

Abstract
The chemotactic migration of eukaryotic ameboid cells up concentration gradients is among the most advanced forms of cellular behavior. Chemotaxis is controlled by a complex network of signaling proteins bound to specific lipids on the cytoplasmic surface of the plasma membrane at the front of the cell, or the leading edge. The central lipid players in this leading edge signaling pathway include the phosphoinositides PI(4,5)P2 (PIP2) and PI(3,4,5)P3 (PIP3), both of which play multiple roles. The products of PI(4,5)P2 hydrolysis, diacylglycerol (DAG) and Ins(1,4,5)P3 (IP3), are also implicated as important players. Together, these leading edge phosphoinositides and their degradation products, in concert with a local Ca2+ signal, control the recruitment and activities of many peripheral membrane proteins that are crucial to the leading edge signaling network. The present critical review summarizes the current molecular understanding of chemotactic signaling at the leading edge, including newly discovered roles of phosphoinositide lipids and Ca2+, while highlighting key questions for future research.
Keywords: Phosphatidylinositol lipid, PH domain, C2 domain, PKC, PI3K, PDK1
1. Introduction
Plasma membrane signaling events play a central role in the directed migration of eukaryotic ameboid cells up concentration gradients of specific attractant molecules. Leukocytes including macrophages and neutrophils possess one of the most highly developed chemotaxis pathways, enabling efficient cell migration toward infection or tissue damage (Jin, 2013; Kolaczkowska and Kubes, 2013; Parent and Weiner, 2013; Weiger and Parent, 2012; Cai and Devreotes, 2011; Hawkins et al., 2010; Collins and Meyer, 2009). Many autoimmune diseases yield hyperactivation of leukocyte chemotaxis, while certain infectious diseases interfere with leukocyte chemotaxis (Kolaczkowska and Kubes, 2013; Fahey et al., 2013). Thus, a molecular understanding of the chemotaxis signaling pathway could facilitate the development of useful therapies targeting inflammation and infection. More broadly, many eukaryotic cells possess the ability to migrate or grow up gradients of attractant molecules using the ubiquitous elements of the widely conserved chemotaxis pathway. In addition, specific cell types possess specialized pathway elements that optimize performance for particular applications. Chemotactic cells in which the chemotaxis pathway has been investigated at the molecular level include leukocytes (Jin, 2013; Kolaczkowska and Kubes, 2013; Weiger and Parent, 2012; Cai and Devreotes, 2011; Hawkins et al., 2010; Collins and Meyer, 2009), fibroblasts (Wei et al., 2012), endothelial cells (Tsai and Meyer, 2012), neurons (Zamburlin et al., 2013; Henle et al., 2011; Waite and Eickholt, 2010), muscle cells (Trovati et al., 2013), mammary epithelial cells (Zhu and Nelson, 2013), cancer cells (Li et al., 2013; Vadas et al., 2011), and free-living amoeboid organisms such as Dictyostelium (Weiger and Parent, 2012; Cai and Devreotes, 2011).
2. Leading edge lipid and Ca2+ signals
The first major step in chemotaxis is the polarization of the cell, which generates an active, ruffling region of plasma membrane, termed the leading edge, at the front end of the cell that largely controls the direction of cell movement (Fig. 1) (Jin, 2013; Weiger and Parent, 2012; Cai and Devreotes, 2011; Hawkins et al., 2010; Swaney et al., 2010; Kolsch et al., 2008; Mortimer et al., 2008; von Philipsborn and Bastmeyer, 2007; Bourne and Weiner, 2002). In general, leading edge formation and maintenance requires the appearance of two localized second messenger signals: the signaling lipid phosphatidylinositol 3,4,5-trisphosphate (PI(3,4,5)P3) (Jin, 2013; Weiger and Parent, 2012; Cai and Devreotes, 2011), and the signaling ion Ca2+ (Collins and Meyer, 2009; Wei et al., 2012; Tsai and Meyer, 2012; Zamburlin et al., 2013; Henle et al., 2011; Tian et al., 2010; Wei et al., 2009; Evans and Falke, 2007). While the importance of PI(3,4,5)P3 at the leading edge membrane has long been recognized, Ca2+ has emerged as an essential leading edge signal more recently. Contemporary leading edge models ignored Ca2+ until local Ca2+ signals at the leading edge were found to be essential for macrophage leading edge stability, dynamics and function (Evans and Falke, 2007), to play a role in directional control of migrating fibroblasts and endothelial cells (Wei et al., 2012; Tsai and Meyer, 2012; Wei et al., 2009), and to be involved in the guidance mechanism of neuronal growth (Zamburlin et al., 2013; Henle et al., 2011; Heckman and Plummer, 2013). Thus in recent years Ca2+ has joined PI(3,4,5)P3 as an essential second messenger in leading edge signaling (Collins and Meyer, 2009; Wei et al., 2012; Tsai and Meyer, 2012; Zamburlin et al., 2013; Henle et al., 2011; Tian et al., 2010; Wei et al., 2009; Evans and Falke, 2007; Heckman and Plummer, 2013; Schafer et al., 2012), with the possible exception of cells that lack constant external Ca2+ levels such as Dictyostelium (Wessels et al., 2012; Traynor et al., 2000). Besides PI(3,4,5)P3 and Ca2+, another second messenger – the signaling lipid diacylglycerol (DAG) and likely its phosphorylation product phosphatidic acid – is indirectly implicated at the leading edge (Tsai and Meyer, 2012; Abramovici et al., 2009). Finally, the constitutive plasma membrane lipid phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) plays crucial roles in the leading edge PI(3,4,5)P3, Ca2+, and putative DAG signals (Jin, 2013; Evans and Falke, 2007; Thapa and Anderson, 2012; Corbalan-Garcia et al., 2007; Corbin et al., 2007; Evans et al., 2006; Luo et al., 2004).
