

Significance
Wood decay fungi have historically been characterized as either white rot, which degrade all components of plant cell walls, including lignin, or brown rot, which leave lignin largely intact. Genomic analyses have shown that white-rot species possess multiple lignin-degrading peroxidases (PODs) and expanded suites of enzymes attacking crystalline cellulose. To test the adequacy of the white/brown-rot categories, we analyzed 33 fungal genomes. Some species lack PODs, and thus resemble brown-rot fungi, but possess the cellulose-degrading apparatus typical of white-rot fungi. Moreover, they appear to degrade lignin, based on decay analyses on wood wafers. Our results indicate that the prevailing paradigm of white rot vs. brown rot does not capture the diversity of fungal wood decay mechanisms.
Keywords: lignocellulose, phylogenomics, bioenergy
Abstract
Basidiomycota (basidiomycetes) make up 32% of the described fungi and include most wood-decaying species, as well as pathogens and mutualistic symbionts. Wood-decaying basidiomycetes have typically been classified as either white rot or brown rot, based on the ability (in white rot only) to degrade lignin along with cellulose and hemicellulose. Prior genomic comparisons suggested that the two decay modes can be distinguished based on the presence or absence of ligninolytic class II peroxidases (PODs), as well as the abundance of enzymes acting directly on crystalline cellulose (reduced in brown rot). To assess the generality of the white-rot/brown-rot classification paradigm, we compared the genomes of 33 basidiomycetes, including four newly sequenced wood decayers, and performed phylogenetically informed principal-components analysis (PCA) of a broad range of gene families encoding plant biomass-degrading enzymes. The newly sequenced Botryobasidium botryosum and Jaapia argillacea genomes lack PODs but possess diverse enzymes acting on crystalline cellulose, and they group close to the model white-rot species Phanerochaete chrysosporium in the PCA. Furthermore, laboratory assays showed that both B. botryosum and J. argillacea can degrade all polymeric components of woody plant cell walls, a characteristic of white rot. We also found expansions in reducing polyketide synthase genes specific to the brown-rot fungi. Our results suggest a continuum rather than a dichotomy between the white-rot and brown-rot modes of wood decay. A more nuanced categorization of rot types is needed, based on an improved understanding of the genomics and biochemistry of wood decay.
Fungi of the phylum Basidiomycota (basidiomycetes) comprise 32% of the described fungi (1) and are important to forestry (2–4), agriculture (5–7), and medicine (8–11). This diverse phylum includes the mushrooms (12–14); pathogens of plants (2), animals (9–11), and other fungi (15); osmotically tolerant molds (16); ectomycorrhizal symbionts like Laccaria bicolor, which are critical for plant growth; plant pathogens, such as rusts and smuts (7); and saprotrophs, including wood-decaying fungi (17).
The 26,000 basidiomycete taxa in the National Center for Biotechnology Information (NCBI) database (18, 19) are divided into three subphyla: Agaricomycotina (∼22,000 taxa), Pucciniomycotina (∼2,300 taxa), and Ustilaginomycotina (∼1,000 taxa). Agaricomycotina includes many decomposers of wood and leaf litter (12, 17, 20–23) that produce lignocellulolytic enzymes that have potential to be used in bioenergy production (24–27). Thus, much of sequencing effort at the US Department of Energy (DOE) Joint Genome Institute (JGI) (http://jgi.doe.gov/fungi) has targeted Agaricomycotina, particularly the Agaricomycetes (mushroom-forming fungi), with the large orders Agaricales (predominantly gilled mushrooms), Polyporales (wood-decaying polypores and others), and Boletales (porcini mushrooms and others) being especially deeply sampled.
A keen focus in the comparative genomics of Basidiomycota has concerned lineages of wood decay fungi (13, 17, 20–23, 28). For decades, two broad categories have been recognized: white rot and brown rot (29–31). During brown rot, cellulose is rapidly depolymerized via oxidative mechanisms, whereas modified lignin remains as a polymeric residue (32–35). In contrast, white-rot fungi use hydrolases that gradually degrade cellulose while lignin is completely mineralized. Lignin degradation involves high-oxidation potential class II peroxidases (PODs) that, on the basis of conserved catalytic and Mn-binding sites, are classified as lignin peroxidase (LiP), manganese peroxidase (MnP), or versatile peroxidase (VP) (36–38). The first genomes of the white-rot fungus Phanerochaete chrysosporium and brown-rot fungus Postia placenta revealed a gene complement consistent with their respective modes of wood decay (22, 23). Further comparative genomics studies of larger sets of wood decay fungi supported a consistent relationship between decay patterns and several enzyme families. Specifically, white-rot fungi had high-oxidation potential PODs for lignin degradation as well as cellobiohydrolases for degrading crystalline cellulose. Classified in glycoside hydrolase (GH) families (39) GH6 and GH7, cellobiohydrolases attack cellulose in a synergistic manner and often carry a cellulose binding module (CBM1). In contrast, the genomes of brown-rot fungi did not encode PODs and the predicted cellobiohydrolase-encoding genes were generally absent or lacking a CBM1 domain.
Here, we present comparative analyses of 33 sequenced basidiomycete genomes (Table S1). Included are 22 wood decayers, of which Galerina marginata, Pleurotus ostreatus, Botryobasidium botryosum, and Jaapia argillacea are newly sequenced. The results call into question the prevailing white-rot/brown-rot dichotomy.
Results and Discussion
Phylogeny and Protein Conservation.
A maximum-likelihood (ML) phylogeny (40) was inferred from protein sequence alignments of 183 conserved gene families (Fig. S1A). Overall, the tree topology is consistent with prior studies using genome-scale datasets (17) as well as phylogenetic analyses using small numbers of genes (41), such as the sister group relationship between Ustilaginomycotina and Agaricomycotina, and the placement of Wallemia sebi (Wallemiomycetes) as the sister group of the rest of the Agaricomycotina (16). Brown-rot fungi are polyphyletic, as shown previously (17), and include species in Polyporales, Boletales, Gloeophyllales, and Dacrymycetales. Some aspects of the phylogeny remain uncertain: our placement of Jaapia argillacea as the sister group of the Gloeophyllales conflicts with a previous study using six genes (42); and Auricularia delicata was inferred as the sister group of Piriformospora indica, differing from multigene phylogenies (41, 43–45), which suggested that these lineages form a paraphyletic assemblage.
