

Abstract

GPR88 is an orphan G-protein-coupled receptor (GPCR) enriched in the striatum. Genetic deletion and gene expression studies have suggested that GPR88 plays an important role in the regulation of striatal functions and is implicated in psychiatric disorders. The signal transduction pathway and receptor functions of GPR88, however, are still largely unknown due to the lack of endogenous and synthetic ligands. In this paper, we report the synthesis of a GPR88 agonist 2-PCCA and its pure diastereomers, which were functionally characterized in both transiently and stably expressing GPR88 HEK293 cells. 2-PCCA inhibited isoproterenol-stimulated cAMP accumulation in a concentration-dependent manner in cells expressing GPR88 but not in the control cells, suggesting that the observed cAMP inhibition is mediated through GPR88 and that GPR88 is coupled to Gαi. 2-PCCA did not induce calcium mobilization in GPR88 cells, indicating no Gαq-mediated response. A structure–activity relationship (SAR) study of 2-PCCA was also conducted to explore the key structural features for GPR88 agonist activity.
Keywords: Orphan GPR88, agonists, 2-PCCA
GPR88 is an orphan G-protein-coupled receptor, which was originally identified as a striatum-specific receptor (designated Strg/GPR88),1 though it is also expressed in other brain regions, including the cerebral cortex, amygdala, and hypothalamus.2 In the striatum, GPR88 is highly expressed in both D1 and D2 receptor-expressing medium spiny neurons (MSNs),2b,3 suggesting the receptor may play a role in regulating dopaminergic activity. GPR88 knockout mice demonstrated disrupted prepulse inhibition of the startle response, a phenotype of schizophrenia, and exhibited D2 receptors hypersensitivity (as evidenced by increased sensitivity to apomorphine-induced climbing and stereotypy, and amphetamine-stimulated locomotor activity).4 In another study of GPR88 knockout mice,3,5 the animals exhibited increased locomotion, and impaired motor coordination and cue-based learning. GPR88 re-expression normalized these impaired behaviors, suggesting that GPR88 dysfunction may contribute to abnormal behaviors observed in neurological and psychiatric diseases.3 In line with these findings from GPR88 knockout studies, transcriptional profiling studies have revealed GPR88 gene expression is altered by treatment or conditions related to schizophrenia,6 bipolar disorder,7 depression,8 and drug addiction.9 Taken together, these studies suggest that GPR88 plays an important role in the regulation of striatal functions and is a promising drug target for treating basal ganglia-associated disorders.
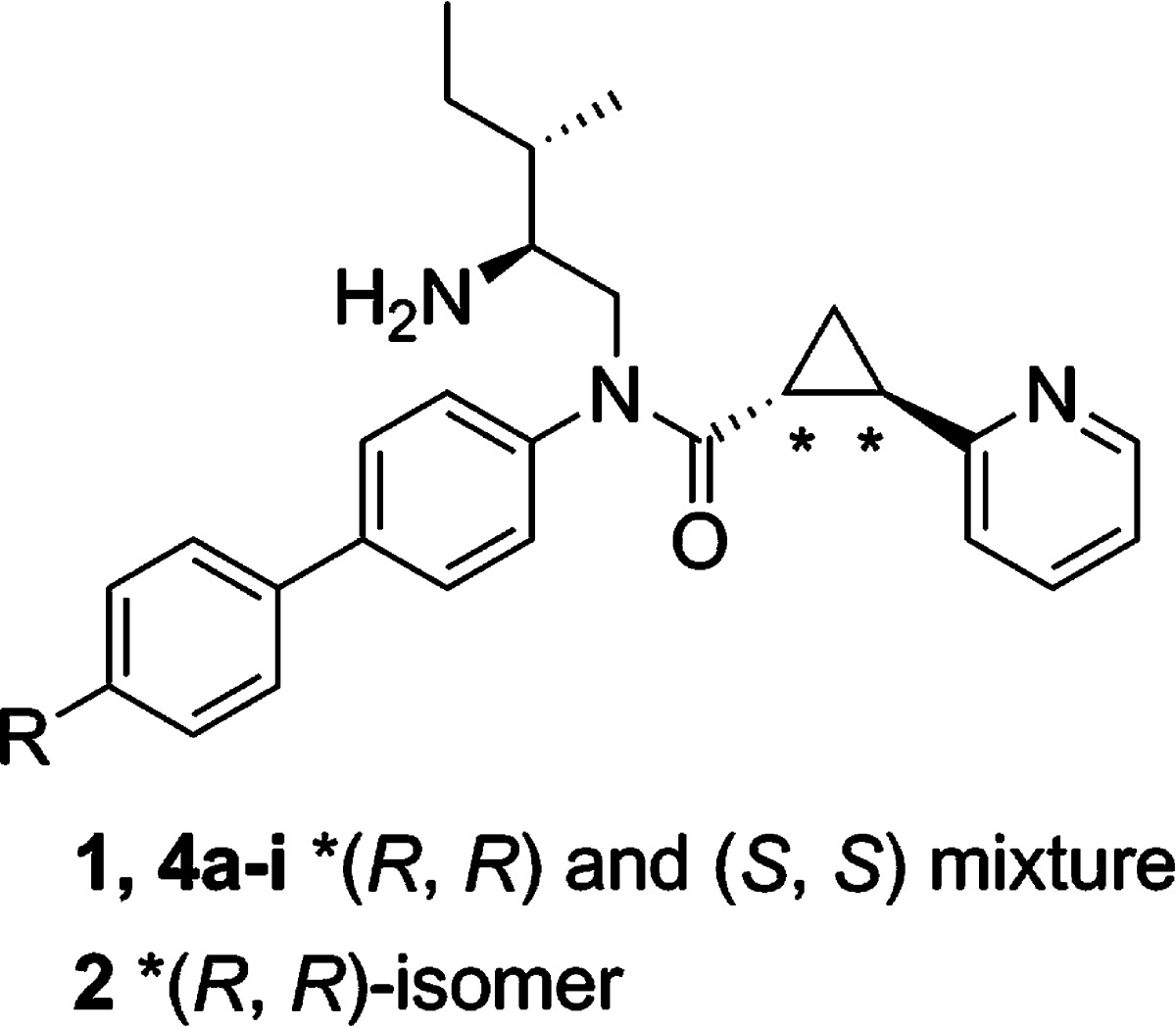
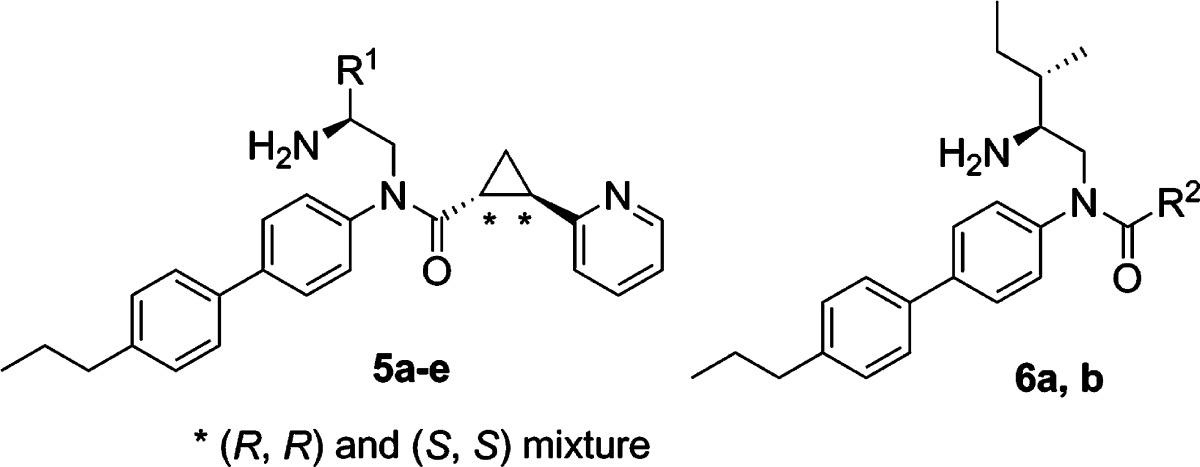
In order to elucidate the biological function of GPR88, selective agonists are required. Recently, a series of surrogate agonists of GPR88 have been reported in the patent literature and were suggested to activate GPR88 coupling to Gαi pathways.10 However, the function and structure–activity relationship (SAR) relative to these compounds are unclear. In this paper, we report the synthesis of a GPR88 ligand 2-PCCA [(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarboxylic acid ((2S,3S)-2-amino-3-methylpentyl)-(4′-propylbiphenyl-4-yl)amide (1); Figure 1] and its pure (1R,2R)- and (1S,2S)-diastereomers (2 and 3, respectively), which were functionally characterized in the GPR88 cell-based cAMP assays. A series of 2-PCCA analogues (4a–i, 5a–e, 6a, and 6b; Figure 2) were also synthesized and examined to explore the SAR of this chemical scaffold at GPR88.
Figure 1.

Structures of 2-PCCA (1), 2, and 3.
Figure 2.

2-PCCA analogues.
Chemistry
2-PCCA (1) and the 4-substituted bisphenyl analogues 4a–i were synthesized by a known procedure,10b outlined in Scheme 1, with some modifications. A diastereomeric mixture was first synthesized to characterize the GPR88 signaling pathways. Asymmetric synthesis of both pure diastereomers 2 and 3 of 2-PCCA was conducted later to determine which isomer is more active. Cyclopropanation of 2-vinylpyridine (8) with tert-butyl diazoacetate under catalytic conditions led to tert-butyl ester 9, which was then treated with 4 M HCl in dioxane to give the racemic (1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarboxylic acid (10) in 56% yield. Reductive amination of aldehyde 12, prepared by Dess-Martin oxidation of commercially available (−)-(2S,3S)-N-Boc-2-amino-3-methyl-1-pentanol with 4-bromoaniline (11) afforded amine 13 in 75% yield. With both building blocks available, amide 14 was synthesized in 72% yield by converting acid 10 into the corresponding acid chloride, followed by reaction with amine 13. Suzuki coupling of 14 with an appropriate arylboronic acid under microwave conditions gave intermediates 15a–j in the range of 65–96% yields. Removal of the Boc protecting group with 4 M HCl in dioxane provided 1 and 4a–i in 90–98% yields. Compounds 1 and 4a–i were determined to be 1:1 diastereomeric mixtures by 1H NMR and HPLC analyses.
Scheme 1.

Synthesis of pure diastereomers 2 and 3 of 2-PCCA is described in Scheme 2. The key intermediate 14, prepared from enantiomerically pure (−)-(2S,3S)-N-Boc-2-amino-3-methyl-1-pentanol as described in Scheme 1, is a 1:1 mixture of (1R,2R)- and (1S,2S)-diastereomers differentiating at the configuration of the trans-substituted cyclopropane ring. Asymmetric synthesis of (1R,2R)-14 was accomplished starting from the preparation of pure enantiomer (1R,2R)-2-(pyridin-2-yl)cyclopropanecarboxylic acid ((1R,2R)-10). Thus, asymmetric cyclopropanation of 8 using the known chiral porphyrin catalyst [Co(3,5-DitBu-ChenPhyrin)]11 afforded the tert-butyl ester (1R,2R)-9 in 97% ee, as determined by chiral HPLC analysis. The chiral porphyrin Co(II) catalysts have been well studied in the asymmetric cyclopropanation of olefins using diazoacetates to give the corresponding cyclopropanes with high diastereoselectivity and enantioselectivity.11 Assignment of the absolute configuration of (1R,2R)-9 was made based on an analogy to the known (1R,2R)-2-phenyl-1-cyclopropanecarboxylic acid tert-butyl ester11a synthesized using the same chiral porphyrin catalyst. Acidic hydrolysis of (1R,2R)-9 led to acid (1R,2R)-10, which was then coupled with amine 13 to provide (1R,2R)-14 in 40% yield over three steps. To obtain the pure diastereomer (1S,2S)-14, the mixture 14 was separated by HPLC using a ChiralPak IA column to afford (1R,2R)-14 and (1S,2S)-14 in 40% and 39% yield, respectively. Suzuki coupling of (1R,2R)-14 and (1S,2S)-14 with 4-propylphenylboronic acid, followed by removal of the Boc protecting group with HCl gave 2 and 3, in 80% and 81% yield, respectively.
Scheme 2.