Fig. 1.

Second messenger signals at the leading edge of polarized ameboid cells (Evans and Falke, 2007). Shown are polarized RAW macrophages illustrating (A) the ruffling leading edge in a phase contrast image, (B) the local Ca2+ signal revealed by YFP-PKCα recruited to the leading edge membrane in a wide field fluorescence image, and (C) the local PI(3,4,5)P3 signal revealed by GFP-PKB/AKT-PH domain recruited to the leading edge membrane in a wide field fluorescence image. (D) Schematic overview of parallel Ca2+ and PI(3,4,5)P3 (PIP3) signals that activate PH and C2 membrane targeting domains, triggering recruitment of signaling proteins to the leading edge membrane (Jin, 2013; Weiger and Parent, 2012; Cai and Devreotes, 2011; Kolsch et al., 2008; von Philipsborn and Bastmeyer, 2007; Evans and Falke, 2007; Corbalan-Garcia et al., 2007; Leonard and Hurley, 2011; Newton, 2009; Lemmon, 2008; Cho, 2001; Nalefski and Falke, 1996). The resulting bilayer-associated signaling pathway includes a positive feedback loop that controls actin and membrane remodeling essential for leading edge structure, stability, dynamics, and directed cell migration (Jin, 2013; Weiger and Parent, 2012; Cai and Devreotes, 2011; Hawkins et al., 2010; Swaney et al., 2010; Kolsch et al., 2008; Mortimer et al., 2008; von Philipsborn and Bastmeyer, 2007; Bourne and Weiner, 2002; Evans and Falke, 2007; Charest and Firtel, 2006).
PI(3,4,5)P3 signals are triggered by attractant binding to G-protein coupled receptors (GPCRs) or receptor tyrosine kinases (RTKs), which activate membrane-targeted enzyme phosphoinositide 3-kinase (PI3K) (Jin, 2013; Vadas et al., 2011). This lipid kinase phosphorylates the constitutive lipid PI(4,5)P2, yielding the signaling lipid PI(3,4,5)P3. Since the substrate PI(4,5)P2 is only ~2 mole % of the total lipid in the cytoplasmic leaflet of the plasma membrane, the PI(3,4,5)P3 product density never exceeds this average level and is often a relatively rare bilayer component even during a peak signal (Corbin et al., 2004). The localized nature of the leading edge PI(3,4,5)P3 signal arises both from the localized recruitment and/or activation of PI3K in this region, and from the suppressed membrane targeting or activity of the lipid phosphatase PTEN (phosphatase and tensin homologue) that hydrolyzes PI(3,4,5)P3 back to PI(4,5)P2 (1,4,5). Outside the leading edge region, active PTEN is bound to the plasma membrane and destroys PI(3,4,5)P3 molecules emerging from the leading edge via diffusion (Cai and Devreotes, 2011).
The leading edge Ca2+ signal has been linked to both influx through plasma membrane Ca2+ channels and efflux from internal Ca2+ stores, although certain cell types may employ only one of these mechanisms (Wei et al., 2012; Tsai and Meyer, 2012; Zamburlin et al., 2013; Henle et al., 2011; Tian et al., 2010; Wei et al., 2009; Evans and Falke, 2007; Heckman and Plummer, 2013; Schafer et al., 2012). Plasma membrane Ca2+ channels including transient receptor potential (TRP) and Orai1 channels, and endoplasmic reticulum Ca2+ channels including ryanodine and IP3 receptors, are proposed to give rise to the leading edge Ca2+ influxes and effluxes, respectively (Wei et al., 2012; Tsai and Meyer, 2012; Zamburlin et al., 2013; Henle et al., 2011; Tian et al., 2010; Wei et al., 2009; Heckman and Plummer, 2013; Schafer et al., 2012). In neurite growth, plasma membrane L- and N-type voltage dependent Ca2+ channels may also play a role (Zamburlin et al., 2013). In general, the signaling events that trigger the local, leading edge Ca2+ signals in chemotaxing cells are not yet well understood, but at least in certain cell types PI(3,4,5)P3 may trigger Ca2+ influx by directly or indirectly opening leading edge TRP channels (Henle et al., 2011). Notably, the leading edge Ca2+ signal is quite low amplitude (Wei et al., 2012; Tsai and Meyer, 2012; Wei et al., 2009; Evans and Falke, 2007) and easily collapsed by presence of high affinity Ca2+ chelators (Evans and Falke, 2007). Besides this modest leading edge Ca2+ signal, migrating cells typically possess a larger Ca2+ signal at the trailing edge of the cell where it is required for retraction from the substrate (Wei et al., 2012, 2009; Yang and Huang, 2005). Thus, although both ends of the cell possess Ca2+ signals, the characteristics and functions of the two polar signals are different.
Further studies are needed to define the molecular steps that couple receptor activation to PI3K activation at the leading edge membrane, and the pathway mechanisms that control the steady state levels of the substrate PI(4,5)P2 and product PI(3,4,5)P3 lipids during a signal. Additional key questions include the relative importance of the different Ca2+ channels and Ca2+ pools implicated in chemotaxis, how the key Ca2+ channels are regulated by the chemotaxis pathway, and whether multiple types of Ca2+ signals exist and play different roles at the leading edge – for example, influx vs efflux, or lamellapodia vs filapodia.