Gene families in 33 basidiomycetes and 30 other fungi (Table S2) with sequenced genomes were inferred by Markov chain (MCL) clustering (46) of all-vs.-all protein BLAST (47) alignments. Analysis of protein conservation suggests a conserved core fungal genome of ∼5,000 genes (Fig. S1C). Roughly one-half of the proteins (49%) in Basidiomycota lack homologs in other groups of fungi, and 23% of Basidiomycota proteins are unique to a single organism (i.e., each of the 33 basidiomycetes analyzed harbored proteins not found in any other sampled species, ranging from 4% to 51%). In contrast, in Ascomycota, 30% of the proteins are phylum specific and 13% are organism specific (using a comparative set of 27 Ascomycota fungi; Table S2). We were able to assign functions from the Eukaryotic Clusters of Orthologs (KOG) (48) to 38% of Basidiomycota proteins (compared with 43% in Ascomycota); 74% of core proteins in both Basidiomycota and Ascomycota; and 17% of Basidiomycota-specific proteins (23% in Ascomycota) (Fig. S2).
Diversity of CAZymes and Associated Activities in Basidiomycota.
Thirty-three basidiomycete genomes were searched for genes whose protein products are implicated in the breakdown of the polysaccharide portion of plant cell walls (cellulose, hemicellulose, pectin) using the CAZy database pipeline (39, 49). The number of genes in each CAZy family, in each organism, is presented in Dataset S1.
Cellulolytic families.
During white-rot decay, cellulose is targeted primarily by hydrolytic enzymes of multiple GH families. Lytic polysaccharide monooxygenases (LPMOs) of the AA9 family (formerly GH61) also participate via oxidative mechanisms. White-rot fungi generally have more cellulolytic genes (both hydrolytic and oxidative) and cellulose binding domains (CBM1) relative to brown-rot fungi (Fig. 1). Cellobiohydrolases of families GH6 and GH7 (which cleave cellulose to the disaccharide cellobiose) are correlated with white rot [r = 0.3 and 0.6, respectively, by the independent contrasts method (50)]. Likely involved in boosting cellulase activity (51–54), LPMOs are abundant in white-rot fungi (r = 0.3) but reduced in brown-rot fungi (r = −0.5). Cellobiose dehydrogenases (family AA3_1), which enhance cellulose degradation (52), are uniformly present in a single copy in all white-rot fungi and absent from the majority of brown-rot fungi (although Gloeophyllum trabeum has one copy, and Boletales Coniophora puteana and Serpula lacrymans each have two copies). Additional cellulolytic families were found in basidiomycetes. For examples, GH5 (containing endo-acting cellulases as well as many other enzymes of differing substrate specificity; Dataset S2), GH12, GH44, and GH45 are expanded in white-rot fungi (r = 0.2, 0.1, 0.4, 0.5, respectively) and diminished in brown-rot fungi (r < −0.2; for all, see also Dataset S1).
Fig. 1.

Lignocellulose-degrading and secondary metabolism in wood-decaying fungi. Organisms use the following abbreviations: Aurde, Auricularia delicata; Botbo, Botryobasidium botryosum; Cersu, Ceriporiopsis subvermispora; Conpu, Coniophora puteana; Dacsp, Dacryopinax sp.; Dicsq, Dichomitus squalens; Fomme, Fomitiporia mediterranea; Fompi, Fomitopsis pinicola; Galma, Galerina marginata; Glotr, Gloeophyllum trabeum; Hetan, Heterobasidion annosum; Jaaar, Jaapia argillacea; Phaca, Phanerochaete carnosa; Phchr, Phanerochaete chrysosporium; Pleos, Pleurotus ostreatus; Pospl, Postia placenta; Punst, Punctularia strigosozonata; Schco, Schizophyllum commune; Serla, Serpula lacrymans; Stehi, Stereum hirsutum; Trave, Trametes versicolor; Wolco, Wolfiporia cocos. Gene number is shaded red/white, and independent-contrasts correlation of enzyme with rot type is shaded orange/blue. Notice a strict white/brown-rot dichotomy with respect to the lignin-attacking PODs and the CAZymes that target crystalline cellulose (CBM1, GH6, and GH7), and a continuum with other lignin-targeting enzymes.
Among the newly sequenced genomes, G. marginata, P. ostreatus, B. botryosum, and J. argillacea all possess diverse CAZymes typical of white-rot fungi. B. botryosum has 32 genes encoding LPMOs, and three encoding cellobiose dehydrogenases, both more than any other wood decay fungus in our set. J. argillacea has 15 genes encoding LPMOs, and one encoding a cellobiose dehydrogenase, both numbers typical of white-rot fungi. A similar pattern is evident when considering families GH6, GH7, and CBM1; B. botryosum and J. argillacea show a genetic complement similar to white-rot fungi.
Hemicellulolytic and pectinolytic families.
Hemicellulose and pectin comprise a variety of linear and branched complex polysaccharides. Hemicellulose includes xylans, xyloglucans, glucuronoxylans, arabinoxylans, mannans, glucomannans, and galactoglucomannans. Pectins include polygalacturonic acid, linear and branched rhamnogalacturonans, and arabinogalactans. Basidiomycetes contain some seven CAZy families that target hemicelluloses, and 11 that target pectins (Table S3). In contrast to the cellulolytic families, there is not a clear dichotomy in which white-rot fungi have more genes encoding hemicellulases and pectinases, and brown-rot fungi have fewer (Dataset S1).
Distribution of Lignin-Degrading Enzymes Blurs the Distinction Between White- and Brown-Rot Fungi.
With respect to PODs, there was a clear dichotomy between white-rot fungi, which have various combinations of MnP, LiP, and VP (r = 0.8), and brown-rot fungi, which lack PODs (r = −0.5). By this measure, the newly sequenced G. marginata and P. ostreatus resemble typical white-rot fungi with 10 and 9 PODs, respectively. In contrast, B. botryosum and J. argillacea resemble typical brown-rot fungi in their lack of PODs. Schizophyllum commune had previously been characterized as a white-rot fungus (55), but it also lacks PODs. The one class II peroxidase in J. argillacea lacks the catalytic Trp and Mn-binding sites, and is therefore classified as a generic peroxidase (56, 57) that is unlikely to directly modify lignin. Affirming the observed genetic complement, extracellular filtrates from J. argillacea and B. botryosum cultures were unable to oxidize veratryl alcohol or 2,6-dimethoxyphenol, standard substrates for high-oxidation potential PODs (58, 59). According to our phylogeny (Fig. S1A), the contraction of ligninolytic PODs in J. argillacea may have arisen before its evolutionary separation from G. trabeum because both species lack these proteins. However, G. trabeum also has a reduced complement of enzymes attacking crystalline cellulose and is a model system for brown rot (60, 61).