Compounds 5a–e were synthesized using the procedure, outlined in Scheme 3, analogous to that used to prepare 1. Reductive amination of an appropriate aldehyde 17a–e with 4-bromoaniline (11) or 4-(4′-propylphenyl)aniline (16) afforded amine 18a–e in 50–86% yields. Amide formation with the acid chloride, prepared from the racemic 10, gave 19a–e in 53–60% yields. Suzuki coupling of 19a, 19b, and 19e with 4-propylphenylboronic acid yielded 53–80% of 20a, 20b, and 20e. Deprotection of the Boc group furnished 5a–e as 1:1 diastereomeric mixtures in 92–98% yields.
Scheme 3.

Synthesis of compounds 6a and 6b started with the reaction of amine 13 with (1R,2R)-2-phenyl-1-cyclopropanecarboxylic acid12 and 2-pyridylacetic acid (Scheme 4), respectively. The resulting amides 21a and 21b were coupled with 4-propylphenylboronic acid, followed by HCl treatment to provide 6a and 6b in 55% and 33% overall yield, respectively. All synthesized compounds were >95% pure as determined by HPLC analyses. The 1H NMR spectra of the target compounds were in agreement with the assigned structures.
Scheme 4.

Results and Discussion
Despite emerging pharmacological implications of the orphan GPR88 receptor, little is known regarding its downstream signaling pathways, as identification of endogenous and synthetic ligands has been elusive. Results from the patent literature10 indicated that GPR88 couples to Gαi proteins and thereby inhibits cAMP production. To develop an appropriate cell based assay system to support drug discovery for the GPR88 receptor, we initially transiently cotransfected HEK 293T cells with the human GPR88 cDNA and a luminescent cAMP biosensor and determined both Gαs and Gαi activations.13 2-PCCA (1) produced no measurable increases in cAMP levels at concentrations up to 30 μM. As a positive control, isoproterenol (ISO) activated the endogenous β2 adrenergic receptors expressed in HEK293T cells and greatly stimulated cAMP production in a concentration-dependent manner. 2-PCCA inhibited ISO-induced cAMP formation with a pEC50 value of 6.06 (EC50 = 877 nM) in GPR88 cells but not in the control cells transiently transfected with the biosensor, demonstrating that 2-PCCA activates GPR88-mediated Gαi signaling (Figure 3). In addition, the (1R,2R)-isomer 2 (EC50 = 373 nM) is approximately 5-fold more potent than the (1S, 2S)-isomer 3. Furthermore, 2-PCCA did not induce calcium mobilization measured by the fluorescent imaging plate reader (FLIPR) calcium assay13b,14 in HEK293T/GPR88 cells (data not shown), indicating that GPR88 is likely not coupled to Gαq proteins in our assay systems.
Figure 3.
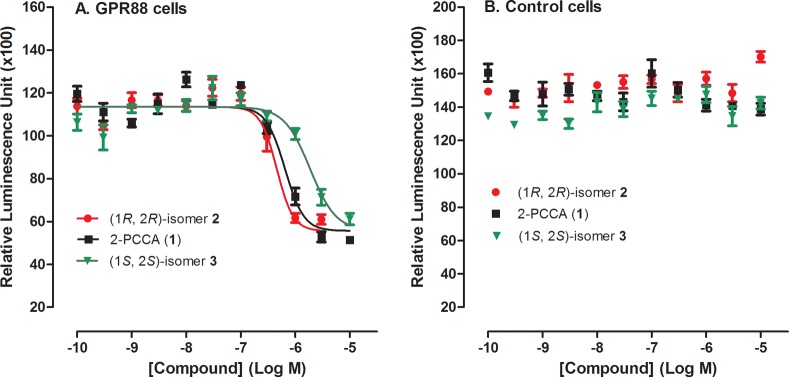
HEK 293T cells were transiently transfected with GPR88 and GloSensor cAMP construct (A) or GloSensor cAMP only (B). 2-PCCA (1), 2, and 3 inhibited isoproterenol-induced cAMP production in GPR88 cells, but not in the control cells. The data are the means of quadruplicate measurements with standard deviation shown as error bars and are representative of at least three independent experiments.
To further characterize the GPR88 in vitro functions and explore the key structural features of 2-PCCA agonist activity, HEK293 cells stably expressing the human GPR88 receptor and the GloSensor-22F cAMP construct were established. In the stable GPR88-22F cells, 2-PCCA and its pure diastereomer 2 had pEC50 values of 6.04 (EC50 = 911 nM) and 6.22 (EC50 = 603 nM), respectively. To explore the SAR of 2-PCCA, substitution effects of the bisphenyl moiety were first examined. As seen in Table 1, unsubstituted analogue 4a was less potent than 2-PCCA. The 4-position tolerated small to medium size of alkyl substitutions with the methyl analogue 4b (EC50 = 845 nM) and cyclohexyl analogue 4e (EC50 = 746 nM) being the most potent compounds in the series. Replacing the methyl group with an electron-withdrawing trifluoromethyl group (4f) markedly reduced activity. The addition of a fluoro or chloro group to the 4-position led to 4g and 4h, respectively, resulting in even lower potency. Somewhat surprisingly, the 4-acetyl analogue 4i possessed a similar potency (EC50 = 923 nM) at GPR88 relative to 2-PCCA.
Table 1. Structures and Activities of Compounds 1, 2, and 4a–i.
| compda | R | pEC50 (EC50, nM)b |
|---|---|---|
| 2-PCCA (1) | Pr | 6.04 ± 0.06 (911) |
| 2 | Pr | 6.22 ± 0.10 (603) |
| 4a | H | 5.48 ± 0.08 (3321) |
| 4b | Me | 6.07 ± 0.14 (845) |
| 4c | Et | 5.70 ± 0.08 (1989) |
| 4d | i-Bu | 5.74 ± 0.06 (1803) |
| 4e | cyclohexyl | 6.13 ± 0.13 (746) |
| 4f | CF3 | 5.49 ± 0.08 (3266) |
| 4g | fluoro | NAc |
| 4h | chloro | NAc |
| 4i | acetyl | 6.03 ± 0.05 (923) |
All compounds were tested as the HCl salt.
pEC50 values are means ± standard error of at least three independent experiments performed in duplicate.
EC50 > 10 μM, tested in two independent experiments performed in duplicate.
Investigation of the substituted ethylamine moiety of 2-PCCA (Table 2) showed that hydrophobic substitutions were well tolerated. The trend of increased potency with large substituents (5a–e) at the ethylamine moiety suggests that a hydrophobic pocket may be present in the GPR88 receptor. A limited examination of the amide carbonyl groups observed that the pyridyl group in 2-PCCA could be replaced with a phenyl group (6a) while causing only a slight decrease in potency. However, replacement of the cyclopropane moiety with a methylene group (6b) resulted in a loss of activity, indicating the central linker of the aromatic and carbonyl moieties is critical for GPR88 recognition.
Table 2. Structures and Activities of Compounds 5a–e, 6a, and 6b.
| compda | R1 | R2 | pEC50 (EC50, nM)b |
|---|---|---|---|
| 2-PCCA (1) | 6.04 ± 0.06 (911) | ||
| 5a | H | 5.27 ± 0.12 (5330) | |
| 5b | Me | 5.42 ± 0.12 (3821) | |
| 5c | i-Pr | 5.80 ± 0.09 (1602) | |
| 5d | i-Bu | 6.00 ± 0.21 (994) | |
| 5e | PhCH2 | 6.05 ± 0.19 (898) | |
| 6a | (1R,2R)-2-phenyl-cyclopropan-1-yl | 5.90 ± 0.05 (1250) | |
| 6b | (pyridin-2-yl)methyl | NAc |
All compounds were tested as the HCl salt.
pEC50 values are means ± standard error of at least three independent experiments performed in duplicate.
EC50 > 10 μM
Conclusions
In summary, we demonstrated that GPR88 couples to the Gαi subunits, and is activated by 2-PCCA in both transient and stable GPR88 expressing cells. In an effort to determine the key structural features for 2-PCCA agonist activity, we designed and synthesized a series of 2-PCCA analogues 4a–i, 5a–e, 6a, and 6b. Further pharmacological evaluation of the (1R,2R)-isomer 2 and phenyl analogue 6a, including receptor specificity and in vivo behavioral studies, are in progress. These studies will facilitate the identification of highly potent, selective ligands for GPR88 and the understanding of its physiological functions in vivo.
Methods
Chemistry
General Methods
Melting points were determined using a MEL-TEMP II capillary melting point apparatus and are uncorrected. Nuclear magnetic resonance (1H NMR and 13C NMR) spectra were obtained on a Bruker Avance DPX-300 MHz NMR spectrometer. Chemical shifts are reported in parts per million (ppm) with reference to internal solvent. 13C NMR data of diastereomeric mixtures were not reported due to the complicity of the spectra. Mass spectra (MS) were run on a PerkinElmer Sciex API 150 EX mass spectrometer. HRMS spectra were run on a Waters Synapt G2 HDMS Q-TOF mass spectrometer, using electrospray ionization in positive ion mode. Optical rotations were measured on an AutoPol III polarimeter, purchased from Rudolf Research. Analytical thin-layer chromatography (TLC) was carried out using EMD silica gel 60 F254 TLC plates. TLC visualization was achieved with a UV lamp or in an iodine chamber. Flash column chromatography was done on a CombiFlash Companion system using Isco prepacked silica gel columns. Unless otherwise stated, reagent-grade chemicals were obtained from commercial sources and were used without further purification. All moisture- and air-sensitive reactions and reagent transfers were carried out under dry nitrogen.
(±)-tert-Butyl (1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarboxylate (9)
A solution of 2-vinylpyridine (8) (0.58 mL, 5.4 mmol), tert-butyl diazoacetate (0.88 mL, 6.4 mmol), and 5,10,15,20-tetraphenyl-21H,23H-porphine cobalt(II) (72 mg, 0.11 mmol) in toluene (25 mL) was heated in a sealed tube at 80 °C for 2 h. After cooling to room temperature, the mixture was concentrated under reduced pressure. Flash column chromatography of the crude product on silica gel using 0–30% EtOAc in hexanes afforded 9 (0.82 g, 69%) as a brown oil: 1H NMR (300 MHz; CDCl3) δ 8.44 (d, J = 6.0 Hz, 1H), 7.56 (td, J = 6.0, 3.0 Hz, 1H)), 7.23 (t, J = 6.0 Hz, 1H), 7.11–6.93 (m, 1H), 2.51 (ddd, J = 9.0, 6.0, 3.0 Hz, 1H), 2.16 (ddd, J = 9.0, 6.0, 3.0 Hz, 1H), 1.57–1.48 (m, 2H), 1.45 (s, 9H); 13C NMR (75 MHz; CDCl3) δ 172.6, 159.3, 149.4, 135.9, 123.3, 121.1, 80.5, 28.2, 26.8, 25.4, 17.2; MS (ESI) m/z 220.4 [M + H]+.
(±)-(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid Hydrochloride (10)
To a solution of 9 (0.80 g, 3.65 mmol) in CH2Cl2 (10 mL) was added 4 M HCl in dioxane (3 mL), and the reaction was stirred at room temperature overnight. The solvent was removed under reduced procedure. The residue was triturated with CH2Cl2/Et2O to afford 10 (0.60 g, 81%) as a greenish solid: mp 141–143 °C; 1H NMR (300 MHz; DMSO-d6) δ 12.00 (br s, 1H), 8.67 (dd, J = 6.0, 3.0 Hz, 1H), 8.30 (td, J = 6.0, 3.0 Hz, 1H), 7.81–7.68 (m, 2H), 2.90 (ddd, J = 9.0, 6.0, 3.0 Hz, 1H), 2.33 (ddd, J = 9.0, 6.0, 3.0 Hz, 1H), 1.80–1.60 (m, 2H); 13C NMR (75 MHz; DMSO-d6) δ 172.5, 156.1, 134.5, 143.0, 123.8, 123.4, 25.0, 23.2, 17.3; MS (ESI) m/z 164.4 [M + H]+.