3. Functions of the leading edge lipid and Ca2+ signals
3.1. Roles of PI(3,4,5)P3 and Ca2+
The primary shared function of the leading edge PI(3,4,5)P3 and Ca2+ signals is to drive the targeting of dozens of signaling proteins from cytoplasm to the leading edge membrane, yielding one of the most dramatic protein recruitments in cell biology (Fig. 1) (Jin, 2013; Weiger and Parent, 2012; Cai and Devreotes, 2011; Kolsch et al., 2008; von Philipsborn and Bastmeyer, 2007; Evans and Falke, 2007). The recruited signaling proteins each recognize and bind specific lipids on the cytoplasmic leaflet of the leading edge plasma membrane.
The diverse signaling proteins recruited from the cytoplasm to the leading edge typically possess a regulated membrane targeting domain. In most cases this targeting domain is either a pleckstrin homology (PH) domain that recognizes and binds the PI(3,4,5)P3 signaling lipid, or a C2 domain that is activated by Ca2+ binding and then docks to lipids on the target membrane surface (Corbalan-Garcia et al., 2007; Leonard and Hurley, 2011; Newton, 2009; Lemmon, 2008; Cho, 2001; Nalefski and Falke, 1996). C2- and PH-driven targeting is a common feature of signaling pathways, and these two ubiquitous targeting motifs are found in over 620 and 330 human proteins, respectively, (Punta et al., 2012).
Typically, the C2 or PH domain-directed membrane targeting reaction serves to activate the signaling protein via enhanced proximity to key reactants in its signaling reaction (Fig. 2) (Corbalan-Garcia et al., 2007; Leonard and Hurley, 2011; Newton, 2009; Lemmon, 2008; Cho, 2001; Nalefski and Falke, 1996). Specifically, the reaction brings the signaling protein to the membrane surface where its effector and substrate molecules are located, yielding faster on-rates for the binding events essential for signaling domain activity. In some cases, direct interactions of the signaling domain with the bilayer may alter the conformation of the signaling protein or the interactions between its domains, providing additional activation.
Fig. 2.
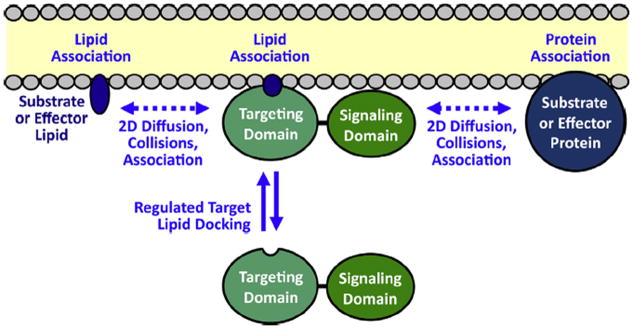
Function of PH and C2 membrane targeting domains in the recruitment and activation of signaling proteins on target membrane surfaces (Corbalan-Garcia et al., 2007; Leonard and Hurley, 2011; Newton, 2009; Lemmon, 2008; Cho, 2001; Nalefski and Falke, 1996). First, a 2° messenger signal triggers the recruitment of the signaling protein targeting domain from the cytoplasm to the bilayer surface: many PH domains are recruited to the plasma membrane by a PI(3,4,5)P3 (PIP3) lipid signal, while the C2 domains of classical protein kinase C isoforms (cPKCs) are activated by Ca2+ binding and then dock to PS and PI(4,5)P2 (PIP2) on the plasma membrane. Second, the signaling domain of each recruited protein is activated by its proximity on the membrane surface to membrane-associated protein and/or lipid molecules that serve as effectors or substrates. At the leading edge membrane, PIP3 and Ca2+ both recruit signaling proteins and establish a signaling network on the bilayer surface. Lateral, 2-dimensional diffusion on the bilayer is required for the signaling proteins to encounter and bind their signaling partners, and for the resulting membrane-bound products to diffuse away. Thus, the lipid bilayer serves as a 2-dimensional platform for the assembly of the dynamic signaling circuit, and for the diffusion of its many components during signaling reactions.
Further studies are needed to investigate isolated reports that certain cell types migrating in specific conditions can utilize an alternative pathway, not involving PI(3,4,5)P3 and/or Ca2+, to control chemotaxis, although the normal pathway involving PI(3,4,5)P3 and Ca2+ appears to be dominant in most cell types and settings (Jin, 2013; Weiger and Parent, 2012). Additional work is also needed to ascertain whether PI(3,4,5)P3 and Ca2+ have roles beyond membrane recruitment at the leading edge, as exemplified by the plausible regulation of Ca2+ influx by PI(3,4,5)P3-gated TRP channels (Henle et al., 2011), and the likely role of Ca2+ in regulating leading edge myosin activity (Tsai and Meyer, 2012).
