency of von Willebrand factor protects mice from ischemic stroke. Blood. 2009;113:3600–3603. doi: 10.1182/blood-2008-09-180695. [DOI] [PubMed] [Google Scholar]
- 46.Zhao B-Q, Chauhan AK, Canault M, Patten IS, Yang JJ, Dockal M, et al. von Willebrand factor-cleaving protease ADAMTS13 reduces ischemic brain injury in experimental stroke. Blood. 2009;114:3329–3334. doi: 10.1182/blood-2009-03-213264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.De Meyer SF, Vandeputte N, Pareyn I, Petrus I, Lenting PJ, Chuah MKL, et al. Restoration of plasma von Willebrand factor deficiency is sufficient to correct thrombus formation after gene therapy for severe von Willebrand disease. Arterioscler Thromb Vasc Biol. 2008;28:1621–1626. doi: 10.1161/ATVBAHA.108.168369. [DOI] [PubMed] [Google Scholar]
- 48.Brill A, Fuchs TA, Chauhan AK, Yang JJ, De Meyer SF, Köllnberger M, et al. von Willebrand factor-mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood. 2011;117:1400–1407. doi: 10.1182/blood-2010-05-287623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chauhan AK, Kisucka J, Lamb CB, Bergmeier W, Wagner DD. von Willebrand factor and factor VIII are independently required to form stable occlusive thrombi in injured veins. Blood. 2007;109:2424–2429. doi: 10.1182/blood-2006-06-028241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ni H, Denis CV, Subbarao S, Degen JL, Sato TN, Hynes RO, et al. Persistence of platelet thrombus formation in arterioles of mice lacking both von Willebrand factor and fibrinogen. J Clin Invest. 2000;106:385–392. doi: 10.1172/JCI9896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fujioka M, Hayakawa K, Mishima K, Kunizawa A, Irie K, Higuchi S, et al. ADAMTS13 gene deletion aggravates ischemic brain damage: a possible neuroprotective role of ADAMTS13 by ameliorating postischemic hypoperfusion. Blood. 2010;115:1650–1653. doi: 10.1182/blood-2009-06-230110. [DOI] [PubMed] [Google Scholar]
- 52.Kleinschnitz C, Pozgajova M, Pham M, Bendszus M, Nieswandt B, Stoll G. Targeting platelets in acute experimental stroke: impact of glycoprotein Ib, VI, and IIb/IIIa blockade on infarct size, functional outcome, and intracranial bleeding. Circulation. 2007;115:2323–2330. doi: 10.1161/CIRCULATIONAHA.107.691279. [DOI] [PubMed] [Google Scholar]
- 53.Kanaji T, Russell S, Ware J. Amelioration of the macrothrombocytopenia associated with the murine Bernard-Soulier syndrome. Blood. 2002;100:2102–2107. doi: 10.1182/blood-2002-03-0997. [DOI] [PubMed] [Google Scholar]
- 54.Elvers M, Stegner D, Hagedorn I, Kleinschnitz C, Braun A, Kuijpers MEJ, et al. Impaired alpha(IIb)beta(3) integrin activation and shear-dependent thrombus formation in mice lacking phospholipase D1. Sci Signal. 2010;3:ra1. doi: 10.1126/scisignal.2000551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.De Meyer SF, De Maeyer B, Deckmyn H, Vanhoorelbeke K. Von Willebrand factor: drug and drug target. Cardiovasc Hematol Disord Drug Targets. 2009;9:9–20. doi: 10.2174/187152909787581327. [DOI] [PubMed] [Google Scholar]
- 56.De Meyer SF, Vanhoorelbeke K, Ulrichts H, Staelens S, Feys HB, Salles I, et al. Development of monoclonal antibodies that inhibit platelet adhesion or aggregation as potential anti-thrombotic drugs. Cardiovasc Hematol Disord Drug Targets. 2006;6:191–207. doi: 10.2174/187152906778249536. [DOI] [PubMed] [Google Scholar]
- 57.Firbas C, Siller-Matula JM, Jilma B. Targeting von Willebrand factor and platelet glycoprotein Ib receptor. Expert Rev Cardiovasc Ther. 2010;8:1689–1701. doi: 10.1586/erc.10.154. [DOI] [PubMed] [Google Scholar]
- 58.Wu D, Vanhoorelbeke K, Cauwenberghs N, Meiring M, Depraetere H, Kotze HF, et al. Inhibition of the von Willebrand (VWF)-collagen interaction by an antihuman VWF monoclonal antibody results in abolition of in vivo arterial platelet thrombus formation in baboons. Blood. 2002;99:3623–3628. doi: 10.1182/blood.v99.10.3623. [DOI] [PubMed] [Google Scholar]