Although PODs have been shown to be important for lignin degradation, numerous other classes of enzymes are capable of degrading, or at least modifying, lignin and about a dozen kinds of enzymes potentially participating in ligninolysis are found in basidiomycetes (Fig. 1). The multicopper oxidases of family AA1 are such an example. Laccases (subfamily AA1_1) may be capable of cleaving lignin bonds in the presence of certain mediators (62) and are more abundant in white-rot fungi than brown-rot fungi (Fig. 1; r = 0.4 for white rot; r = −0.3 for brown rot). In the cases of J. argillacea and B. botryosum, no laccase activity was detected in concentrated culture filtrates (63). Nevertheless, the white-rot fungi P. chrysosporium, Phanerochaete carnosa, and A. delicata have no basidiomycete-type laccases sensu stricto (17). These three fungi instead have four to eight genes each of the related AA1_dist subfamily of multicopper oxidases (which, predictably, are reduced in brown-rot fungi at 0 to two genes), reflecting the diversity of chemistries used in lignin degradation.
Correlations Between Decay Mode and Secondary Metabolism.
Secondary metabolites include polyketides, nonribosomal peptides, and terpenoids, among many other substances that are not considered to be critically involved in an organism’s growth, development, and reproduction, but that typically confer some advantage in the never-ending competition with other organisms (64). To assess the diversity of secondary metabolism in basidiomycetes, we characterized fatty acid synthases (FASs), nonribosomal peptide synthases (NRPSs), polyketide synthases (PKSs), and terpene synthases (TSs). PKSs were additionally subdivided into reducing and nonreducing (R-PKS and NR-PKS), each of which was further divided into subfamilies (Fig. S3). Secondary metabolism enzymes are summarized in the lower panel of Fig. 1.
Wood-decaying basidiomycetes tend to have similar numbers of FAS and NR-PKS genes (approximately one gene each), with the exception of B. botryosum, which has five NR-PKS genes. NRPSs vary in gene number (0 to nine genes), with no apparent correlation with lifestyle. TS genes, as well, vary in number (1–13 genes), with no apparent lifestyle correlation.
R-PKSs are expanded in the brown-rot fungi, with all but the phylogenetically basal Dacryopinax sp. possessing 4–14 R-PKS genes. In contrast, the white-rot fungi have generally fewer R-PKS genes and many taxa lack them entirely. R-PKS gene number correlates with brown rot (independent-contrasts r = 0.8). Although it was previously suggested that secondary metabolism could play a role in wood decay by the brown-rot fungus Serpula lacrymans (20), a widespread pattern of secondary metabolism expansion has not yet been reported for brown-rot fungi in general. The observation that the number of R-PKS genes is expanded in three phylogenetically distinct clades of brown-rot fungi: the Boletales (S. lacrymans and C. puteana), the Polyporales (Wolfiporia cocos, P. placenta, and Fomitopsis pinicola), and Gloeophyllales (G. trabeum), suggests that R-PKSs have a possible role in the brown-rot lifestyle, and that the apparent correlation is not an artifact of phylogenetic sampling. The exact biochemical role of R-PKSs in wood rot in general is unknown. However, it has been suggested that secondary metabolites including certain catechols and quinones can serve as extracellular Fe3+ reductants in G. trabeum, S. lacrymans, P. placenta, and W. cocos. Together with H2O2, the reduced iron is thought to drive a nonenzymatic Fenton reaction in which highly reactive hydroxyl radicals depolymerize cellulose (20, 65–68).
Secondary metabolism plays a critical role for fungal survival and success, including Basidiomycota (69), and a rich array of secondary metabolism genes has been reported in Serpula lacrymans and Postia placenta (20), and Omphalotus olearius (70). It may be that secondary metabolites, widely considered to confer advantage by inhibiting competing microorganisms, are more important in the ecological niches occupied by brown-rot fungi, compared with white-rot fungi. Indeed, our observation that expansions of R-PKSs are correlated with brown rot in multiple polyphyletic lineages highlights gaps in our knowledge of the mechanisms of wood decay even in well-studied species of Boletales, Polyporales, and Gloeophyllales.
Insights into Diversity of Decay Mechanisms from Phylogenetically Informed Principal-Components Analyses of Decay-Related Gene Families.
The expanded sampling of basidiomycete genomes presented here includes species (B. botryosum, J. argillacea, and S. commune) that deviate from the classical models of white rot vs. brown rot, in that they lack PODs (like brown-rot fungi) but possess diverse arrays of enzymes acting on crystalline cellulose (like white-rot fungi). To visualize the diversity of decay chemistries in basidiomycetes, we applied phylogenetically informed principal-components analysis (PCA) (71) to the set of predicted carbohydrate- and lignin-active enzymes (Dataset S1). We hypothesized that organisms should cluster with others of similar nutritional mode, perhaps implying nutritional mode for B. botryosum and J. argillacea. Fig. 2 shows the wood-decaying fungi plotted on the first two principal components, which together explain 50% of the variability in the data. For this analysis, we again relied on categorization of fungi as producing either white or brown rot whenever possible. Clear separation of known white- and brown-rotting fungi is seen along PC1. B. botryosum and J. argillacea group with the white-rot fungi on PC1 and are closest to the model white-rot fungus, P. chrysosporium. In contrast, S. commune is intermediate between white-rot and brown-rot species along PC1. Both white-rot and brown-rot species are widely distributed along PC2, which suggests heterogeneity in modes of wood degradation within these functional classes. Despite the lack of PODs (Fig. 1), PCA analysis suggests that B. botryosum and J. argillacea have wood decay properties that are similar to certain white-rot fungi.
Fig. 2.

Wood-decaying basidiomycetes plotted on the first two principal components from phylogenetic PCA of CAZymes (including lignin-related auxiliary activities) of the organisms.
Decay Assays in Wood Substrates.