tert-Butyl {(2S,3S)-1-[(4-Bromophenyl)amino]-3-methylpentan-2-yl}carbamate (13)
To a solution of (−)-(2S,3S)-N-Boc-2-amino-3-methyl-1-pentanol (2.17 g, 10.0 mmol) in water-saturated CH2Cl2 (10 mL) at room temperature was added Dess-Martin reagent (8.90 g, 21.0 mmol), and the reaction was stirred for 1 h. Additional water-saturated CH2Cl2 (5 mL) was added every 15 min during the reaction time. The mixture was diluted with Et2O (100 mL) and poured into a solution of Na2S2O3 (17 g) in 80% saturated NaHCO3 (100 mL). After stirring for 10 min, the layers were separated and the aqueous layer was extracted with Et2O (100 mL). The combined organic layers were washed with ice-cold saturated NaHCO3 (30 mL) and water (30 mL). The solution was dried (Na2SO4) and concentrated under reduced pressure to give the crude aldehyde 12. To a solution of 4-bromoaniline (11) (1.72 g, 10.0 mmol) in dichloroethane (60 mL) was added the above crude aldehyde, followed by NaBH(OAc)3 (4.24 g, 20.0 mmol). The mixture was stirred at room temperature overnight. Saturated NaHCO3 (20 mL) was added, and the layers were separated. The aqueous layer was extracted with CH2Cl2 (2 × 30 mL). The combined organic layers were washed with brine (3 × 30 mL), dried (Na2SO4) and concentrated under reduced pressure. Flash column chromatography of the crude product on silica gel using 0–30% EtOAc in hexanes afforded 13 (2.78 g, 75%) as a white solid: mp 103–105 °C; [α]23D +11.4° (c 0.52, CH3OH); 1H NMR (300 MHz; CDCl3) δ 7.24 (d, J = 9.0 Hz, 2H), 6.45 (d, J = 9.0 Hz, 2H), 4.52 (d, J = 9.0 Hz, 1H), 4.20 (br s, 1H), 3.82–3.65 (m, 1H), 3.30–3.14 (m, 1H), 3.05–2.89 (m, 1H), 1.60–1.47 (m, 1H), 1.44 (s, 9H), 1.23–1.10 (m, 1H), 0.95 (d, J = 6.0 Hz, 3H), 0.95 (t, J = 7.5 Hz, 3H); 13C NMR (75 MHz; CDCl3) δ 154.9, 145.7, 130.1, 112.4, 106.9, 77.9, 52.9, 45.0, 35.7, 26.6, 23.6, 13.8, 9.9; MS (ESI) m/z 371.3 [M + H]+ (79Br), 373.3 [M + H]+ (81Br).
tert-Butyl [(2S,3S)-1-{4-Bromophenyl-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]carbamate (14)
To a solution of 10 (0.40 g, 2.0 mmol) in CH2Cl2 (20 mL) at room temperature was added oxalyl chloride (0.35 mL, 4.0 mmol) and DMF (50 μL). The mixture was stirred at 40 °C for 2 h and then cooled to room temperature and concentrated under reduced pressure. The residue was dissolved in CH2Cl2 (20 mL) and treated with 13 (0.74 g, 2.0 mmol) and Et3N (1.1 mL, 8.0 mmol). The resulting solution was stirred at room temperature overnight. Saturated NaHCO3 (10 mL) was added, and the layers were separated. The aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with brine (3 × 20 mL), dried (Na2SO4) and concentrated under reduced pressure. Flash column chromatography of the crude product on silica gel using 0–25% EtOAc in hexanes afforded 14 (0.74 g, 72%, 1:1 diastereomeric mixture) as a light yellow foam: 1H NMR (300 MHz; CDCl3) δ 8.30 (d, J = 6.0 Hz, 1H), 7.58–7.34 (m, 3H), 7.22–6.98 (m, 4H), 4.96 (d, J = 9.0 Hz, 1H), 4.45–4.25 (m, 1H), 3.80–3.78 (m, 1H), 3.21–3.06 (m, 1H), 2.71–2.62 (m, 0.5 H), 2.58–2.48 (m, 0.5 H), 1.98–1.86 (m, 1H), 1.75–1.62 (m, 1H), 1.60–1.35 (m, 3H), 1.45 and 1.40 (2s, 9H), 1.15–1.02 (m, 1H), 0.92–0.80 (m, 6H); MS (ESI) m/z 516.7 [M + H]+ (79Br), 518.6 [M + H]+ (81Br).
tert-Butyl [(2S,3S)-1-{(4′-Propylbiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]carbamate (15a)
A mixture of 14 (0.48 g, 0.92 mmol), 4-propylphenylboronic acid (0.25 g, 1.4 mmol), Pd(dppf)Cl2·CH2Cl2 (70 mg, 0.092 mmol), and K3PO4 (0.58 g, 2.7 mmol) in dimethoxyethane (10.5 mL) and water (3.5 mL) was heated in a sealed vessel by microwave irradiation at 160 °C for 6 min. The resulting mixture was poured into 1 N NaOH solution (30 mL) and extracted with CH2Cl2 (3 × 30 mL). The combined organic layers were dried (Na2SO4) and concentrated under reduced pressure. Flash column chromatography of the crude product on silica gel using 0–20% EtOAc in hexanes afforded 15a (0.49 g, 96%, 1:1 diastereomeric mixture) as a white foam: 1H NMR (300 MHz; CDCl3) δ 8.28–8.22 (m, 1H), 7.58–7.38 (m, 5H), 7.32–7.13 (m, 5H), 7.03–6.96 (m, 1H), 5.12–5.05 (m, 1H), 4.48–4.36 (m, 1H), 3.85–3.56 (m, 1H), 3.24–3.13 (m, 1H), 2.71–2.50 (m, 3H), 2.05–1.95 (m, 1H), 1.75–1.58 (m, 3H), 1.51–1.38 (m, 3H), 1.47 and 1.43 (2s, 9H), 1.56–1.40 (m, 1H), 0.97 (t, J = 7.5 Hz, 3H), 0.92–0.80 (m, 6H); MS (ESI) m/z 557.0 [M + H]+.
tert-Butyl [(2S,3S)-1-{(Biphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]carbamate (15b)
The procedure for 15a was followed using 30 mg (0.058 mmol) of 14 and 11 mg (0.087 mmol) of phenylboronic acid to give 24 mg (81%) of 15b as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.28–8.22 (m, 1H), 7.59–7.38 (m, 6H), 7.37–7.14 (m, 5H), 7.12–6.95 (m, 1H), 5.14–5.07 (m, 1H), 4.49–4.36 (m, 1H), 3.85–3.68 (m, 1H), 3.26–3.14 (m, 1H), 2.75–2.66 (m, 0.5 H), 2.60–2.50 (m, 0.5H), 2.08–1.98 (m, 1H), 1.75–1.66 (m, 0.5H), 1.65–1.57 (m, 0.5H), 1.55–1.36 (m, 3H), 1.47 and 1.43 (2s, 9H), 1.18–1.02 (m, 1H), 0.94–0.80 (m, 6H); MS (ESI) m/z 514.7 [M + H]+.
tert-Butyl [(2S,3S)-1-{(4′-Methylbiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]carbamate (15c)
The procedure for 15a was followed using 30 mg (0.058 mmol) of 14 and 12 mg (0.087 mmol) of 4-methylphenylboronic acid to give 20 mg (65%) of 15c as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.29–8.22 (m, 1H), 7.58–7.38 (m, 5H), 7.37–7.16 (m, 5H), 7.02–6.92 (m, 1H), 5.13–5.06 (m, 1H), 4.48–4.37 (m, 1H), 3.83–3.68 (m, 1H), 3.24–3.12 (m, 1H), 2.73–2.65 (m, 0.5 H), 2.60–2.50 (m, 0.5H), 2.39 (s, 3H), 2.08–1.96 (m, 1H), 1.76–1.55 (m, 1H), 1.54–1.37 (m, 3H), 1.47 and 1.43 (2s, 9H), 1.17–1.00 (m, 1H), 0.92–0.78 (m, 6H); MS (ESI) m/z 528.7 [M + H]+.
tert-Butyl [(2S,3S)-1-{(4′-Ethylbiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]carbamate (15d)
The procedure for 15a was followed using 30 mg (0.058 mmol) of 14 and 13 mg (0.087 mmol) of 4-ethylphenylboronic acid to give 25 mg (80%) of 15d as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.28–8.23 (m, 1H), 7.58–7.38 (m, 5H), 7.37–7.15 (m, 5H), 7.04–6.95 (m, 1H), 5.15–5.05 (m, 1H), 4.50–4.37 (m, 1H), 3.85–3.68 (m, 1H), 3.25–3.14 (m, 1H), 2.76–2.66 (m, 2.5 H), 2.60–2.48 (m, 0.5H), 2.10–1.96 (m, 1H), 1.76–1.54 (m, 1H), 1.53–1.37 (m, 3H), 1.47 and 1.43 (2s, 9H), 1.28 (t, J = 7.5 Hz, 3H), 1.17–1.00 (m, 1H), 0.93–0.78 (m, 6H); MS (ESI) m/z 542.6 [M + H]+.
tert-Butyl [(2S,3S)-1-{(4′-Isobutylbiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]carbamate (15e)
The procedure for 15a was followed using 30 mg (0.058 mmol) of 14 and 16 mg (0.087 mmol) of 4-isobutylphenylboronic acid to give 23 mg (77%) of 15e as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.28–8.23 (m, 1H), 7.60–7.38 (m, 5H), 7.34–7.15 (m, 5H), 7.04–6.95 (m, 1H), 5.14–5.05 (m, 1H), 4.50–4.36 (m, 1H), 3.86–3.68 (m, 1H), 3.24–3.14 (m, 1H), 2.74–2.65 (m, 0.5 H), 2.60–2.46 (m, 2.5H), 2.10–1.82 (m, 2H), 1.76–1.54 (m, 1H), 1.53–1.36 (m, 3H), 1.47 and 1.43 (2s, 9H), 1.16–1.00 (m, 1H), 0.98–0.78 (m, 12H); MS (ESI) m/z 570.6 [M + H]+.
tert-Butyl [(2S,3S)-1-{(4′-Cyclohexylbiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]carbamate (15f)
The procedure for 15a was followed using 30 mg (0.058 mmol) of 14 and 18 mg (0.087 mmol) of 4-cyclohexylphenylboronic acid to give 28 mg (81%) of 15f as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.28–8.21 (m, 1H), 7.58–7.40 (m, 5H), 7.32–7.12 (m, 5H), 7.02–6.92 (m, 1H), 5.15–5.05 (m, 1H), 4.50–4.36 (m, 1H), 3.88–3.68 (m, 1H), 3.26–3.13 (m, 1H), 2.75–2.64 (m, 0.5 H), 2.60–2.48 (m, 1.5H), 2.05–1.71 (m, 7H), 1.69–1.25 (m, 8H), 1.47 and 1.43 (2s, 9H), 1.17–1.00 (m, 1H), 0.96–0.80 (m, 6H); MS (ESI) m/z 596.9 [M + H]+.
tert-Butyl [(2S,3S)-1-{(4′-Trifluoromethylbiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]carbamate (15g)
The procedure for 15a was followed using 30 mg (0.058 mmol) of 14 and 17 mg (0.087 mmol) of 4-trifluoromethylphenylboronic acid to give 25 mg (74%) of 15g as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.30–8.22 (m, 1H), 7.72–7.40 (m, 7H), 7.38–7.16 (m, 3H), 7.02–6.93 (m, 1H), 5.13–5.02 (m, 1H), 4.50–4.38 (m, 1H), 3.84–3.65 (m, 1H), 3.28–3.15 (m, 1H), 2.76–2.65 (m, 0.5 H), 2.61–2.48 (m, 0.5H), 2.08–1.96 (m, 1H), 1.76–1.55 (m, 1H), 1.54–1.37 (m, 3H), 1.47 and 1.43 (2s, 9H), 1.18–1.00 (m, 1H), 0.94–0.78 (m, 6H); MS (ESI) m/z 582.7 [M + H]+.
tert-Butyl [(2S,3S)-1-{(4′-Fluorobiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]carbamate (15h)
The procedure for 15a was followed using 30 mg (0.058 mmol) of 14 and 12 mg (0.087 mmol) of 4-fluorophenylboronic acid to give 20 mg (74%) of 15h as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.30–8.22 (m, 1H), 7.56–7.48 (m, 4H), 7.30–7.18 (m, 6H), 7.02–7.92 (m, 1H), 5.12–5.05 (m, 1H), 4.52–4.38 (m, 1H), 3.82–3.65 (m, 1H), 3.25–3.12 (m, 1H), 2.76–2.65 (m, 0.5 H), 2.61–2.48 (m, 0.5H), 2.08–1.95 (m, 1H), 1.78–1.55 (m, 1H), 1.54–1.37 (m, 3H), 1.47 and 1.43 (2s, 9H), 1.18–1.00 (m, 1H), 0.95–0.80 (m, 6H); MS (ESI) m/z 532.5 [M + H]+.