- 59.Staelens S, Desmet J, Ngo TH, Vauterin S, Pareyn I, Barbeaux P, et al. Humanization by variable domain resurfacing and grafting on a human IgG4, using a new approach for determination of non-human like surface accessible framework residues based on homology modelling of variable domains. Mol Immunol. 2006;43:1243–1257. doi: 10.1016/j.molimm.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 60.Kageyama S, Yamamoto H, Nagano M, Arisaka H, Kayahara T, Yoshimoto R. Anti-thrombotic effects and bleeding risk of AJvW-2, a monoclonal antibody against human von Willebrand factor. Br J Pharmacol. 1997;122:165–171. doi: 10.1038/sj.bjp.0701354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kageyama S, Yamamoto H, Nakazawa H, Yoshimoto R. Anti-human vWF monoclonal antibody, AJvW-2 Fab, inhibits repetitive coronary artery thrombosis without bleeding time prolongation in dogs. Thromb Res. 2001;101:395–404. doi: 10.1016/s0049-3848(00)00430-8. [DOI] [PubMed] [Google Scholar]
- 62.Kageyama S, Yamamoto H, Yoshimoto R. Anti-human von willebrand factor monoclonal antibody AJvW-2 prevents thrombus deposition and neointima formation after balloon injury in guinea pigs. Arterioscler Thromb Vasc Biol. 2000;20:2303–2308. doi: 10.1161/01.atv.20.10.2303. [DOI] [PubMed] [Google Scholar]
- 63.Cauwenberghs N, Meiring M, Vauterin S, van Wyk V, Lamprecht S, Roodt JP, et al. Antithrombotic effect of platelet glycoprotein Ib-blocking monoclonal antibody Fab fragments in nonhuman primates. Arterioscler Thromb Vasc Biol. 2000;20:1347–1353. doi: 10.1161/01.atv.20.5.1347. [DOI] [PubMed] [Google Scholar]
- 64.Fontayne A, Meiring M, Lamprecht S, Roodt J, Demarsin E, Barbeaux P, et al. The humanized anti-glycoprotein Ib monoclonal antibody h6B4-Fab is a potent and safe antithrombotic in a high shear arterial thrombosis model in baboons. Thromb Haemost. 2008;100:670–677. [PubMed] [Google Scholar]
- 65.Fontayne A, Vanhoorelbeke K, Pareyn I, Van Rompaey I, Meiring M, Lamprecht S, et al. Rational humanization of the powerful antithrombotic anti-GPIbalpha antibody: 6B4. Thromb Haemost. 2006;96:671–684. [PubMed] [Google Scholar]
- 66.Wu D, Meiring M, Kotze HF, Deckmyn H, Cauwenberghs N. Inhibition of platelet glycoprotein Ib, glycoprotein IIb/IIIa, or both by monoclonal antibodies prevents arterial thrombosis in baboons. Arterioscler Thromb Vasc Biol. 2002;22:323–328. doi: 10.1161/hq0202.102321. [DOI] [PubMed] [Google Scholar]
- 67.Kageyama S, Yamamoto H, Nakazawa H, Matsushita J, Kouyama T, Gonsho A, et al. Pharmacokinetics and pharmacodynamics of AJW200, a humanized monoclonal antibody to von Willebrand factor, in monkeys. Arterioscler Thromb Vasc Biol. 2002;22:187–192. doi: 10.1161/hq0102.101520. [DOI] [PubMed] [Google Scholar]
- 68.Machin SJ, Clarke C, Ikemura O, Kageyama S, Mackie IJ, Talbot JA, et al. A humanized monoclonal antibody against VWF A1 domain inhibits VWF:RiCof activity and platelet adhesion in human volunteers. J Thromb Haemost. 2003;1 Abstract OC328. [Google Scholar]
- 69.Hennan JK, Swillo RE, Morgan GA, Leik CE, Brooks JM, Shaw GD, et al. Pharmacologic inhibition of platelet vWF-GPIb alpha interaction prevents coronary artery thrombosis. Thromb Haemost. 2006;95:469–475. doi: 10.1160/TH05-09-0640. [DOI] [PubMed] [Google Scholar]
- 70.Wadanoli M, Sako D, Shaw GD, Schaub RG, Wang Q, Tchernychev B, et al. The von Willebrand factor antagonist (GPG-290) prevents coronary thrombosis without prolongation of bleeding time. Thromb Haemost. 2007;98:397–405. [PubMed] [Google Scholar]
- 71.Keefe AD, Schaub RG. Aptamers as candidate therapeutics for cardiovascular indications. Curr Opin Pharmacol. 2008;8:147–152. doi: 10.1016/j.coph.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 72.Diener JL, Daniel Lagassé HA, Duerschmied D, Merhi Y, Tanguay J-F, Hutabarat R, et al. Inhibition of von Willebrand factor-mediated platelet activation and thrombosis by the anti-von Willebrand factor A1-domain aptamer ARC1779. J Thromb Haemost. 2009;7:1155–1162. doi: 10.1111/j.1538-7836.2009.03459.x. [DOI] [PubMed] [Google Scholar]