To assess the extent to which B. botryosum and J. argillacea are able to degrade all components of wood, we performed experiments using single-spore isolates of each fungus, on both pine and aspen wood. Both species readily colonized both types of wood, but caused only superficial degradation of the wood surfaces. Nevertheless, in localized areas B. botryosum eroded all cell wall layers, indicating degradation and disruption of all cell wall polymers. Hyphae of B. botryosum were visible growing in the voids where cell wall degradation had taken place (Fig. 3). The removal of cellulose, hemicellulose, and lignin in these areas is consistent with white rot, whereas in brown rot we would observe residual lignin remaining after a diffuse depolymerization of the cellulose. Similarly, J. argillacea degraded a localized area of the wood cells in which all of the cell wall layers were degraded and replaced with the mycelia of the fungus. Once again, complete degradation of all cell wall layers implies all of the wall components have been degraded, as is characteristic of white rot.
Fig. 3.
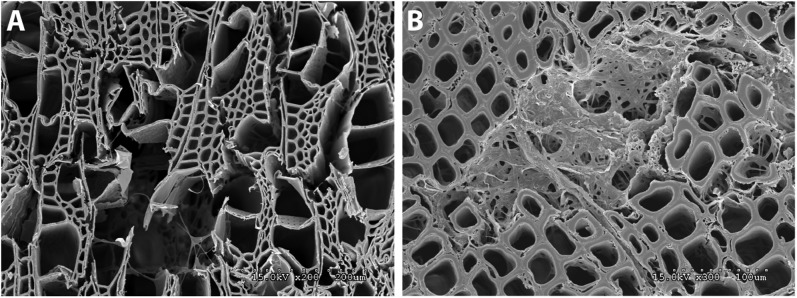
Wood decay experiments indicating mode of decay by Botryobasidium botryosum and Jaapia argillacea. (A) Micrograph of B. botryosum on aspen wood with vessel, fiber, and parenchyma cell walls degraded. Mycelia are visible growing through the voids. (B) Micrograph of J. argillacea on pine showing an area within the wood where the fungus has caused a localized simultaneous decay of the cells. Residual cell wall material and mycelia fill the degraded zone.
We did not include S. commune in decay assays as previous studies have shown that this species, although classified as a white-rot species, lacks the ability to degrade lignin appreciably in vitro (55). The S. commune genome lacks PODs and has a reduced complement of enzymes bearing cellulose-binding modules (CBM1), but it retains 22 genes encoding LPMOs. Thus, S. commune likely has a very different mode of decay than B. botryosum and J. argillacea (with which it shares the absence of PODs), which is consistent with its isolated position, intermediate between white- and brown-rot species, in the PCA (Fig. 2).
Need for a New Classification of Wood Decay Modes.
Discussions of the genomic correlates of the white- vs. brown-rot modes of wood decay have typically focused on the lignin-degrading PODs and the hydrolytic and oxidative enzymes involved in attack of crystalline cellulose (17, 23, 25, 72). However, it is also accepted that other enzymes, such as laccases, cellobiose dehydrogenases, and potentially many others, can contribute to white rot (62, 73, 74); accordingly, a database of functional annotations has been dedicated to cataloging lignin-degrading enzymes (49) (Figs. S4 and S5). Our results indicate that the simple dichotomy of white rot vs. brown rot does not adequately reflect the diversity of mechanisms by which wood-rotting fungi obtain nutrition. Specifically, B. botryosum and J. argillacea show similarities to white-rot fungi in PCA of all predicted carbohydrate- and lignin-active enzymes and can degrade all components of wood, but they do so without the PODs that are a hallmark of white rot. S. commune is another putative white-rot fungus (55) that lacks PODs, but it is quite distinct from B. botryosum and J. argillacea in the PCA (and presumably also in its mode of wood decay). Therefore, we suggest that a more nuanced categorization scheme is needed to describe wood decay by species that degrade all cell wall polymers, including lignin, but lack PODs. As our discovery of the potential role of R-PKSs in brown rot indicates, other functional considerations beyond lignin-degrading PODs and CAZymes may be important for a biologically descriptive classification of decay modes. Finally, we suggest restricting the term “white rot” to only those fungi that degrade all cell wall polymers through the action of the lignin-degrading PODs in concert with enzymes that attack crystalline cellulose.
Methods
Data Access.
Genome assemblies and annotations for the organisms used in this study are available via the JGI Genome Portal MycoCosm (http://jgi.doe.gov/fungi; see also Table S1). In addition, the newly sequenced genome assemblies and annotations have been deposited to GenBank under the following accession numbers: B. botryosum: AYEP00000000; G. marginata: AYUM00000000; J. argillacea: AYUL00000000; P. ostreatus: AYUK00000000. The versions described in this paper are AYEP01000000, AYUM01000000, AYUL01000000, and AYUK01000000, respectively.
Sequencing, Assembly, and Annotation.
The B. botryosum, G. marginata, and J. argillacea genome assemblies were produced using shredded consensi from Velvet-assembled (75) Illumina paired-end data combined with Roche (454) data assembled with Newbler (www.454.com). P. ostreatus was sequenced with the Sanger whole-genome shotgun approach and assembled with Arachne (76). All genomes were annotated using the JGI Annotation Pipeline (77), which combines several gene prediction and annotation methods with transcriptomics data, and integrates the annotated genomes into MycoCosm (78), a Web-based fungal resource for comparative analysis.
Protein Sequence Clustering.
Predicted protein sequences were clustered using the MCL clustering algorithm (46), with an inflation parameter of 2.0. Two clustering runs were performed: a first one using 63 organisms (33 basidiomycetes, 27 ascomycetes, and 3 from other fungal phyla), which was used for core genes and protein conservation analysis; and a second using 39 organisms (33 basidiomycetes and 4 ascomycetes: Aspergillus nidulans, Pichia stipitis, Stagonospora nodorum, and Trichoderma reesei) as outgroups, which was used for generating the phylogenetic tree (see SI Text and Dataset S3 for further details).
Phylogeny.
Protein sequences from 183 single-copy clusters were concatenated and multiple-aligned using MAFFT (79) with default parameters, resulting in an alignment with 163,105 amino acid positions. Gblocks (80) was run with the options t = p and −b4 = 5 to remove poorly aligned regions, resulting in 38,423 positions. A ML tree was then inferred using RAxML (40) on the Gblocks-trimmed alignment with the PROTGAMMAWAG model, 100 rounds of bootstrapping, and the four Ascomycete organisms A. nidulans, P. stipitis, S. nodorum, and T. reesei specified to the program as outgroups. The resulting tree was used for subsequent analyses.
Annotation of Carbohydrate-Active and Auxiliary Redox Enzymes.
The carbohydrate-active enzymes (CAZy) and auxiliary redox enzymes (AA) were annotated as in refs. 39 and 49. A manually curated subset of the AA2 family was created, limited to genes predicted to encode high-oxidation potential peroxidases: LiP, MnP, and VP. These are referred to in this work as “POD.”