3.2. Roles of PI(4,5)P2 and DAG
While not a signaling molecule itself, the constitutive plasma membrane lipid PI(4,5)P2 is essential as the substrate for production of PI(3,4,5)P3 by PI3K (Jin, 2013; Vadas et al., 2011). PI(4,5)P2 also plays a major role as a target lipid for the plasma membrane recruitment of various signaling proteins including the classical isoforms of protein kinase C (cPKCs) (Evans and Falke, 2007; Thapa and Anderson, 2012; Corbalan-Garcia et al., 2007; Corbin et al., 2007; Evans et al., 2006), and serves as an attachment point for cytoskeletal proteins involved in leading edge dynamics (Thapa and Anderson, 2012; Golebiewska et al., 2008). For example, the cPKC isoform PKCα is known to be recruited by the local Ca2+ signal to the leading edge, where its C2 domain must bind both PS and PI(4,5)P2 to ensure the high affinity, specific plasma membrane specificity while awaiting the appearance of a DAG signal (Evans and Falke, 2007; Corbalan-Garcia et al., 2007; Corbin et al., 2007; Evans et al., 2006).
In certain cases, mutations that enhance PI(4,5)P2 targeting can cause human disease. In targeting reactions controlled by PI(3,4,5)P3-specific PH domains, the PH domain must ignore the constitutive PI(4,5)P2 lipid while recognizing and binding to the rare PI(3,4,5)P3 signaling lipid. Certain PH domain mutations are known to convert the highly specific PI(3,4,5)P3 binding site into a dual specificity PI(4,5)P2/PI(3,4,5)P3 binding site, causing constant PH domain targeting to plasma membrane PI(4,5)P2 (Pilling et al., 2011; Landgraf et al., 2008; Carpten et al., 2007). The resulting constitutive membrane targeting yields superactivation of the signaling protein and often triggers disease. For example, the E17K mutation at the PI(3,4,5)P3 binding site of PKB/AKT PH domain generates dual PI(4,5)P2/PI(3,4,5)P3 specificity, yielding constitutive plasma membrane targeting and superactivation of the PKB/AKT kinase domain, thereby greatly increasing the incidence of multiple human cancers and Proteus syndrome (Landgraf et al., 2008; Carpten et al., 2007; Lindhurst et al., 2011).
If phospholipase C (PLC) activity is present at the leading edge, the resulting PI(4,5)P2 hydrolysis would produce both the soluble second messenger Ins(1,4,5)P3 and the signaling lipid DAG (Tsai and Meyer, 2012; Abramovici et al., 2009; Thapa and Anderson, 2012). In turn, Ins(1,4,5)P3 could open the IP3-sensitive Ca2+ channels of internal stores, contributing to the local Ca2+ signal.
Furthermore, the DAG resulting from PLC hydrolysis of PI(4,5)P2 at the leading edge would likely activate cPKCs. At least one isoform, PKCα, is known to be recruited to the macrophage leading edge by its C2 domain that associates with Ca2+, PS and PI(4,5)P2 (Evans and Falke, 2007; Corbin et al., 2007; Evans et al., 2006). The binding of DAG to the C1 domain is essential for full activation of the cPKC kinase domain (Newton, 2009), and for its proposed role in generating free PI(4,5)P2 by phosphorylating MARCKS (see 4.4 below). In short, while direct detection of Ins(1,4,5)P3 and DAG signals has not yet been achieved at the leading edge, such signals are likely to be present and essential.
Further studies are needed to test the prediction that PLC activity and localized accumulations of Ins(1,4,5)P3 and DAG are essential players in leading edge signaling, and to determine which PLC isoforms are involved in leading edge PI(4,5)P2 hydrolysis and their mechanisms of regulation.
4. The assembled leading edge signaling circuit
4.1. Role of surface binding and 2-D diffusion
Together the dozens of signaling proteins recruited to the leading edge by PI(3,4,5)P3 and Ca2+ signals form a complex, dynamic, signaling network on the membrane surface. The lipid bilayer plays a central role in this network by serving as a shared platform for two-dimensional diffusion, thereby enabling the membrane-bound signaling proteins to encounter and bind their membrane-associated, lipid and/or protein effectors and substrates (Ziemba et al., 2013; Ziemba and Falke, 2013; Ziemba et al., 2012; Knight et al., 2010; Knight and Falke, 2009). The resulting, membrane-localized signaling circuit controls actin polymerization, membrane remodeling, and other dynamic processes essential for growth of the leading edge up attractant gradients. The many important components of this network are too numerous to consider herein, but the reader is referred to extensive reviews (Jin, 2013; Weiger and Parent, 2012; Cai and Devreotes, 2011; Kolsch et al., 2008; von Philipsborn and Bastmeyer, 2007; Evans and Falke, 2007).
4.2. An essential leading edge positive feedback loop
A key feature of the leading edge signaling circuit is a positive feedback loop formed by a subset of the signaling proteins recruited to the leading edge (Jin, 2013; Weiger and Parent, 2012; Cai and Devreotes, 2011; Hawkins et al., 2010; Swaney et al., 2010; Kolsch et al., 2008; Mortimer et al., 2008; von Philipsborn and Bastmeyer, 2007; Bourne and Weiner, 2002; Evans and Falke, 2007; Charest and Firtel, 2006). After this positive feedback loop is established it amplifies both activating and inhibitory signals, such that the activation of an individual loop component yields upregulation of all other loop components. At the other extreme, inhibition of any loop component yields downregulation of all other loop components. The positive feedback loop has been shown to be essential for maintenance of a stable, ruffling leading edge possessing a very high density of signaling proteins, actin mesh and other cytoskeletal elements. The resulting active leading edge region often becomes a semi-permanent structural and functional region of the cell, approaching an organelle in its complexity and stability, but of course lacking a complete membrane boundary.