tert-Butyl [(2S,3S)-1-{(4′-Chlorobiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]carbamate (15i)
The procedure for 15a was followed using 30 mg (0.058 mmol) of 14 and 14 mg (0.087 mmol) of 4-chlorophenylboronic acid to give 22 mg (74%) of 15i as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.30–8.21 (m, 1H), 7.55–7.36 (m, 6H), 7.30–7.15 (m, 4H), 7.02–7.95 (m, 1H), 5.10–5.00 (m, 1H), 4.50–4.35 (m, 1H), 3.80–3.62 (m, 1H), 3.26–3.15 (m, 1H), 2.76–2.65 (m, 0.5 H), 2.61–2.48 (m, 0.5H), 2.08–1.95 (m, 1H), 1.75–1.55 (m, 1H), 1.54–1.37 (m, 3H), 1.47 and 1.42 (2s, 9H), 1.16–1.00 (m, 1H), 0.95–0.80 (m, 6H); MS (ESI) m/z 548.5 [M + H]+.
tert-Butyl [(2S,3S)-1-{(4′-Acetylbiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]carbamate (15j)
The procedure for 15a was followed using 30 mg (0.058 mmol) of 14 and 14 mg (0.087 mmol) of 4-acetylphenylboronic acid to give 25 mg (76%) of 15j as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.30–8.21 (m, 1H), 8.06–7.98 (m, 2H), 7.68–7.48 (m, 5H), 7.38–7.15 (m, 3H), 7.02–7.93 (m, 1H), 5.10–5.00 (m, 1H), 4.50–4.36 (m, 1H), 3.80–3.62 (m, 1H), 3.28–3.15 (m, 1H), 2.75–2.65 (m, 0.5 H), 2.64 (s, 3H), 2.61–2.48 (m, 0.5H), 2.08–1.95 (m, 1H), 1.76–1.55 (m, 1H), 1.54–1.37 (m, 3H), 1.47 and 1.43 (2s, 9H), 1.16–0.98 (m, 1H), 0.92–0.78 (m, 6H); MS (ESI) m/z 557.2 [M + H]+.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-propylbiphenyl-4-yl)amide Dihydrochloride (1)
A solution of 15a (150 mg, 0.27 mmol) and 4 M HCl in dioxane (2 mL) in CH2Cl2 (5 mL) was stirred at room temperature for 6 h. The solvent was removed under reduced pressure. The resulting residue was triturated with Et2O to give 1 (135 mg, 95%, 1:1 diastereomeric mixture) as a white solid: 1H NMR (300 MHz; CD3OD) δ 8.66–8.54 (m, 1H), 8.37–8.23 (m, 1H), 7.82–7.44 (m, 8H), 7.32–7.22 (m, 2H), 4.43 (dd, J = 15.0, 9.0 Hz, 0.5H), 4.28 (dd, J = 15.0, 9.0 Hz, 0.5H), 3.82 (d, J = 15.0 Hz, 0.5H), 3.68 (d, J = 15.0 Hz, 0.5H), 3.46–3.35 (m, 1H), 3.11–3.02 (m, 0.5H), 3.00–2.90 (m, 0.5H), 2.62 (t, J = 7.5 Hz, 2H), 2.22–2.08 (m, 1H), 2.02–1.88 (m, 1H), 1.87–1.62 (m, 4H), 1.50–1.12 (m, 2H), 1.02–0.78 (m, 9H); HRMS (ESI) calcd. for C30H37N3O [M + H]+: 456.3009. Found: 456.3020.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(biphenyl-4-yl)amide Dihydrochloride (4a)
The procedure for 1 was followed using 20 mg (0.039 mmol) of 15b and 1 mL of 4 M HCl in dioxane to give 17 mg (90%) of 4a as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.28–8.21 (m, 1H), 7.72–7.52 (m, 5H), 7.48–7.32 (m, 5H), 7.30–7.23 (m, 1H), 7.19–7.10 (m, 1H), 4.40–4.22 (m, 1H), 3.80–3.66 (m, 1H), 3.40–3.22 (m, 1H), 2.70–2.53 (m, 1H), 2.05–1.90 (m, 1H), 1.84–1.62 (m, 2H), 1.50–1.15 (m, 3H), 0.99 and 0.97 (2d, J = 6.0 Hz, 3H), 0.90–0.80 (m, 3H); HRMS (ESI) calcd. for C27H31N3O [M + H]+: 414.2541. Found: 414.2540.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-methylbiphenyl-4-yl)amide Dihydrochloride (4b)
The procedure for 1 was followed using 20 mg (0.038 mmol) of 15c and 1 mL of 4 M HCl in dioxane to give 18 mg (95%) of 4b as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.68–8.55 (m, 1H), 8.42–8.30 (m, 1H), 7.88–7.40 (m, 8H), 7.30–7.20 (m, 2H), 4.50–4.40 (m, 0.5H), 4.40–4.28 (m, 0.5H), 3.88–3.56 (m, 1H), 3.48–3.38 (m, 1H), 3.12–3.05 (m, 0.5H), 3.05–2.96 (m, 0.5H), 2.37 (s, 3H), 2.22–2.10 (m, 1H), 2.10–1.88 (m, 1H), 1.87–1.60 (m, 2H), 1.48–1.15 (m, 2H), 1.06–0.80 (m, 6H); HRMS (ESI) calcd. for C28H33N3O [M + H]+: 428.2696. Found: 428.2701.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-ethylbiphenyl-4-yl)amide Dihydrochloride (4c)
The procedure for 1 was followed using 20 mg (0.037 mmol) of 15d and 1 mL of 4 M HCl in dioxane to give 18 mg (95%) of 4c as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.70–8.58 (m, 1H), 8.45–8.30 (m, 1H), 7.88–7.45 (m, 8H), 7.32–7.20 (m, 2H), 4.52–4.40 (m, 0.5H), 4.38–4.28 (m, 0.5H), 3.88–3.56 (m, 1H), 3.48–3.35 (m, 1H), 3.15–3.05 (m, 0.5H), 3.05–2.95 (m, 0.5H), 2.80–2.60 (m, 2H), 2.25–2.10 (m, 1H), 2.10–1.90 (m, 1H), 1.87–1.60 (m, 2H), 1.50–1.10 (m, 5H), 1.08–0.80 (m, 6H); HRMS (ESI) calcd. for C29H35N3O [M + H]+: 442.2853. Found: 442.2867.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-isobutylbiphenyl-4-yl)amide Dihydrochloride (4d)
The procedure for 1 was followed using 20 mg (0.035 mmol) of 15e and 1 mL of 4 M HCl in dioxane to give 17 mg (90%) of 4d as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.68–8.58 (m, 1H), 8.40–8.28 (m, 1H), 7.92–7.43 (m, 8H), 7.30–7.20 (m, 2H), 4.50–4.40 (m, 0.5H), 4.38–4.25 (m, 0.5H), 3.88–3.60 (m, 1H), 3.50–3.38 (m, 1H), 3.13–3.05 (m, 0.5H), 3.05–2.90 (m, 0.5H), 2.51 (d, J = 6.0 Hz, 2H), 2.25–2.10 (m, 1H), 2.05–1.60 (m, 4H), 1.50–1.20 (m, 2H), 1.10–0.76 (m, 12H); HRMS (ESI) calcd. for C31H39N3O [M + H]+: 470.3166. Found: 470.3178.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-cyclohexylbiphenyl-4-yl)amide Dihydrochloride (4e)
The procedure for 1 was followed using 20 mg (0.034 mmol) of 15f and 1 mL of 4 M HCl in dioxane to give 19 mg (98%) of 4e as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.55–8.40 (m, 1H), 8.15–8.00 (m, 1H), 7.75–7.40 (m, 8H), 7.35–7.20 (m, 2H), 4.48–4.20 (m, 1H), 3.88–3.68 (m, 1H), 3.42–3.35 (m, 1H), 2.98–2.88 (m, 1H), 2.68–2.48 (m, 1H), 2.20–2.00 (m, 1H), 1.98–1.70 (m, 7H), 1.66–1.10 (m, 8H), 1.05–0.78 (m, 6H); HRMS (ESI) calcd. for C33H41N3O [M + H]+: 496.3322. Found: 496.3323.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-trifluoromethylbiphenyl-4-yl)amide Dihydrochloride (4f)
The procedure for 1 was followed using 20 mg (0.034 mmol) of 15g and 1 mL of 4 N HCl in dioxane to give 18 mg (95%) of 4f as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.70–8.58 (m, 1H), 8.42–8.30 (m, 1H), 7.90–7.50 (m, 10H), 4.55–4.40 (m, 0.5H), 4.40–4.26 (m, 0.5H), 3.90–3.60 (m, 1H), 3.50–3.38 (m, 1H), 3.15–3.05 (m, 0.5H), 3.05–2.92 (m, 0.5H), 2.25–2.10 (m, 1H), 2.10–1.92 (m, 1H), 1.92–1.62 (m, 2H), 1.50–1.15 (m, 2H), 1.08–0.80 (m, 6H); HRMS (ESI) calcd. for C28H30F3N3O [M + H]+: 482.2414. Found: 482.2416.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-fluorobiphenyl-4-yl)amide Dihydrochloride (4g)
The procedure for 1 was followed using 20 mg (0.038 mmol) of 15h and 1 mL of 4 M HCl in dioxane to give 18 mg (94%) of 4g as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.70–8.58 (m, 1H), 8.45–8.35 (m, 1H), 7.90–7.50 (m, 8H), 7.25–7.10 (m, 2H), 4.52–4.40 (m, 0.5H), 4.40–4.25 (m, 0.5H), 3.90–3.60 (m, 1H), 3.52–3.40 (m, 1H), 3.16–3.06 (m, 0.5H), 3.05–2.92 (m, 0.5H), 2.25–2.10 (m, 1H), 2.10–1.92 (m, 1H), 1.92–1.60 (m, 2H), 1.50–1.18 (m, 2H), 1.10–0.80 (m, 6H); HRMS (ESI) calcd. for C27H30FN3O [M + H]+: 432.2446. Found: 432.2453.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-chlorobiphenyl-4-yl)amide Dihydrochloride (4h)
The procedure for 1 was followed using 20 mg (0.036 mmol) of 15i and 1 mL of 4 M HCl in dioxane to give 18 mg (96%) of 4h as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.72–8.58 (m, 1H), 8.45–8.35 (m, 1H), 7.90–7.55 (m, 8H), 7.55–7.40 (m, 2H), 4.55–4.42 (m, 0.5H), 4.40–4.25 (m, 0.5H), 3.90–3.60 (m, 1H), 3.50–3.40 (m, 1H), 3.18–3.07 (m, 0.5H), 3.06–2.96 (m, 0.5H), 2.25–2.10 (m, 1H), 2.08–1.90 (m, 1H), 1.90–1.68 (m, 2H), 1.58–1.12 (m, 2H), 1.10–0.80 (m, 6H); HRMS (ESI) calcd. for C27H30ClN3O [M + H]+: 448.2150. Found: 448.2162.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-acetylbiphenyl-4-yl)amide Dihydrochloride (4i)
The procedure for 1 was followed using 20 mg (0.036 mmol) of 15j and 1 mL of 4 M HCl in dioxane to give 17 mg (90%) of 4i as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.35–8.22 (m, 1H), 8.05–7.92 (m, 2H), 7.90–7.75 (m, 1H), 7.72–7.60 (m, 4H), 7.52–7.40 (m, 2H), 7.35–7.22 (m, 2H), 4.38–4.16 (m, 1H), 3.80–3.55 (m, 1H), 3.38–3.20 (m, 1H), 2.80–2.60 (m, 1H), 2.54 (s, 3H), 2.00–1.88 (m, 1H), 1.78–1.60 (m, 2H), 1.50–1.10 (m, 3H), 0.95–0.70 (m, 6H); HRMS (ESI) calcd. for C29H33N3O2 [M + H]+: 456.2646. Found: 456.2658.
tert-Butyl (1R,2R)-2-(Pyridin-2-yl)cyclopropanecarboxylate ((1R,2R)-9)
An oven-dried Schlenk tube, that was previously charged with chiral porphyrin catalyst [Co(3,5-DitBu-ChenPhyrin)]11 (3.4 mg, 0.0025 mmol) and DMAP (15 mg, 0.13 mmol), was evacuated and backfilled with nitrogen gas. Toluene (0.5 mL) and 2-vinylpyridine (8) (26 mg, 0.25 mmol) were added, followed by the remaining solvent (total 1.0 mL). The Schlenk tube was then placed in a −20 °C cooling bath, and tert-butyl diazoacetate (41 μL, 0.3 mmol) was added dropwise. After addition, the tube was purged with nitrogen for 2 min and the mixture was stirred at −20 °C for 12 h, then at 0 °C for 12 h. After warming to room temperature, the reaction was continued for another 24 h. Flash column chromatography of the crude mixture on silica gel using 10% EtOAc in hexanes afforded (1R,2R)-9 (35 mg, 64%) as a colorless oil. Assignment of the absolute configuration was made based on the analogy to similar compounds with known absolute configurations synthesized using the same chiral catalyst.11 The enantiomeric excess (97% ee) was determined by HPLC (ChiralPak AD-H column; 0.5% isopropanol/hexanes; flow rate 0.8 mL/min; detection 254 nm; retention time 10.6 min).