Annotation of Secondary Metabolism Enzymes.
Proteins with an AMP-binding domain (PF00501), a phosphopantetheinyl-binding domain (PF00550), and a condensation domain (PF00668) were designated as NRPS. Proteins with a terpene synthase domain (PF03936) were designated as TS. Proteins with a ketoacyl-synthase (KS) domain (PF00109 and PF02801) were divided into three categories: FAS, NR-PKS, and R-PKS based on their phylogenetic relationships. A gene genealogy was constructed by maximum parsimony analysis in PAUP* 4.0b10 (81) using a ClustalW+ (82) alignment of deduced amino acid sequences. Statistical support for branches was generated by bootstrap analysis with 1,000 pseudoreplications. Putative FASs were grouped in a clade (100% support) that included an Ascomycete KS with significant identity to a FAS from Aspergillus nidulans (83). Putative NR-PKSs were grouped in a clade (90% support) that included an Ascomycete NR-PKS involved in the synthesis of a nonreduced polyketide (84). Putative R-PKSs were grouped in three clades (83%, 100%, and 64% support), of which a majority included a ketoreductase (KR) domain (PF08659) (85).
Correlation of Genomic Features and Lifestyle.
To infer correlations between genomic features and phenotypes (e.g., repeat content with genome size, or CAZymes with white or brown rot), we used Felsenstein’s method of independent contrasts (50) as implemented in the contrast program from Phylip, version 3.62 downloaded from http://evolution.genetics.washington.edu/phylip (86).
Phylogenetic PCA.
Phylogenetic PCA was implemented using the function phyl.pca from the R package phytools (www.phytools.org/). The algorithm corrects the covariance/correlation matrices for nonindependence among the observations for species, by additionally taking into account the expected covariances/correlations due to pure phylogenetic relatedness of species (71). A matrix of CAZyme gene number in each organism (Dataset S1), restricted to the wood decay fungi (Aurde, Botbo, Cersu, Conpu, Dacsp, Dicsq, Fomme, Fompi, Galma, Glotr, Hetan, Jaaar, Phaca, Phchr, Pleos, Pospl, Punst, Schco, Serla, Stehi, Trave, and Wolco), and the ML tree, were used as input.
Wood Decay Assays.
Micromorphological observations of degraded aspen (Populus sp.) and pine (Pinus sp.) wood were used to elucidate the type of decay resulting after inoculation with B. botryosum or J. argillacea. Physical aspects of decay were investigated using scanning electron microscopy on wood wafers after decay using the same single spore isolate of each fungus that was used for genome sequencing. Dried wood wafers from the sapwood of aspen and pine were cut to 10 × 10 × 1 mm, hydrated to 80–100% moisture, and autoclaved for 60 min at 120 °C. Wood wafers were placed on inoculated Petri plates with malt extract agar (15 g of malt extract, 15 g of agar, 2 g of yeast extract, and 1,000 mL of water). Six plates of each wood type were inoculated with each fungus by placing a 2-mm plug of the assay fungus at the center of a Petri plate 14 d before wafers were added. Wafers were removed 90 d after inoculation and prepared for scanning electron microscopy as previously described (87). Samples were examined and photographed using a Hitachi S3500 N (Hitachi) scanning electron microscope.
Culture Conditions and Enzyme Assays.
Standard media included carbon- and nitrogen-starved B3 (88, 89) as well as a more complex medium containing 0.5% (wt/vol) Wiley-mill ground aspen as sole carbon source (90). Culture filtrates were concentrated 6- to 10-fold with 10-kDa spin concentrators (Pall). Protein concentration was determined by the Bradford assay (Sigma-Aldrich). Measurement of LiP, MnP, laccase, and glyoxal oxidase involved oxidation of veratryl alcohol (Sigma-Aldrich) (58), 2,6-dimethoxyphenol (59), 2,2-azonodi-3-ethylbenzothiazoline-6-sulfuric acid (Boehringer), and methyl glyoxal as substrate (91), respectively (see SI Text and Table S4 for further details).
Supplementary Material
Acknowledgments
We thank Jill Gaskell (Forest Products Laboratory) for assistance with cultures and enzyme assays. The work conducted by the US Department of Energy (DOE) Joint Genome Institute is supported by the Office of Science of the DOE under Contract DE-AC02-05CH11231. J.D.W. and D.J. were supported by the DOE Great Lakes Bioenergy Research Center (DOE Office of Science Biological and Environmental Research Contract DE-FC02-07ER64494). B.H. is Honorary Professor at the Faculty of Health and Medical Sciences, University of Copenhagen. F.M.’s research group is part of the Laboratory of Excellence for Advanced Research on the Biology of Forest Ecosystems (ANR-11-LABX-0002-01).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The sequences reported in this paper have been deposited in the GenBank database [AYEP00000000 (Botryobasidium botryosum), AYUM00000000 (Galerina marginata), AYUL00000000 (Jaapia argillacea), and AYUK00000000 (Pleurotus ostreatus)].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1400592111/-/DCSupplemental.