It is hypothesized that the positive feedback loop plays a central role in the compass that determines the direction of leading edge movement up the attractant gradient (Cai and Devreotes, 2011; Evans and Falke, 2007; Charest and Firtel, 2006). In this model, when a filopodium grows up the attractant gradient the positive feedback loop amplifies the growing attractant signal and speeds filopodial growth in this favorable direction. By contrast, when a filopodium grows perpendicular to the gradient or down the gradient, the positive feedback loop detects either no attractant change or an attractant decrease, and responds by ignoring or perhaps even slowing filopodial growth in this counterproductive direction.
4.3. Elements of the leading edge positive feedback loop
It has become apparent that the positive feedback loop has many essential components. Strong evidence exists that PI3K, PI(3,4,5)P3, filamentous actin, Ca2+, PKCα, Ras GTPase and Rho GTPase are central components (Jin, 2013; Weiger and Parent, 2012; Cai and Devreotes, 2011; Hawkins et al., 2010; Collins and Meyer, 2009; Wei et al., 2012; Tsai and Meyer, 2012; Zamburlin et al., 2013; Henle et al., 2011; Swaney et al., 2010; Kolsch et al., 2008; Mortimer et al., 2008; von Philipsborn and Bastmeyer, 2007; Tian et al., 2010; Wei et al., 2009; Evans and Falke, 2007). The Ca2+ signal, missing in earlier models, is now a central feature. Fig. 3 presents both confirmed components and unconfirmed, but plausible, elements in a preliminary working model for the leading edge pathway and its positive feedback circuit.
Fig. 3.

Schematic, partial model of the intricate leading edge signaling circuit, illustrating known and hypothesized components and their interactions (Jin, 2013; Weiger and Parent, 2012; Cai and Devreotes, 2011; Hawkins et al., 2010; Collins and Meyer, 2009; Wei et al., 2012; Tsai and Meyer, 2012; Zamburlin et al., 2013; Henle et al., 2011; Swaney et al., 2010; Kolsch et al., 2008; Mortimer et al., 2008; von Philipsborn and Bastmeyer, 2007; Tian et al., 2010; Wei et al., 2009; Evans and Falke, 2007). Attractant signals switch on receptors and the Ras protein, which recruit and activate the lipid kinase activity of PI3K that phosphorylates the constitutive lipid PI(4,5)P2 (PIP2) to generate the signaling lipid PI(3,4,5)P3 (PIP3). Attractant signals may also activate plasma membrane Ca2+ channels that generate an essential Ca2+ influx at the leading edge. The PI(3,4,5)P3 and Ca2+ second messengers are essential players in the leading edge positive feedback loop (dashed line) along with one or more small GTPases of the Rho family, filamentous actin and PKCα. The PI(3,4,5)P3 signal recruits PDK1 to the leading edge where it is activated and stimulates branches leading to membrane and actin remodeling needed for leading edge expansion up the attractant gradient. The Ca2+ signal activates and recruits PKCα to the leading edge membrane where it is proposed to phosphorylate the PI(4,5)P2-sequestration region of MARCKS protein, which in turn is predicted to increase the free PI(4,5)P2 density and thereby activate PI3K and self-activate PKCα. Ca2+ may also activate PLC to generate a putative DAG lipid signal. Finally, PDK1 phosphorylation of PKCα in solution is essential for maturation and activation of the latter. On the membrane surface PDK1 and PKCα are hypothesized to combine and form a stable, inactive complex that could act as a brake to prevent runaway positive feedback. Proteins in boxes contain a PH domain (black box) or a C2 domain (white box). Second messengers are also highlighted (blue box).
Further studies are needed to test and fill in the gaps of this working circuit diagram (Fig. 3), which remains speculative and is undoubtedly missing important components and regulatory interactions. For example, much has been learned about the proteins that regulate actin remodeling (Heckman and Plummer, 2013; Insall and Machesky, 2009), but less is known about the machinery that controls membrane remodeling. The connections of the feedback loop to these remodeling systems must be defined and incorporated into the model.
4.4. Working model for the regulation of PKCα, PI3K and PDK1 in the leading edge positive feedback loop
Beyond the circuit diagram of the positive feedback loop, much remains to be learned about the structural and mechanistic underpinnings of information flow and regulation within the loop. Fig. 4 illustrates a working model proposing plausible, but largely untested, regulatory interactions between the three master kinases PKCα, PI3K and PDK1. These interactions are hypothesized to provide both positive feedback and an internal brake that prevents runaway loop activation.
Fig. 4.