(1R,2R)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid Hydrochloride ((1R,2R)-10)
To a solution of (1R,2R)-9 (110 mg, 0.50 mmol) in CH2Cl2 (5 mL) was added 4 M HCl in dioxane (2 mL), and the reaction was stirred at room temperature for 5 h. The solvent was removed under reduced procedure. The residue was triturated with CH2Cl2/Et2O to afford (1R,2R)-10 (95 mg, 95%) as a white solid: mp 155–157 °C; [α]23D −206.8° (c 0.5, CH3OH).
tert-Butyl [(2S,3S)-1-{4-Bromophenyl-[(1R,2R)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]carbamate ((1R,2R)-14)
The procedure for 14 was followed using 108 mg (0.29 mmol) of 13 and 70 mg (0.35 mmol) of (1R,2R)-10 to give 97 mg (65%) of (1R,2R)-14 as a white foam: [α]23D −21.0° (c 0.58, CH3OH); 1H NMR (300 MHz; CDCl3) δ 8.30 (d, J = 6.0 Hz, 1H), 7.56–7.40 (m, 3H), 7.22–7.06 (m, 3H), 7.06–6.98 (m, 1H), 4.96 (d, J = 9.0 Hz, 1H), 4.36 (t, J = 12.0 Hz, 1H), 3.78–3.65 (m, 1H), 3.12 (dd, J = 12.0, 3.0 Hz, 1H), 2.67 (ddd, J = 9.0, 6.0, 3.0 Hz, 1H), 1.93–1.86 (m, 1H), 1.61–1.53 (m, 1H), 1.52–1.41 (m, 3H), 1.45 (s, 9H), 1.15–1.02 (m, 1H), 0.87 (d, J = 6.0 Hz, 3H), 0.85 (t, J = 7.5 Hz, 3H); 13C NMR (75 MHz; CDCl3) δ 173.2, 159.3, 156.4, 149.4, 141.3, 136.0, 133.1, 130.1, 122.7, 121.9, 121.2, 79.0, 54.0, 50.3, 38.3, 28.6, 27.5, 25.3, 24.7, 18.3, 15.2, 11.9; MS (ESI) m/z 516.5 [M + H]+ (79Br), 518.4 [M + H]+ (81Br). The diastereomeric excess (>97% de) was determined by HPLC (ChiralPak IA column; 5% EtOH/hexanes; flow rate 1 mL/min; detection 220 nm; retention time 9.1 min).
tert-Butyl [(2S,3S)-1-{4-Bromophenyl-[(1S,2S)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]carbamate ((1S,2S)-14)
The diastereomeric mixture 14 (350 mg) was separated to (1R,2R)-14 (140 mg) and (1S,2S)-14 (137 mg) by preparative HPLC using ChiralPak IA column: mobile phase, 5% EtOH/hexanes; flow rate, 5 mL/min; detection 220 nm. The diastereomeric excess (de) of both of separated compounds was determined to be >99% by HPLC (ChiralPak IA column; 5% EtOH/hexanes; flow rate 1 mL/min; detection 220 nm; retention time, (1R,2R)-14: 9.1 min, (1S,2S)-14: 11.5 min). (1S,2S)-14: White foam; [α]23D +52.7° (c 0.55, CH3OH); 1H NMR (300 MHz; CDCl3) δ 8.30 (d, J = 6.0 Hz, 1H), 7.52 (td, J = 9.0, 3.0 Hz, 1H), 7.40 (d, J = 6.0 Hz, 2H), 7.16 (d, J = 9.0 Hz, 1H), 7.12–6.99 (m, 3H), 4.96 (d, J = 9.0 Hz, 1H), 4.38 (dd, J = 15.0, 12.0 Hz, 1H), 3.72–3.58 (m, 1H), 3.15 (dd, J = 15.0, 3.0 Hz, 1H), 2.52 (ddd, J = 9.0, 6.0, 3.0 Hz, 1H), 1.97–1.90 (m, 1H), 1.70–1.63 (m, 1H), 1.54–1.38 (m, 3H), 1.40 (s, 9H), 1.14–1.02 (m, 1H), 0.87 (d, J = 6.0 Hz, 3H), 0.84 (t, J = 7.5 Hz, 3H); 13C NMR (75 MHz; CDCl3) δ 173.1, 159.2, 156.2, 149.5, 141.1, 135.9, 132.9, 130.1, 122.5, 121.8, 121.2, 78.9, 53.8, 50.0, 38.2, 28.6, 28.1, 25.3, 24.7, 17.7, 15.2, 11.9; MS (ESI) m/z 516.5 [M + H]+ (79Br), 518.4 [M + H]+ (81Br).
(1R,2R)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-propylbiphenyl-4-yl)amide Dihydrochloride (2)
The procedure for 15a was followed using 120 mg (0.23 mmol) of (1R,2R)-14, followed by deprotection of the Boc protecting group with 4 M HCl in dioxane, to give 97 mg (80% over two steps) of 2 as a white solid: mp 125 °C (fusion); [α]23D +6.3° (c 1, CH3OH); 1H NMR (300 MHz; CD3OD) δ 8.58 (d, J = 6.0 Hz, 1H), 8.28 (t, J = 9.0 Hz, 1H), 7.78–7.52 (m, 6H), 7.51 (d, J = 9.0 Hz, 2H), 7.27 (d, J = 9.0 Hz, 2H), 4.29 (dd, J = 15.0, 9.0 Hz, 1H), 3.83 (d, J = 12.0 Hz, 1H), 3.48–3.38 (m, 1H), 3.01–2.90 (m, 1H), 2.62 (t, J = 7.5 Hz, 2H), 2.22–2.12 (m, 1H), 2.02–1.92 (m, 1H), 1.90–1.75 (M, 1H), 1.75–1.60 (m, 3H), 1.50–1.35 (m, 1H), 1.35–1.15 (m, 1H), 1.05–0.90 (m, 6H), 0.86 (t, J = 6.0 Hz, 3H); 13C NMR (75 MHz; CD3OD) δ 173.4, 157.6, 146.6, 143.9, 143.3, 142.9, 141.6, 138.2, 130.2, 129.8, 129.5, 127.9, 125.5, 125.1, 56.6, 50.9, 38.7, 37.2, 27.4, 26.5, 25.7, 25.2, 18.1, 14.2, 14.2, 11.7; HRMS (ESI) calcd. for C30H37N3O [M + H]+: 456.3009. Found: 456.3023. The diastereomeric excess (>99% de) was determined by HPLC (XTerra MS C-18 column; gradient 40–60% of (0.1%TFA/MeCN)/(0.1%TFA/water); flow rate 1 mL/min; detection 254 nm; retention time 5.66 min).
(1S,2S)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-propylbiphenyl-4-yl)amide Dihydrochloride (3)
The procedure for 15a was followed using 120 mg (0.23 mmol) of (1S,2S)-14, followed by deprotection of the Boc protecting group with 4 M HCl in dioxane, to give 98 mg (81% over two steps) of 3 as a white solid: mp 122 °C (fusion); [α]23D −91.3° (c 1, CH3OH); 1H NMR (300 MHz; CD3OD) δ 8.62 (br d, J = 3.0 Hz, 1H), 8.31 (br t, J = 7.5 Hz, 1H), 7.80–7.58 (m, 6H), 7.49 (d, J = 9.0 Hz, 2H), 7.26 (d, J = 9.0 Hz, 2H), 4.46 (dd, J = 15.0, 9.0 Hz, 1H), 3.68 (d, J = 15.0 Hz, 1H), 3.46–3.38 (m, 1H), 3.12–3.02 (m, 1H), 2.61 (t, J = 7.5 Hz, 2H), 2.25–2.05 (m, 1H), 1.98–1.86 (m, 1H), 1.86–1.60 (m, 4H), 1.45–1.15 (m, 2H), 1.10–0.90 (m, 6H), 0.82 (t, J = 6.0 Hz, 3H); 13C NMR (75 MHz; CD3OD) δ 173.3, 157.5, 146.7, 143.9, 143.3, 142.9, 141.4, 138.2, 130.2, 130.0, 129.4, 127.9, 125.6, 125.3, 56.5, 50.6, 38.7, 37.4, 27.0, 26.7, 25.7, 25.6, 17.2, 14.1, 11.8; HRMS (ESI) calcd. for C30H37N3O [M + H]+: 456.3009. Found: 456.3018. The diastereomeric excess (>99% de) was determined by HPLC (XTerra MS C-18 column; gradient 40–60% of (0.1%TFA/MeCN)/(0.1%TFA/water); flow rate 1 mL/min; detection 254 nm; retention time 6.35 min).
tert-Butyl {2-[(4-Bromophenyl)amino]ethyl}carbamate (18a)
The procedure for 13 was followed using 1.03 g (5.96 mmol) of 11 and 0.95 g (5.96 mmol) of N-Boc-2-aminoacetaldehyde (17a) to give 1.38 g (50%) of 18a as a yellow oily residue: 1H NMR (300 MHz; CDCl3) δ 7.23 (d, J = 9.0 Hz, 2H), 6.48 (d, J = 9.0 Hz, 2H), 4.80 (br s, 1H), 4.10 (br s, 1H), 3.41–3.30 (m, 2H), 3.30–3.26 (m, 2H), 1.45 (s, 9H); 13C NMR (75 MHz; CDCl3) δ 156.5, 147.1, 131.9, 114.2, 108.8, 79.6, 44.4, 40.0, 28.4; MS (ESI) m/z 315.1 [M + H]+ (79Br), 317.2 [M + H]+ (81Br).
tert-Butyl {(2S)-1-[(4-Bromophenyl)amino]propan-2-yl}carbamate (18b)
The procedure for 13 was followed using 247 mg (1.44 mmol) of 11 and 250 mg (1.44 mmol) of N-Boc-l-alaninal (17b) to give 410 mg (86%) of 18b as a white solid: mp 115–117 °C; [α]23D +3.1° (c 0.52, CH3OH); 1H NMR (300 MHz; CDCl3) δ 7.23 (d, J = 9.0 Hz, 2H), 6.48 (d, J = 9.0 Hz, 2H), 4.48 (br s, 1H), 4.12 (br s, 1H), 4.00–3.86 (m, 1H), 3.18–2.98 (m, 2H), 1.45 (s, 9H), 1.21 (d, J = 6.0 Hz, 3H); 13C NMR (75 MHz; CDCl3) δ 156.0, 147.3, 131.9, 114.1, 108.7, 79.7, 50.6, 46.3, 28.4, 19.0; MS (ESI) m/z 329.3 [M + H]+ (79Br), 331.2 [M + H]+ (81Br).
tert-Butyl {(2S)-1-[(4′-Propylbiphenyl-4-yl)amino]-3-methylbutan-2-yl}carbamate (18c)
The procedure for 13 was followed using 150 mg (0.71 mmol) of 4-(4′-propylphenyl)aniline (16) and 143 mg (0.71 mmol) of aldehyde 17c, prepared by oxidation of N-Boc-l-valinol, to give 185 mg (66%) of 18c as a white solid: mp 83–85 °C; [α]23D +22.2° (c 0.54, CH3OH); 1H NMR (300 MHz; CDCl3) δ 7.45 (d, J = 9.0 Hz, 2H), 7.42 (d, J = 9.0 Hz, 2H), 7.20 (d, J = 9.0 Hz, 2H), 6.65 (d, J = 9.0 Hz, 2H), 4.51 (d, J = 6.0 Hz, 1H), 4.12 (br s, 1H), 3.80–3.65 (m, 1H), 3.35–3.20 (m, 1H), 3.12–3.00 (m, 1H), 2.60 (t, J = 7.5 Hz, 2H), 1.95–1.80 (m, 1H), 1.75–1.58 (m, 2H), 1.45 (s, 9H), 1.05–0.90 (m, 9H); 13C NMR (75 MHz; CDCl3) δ 156.8, 147.8, 140.5, 138.8, 130.3, 128.9, 127.8, 126.2, 113.0, 79.5, 55.7, 47.1, 37.8, 30.6, 28.5, 24.7, 19.6, 18.2, 14.0; MS (ESI) m/z 397.5 [M + H]+.