- 73.Rottman JB, Gilbert M, Marsh HN, Boomer RM, Fraone JM, Makim A, et al. An anti-von Willebrand’s factor A1 domain aptamer inhibits arterial thrombosis induced by electrical injury in cynomolgus macaques. J Thromb Haemost. 2007;5 Abstract P-S-664. [Google Scholar]
- 74.Cataland SR, Peyvandi F, Mannucci PM, Lammle B, Hovinga JAK, Machin SJ, et al. A Randomized, Double-Blind, Placebo-Controlled, Clinical Outcome Study of ARC1779 In Patients with Thrombotic Thrombocytopenic Purpura (TTP) Blood. 2010;116 doi: 10.1002/ajh.23106. Abstract 726. [DOI] [PubMed] [Google Scholar]
- 75.Jilma-Stohlawetz P, Gorczyca ME, Jilma B, Siller-Matula J, Gilbert JC, Knöbl P. Inhibition of von Willebrand factor by ARC1779 in patients with acute thrombotic thrombocytopenic purpura. Thromb Haemost. 2011;105:545–552. doi: 10.1160/TH10-08-0520. [DOI] [PubMed] [Google Scholar]
- 76.Knöbl P, Jilma B, Gilbert JC, Hutabarat RM, Wagner PG, Jilma-Stohlawetz P. Anti-von Willebrand factor aptamer ARC1779 for refractory thrombotic thrombocytopenic purpura. Transfusion. 2009;49:2181–2185. doi: 10.1111/j.1537-2995.2009.02232.x. [DOI] [PubMed] [Google Scholar]
- 77.Jilma B, Paulinska P, Jilma-Stohlawetz P, Gilbert JC, Hutabarat R, Knöbl P. A randomised pilot trial of the anti-von Willebrand factor aptamer ARC1779 in patients with type 2b von Willebrand disease. Thromb Haemost. 2010;104:563–570. doi: 10.1160/TH10-01-0027. [DOI] [PubMed] [Google Scholar]
- 78.Woelfel M, De Meyer SF, Wagner P, McGinness KE, Wagner DD, Schaub R. The Generation of An Aptamer Inhibitor of Murine Von Willebrand Factor (VWF) Mediated Platelet Aggregation. Blood. 2010;116 Abstract 4312. [Google Scholar]
- 79.Markus HS, McCollum C, Imray C, Goulder MA, Gilbert J, King A. The von Willebrand Inhibitor ARC1779 Reduces Cerebral Embolization After Carotid Endarterectomy: A Randomized Trial. Stroke. 2011;42:2149–2153. doi: 10.1161/STROKEAHA.111.616649. [DOI] [PubMed] [Google Scholar]
- 80.Ulrichts H, Silence K, Schoolmeester A, de Jaegere P, Rossenu S, Roodt J, et al. Antithrombotic drug candidate ALX-0081 shows superior preclinical efficacy and safety compared to currently marketed antiplatelet drugs. Blood. 2011;118:757–765. doi: 10.1182/blood-2010-11-317859. [DOI] [PubMed] [Google Scholar]
- 81.van Loon JE, de Jaegere PP, Ulrichts H, van Vliet HH, de Maat MP, de Groot PG, et al. The in vitro effect of the new antithrombotic drug candidate ALX-0081 on blood samples of patients undergoing percutaneous coronary intervention. Thromb Haemost. 2011;106:165–171. doi: 10.1160/TH10-12-0804. [DOI] [PubMed] [Google Scholar]
- 82.Kidwell CS, Saver JL, Starkman S, Duckwiler G, Jahan R, Vespa P, et al. Late secondary ischemic injury in patients receiving intraarterial thrombolysis. Ann Neurol. 2002;52:698–703. doi: 10.1002/ana.10380. [DOI] [PubMed] [Google Scholar]
- 83.del Zoppo GJ, Koziol JA. Recanalization and stroke outcome. Circulation. 2007;115:2602–2605. doi: 10.1161/CIRCULATIONAHA.107.698225. [DOI] [PubMed] [Google Scholar]
- 84.Sandercock PA, Counsell C, Kamal AK. Anticoagulants for acute ischaemic stroke. Cochrane Database Syst Rev. 2008:CD000024. doi: 10.1002/14651858.CD000024.pub3. [DOI] [PubMed] [Google Scholar]
- 85.Adams HP, Jr, del Zoppo G, Alberts MJ, Bhatt DL, Brass L, Furlan A, et al. Guidelines for the early management of adults with ischemic stroke: a guideline from the American Heart Association/American Stroke Association Stroke Council, Clinical Cardiology Council, Cardiovascular Radiology and Intervention Council, and the Atherosclerotic Peripheral Vascular Disease and Quality of Care Outcomes in Research Interdisciplinary Working Groups: the American Academy of Neurology affirms the value of this guideline as an educational tool for neurologists. Stroke. 2007;38:1655–1711. doi: 10.1161/STROKEAHA.107.181486. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.