References
- 1.Kirk PMCP, Minter DW, Stalpers JA. Dictionary of the Fungi. 10th Ed. Wallingford, UK: CAB International; 2008. p. 72. [Google Scholar]
- 2.Johansson M, Stenlid J. Infection of roots of Norway spruce (Picea abies) by Heterobasidion Annosum: Initial reactions in sapwood by wounding and infection. Eur J Forest Pathol. 1985;15(1):32–45. [Google Scholar]
- 3.Otrosina WJ, Chase TE, Cobb FW, Korhonen K. Population structure of Heterobasidion annosum from North America and Europe. Can J Bot. 1993;71(8):1064–1071. [Google Scholar]
- 4.Martin F, et al. The genome of Laccaria bicolor provides insights into mycorrhizal symbiosis. Nature. 2008;452(7183):88–92. doi: 10.1038/nature06556. [DOI] [PubMed] [Google Scholar]
- 5.Jin Y, et al. Characterization of seedling infection types and adult plant infection responses of monogenic Sr gene lines to race TTKS of Puccinia graminis f. sp tritici. Plant Dis. 2007;91(9):1096–1099. doi: 10.1094/PDIS-91-9-1096. [DOI] [PubMed] [Google Scholar]
- 6.Jin Y, Wanyera R, Kinyua M, Jin Y, Singh R. The spread of Puccinia graminis f. sp tritici with broad virulence in eastern Africa. Phytopathology. 2005;95(6):S49. [Google Scholar]
- 7.Duplessis S, et al. Obligate biotrophy features unraveled by the genomic analysis of rust fungi. Proc Natl Acad Sci USA. 2011;108(22):9166–9171. doi: 10.1073/pnas.1019315108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rihs JD, Padhye AA, Good CB. Brain abscess caused by Schizophyllum commune: An emerging basidiomycete pathogen. J Clin Microbiol. 1996;34(7):1628–1632. doi: 10.1128/jcm.34.7.1628-1632.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lacaz CdaS, Heins-Vaccari EM, De Melo NT, Hernandez-Arriagada GL. Basidiomycosis: A review of the literature. Rev Inst Med Trop Sao Paulo. 1996;38(5):379–390. doi: 10.1590/s0036-46651996000500011. [DOI] [PubMed] [Google Scholar]
- 10.Brown SM, Campbell LT, Lodge JK. Cryptococcus neoformans, a fungus under stress. Curr Opin Microbiol. 2007;10(4):320–325. doi: 10.1016/j.mib.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dawson TL., Jr Malassezia globosa and restricta: Breakthrough understanding of the etiology and treatment of dandruff and seborrheic dermatitis through whole-genome analysis. J Investig Dermatol Symp Proc. 2007;12(2):15–19. doi: 10.1038/sj.jidsymp.5650049. [DOI] [PubMed] [Google Scholar]
- 12.Morin E, et al. Genome sequence of the button mushroom Agaricus bisporus reveals mechanisms governing adaptation to a humic-rich ecological niche. Proc Natl Acad Sci USA. 2012;109(43):17501–17506. doi: 10.1073/pnas.1206847109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohm RA, et al. Genome sequence of the model mushroom Schizophyllum commune. Nat Biotechnol. 2010;28(9):957–963. doi: 10.1038/nbt.1643. [DOI] [PubMed] [Google Scholar]
- 14.Stajich JE, et al. Insights into evolution of multicellular fungi from the assembled chromosomes of the mushroom Coprinopsis cinerea (Coprinus cinereus) Proc Natl Acad Sci USA. 2010;107(26):11889–11894. doi: 10.1073/pnas.1003391107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Griffith NT, Barnett HL. Mycoparasitism by Basidiomycetes in culture. Mycologia. 1967;59(1):149–154. [PubMed] [Google Scholar]
- 16.Padamsee M, et al. The genome of the xerotolerant mold Wallemia sebi reveals adaptations to osmotic stress and suggests cryptic sexual reproduction. Fungal Genet Biol. 2012;49(3):217–226. doi: 10.1016/j.fgb.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 17.Floudas D, et al. The Paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science. 2012;336(6089):1715–1719. doi: 10.1126/science.1221748. [DOI] [PubMed] [Google Scholar]
- 18.Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res. 2009;37(Database issue):D26–D31. doi: 10.1093/nar/gkn723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sayers EW, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2009;37(Database issue):D5–D15. doi: 10.1093/nar/gkn741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eastwood DC, et al. The plant cell wall-decomposing machinery underlies the functional diversity of forest fungi. Science. 2011;333(6043):762–765. doi: 10.1126/science.1205411. [DOI] [PubMed] [Google Scholar]
- 21.Fernandez-Fueyo E, et al. Comparative genomics of Ceriporiopsis subvermispora and Phanerochaete chrysosporium provide insight into selective ligninolysis. Proc Natl Acad Sci USA. 2012;109(14):5458–5463. doi: 10.1073/pnas.1119912109. and correction (2012) 109(21):8352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martinez D, et al. Genome, transcriptome, and secretome analysis of wood decay fungus Postia placenta supports unique mechanisms of lignocellulose conversion. Proc Natl Acad Sci USA. 2009;106(6):1954–1959. doi: 10.1073/pnas.0809575106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martinez D, et al. Genome sequence of the lignocellulose degrading fungus Phanerochaete chrysosporium strain RP78. Nat Biotechnol. 2004;22(6):695–700. doi: 10.1038/nbt967. [DOI] [PubMed] [Google Scholar]
- 24.Baldrian P, Valásková V. Degradation of cellulose by basidiomycetous fungi. FEMS Microbiol Rev. 2008;32(3):501–521. doi: 10.1111/j.1574-6976.2008.00106.x. [DOI] [PubMed] [Google Scholar]
- 25.Lundell TK, Mäkelä MR, Hildén K. Lignin-modifying enzymes in filamentous basidiomycetes—ecological, functional and phylogenetic review. J Basic Microbiol. 2010;50(1):5–20. doi: 10.1002/jobm.200900338. [DOI] [PubMed] [Google Scholar]
- 26.Morita T, Fukuoka T, Imura T, Kitamoto D. Production of glycolipid biosurfactants by basidiomycetous yeasts. Biotechnol Appl Biochem. 2009;53(Pt 1):39–49. doi: 10.1042/BA20090033. [DOI] [PubMed] [Google Scholar]
- 27.Rasmussen ML, Shrestha P, Khanal SK, Pometto AL, 3rd, Hans van Leeuwen J. Sequential saccharification of corn fiber and ethanol production by the brown rot fungus Gloeophyllum trabeum. Bioresour Technol. 2010;101(10):3526–3533. doi: 10.1016/j.biortech.2009.12.115. [DOI] [PubMed] [Google Scholar]
- 28.Grigoriev IV, et al. Fueling the future with fungal genomics. Mycology. 2011;2(3):192–209. [Google Scholar]
- 29.Daniel G. Use of electron microscopy for aiding our understanding of wood biodegradation. FEMS Microbiol Rev. 1994;13:199–233. [Google Scholar]