tert-Butyl {(2S)-1-[(4′-Propylbiphenyl-4-yl)amino]-4-methylpentan-2-yl}carbamate (18d)
The procedure for 13 was followed using 90 mg (0.43 mmol) of 16 and 93 mg (0.43 mmol) of aldehyde 17d, prepared by oxidation of N-Boc-l-leucinol, to give 120 mg (68%) of 18d as a white solid: mp 98–100 °C; [α]23D +2.4° (c 0.54, CH3OH); 1H NMR (300 MHz; CDCl3) δ 7.45 (d, J = 9.0 Hz, 2H), 7.42 (d, J = 9.0 Hz, 2H), 7.22 (d, J = 9.0 Hz, 2H), 6.66 (d, J = 9.0 Hz, 2H), 4.41 (br s, 1H), 4.21 (br s, 1H), 4.00–3.82 (m, 1H), 3.32–3.20 (m, 1H), 3.12–3.00 (m, 1H), 2.62 (t, J = 7.5 Hz, 2H), 1.80–1.60 (m, 3H), 1.45 (s, 9H), 1.43–1.35 (m, 2H), 1.05–0.90 (m, 9H); 13C NMR (75 MHz; CDCl3) δ 156.3, 147.7, 140.5, 138.7, 130.2, 128.7, 127.8, 126.1, 112.9, 79.5, 49.9, 49.0, 42.5, 37.7, 28.4, 25.0, 24.6, 23.1, 22.1, 13.9; MS (ESI) m/z 411.5 [M + H]+.
tert-Butyl {(2S)-1-[(4-Bromophenyl)amino]-3-phenylpropan-2-yl}carbamate (18e)
The procedure for 13 was followed using 172 mg (1.00 mmol) of 11 and 250 mg (1.00 mmol) of N-Boc-l-phenylalaninal (17e) to give 350 mg (86%) of 18e as a white solid: mp 124–126 °C; [α]23D +12.2° (c 0.53, CH3OH); 1H NMR (300 MHz; CDCl3) δ 7.36–7.15 (m, 7H), 6.43 (d, J = 9.0 Hz, 2H), 4.51 (br s, 1H), 4.15–3.98 (m, 2H), 3.26–3.15 (m, 1H), 3.10–2.92 (m, 1H), 2.96–1.80 (m, 2H), 1.41 (s, 9H); 13C NMR (75 MHz; CDCl3) δ 156.0, 147.1, 137.3, 131.9, 129.2, 128.7, 126.8, 114.3, 109.0, 79.8, 51.4, 47.9, 39.2, 28.3; MS (ESI) m/z 405.3 [M + H]+ (79Br), 407.3 [M + H]+ (81Br).
tert-Butyl (2-{4-Bromophenyl-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}ethyl)carbamate (19a)
The procedure for 14 was followed using 180 mg (0.57 mmol) of 18a and 136 mg (0.68 mmol) of racemic 10 to give 140 mg (53%) of 19a as a yellow oil: 1H NMR (300 MHz; CDCl3) δ 8.31 (d, J = 6.0 Hz, 1H), 7.60–7.50 (m, 1H), 7.43 (d, J = 9.0 Hz, 2H), 7.21–7.00 (m, 4H), 5.05 (br s, 1H), 3.92–3.80 (m, 2H), 3.40–3.25 (m, 2H), 2.68–2.58 (m, 1H), 2.00–1.90 (m, 1H), 1.70–1.58 (m, 1H), 1.41 (s, 9H), 1.20–1.05 (m, 1H); MS (ESI) m/z 460.3 [M + H]+ (79Br), 462.1 [M + H]+ (81Br). This product contained impurity as judged by 1H NMR analysis, which was used in the next step without further purification.
tert-Butyl [(2S)-1-{4-Bromophenyl-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}propan-2-yl]carbamate (19b)
The procedure for 14 was followed using 99 mg (0.30 mmol) of 18b and 72 mg (0.36 mmol) of racemic 10 to give 85 mg (60%) of 19b as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.30 (d, J = 6.0 Hz, 1H), 7.58–7.36 (m, 3H), 7.20–6.95 (m, 4H), 4.98 (d, J = 6.0 Hz, 1H), 4.22–4.05 (m, 1H), 3.92–3.70 (m, 1H), 3.32–3.18 (m, 2H), 2.68–2.60 (m, 0.5H), 2.60–2.46 (m, 0.5H), 1.95–1.80 (m, 1H), 1.68–1.52 (m, 1H), 1.43 and 1.41 (2s, 9H), 1.10 (d, J = 6.0 Hz, 3H); MS (ESI) m/z 474.4 [M + H]+ (79Br), 476.5 [M + H]+ (81Br).
tert-Butyl (2S)-1-{(4′-Propylbiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylbutan-2-yl}carbamate (19c)
The procedure for 14 was followed using 120 mg (0.34 mmol) of 18c and 81 mg (0.40 mmol) of racemic 10 to give 110 mg (60%) of 19c as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.26–8.18 (m, 1H), 7.55–7.34 (m, 5H), 7.30–7.21 (m, 5H), 6.98–6.90 (m, 1H), 5.08–4.92 (m, 1H), 4.45–4.32 (m, 1H), 3.80–3.60 (m, 1H), 3.25–3.12 (m, 1H), 2.74–2.48 (m, 3H), 2.08–1.92 (m, 1H), 1.78–1.52 (m, 5H), 1.47 and 1.43 (2s, 9H), 1.00–0.80 (m, 9H); MS (ESI) m/z 542.6 [M + H]+.
tert-Butyl (2S)-1-{(4′-Propylbiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-4-methylpentan-2-yl}carbamate (19d)
The procedure for 14 was followed using 90 mg (0.22 mmol) of 18d and 53 mg (0.26 mmol) of racemic 10 to give 65 mg (53%) of 19d as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.28–8.20 (m, 1H), 7.55–7.35 (m, 5H), 7.32–7.12 (m, 5H), 7.00–6.90 (m, 1H), 4.90–4.80 (m, 1H), 4.28–4.12 (m, 1H), 4.00–3.80 (m, 1H), 3.38–3.20 (m, 1H), 2.75–2.50 (m, 3H), 2.10–1.90 (m, 1H), 1.76–1.52 (m, 5H), 1.45 and 1.41 (2s, 9H), 1.40–1.12 (m, 2H), 1.02–0.80 (m, 9H); MS (ESI) m/z 556.8 [M + H]+.
tert-Butyl [(2S)-1-{4-Bromophenyl-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-phenylpropan-2-yl]carbamate (19e)
The procedure for 14 was followed using 99 mg (0.30 mmol) of 18e and 72 mg (0.36 mmol) of racemic 10 to give 85 mg (60%) of 19e as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.29 (d, J = 6.0 Hz, 1H), 7.55–7.30 (m, 3H), 7.30–6.90 (m, 9H), 5.05–4.92 (m, 1H), 4.28–3.90 (m, 2H), 3.30–3.20 (m, 1H), 2.88–2.48 (m, 3H), 1.95–1.82 (m, 1H), 1.70–1.50 (m, 1H), 1.39 and 1.37 (2s, 9H), 1.32–1.20 (m, 1H); MS (ESI) m/z 550.4 [M + H]+ (79Br), 552.5 [M + H]+ (81Br).
tert-Butyl (2-{(4′-Propylbiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}ethyl)carbamate (20a)
The procedure for 15a was followed using 140 mg (0.30 mmol) of 19a and 83 mg (0.47 mmol) of 4-propylphenylboronic acid to give 80 mg (53%) of 20a: 1H NMR (300 MHz; CDCl3) δ 8.22 (d, J = 6.0 Hz, 1H), 7.55–7.40 (m, 5H), 7.25–7.12 (m, 5H), 7.00–6.90 (m, 1H), 5.17 (br s, 1H), 3.90 (t, J = 6.0 Hz, 2H), 3.42–3.25 (m, 3H), 2.62 (t, J = 7.5 Hz, 2H), 2.10–1.98 (m, 1H), 1.75–1.60 (m, 2H), 1.55–1.35 (m, 2H), 1.42 (s, 9H), 0.97 (t, J = 6.0 Hz, 3H); 13C NMR (75 MHz; CDCl3) δ 172.9, 159.4, 156.1, 149.3, 142.3, 141.0, 137.4, 135.8, 129.7, 129.0, 128.2, 128.1, 126.9, 122.4, 121.0, 79.1, 49.2, 39.7, 37.7, 28.4, 27.7, 24.9, 24.5, 17.7, 13.9; MS (ESI) m/z 500.8 [M + H]+.
tert-Butyl [(2S)-1-{(4′-Propylbiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}propan-2-yl]carbamate (20b)
The procedure for 15a was followed using 50 mg (0.11 mmol) of 19b and 29 mg (0.16 mmol) of 4-propylphenylboronic acid to give 45 mg (80%) of 20b as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.28–8.18 (m, 1H), 7.56–7.38 (m, 5H), 7.28–7.10 (m, 5H), 7.00–6.90 (m, 1H), 5.20–5.06 (m, 1H), 4.32–4.15 (m, 1H), 3.96–3.78 (m, 1H), 3.38–3.22 (m, 1H), 2.70–2.48 (m, 3H), 2.06–1.90 (m, 2H), 1.78–1.58 (m, 3H), 1.45 and 1.43 (2s, 9H), 1.12 (d, J = 6.0 Hz, 3H), 0.97 (t, J = 7.5 Hz, 3H); MS (ESI) m/z 514.6 [M + H]+.
tert-Butyl [(2S)-1-{(4′-Propylbiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-phenylpropan-2-yl]carbamate (20e)
The procedure for 15a was followed using 90 mg (0.16 mmol) of 19e and 44 mg (0.24 mmol) of 4-propylphenylboronic acid to give 75 mg (80%) of 20e as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.26–8.15 (m, 1H), 7.55–7.38 (m, 6H), 7.28–7.05 (m, 9H), 7.00–6.90 (m, 1H), 5.16–5.05 (m, 1H), 4.35–4.18 (m, 1H), 4.15–3.95 (m, 1H), 3.34–3.22 (m, 1H), 2.88–2.45 (m, 5H), 2.05–1.90 (m, 1H), 1.75–1.50 (m, 3H), 1.41 and 1.40 (2s, 9H), 1.30–1.20 (m, 1H), 0.97 (t, J = 7.5 Hz, 3H); MS (ESI) m/z 590.8 [M + H]+.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [2-Aminoethyl-(4′-propylbiphenyl-4-yl)amide Dihydrochloride (5a)
The procedure for 1 was followed using 60 mg (0.12 mmol) of 20a and 1 mL of 4 M HCl in dioxane to give 52 mg (92%) of 5a as a light yellow foam: 1H NMR (300 MHz; CD3OD) δ 8.56 (br s, 1H), 8.23 (br t, 1H), 7.50–7.63 (m, 3H), 7.62–7.43 (m, 5H), 7.27 (d, J = 9.0 Hz, 2H), 4.25–3.98 (m, 2H), 3.22–3.10 (m, 2H), 3.00–2.78 (m, 1H), 2.63 (t, J = 7.5 Hz, 2H), 2.18–2.08 (m, 1H), 1.94–182 (m, 1H), 1.72–1.60 (m, 3H), 0.96 (t, J = 7.5 Hz, 3H); 13C NMR (75 MHz; CD3OD) δ 173.1, 157.9, 146.2, 144.1, 143.9, 143.0, 141.4, 138.2, 130.2, 130.0, 129.6, 128.0, 125.5, 125.3, 39.9, 38.7, 27.1, 25.8, 25.7, 18.0, 14.1; HRMS (ESI) calcd. for C26H29N3O [M + H]+: 400.2383. Found: 400.2388.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S)-2-Aminopropyl-(4′-propylbiphenyl-4-yl)amide Dihydrochloride (5b)
The procedure for 1 was followed using 40 mg (0.078 mmol) of 20b and 1 mL of 4 M HCl in dioxane to give 37 mg (98%) of 5b as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.70–8.58 (m, 1H), 8.40–8.28 (m, 1H), 7.85–7.40 (m, 8H), 7.30–7.20 (m, 2H), 4.42–4.26 (m, 0.5H), 4.25–4.10 (m, 0.5H), 3.95–3.82 (m, 0.5H), 3.78–3.48 (m, 1.5H), 3.10–2.90 (m, 1H), 2.62 (t, J = 7.5 Hz, 2H), 2.22–2.06 (m, 1H), 2.05–1.85 (m, 1H), 1.78–1.68 (m, 3H), 1.42–1.20 (m, 3H), 0.95 (t, J = 6.0 Hz, 3H); HRMS (ESI) calcd. for C27H31N3O [M + H]+: 414.2540. Found: 414.2547.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S)-2-Amino-3-methylbutyl]-(4′-propylbiphenyl-4-yl)amide Dihydrochloride (5c)
The procedure for 1 was followed using 60 mg (0.11 mmol) of 19c and 1 mL of 4 M HCl in dioxane to give 54 mg (95%) of 5c as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.55–8.38 (m, 1H), 8.22–8.10 (m, 1H), 7.70–7.30 (m, 8H), 7.25–7.10 (m, 2H), 4.38–4.25 (m, 0.5H), 4.24–4.10 (m, 0.5H), 3.88–3.40 (m, 2H), 3.30–3.10 (m, 1H), 2.98–2.80 (m, 1H), 2.52 (t, J = 7.5 Hz, 2H), 2.16–1.78 (m, 3H), 1.68–1.46 (m, 2H), 1.00–0.75 (m, 9H); HRMS (ESI) calcd. for C29H35N3O [M + H]+: 442.2853. Found: 442.2856.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S)-2-Amino-4-methylpentyl]-(4′-propylbiphenyl-4-yl)amide Dihydrochloride (5d)
The procedure for 1 was followed using 50 mg (0.09 mmol) of 19d and 1 mL of 4 M HCl in dioxane to give 46 mg (97%) of 5d as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.60–8.50 (m, 1H), 8.30–8.18 (m, 1H), 7.78–7.40 (m, 8H), 7.27 (d, J = 9.0 Hz, 2H), 4.36–4.20 (m, 0.5H), 4.20–4.18 (m, 0.5H), 4.05–3.90 (m, 0.5H), 3.88–3.60 (m, 1.5H), 3.52–3.40 (m, 1H), 3.10–2.90 (m, 1H), 2.62 (t, J = 7.5 Hz, 2H), 2.22–2.08 (m, 1H), 2.00–1.88 (m, 1H), 1.76–1.42 (m, 5H), 1.02–0.75 (m, 9H); HRMS (ESI) calcd. for C30H37N3O [M + H]+: 456.3009. Found: 456.3017.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S)-2-Amino-3-phenylpropyl]-(4′-propylbiphenyl-4-yl)amide Dihydrochloride (5e)
The procedure for 1 was followed using 70 mg (0.12 mmol) of 20e and 1 mL of 4 M HCl in dioxane to give 65 mg (96%) of 5e as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.65–8.58 (m, 1H), 8.40–8.26 (m, 1H), 7.82–7.70 (m, 1H), 7.70–7.36 (m, 7H), 7.35–7.05 (m, 7H), 4.45–4.30 (m, 0.5H), 4.25–4.10 (m, 0.5H), 4.00–3.86 (m, 0.5H), 3.85–3.55 (m, 1.5H), 3.16–2.88 (m, 3H), 2.69 (t, J = 7.5 Hz, 2H), 2.22–2.05 (m, 1H), 2.05–1.88 (m, 1H), 1.76–1.58 (m, 3H), 0.95 (t, J = 7.5 Hz, 3H); HRMS (ESI) calcd. for C33H35N3O [M + H]+: 490.2853. Found: 490.2859.