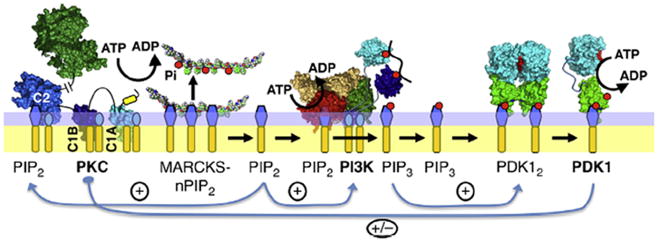
Working model for the molecular interactions between the activities of PKCα, PI3K and PDK1 in the leading edge positive feedback loop. Shown are the three master kinases and their effector lipids on the cytoplasmic leaflet of the leading edge membrane. Kinase-active PKCα is bound via its Ca2+-occupied C2 domain to PS and PI(4,5)P2 (PIP2), and via its C1A and C1B domains to PS and DAG (Evans and Falke, 2007; Corbalan-Garcia et al., 2007; Corbin et al., 2007; Evans et al., 2006; Newton, 2009). This active PKCα is proposed to phosphorylate the PI(4,5)P2-sequestration region of MARCKS protein, thereby releasing PI(4,5)P2 from MARCKS and increasing the local free PI(4,5)P2 density at the leading edge (Golebiewska et al., 2008; McLaughlin et al., 2002; Wang et al., 2004; Gambhir et al., 2004). The newly released PIP2 helps counteract the loss of PI(4,5)P2 due to the enzymatic activities of PI3K and PLC (Jin, 2013; Wei et al., 2012; Tsai and Meyer, 2012; Vadas et al., 2011; Wei et al., 2009; Evans and Falke, 2007), and this PI(4,5)P2 release activates both PI3K that requires PI(4,5)P2 as a substrate, and PKCα that requires PI(4,5)P2 for stable plasma membrane association. PI3K converts PI(4,5)P2 to the signaling lipid PI(3,4,5)P3 (PIP3), which recruits many signaling proteins including PDK1 to the leading edge membrane. PDK1 is an inactive dimer that is converted on the membrane surface to an active monomer (Ziemba et al., 2013; Masters et al., 2010). The resulting PDK1 monomer may form an inhibitory complex with PKCα that has been isolated from cytoplasm but not yet investigated on a membrane surface (Le Good et al., 1998). Further studies are needed to test the proposed interactions between the three master kinases, and the roles of these interactions in leading edge positive feedback and signal amplification.
This small section of the feedback loop is proposed to begin with Ca2+- and DAG-activated, membrane-bound PKCα docked to target lipids PS, PI(4,5)P2 and DAG (Evans and Falke, 2007; Corbalan-Garcia et al., 2007; Corbin et al., 2007; Evans et al., 2006; Newton, 2009). PKCα is known to phosphorylate the membrane binding peptide of the MARCKS protein (McLaughlin et al., 2002). Unphosphorylated MARCKS tightly binds multiple (~3) PI(4,5)P2 molecules and effectively sequesters them. The model supposes that unmodified MARCKS maintains the density of free, leading edge PI(4,5)P2 at a level sufficient to recruit PKCα via its C2 domain, but low enough to limit the activity of the lipid kinase PI3K and the lipid phosphatase PTEN which require PI(4,5)P2 as a substrate and a target lipid, respectively. When PKCα modifies MARCKS, each phosphorylation event is believed to release a free PI(4,5)P2 molecule in a stepwise fashion (Golebiewska et al., 2008; McLaughlin et al., 2002; Wang et al., 2004; Gambhir et al., 2004). The resulting rise in free PI(4,5)P2 levels increases the substrate availability and lipid kinase activity of PI3K, which phosphorylates PI(4,5)P2 yielding PI(3,4,5)P3. This PI3K activity decreases the leading edge PI(4,5)P2, which can also be degraded by phospholipase C activity to generate leading edge DAG and Ins(1,4,5)P3 (Jin, 2013; Wei et al., 2012; Tsai and Meyer, 2012; Vadas et al., 2011; Wei et al., 2009; Evans and Falke, 2007). MARCKS is proposed to buffer the PI(4,5)P2, thereby inhibiting these degradation reactions while being fully reversible by PKCα activity.
Following activation of the PI3K, the product PI(3,4,5)P3 diffuses away from the PI3K and recruits PH domains to the plasma membrane. One such PH domain is that of PDK1, which is recruited by the PI(3,4,5)P3 signal to the leading edge membrane. Subsequently, the PI(3,4,5)P3-occupied, membrane-bound PH domain plays a central role in the activation of the PDK1 catalytic domain and the stimulation of its protein kinase activity (Ziemba et al., 2013; Masters et al., 2010).
The indicated flow of information can contribute to positive feedback since the first and last steps are cascade-type activation events: (i) a single activated PKCα protein kinase molecule generates many free PI(4,5)P2 molecules (amplifying cascade), then (ii) each PI(4,5)P2 molecule can be converted by PI3K into a PI(3,4,5)P3 molecule (no amplification), then (iii) each PI(3,4,5)P3 molecule can recruit a PDK1 molecule to the membrane (no amplification), and finally (iv) each membrane-activated PDK1 kinase molecule phosphorylates many substrate proteins (amplifying cascade).
It is hypothesized that membrane-bound, active PKCα and PDK1 form a stable, membrane-associated complex that may inhibit both enzymes, thereby yielding an activity brake ensuring that the positive feedback signal does not grow in an uncontrolled way, ultimately yielding a runaway pathway. The activation loops of cPKCs are phosphorylated by PDK1 early in their cellular processing, thus PKCα bound to leading edge is already phosphorylated by PDK1 (Newton, 2010). In the cytoplasm, both unphosphorylated and phosphorylated PKCα form inactive complexes with PDK1 that are sufficiently stable to be isolated by immunoprecipitation (Le Good et al., 1998). The hypothesized membrane-bound, PKCα-PDK1 complex has not yet been detected but is plausible given the stability of the soluble complex.
Further studies are needed to test and further refine the proposed protein regulatory interactions in the PKCα – PI3K – PDK1 branch of the leading edge positive feedback loop. The current working model (Fig. 4) is presented as a starting point to facilitate such testing.