- 30.Blanchette R. Delignification by wood-decay fungi. Annu Rev Phytopathol. 1991;29:381–398. [Google Scholar]
- 31.Eriksson K-EL, Blanchette RA, Ander P. Microbial and Enzymatic Degradation of Wood and Wood Components. Berlin: Springer; 1990. [Google Scholar]
- 32.Blanchette R. Degradation of the lignocellulose complex in wood. Can J Bot. 1995;73(Suppl 1):999–1010. [Google Scholar]
- 33.Worrall JJ, Anagnost SE, Zabel RA. Comparison of wood decay among diverse lignicolous fungi. Mycologia. 1997;89:199–219. [Google Scholar]
- 34.Yelle DJ, Ralph J, Lu F, Hammel KE. Evidence for cleavage of lignin by a brown rot basidiomycete. Environ Microbiol. 2008;10(7):1844–1849. doi: 10.1111/j.1462-2920.2008.01605.x. [DOI] [PubMed] [Google Scholar]
- 35.Niemenmaa O, Uusi-Rauva A, Hatakka A. Demethoxylation of [O14CH3]-labelled lignin model compounds by the brown-rot fungi Gloeophyllum trabeum and Poria (Postia) placenta. Biodegradation. 2008;19(4):555–565. doi: 10.1007/s10532-007-9161-3. [DOI] [PubMed] [Google Scholar]
- 36.Kirk TK, Farrell RL. Enzymatic “combustion”: The microbial degradation of lignin. Annu Rev Microbiol. 1987;41:465–505. doi: 10.1146/annurev.mi.41.100187.002341. [DOI] [PubMed] [Google Scholar]
- 37.Hammel KE, Cullen D. Role of fungal peroxidases in biological ligninolysis. Curr Opin Plant Biol. 2008;11(3):349–355. doi: 10.1016/j.pbi.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 38.Martinez AT. Molecular biology and structure-function of lignin-degrading heme peroxidases. Enzyme Microb Technol. 2002;30:425–444. [Google Scholar]
- 39.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42(Database issue):D490–D495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stamatakis A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22(21):2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 41.Matheny PB, et al. Contributions of rpb2 and tef1 to the phylogeny of mushrooms and allies (Basidiomycota, Fungi) Mol Phylogenet Evol. 2007;43(2):430–451. doi: 10.1016/j.ympev.2006.08.024. [DOI] [PubMed] [Google Scholar]
- 42.Binder M, Larsson KH, Matheny PB, Hibbett DS. Amylocorticiales ord. nov. and Jaapiales ord. nov.: Early diverging clades of agaricomycetidae dominated by corticioid forms. Mycologia. 2010;102(4):865–880. doi: 10.3852/09-288. [DOI] [PubMed] [Google Scholar]
- 43.Hibbett DS. A phylogenetic overview of the Agaricomycotina. Mycologia. 2006;98(6):917–925. doi: 10.3852/mycologia.98.6.917. [DOI] [PubMed] [Google Scholar]
- 44.Hibbett DS, et al. A higher-level phylogenetic classification of the Fungi. Mycol Res. 2007;111(Pt 5):509–547. doi: 10.1016/j.mycres.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 45.Matheny PB, Gossmann JA, Zalar P, Kumar TKA, Hibbett DS. Resolving the phylogenetic position of the Wallemiomycetes: An enigmatic major lineage of Basidiomycota. Can J Bot. 2006;84(12):1794–1805. [Google Scholar]
- 46.Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30(7):1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 48.Koonin EV, et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004;5(2):R7. doi: 10.1186/gb-2004-5-2-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Levasseur A, Drula E, Lombard V, Coutinho PM, Henrissat B. Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol Biofuels. 2013;6(1):41. doi: 10.1186/1754-6834-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Felsenstein J. Phylogenies and the comparative method. Am Nat. 1985;125(1):1–15. doi: 10.1086/703055. [DOI] [PubMed] [Google Scholar]
- 51.Harris PV, et al. Stimulation of lignocellulosic biomass hydrolysis by proteins of glycoside hydrolase family 61: Structure and function of a large, enigmatic family. Biochemistry. 2010;49(15):3305–3316. doi: 10.1021/bi100009p. [DOI] [PubMed] [Google Scholar]
- 52.Langston JA, et al. Oxidoreductive cellulose depolymerization by the enzymes cellobiose dehydrogenase and glycoside hydrolase 61. Appl Environ Microbiol. 2011;77(19):7007–7015. doi: 10.1128/AEM.05815-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Quinlan RJ, et al. Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc Natl Acad Sci USA. 2011;108(37):15079–15084. doi: 10.1073/pnas.1105776108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vaaje-Kolstad G, et al. An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science. 2010;330(6001):219–222. doi: 10.1126/science.1192231. [DOI] [PubMed] [Google Scholar]
- 55.Schmidt O, Liese W. Variability of wood degrading enzymes of Schizophyllum commune. Holzforschung. 1980;34(2):67–72. [Google Scholar]
- 56.Blodig W, et al. Autocatalytic formation of a hydroxy group at C beta of trp171 in lignin peroxidase. Biochemistry. 1998;37(25):8832–8838. doi: 10.1021/bi9727186. [DOI] [PubMed] [Google Scholar]
- 57.Poulos TL, Edwards SL, Wariishi H, Gold MH. Crystallographic refinement of lignin peroxidase at 2 Å. J Biol Chem. 1993;268(6):4429–4440. doi: 10.2210/pdb1lga/pdb. [DOI] [PubMed] [Google Scholar]
- 58.Tien M, Kirk TK. Lignin-degrading enzyme from Phanerochaete chrysosporium: Purification, characterization, and catalytic properties of a unique H2O2-requiring oxygenase. Proc Natl Acad Sci USA. 1984;81(8):2280–2284. doi: 10.1073/pnas.81.8.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wariishi H, Valli K, Gold MH. Manganese(II) oxidation by manganese peroxidase from the basidiomycete Phanerochaete chrysosporium. Kinetic mechanism and role of chelators. J Biol Chem. 1992;267(33):23688–23695. [PubMed] [Google Scholar]
- 60.Kerem Z, hammel, Hammel KE. Biodegradative mechanism of the brown rot basidiomycete Gloeophyllum trabeum: Evidence for an extracellular hydroquinone-driven fenton reaction. FEBS Lett. 1999;446(1):49–54. doi: 10.1016/s0014-5793(99)00180-5. [DOI] [PubMed] [Google Scholar]