tert-Butyl [(2S,3S)-1-{4-Bromophenyl-[(1R,2R)-2-phenyl-1-cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]carbmate (21a)
A solution of (1R,2R)-2-phenyl-1-cyclopropanecarboxylic acid12 (97 mg, 0.6 mmol) in thionyl chloride (3 mL) was refluxed overnight. After cooling to room temperature, the mixture was concentrated under reduced pressure. The resulting residue was dissolved in CH2Cl2 (5 mL) and treated with 13 (150 mg, 0.4 mmol) and Et3N (0.17 mL, 1.2 mmol). The reaction mixture was stirred at room temperature overnight. Saturated NaHCO3 (5 mL) was added and the layers were separated. The aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with brine (3 × 10 mL), dried (Na2SO4), and concentrated under reduced pressure. Flash column chromatography of the crude product on silica gel using 0–25% EtOAc in hexanes afforded 21a (155 mg, 75%) as a white foam: [α]23D +35.2° (c 0.52, CH3OH); 1H NMR (300 MHz; CDCl3) δ 7.49 (d, J = 9.0 Hz, 2H), 7.25–7.10 (m, 5H), 6.94 (d, J = 9.0 Hz, 2H), 4.96 (d, J = 12.0 Hz, 1H), 4.37 (dd, J = 12.0, 10.5 Hz, 1H), 3.80–3.66 (m, 1H), 3.13 (dd, J = 12.0, 3.0 Hz, 1H), 2.66–2.55 (m, 1H), 1.61–1.48 (m, 4H), 1.45 (s, 9H), 1.16–1.10 (m, 2H), 0.91–0.82 (m, 6H); 13C NMR (75 MHz; CDCl3) δ 173.3, 156.4, 141.4, 140.4, 133.1, 130.2, 128.5, 126.4, 122.0, 79.0, 54.1, 50.3, 38.3, 28.6, 26.4, 25.4, 24.1, 17.8, 15.3, 11.9; MS (ESI) m/z 515.4 [M + H]+ (79Br), 517.4 [M + H]+ (81Br).
tert-Butyl [(2S,3S)-1-{4-Bromophenyl-[(pyridin-2-yl)methylcarbonyl]amino}-3-methylpentan-2-yl]carbamate (21b)
To a solution of 13 (200 mg, 0.54 mmol) in CH3CN (10 mL) at room temperature were added 2-pyridylacetic acid hydrochloride (113 mg, 0.65 mmol), Et3N (0.23 mL, 1.6 mmol), and HBTU (246 mg, 0.65 mmol). The reaction mixture was stirred at room temperature overnight. The solution was diluted with EtOAc (50 mL) and washed with saturated NaHCO3 (10 mL) and brine (10 mL). The organic phase was dried (Na2SO4) and concentrated under reduced pressure. Flash column chromatography of the crude product on silica gel using 0–25% EtOAc in hexanes afforded 21b (170 mg, 64%) as a brown residue: 1H NMR (300 MHz; CDCl3) δ 8.40 (d, J = 3.0 Hz, 1H), 7.56–7.48 (m, 1H), 7.47 (d, J = 9.0 Hz, 2H), 7.13–6.98 (m, 4H), 4.89 (d, J = 9.0 Hz, 1H), 4.30 (t, J = 12.0 Hz, 1H), 3.70–3.55 (m, 1H), 3.54 (s, 2H), 3.06 (dd, J = 15.0, 3.0 Hz, 1H), 1.50–1.42 (m, 2H), 1.41 (s, 9H), 1.10–0.92 (m, 1H), 0.83–0.70 (m, 6H); 13C NMR (75 MHz; CDCl3) δ 169.9, 155.1, 154.5, 148.3, 140.1, 135.3, 132.0, 129.2, 122.8, 121.3, 120.7, 77.9, 52.5, 49.0, 42.9, 37.0, 27.4, 24.2, 14.0, 10.7; MS (ESI) m/z 490.8 [M + H]+ (79Br), 492.6 [M + H]+ (81Br).
tert-Butyl [(2S,3S)-1-{(4′-Propylbiphenyl-4-yl)-[(1R,2R)-2-phenyl-1-cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]carbamate (22a)
The procedure for 15a was followed using 100 mg (0.19 mmol) of 21a and 54 mg (0.32 mmol) of 4-propylphenylboronic acid to give 80 mg (76%) of 22a as a white foam: [α]23D +71.4° (c 0.52, CH3OH); 1H NMR (300 MHz; CDCl3) δ 7.56 (d, J = 9.0 Hz, 2H), 7.48 (d, J = 9.0 Hz, 2H), 7.36–7.22 (m, 4H), 7.21–7.01 (m, 3H), 6.94 (d, J = 9.0 Hz, 2H), 5.08 (d, J = 12.0 Hz, 1H), 4.43 (t, J = 12.0 Hz, 1H), 3.86–3.72 (m, 1H), 3.19 (dd, J = 13.5, 4.5 Hz, 1H), 2.70–2.56 (m, 3H), 1.76–1.55 (m, 6H), 1.47 (s, 9H), 1.18–1.04 (m, 2H), 1.00 (t, J = 7.5 Hz, 3H), 0.93–0.82 (m, 6H); 13C NMR (75 MHz; CDCl3) δ 173.5, 156.4, 142.3, 141.0, 140.7, 140.5, 137.3, 129.0, 128.6, 128.3, 128.1, 126.9, 126.4, 126.2, 78.7, 54.1, 50.0, 38.2, 37.7, 28.5, 26.1, 25.3, 24.5, 23.9, 17.6, 15.1, 13.8, 11.8; MS (ESI) m/z 555.8 [M + H]+.
tert-Butyl [(2S,3S)-1-{(4′-Propylbiphenyl-4-yl)[(pyridin-2-yl)methylcarbonyl]amino}-3-methylpentan-2-yl]carbamate (22b)
The procedure for 15a was followed using 170 mg (0.35 mmol) of 21b and 96 mg (0.54 mmol) of 4-propylphenylboronic acid to give 101 mg (55%) of 22b as an oil: 1H NMR (300 MHz; CDCl3) δ 8.47 (d, J = 3.0 Hz, 1H), 765–7.56 (m, 3H), 7.50 (d, J = 9.0 Hz, 2H), 7.42–7.08 (m, 6H), 5.08 (d, J = 9.0 Hz, 1H), 4.44 (t, J = 12.0 Hz, 1H), 3.86–3.74 (m, 1H), 3.69 (s, 2H), 3.08 (dd, J = 15.0, 3.0 Hz, 1H), 2.63 (t, J = 7.5 Hz, 2H), 1.76–1.65 (m, 2H), 1.64–1.47 (m, 2H), 1.45 (s, 9H), 1.16–1.02 (m, 1H), 0.97 (t, J = 7.5 Hz, 3H), 0.92–0.78 (m, 6H); 13C NMR (75 MHz; CDCl3) δ 171.3, 156.2, 155.9, 149.2, 142.4, 141.2, 140.9, 137.3, 130.2, 129.0, 128.7, 128.3, 126.9, 123.9, 121.6, 78.8, 53.7, 49.9, 43.9, 38.0, 37.7, 28.5, 25.3, 24.5, 15.0, 13.8, 11.8; MS (ESI) m/z 530.9 [M + H]+.
(1R,2R)-2-Phenyl-1-cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-propylbiphenyl-4-yl)amide Hydrochloride (6a)
The procedure for 1 was followed using 60 mg (0.11 mmol) of 22a and 1 mL of 4 M HCl in dioxane to give 52 mg (96%) of 6a as a white solid: mp 112 °C (fusion); [α]23D −5.7° (c 0.53, CH3OH); 1H NMR (300 MHz; CD3OD) δ 7.65 (br d, J = 6.0 Hz, 2H), 7.56–7.38 (m, 4H), 7.26 (d, J = 9.0 Hz, 2H), 7.20–7.06 (m, 3H), 6.93 (d, J = 9.0 Hz, 2H), 4.38–4.22 (m, 1H), 3.83–3.60 (m, 1H), 3.44–3.33 (m, 1H), 2.64 (d, J = 7.5 Hz, 2H), 2.54–2.42 (m, 1H), 1.85–1.60 (m, 4H), 1.50–1.16 (m, 4H), 1.03–0.93 (m, 6H), 0.87 (t, J = 6.0 Hz, 3H); 13C NMR (75 MHz; CD3OD) δ 176.0, 143.8, 142.7, 142.0, 141.5, 138.4, 130.3, 129.8, 129.6, 129.5, 128.0, 127.5, 127.2, 57.0, 50.7, 38.7, 37.3, 28.1, 26.5, 26.0, 25.7, 17.9, 14.4, 14.2, 11.9; HRMS (ESI) calcd. for C31H38N2O [M + H]+: 455.3057. Found: 455.3061.
2-Pyridylacetic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-propylbiphenyl-4-yl)amide Dihydrochloride (6b)
The procedure for 1 was followed using 90 mg (0.17 mmol) of 22b and 2 mL of 4 M HCl in dioxane to give 80 mg (94%) of 6b as a white foam: 1H NMR (300 MHz; CD3OD) δ 8.76 (br d, J = 6.0 Hz, 1H), 8.41 (br t, J = 7.5 Hz, 1H), 7.92–7.50 (m, 8H), 7.30 (d, J = 6.0 Hz, 2H), 4.50–4.38 (m, 1H), 3.75–3.56 (m, 1H), 3.52–3.38 (m, 1H), 2.64 (d, J = 7.5 Hz, 2H), 1.90–1.60 (m, 3H), 1.50–1.15 (m, 2H), 1.02–0.90 (m, 6H), 0.84 (t, J = 6.0 Hz, 3H) (Methylene protons overlapped with methanol solvent peak); 13C NMR (75 MHz; CDCl3) δ 169.6, 150.2, 146.5, 142.7, 142.2, 141.6, 139.2, 136.8, 129.6, 129.1, 128.9, 126.9, 125.4, 55.2, 48.9, 39.8, 37.7, 35.8, 26.2, 24.4, 14.2, 13.8, 11.4; HRMS (ESI) calcd. for C28H35N3O [M + H]+: 430.2853. Found: 430.2860.