5. Direct PIP lipid-Ca2+ interactions and PIP lipid clustering
Multiple studies, reviewed in this issue by Janmey et al. (Wang et al., 2013), have suggested that sufficient mole densities of PI(4,5)P2 or PI(3,4,5)P3 lipid in a bilayer, together with sufficient concentrations of Ca2+, can lead to a direct interaction between multiple PIP lipids and Ca2+. Under certain conditions, this direct interaction can lead to Ca2+-induced clustering of PIP lipids in bilayers. To facilitate detection of clusters in vitro, the PIP lipid mole densities and free Ca2+ concentrations typically employed in such studies are significantly higher than physiological levels. Much remains to be learned about the prevalence and significance of Ca2+-induced PIP clusters in cells.
In principle, specific protein–lipid interactions could stabilize Ca2+-induced, PIP lipid clusters, enabling their formation at more physiological lipid and Ca2+ levels. For example, the PI(4,5)P2 binding site of PKCα can bind a single PI(4,5)P2 molecule, but when saturated is occupied by two PI(4,5)P2 molecules (Lai et al., 2010; Corbalan-Garcia et al., 2003). In the latter case, MD simulations indicate the two highly anionic PI(4,5)P2 molecules are bound side-by-side in a pairwise cluster stabilized by Ca2+ and multiple positive charges provided by the protein (Lai et al., 2010). Thus, during a cellular Ca2+ signal a portion of the membrane-bound PKCα population will be associated with Ca2+-stabilized PI(4,5)P2 pairs. If physiological conditions produce membrane regions containing both PI(4,5)P2 clusters and PS, such regions could exhibit increased affinity for cPKC C2 domains that require both PI(4,5)P2 and PS for optimal target membrane affinity and specificity (Evans and Falke, 2007; Corbalan-Garcia et al., 2007; Corbin et al., 2007; Evans et al., 2006). Similarly, PH domains require both PI(3,4,5)P3 and PS for high affinity, rapid target membrane acquisition (Corbin et al., 2004; Knight and Falke, 2009; Lai et al., 2013), thus their targeting reactions could be facilitated by regions containing both PI(3,4,5)P3 clusters and PS.
Further studies are needed to ascertain whether Ca2+-induced, PIP lipid clusters occur in cells, to determine their sizes and lipid compositions, and to elucidate their effects on the crucial C2- and PH-domain driven membrane targeting reactions that control cell migration and many other cellular processes.
Acknowledgments
The authors acknowledge the wealth of important papers in the field, and apologize for the unavoidable omission of many relevant papers.
Abbreviations
- PH domain
pleckstrin homology domain
- C1A and C1B
first conserved domains of classical protein kinase C
- C2
second conserved domain of classical protein kinase C
- PKC
protein kinase C
- cPKC
one of the classical isoforms of protein kinase C
- PI3K
phosphoinositide 3-kinase
- f-Actin
filamentous actin
- PDK1
phosphoinositide-dependent kinase isoform 1
- PLC
phospholipase C
- Ras
a small GTPase or G-protein of the Ras family
- Rho
a small GTPase or G-protein of the Rho family
- GRP1
general receptor for phosphoinositides isoform 1
- PKB/AKT
protein kinase B, also known as AKT
- ARF6
ADP ribosylation factor 6, a small G-protein
- MARCKS
myristoylated alanine-rich C-kinase substrate
- PTEN
phosphatase and tensin homologue
- GPCR
G-protein coupled receptor
- RTK
receptor tyrosine kinase
- PS
phosphatidylserine
- PI(4,5)P2 or PIP2
phosphatidylinositol 4,5-bisphosphate
- PI(3,4,5)P3 or PIP3
phosphatidylinositol 3,4,5-trisphosphate
- Ins(1,4,5)P3 or IP3
inositol 1,4,5-trisphosphate
- DAG
diacylglycerol
Footnotes
Support provided by NIH R01 GM-063235 (to JJF).
References
- Abramovici H, Mojtabaie P, Parks RJ, Zhong XP, Koretzky GA, Topham MK, Gee SH. Diacylglycerol kinase zeta regulates actin cytoskeleton reorganization through dissociation of Rac1 from RhoGDI. Mol Biol Cell. 2009;20:2049–2059. doi: 10.1091/mbc.E07-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne HR, Weiner O. A chemical compass. Nature. 2002;419:21. doi: 10.1038/419021a. [DOI] [PubMed] [Google Scholar]
- Cai H, Devreotes PN. Moving in the right direction: how eukaryotic cells migrate along chemical gradients. Semin Cell Dev Biol. 2011;22:834–841. doi: 10.1016/j.semcdb.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, Hostetter G, Boguslawski S, Moses TY, Savage S, Uhlik M, Lin A, Du J, Qian YW, Zeckner DJ, Tucker-Kellogg G, Touchman J, Patel K, Mousses S, Bittner M, Schevitz R, Lai MH, Blanchard KL, Thomas JE. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439–444. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- Charest PG, Firtel RA. Feedback signaling controls leading-edge formation during chemotaxis. Curr Opin Genet Dev. 2006;16:339–347. doi: 10.1016/j.gde.2006.06.016. [DOI] [PubMed] [Google Scholar]
- Cho W. Membrane targeting by C1 and C2 domains. J Biol Chem. 2001;276:32407–32410. doi: 10.1074/jbc.R100007200. [DOI] [PubMed] [Google Scholar]