- 61.Jensen KA, Jr, Houtman CJ, Ryan ZC, Hammel KE. Pathways for extracellular Fenton chemistry in the brown rot basidiomycete Gloeophyllum trabeum. Appl Environ Microbiol. 2001;67(6):2705–2711. doi: 10.1128/AEM.67.6.2705-2711.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Eggert C, Temp U, Eriksson KEL. The ligninolytic system of the white rot fungus Pycnoporus cinnabarinus: Purification and characterization of the laccase. Appl Environ Microbiol. 1996;62(4):1151–1158. doi: 10.1128/aem.62.4.1151-1158.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Niku-Paavola ML, Karhunen E, Salola P, Raunio V. Ligninolytic enzymes of the white-rot fungus Phlebia radiata. Biochem J. 1988;254(3):877–883. doi: 10.1042/bj2540877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Keller NP, Turner G, Bennett JW. Fungal secondary metabolism—from biochemistry to genomics. Nat Rev Microbiol. 2005;3(12):937–947. doi: 10.1038/nrmicro1286. [DOI] [PubMed] [Google Scholar]
- 65.Arantes V, Milagres AM, Filley TR, Goodell B. Lignocellulosic polysaccharides and lignin degradation by wood decay fungi: The relevance of nonenzymatic Fenton-based reactions. J Ind Microbiol Biotechnol. 2011;38(4):541–555. doi: 10.1007/s10295-010-0798-2. [DOI] [PubMed] [Google Scholar]
- 66.Cohen R, Jensen KA, Houtman CJ, Hammel KE. Significant levels of extracellular reactive oxygen species produced by brown rot basidiomycetes on cellulose. FEBS Lett. 2002;531(3):483–488. doi: 10.1016/s0014-5793(02)03589-5. [DOI] [PubMed] [Google Scholar]
- 67.Korripally P, Timokhin VI, Houtman CJ, Mozuch MD, Hammel KE. Evidence from Serpula lacrymans that 2,5-dimethoxyhydroquinone is a lignocellulolytic agent of divergent brown rot basidiomycetes. Appl Environ Microbiol. 2013;79(7):2377–2383. doi: 10.1128/AEM.03880-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shimokawa T, Nakamura M, Hayashi N, Ishihara M. Production of 2,5-dimethoxyhydroquinone by the brown-rot fungus Serpuyla lacrymans to drive extracellular Fenton reaction. Holzforschung. 2004;58:305–310. [Google Scholar]
- 69.Lackner G, Misiek M, Braesel J, Hoffmeister D. Genome mining reveals the evolutionary origin and biosynthetic potential of basidiomycete polyketide synthases. Fungal Genet Biol. 2012;49(12):996–1003. doi: 10.1016/j.fgb.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 70.Wawrzyn GT, Quin MB, Choudhary S, López-Gallego F, Schmidt-Dannert C. Draft genome of Omphalotus olearius provides a predictive framework for sesquiterpenoid natural product biosynthesis in Basidiomycota. Chem Biol. 2012;19(6):772–783. doi: 10.1016/j.chembiol.2012.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Revell LJ. Size-correction and principal components for interspecific comparative studies. Evolution. 2009;63(12):3258–3268. doi: 10.1111/j.1558-5646.2009.00804.x. [DOI] [PubMed] [Google Scholar]
- 72.Gold MH, Alic M. Molecular biology of the lignin-degrading basidiomycete Phanerochaete chrysosporium. Microbiol Rev. 1993;57(3):605–622. doi: 10.1128/mr.57.3.605-622.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ander P, Eriksson KE. Importance of phenol oxidase activity in lignin degradation by white-rot fungus Sporotrichum pulverulentum. Arch Microbiol. 1976;109(1-2):1–8. [Google Scholar]
- 74.Hiroi T, Eriksson KE. Microbiological degradation of lignin: Influence of cellulose on degradation of lignins by white-rot fungus Pleurotus ostreatus. Sven Papperstidn. 1976;79(5):157–161. [Google Scholar]
- 75.Zerbino DR, Birney E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18(5):821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jaffe DB, et al. Whole-genome sequence assembly for mammalian genomes: Arachne 2. Genome Res. 2003;13(1):91–96. doi: 10.1101/gr.828403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Grigoriev IV, Martinez DA, Salamov AA. Fungal genomic annotation. Appl Mycol Biotechnol. 2006;6:123–142. [Google Scholar]
- 78.Grigoriev IV, et al. MycoCosm portal: Gearing up for 1000 fungal genomes. Nucleic Acids Res. 2014;42(Database issue):D699–704. doi: 10.1093/nar/gkt1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30(14):3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17(4):540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 81.Swofford DL. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4. Sunderland, MA: Sinauer Associates; 2003. [Google Scholar]
- 82.Larkin MA, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 83.Brown DW, Adams TH, Keller NP. Aspergillus has distinct fatty acid synthases for primary and secondary metabolism. Proc Natl Acad Sci USA. 1996;93(25):14873–14877. doi: 10.1073/pnas.93.25.14873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li Y, Chooi YH, Sheng Y, Valentine JS, Tang Y. Comparative characterization of fungal anthracenone and naphthacenedione biosynthetic pathways reveals an α-hydroxylation-dependent Claisen-like cyclization catalyzed by a dimanganese thioesterase. J Am Chem Soc. 2011;133(39):15773–15785. doi: 10.1021/ja206906d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hopwood DA, Sherman DH. Molecular genetics of polyketides and its comparison to fatty acid biosynthesis. Annu Rev Genet. 1990;24:37–66. doi: 10.1146/annurev.ge.24.120190.000345. [DOI] [PubMed] [Google Scholar]
- 86.Felsenstein J. 2005. PHYLIP (Phylogeny Inference Package) Distributed by the author. (Department of Genome Sciences, University of Washington, Seattle), Version 3.6.
- 87.Blanchette RA, et al. An Antarctic hot spot for fungi at Shackleton’s historic hut on Cape Royds. Microb Ecol. 2010;60(1):29–38. doi: 10.1007/s00248-010-9664-z. [DOI] [PubMed] [Google Scholar]
- 88.Brown A, Sims PFG, Raeder U, Broda P. Multiple ligninase-related genes from Phanerochaete chrysosporium. Gene. 1988;73(1):77–85. doi: 10.1016/0378-1119(88)90314-9. [DOI] [PubMed] [Google Scholar]
- 89.Kirk TK, Schultz E, Conners WJ, Lorentz LF, Zeikus JG. Influence of culture parameters on lignin metabolism by Phanerochaete chrysosporium. Arch Microbiol. 1978;117:277–285. [Google Scholar]
- 90.Hori C, et al. Genomewide analysis of polysaccharides degrading enzymes in 11 white- and brown-rot Polyporales provides insight into mechanisms of wood decay. Mycologia. 2013;105(6):1412–1427. doi: 10.3852/13-072. [DOI] [PubMed] [Google Scholar]
- 91.Kersten PJ, Kirk TK. Involvement of a new enzyme, glyoxal oxidase, in extracellular H2O2 production by Phanerochaete chrysosporium. J Bacteriol. 1987;169(5):2195–2201. doi: 10.1128/jb.169.5.2195-2201.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.