Pharmacology
Materials
Isoproterenol was purchased from Sigma-Aldrich, and cell culture reagents (media, supplements, antibiotics, etc.) were purchased from Fisher Scientific. Human GPR88 cDNA was purchased from Missouri S&T cDNA Resource Center. The pGloSensor-22F plasmid was purchased from Promega.
Transient Transfection and cAMP Assay
HEK293T cells were transfected with human GPR88 cDNA and pGloSensor-22F overnight and plated in the poly-l-lysine coated 384-well white clear bottom cell culture plates using DMEM supplemented with 1% dialyzed fetal bovine serum at a density of 15,000 cells in 40 μL medium per well. The cell plates were incubated for 6–20 h before being used for assays. To measure receptor mediated Gαi-activation, culture medium was removed and assay buffer (20 μL per well of 20 mM HEPES, 1× HBSS, pH 7.4) was added, followed by addition of 10 μL of test compound solution at serially diluted concentrations for 15 min at room temperature. After addition of 15 μL of Luciferin (4 mM final) and isoproterenol (200 nM final) and incubation for another 15 min, luminescence was read on the TriLux microbeta counter (PerkinElmer). To measure receptor mediated Gαs-activation, cells were first incubated with test compound for 15 min followed by 15 μL of 4 mM Luciferin for another 15 min, and then luminescence counted as above.
GloSensor cAMP Assay Using Stable GPR88-22F HEK293 Cells
A GPR88-22F stable cell line was created by overexpressing the human GPR88 receptor and the pGloSensor-22F biosensor in HEK293 cells. The day before the assay, GPR88-22F cells were plated into 96-well white-walled assay plates at a density of 40 000 cells per well in culture medium (DMEM-HG supplemented with 10% FBS, 15 mM HEPES, and 100 units of penicillin/streptomycin). The plated cells were incubated overnight at 37 °C, 5% CO2. The next day, the culture medium was gently removed and 100 μL of equilibration medium was gently added per the manufacturer’s instructions (88% CO2-independent medium, 10% FBS, 2% GloSensor cAMP reagent). The cells were incubated in the equilibration medium for 2 h at room temperature in the dark. Test compound dilutions were prepared at 11× concentration in 1× PBS and 10 μL was added to each appropriate well. Following 10 min at room temperature in the dark, 10 μL of 100 nM (final) isoproterenol (prepared at 12× concentration in 1× PBS) was added to each appropriate well. Following 30 min at room temperature in the dark, luminescence was read on the FlexStation III (1000 ms integration time, Molecular Devices).
Data Analysis
Relative luminescence units (RLU) were recorded and plotted against compound concentration. Data were fit to a three-parameter logistic function to generate EC50 values using GraphPad Prism software (San Diego, CA).
Acknowledgments
We thank Dr. Danni Harris for valuable discussions during the course of this work.
Supporting Information Available
Copies of HPLC results of 1–3, 4a–i, 5a–e, 6a, and 6b. This material is available free of charge via the Internet at http://pubs.acs.org.
We thank the National Institute of Mental Health (NIMH) for supporting the research at Psychoactive Drug Screening Program (PDSP). XPZ is grateful for financial support by NSF (CHE-1152767) and NIH (R01-GM098777).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Mizushima K.; Miyamoto Y.; Tsukahara F.; Hirai M.; Sakaki Y.; Ito T. (2000) A novel G-protein-coupled receptor gene expressed in striatum. Genomics 69, 314–321. [DOI] [PubMed] [Google Scholar]
- a Ghate A.; Befort K.; Becker J. A.; Filliol D.; Bole-Feysot C.; Demebele D.; Jost B.; Koch M.; Kieffer B. L. (2007) Identification of novel striatal genes by expression profiling in adult mouse brain. Neuroscience 146, 1182–1192. [DOI] [PubMed] [Google Scholar]; b Massart R.; Guilloux J. P.; Mignon V.; Sokoloff P.; Diaz J. (2009) Striatal GPR88 expression is confined to the whole projection neuron population and is regulated by dopaminergic and glutamatergic afferents. Eur. J. Neurosci. 30, 397–414. [DOI] [PubMed] [Google Scholar]; c Van Waes V.; Tseng K. Y.; Steiner H. (2011) GPR88 - a putative signaling molecule predominantly expressed in the striatum: Cellular localization and developmental regulation. Basal Ganglia 1, 83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Becker J. A.; Befort K.; Blad C.; Filliol D.; Ghate A.; Dembele D.; Thibault C.; Koch M.; Muller J.; Lardenois A.; Poch O.; Kieffer B. L. (2008) Transcriptome analysis identifies genes with enriched expression in the mouse central extended amygdala. Neuroscience 156, 950–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana A.; Sanz E.; Wang W.; Storey G. P.; Guler A. D.; Wanat M. J.; Roller B. A.; La Torre A.; Amieux P. S.; McKnight G. S.; Bamford N. S.; Palmiter R. D. (2012) Lack of GPR88 enhances medium spiny neuron activity and alters motor- and cue-dependent behaviors. Nat. Neurosci. 15, 1547–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue S. F.; Grauer S. M.; Paulsen J.; Graf R.; Taylor N.; Sung M. A.; Zhang L.; Hughes Z.; Pulito V. L.; Liu F.; Rosenzweig-Lipson S.; Brandon N. J.; Marquis K. L.; Bates B.; Pausch M. (2009) The orphan GPCR, GPR88, modulates function of the striatal dopamine system: a possible therapeutic target for psychiatric disorders?. Mol. Cell Neurosci. 42, 438–447. [DOI] [PubMed] [Google Scholar]
- Lovinger D. M. (2012) New twist on orphan receptor GPR88 function. Nat. Neurosci. 15, 1469–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka T.; Tsunoda M.; Sumiyoshi T.; Takasaki I.; Tabuchi Y.; Seo T.; Tanaka K.; Uehara T.; Itoh H.; Suzuki M.; Kurachi M. (2008) Effect of MK-801 on gene expressions in the amygdala of rats. Synapse 62, 1–7. [DOI] [PubMed] [Google Scholar]
- a Ogden C. A.; Rich M. E.; Schork N. J.; Paulus M. P.; Geyer M. A.; Lohr J. B.; Kuczenski R.; Niculescu A. B. (2004) Candidate genes, pathways and mechanisms for bipolar (manic-depressive) and related disorders: an expanded convergent functional genomics approach. Mol. Psychiatry 9, 1007–1029. [DOI] [PubMed] [Google Scholar]; b Brandish P. E.; Su M.; Holder D. J.; Hodor P.; Szumiloski J.; Kleinhanz R. R.; Forbes J. E.; McWhorter M. E.; Duenwald S. J.; Parrish M. L.; Na S.; Liu Y.; Phillips R. L.; Renger J. J.; Sankaranarayanan S.; Simon A. J.; Scolnick E. M. (2005) Regulation of gene expression by lithium and depletion of inositol in slices of adult rat cortex. Neuron 45, 861–872. [DOI] [PubMed] [Google Scholar]
- Conti B.; Maier R.; Barr A. M.; Morale M. C.; Lu X.; Sanna P. P.; Bilbe G.; Hoyer D.; Bartfai T. (2007) Region-specific transcriptional changes following the three antidepressant treatments electro convulsive therapy, sleep deprivation and fluoxetine. Mol. Psychiatry 12, 167–189. [DOI] [PubMed] [Google Scholar]
- Befort K.; Filliol D.; Ghate A.; Darcq E.; Matifas A.; Muller J.; Lardenois A.; Thibault C.; Dembele D.; Le Merrer J.; Becker J. A.; Poch O.; Kieffer B. L. (2008) Mu-opioid receptor activation induces transcriptional plasticity in the central extended amygdala. Eur. J. Neurosci. 27, 2973–2984. [DOI] [PubMed] [Google Scholar]
- a Bi Y., Dzierba C. D., Bronson J. J., Carson K., Cianchetta G., Dong L., Fink C., Green M., Kimball D., Macor J. E., Kwon S., Wang J., Zhang Y., Zipp G. Modulators of G protein-coupled receptor 88. U.S. Patent Application 2011/0245264, 2011;.; b Bi Y., Dzierba C. D., Bronson J. J., Fink C., Green M., Kimball D., Macor J. E., Kwon S., Zhang Y., and Zipp G. (2011) Modulators of G protein-coupled receptor 88. U.S. Patent Application 2011/0251204.; c Dzierba C. D., Hartz R. A., Bi Y., Ahuja V. T., Bronson J. J., Carson K., Cianchetta G., Green M., Kimball D., Kimura S. R., Kwon S., Macor J. E., Zhang Y., and Zipp G. (2011) Modulators of G protein-coupled receptor 88. U.S. Patent Application 2011/0251196.
- a Chen Y.; Fields K. B.; Zhang X. P. (2004) Bromoporphyrins as versatile synthons for modular construction of chiral porphyrins: cobalt-catalyzed highly enantioselective and diastereoselective cyclopropanation. J. Am. Chem. Soc. 126, 14718–14719. [DOI] [PubMed] [Google Scholar]; b Chen Y.; Zhang X. P. (2007) Asymmetric cyclopropanation of styrenes catalyzed by metal complexes of D2-symmetrical chiral porphyrin: superiority of cobalt over iron. J. Org. Chem. 72, 5931–5934. [DOI] [PubMed] [Google Scholar]; c Chen Y.; Ruppel J. V.; Zhang X. P. (2007) Cobalt-catalyzed asymmetric cyclopropanation of electron-deficient olefins. J. Am. Chem. Soc. 129, 12074–12075. [DOI] [PubMed] [Google Scholar]; d Dzik W. I.; Xu X.; Zhang X. P.; Reek J. N.; de Bruin B. (2010) “Carbene radicals” in Co(II)(por)-catalyzed olefin cyclopropanation. J. Am. Chem. Soc. 132, 10891–10902. [DOI] [PubMed] [Google Scholar]; e Lu H.; Dzik W. I.; Xu X.; Wojtas L.; de Bruin B.; Zhang X. P. (2011) Experimental evidence for cobalt(II)-carbene radicals: key intermediates in cobalt(II)-based metalloradical cyclopropanation. J. Am. Chem. Soc. 133, 8518–8521. [DOI] [PubMed] [Google Scholar]
- Cheng K.; Lee Y. S.; Rothman R. B.; Dersch C. M.; Bittman R. W.; Jacobson A. E.; Rice K. C. (2011) Probes for narcotic receptor mediated phenomena. 41. Unusual inverse μ-agonists and potent μ-opioid antagonists by modification of the N-substituent in enantiomeric 5-(3-hydroxyphenyl)morphans. J. Med. Chem. 54, 957–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Allen J. A.; Yost J. M.; Setola V.; Chen X.; Sassano M. F.; Chen M.; Peterson S.; Yadav P. N.; Huang X. P.; Feng B.; Jensen N. H.; Che X.; Bai X.; Frye S. V.; Wetsel W. C.; Caron M. G.; Javitch J. A.; Roth B. L.; Jin J. (2011) Discovery of β-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proc. Natl. Acad. Sci. U.S.A. 108, 18488–18493. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Besnard J.; Ruda G. F.; Setola V.; Abecassis K.; Rodriguiz R. M.; Huang X. P.; Norval S.; Sassano M. F.; Shin A. I.; Webster L. A.; Simeons F. R.; Stojanovski L.; Prat A.; Seidah N. G.; Constam D. B.; Bickerton G. R.; Read K. D.; Wetsel W. C.; Gilbert I. H.; Roth B. L.; Hopkins A. L. (2012) Automated design of ligands to polypharmacological profiles. Nature 492, 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D.; Wang C.; Katritch V.; Han G. W.; Huang X. P.; Vardy E.; McCorvy J. D.; Jiang Y.; Chu M.; Siu F. Y.; Liu W.; Xu H. E.; Cherezov V.; Roth B. L.; Stevens R. C. (2013) Structural features for functional selectivity at serotonin receptors. Science 340, 615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.