- Collins SR, Meyer T. Calcium flickers lighting the way in chemotaxis? Dev Cell. 2009;16:160–161. doi: 10.1016/j.devcel.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbalan-Garcia S, Garcia-Garcia J, Rodriguez-Alfaro JA, Gomez-Fernandez JC. A new phosphatidylinositol 4,5-bisphosphate-binding site located in the C2 domain of protein kinase Calpha. J Biol Chem. 2003;278:4972–4980. doi: 10.1074/jbc.M209385200. [DOI] [PubMed] [Google Scholar]
- Corbalan-Garcia S, Guerrero-Valero M, Marin-Vicente C, Gomez-Fernandez JC. The C2 domains of classical/conventional PKCs are specific PtdIns(4,5)P(2)-sensing domains. Biochem Soc Trans. 2007;35:1046–1048. doi: 10.1042/BST0351046. [DOI] [PubMed] [Google Scholar]
- Corbin JA, Dirkx RA, Falke JJ. GRP1 pleckstrin homology domain: activation parameters and novel search mechanism for rare target lipid. Biochemistry. 2004;43:16161–16173. doi: 10.1021/bi049017a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbin JA, Evans JH, Landgraf KE, Falke JJ. Mechanism of specific membrane targeting by C2 domains: localized pools of target lipids enhance Ca2+ affinity. Biochemistry. 2007;46:4322–4336. doi: 10.1021/bi062140c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans JH, Falke JJ. Ca2+ influx is an essential component of the positive-feedback loop that maintains leading-edge structure and activity in macrophages. Proc Natl Acad Sci U S A. 2007;104:16176–16181. doi: 10.1073/pnas.0707719104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans JH, Murray D, Leslie CC, Falke JJ. Specific translocation of protein kinase Calpha to the plasma membrane requires both Ca2+ and PIP2 recognition by its C2 domain. Mol Biol Cell. 2006;17:56–66. doi: 10.1091/mbc.E05-06-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahey S, Dempsey E, Long A. The role of chemokines in acute and chronic hepatitis C infection. Cell Mol Immunol. 2013;11:25–40. doi: 10.1038/cmi.2013.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambhir A, Hangyas-Mihalyne G, Zaitseva I, Cafiso DS, Wang J, Murray D, Pentyala SN, Smith SO, McLaughlin S. Electrostatic sequestration of PIP2 on phospholipid membranes by basic/aromatic regions of proteins. Biophys J. 2004;86:2188–2207. doi: 10.1016/S0006-3495(04)74278-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golebiewska U, Nyako M, Woturski W, Zaitseva I, McLaughlin S. Diffusion coefficient of fluorescent phosphatidylinositol 4,5-bisphosphate in the plasma membrane of cells. Mol Biol Cell. 2008;19:1663–1669. doi: 10.1091/mbc.E07-12-1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins PT, Stephens LR, Suire S, Wilson M. PI3K signaling in neutrophils. Curr Top Microbiol Immunol. 2010;346:183–202. doi: 10.1007/82_2010_40. [DOI] [PubMed] [Google Scholar]
- Heckman CA, Plummer HK., 3rd Filopodia as sensors. Cell Signal. 2013;25:2298–2311. doi: 10.1016/j.cellsig.2013.07.006. [DOI] [PubMed] [Google Scholar]
- Henle SJ, Wang G, Liang E, Wu M, Poo MM, Henley JR. Asymmetric PI(3,4,5)P3 and Akt signaling mediates chemotaxis of axonal growth cones. J Neurosci. 2011;31:7016–7027. doi: 10.1523/JNEUROSCI.0216-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insall RH, Machesky LM. Actin dynamics at the leading edge: from simple machinery to complex networks. Dev Cell. 2009;17:310–322. doi: 10.1016/j.devcel.2009.08.012. [DOI] [PubMed] [Google Scholar]
- Jin T. Gradient sensing during chemotaxis. Curr Opin Cell Biol. 2013;25:532–537. doi: 10.1016/j.ceb.2013.06.007. [DOI] [PubMed] [Google Scholar]
- Knight JD, Falke JJ. Single-molecule fluorescence studies of a PH domain: new insights into the membrane docking reaction. Biophys J. 2009;96:566–582. doi: 10.1016/j.bpj.2008.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight JD, Lerner MG, Marcano-Velazquez JG, Pastor RW, Falke JJ. Single molecule diffusion of membrane-bound proteins: window into lipid contacts and bilayer dynamics. Biophys J. 2010;99:2879–2887. doi: 10.1016/j.bpj.2010.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13:159–175. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- Kolsch V, Charest PG, Firtel RA. The regulation of cell motility and chemotaxis by phospholipid signaling. J Cell Sci. 2008;121:551–559. doi: 10.1242/jcs.023333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CL, Landgraf KE, Voth GA, Falke JJ. Membrane docking geometry and target lipid stoichiometry of membrane-bound PKCα C2 domain: a combined molecular dynamics and experimental study. J Mol Biol. 2010;402:301–310. doi: 10.1016/j.jmb.2010.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CL, Srivastava A, Pilling C, Chase AR, Falke JJ, Voth GA. Molecular mechanism of membrane binding of the GRP1 PH domain. J Mol Biol. 2013;425:3073–3090. doi: 10.1016/j.jmb.2013.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landgraf KE, Pilling C, Falke JJ. Molecular mechanism of an oncogenic mutation that alters membrane targeting: Glu17Lys modifies the PIP lipid specificity of the AKT1 PH domain. Biochemistry. 2008;47:12260–12269. doi: 10.1021/bi801683k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Good JA, Ziegler WH, Parekh DB, Alessi DR, Cohen P, Parker PJ. Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science. 1998;281:2042–2045. doi: 10.1126/science.281.5385.2042. [DOI] [PubMed] [Google Scholar]
- Lemmon MA. Membrane recognition by phospholipid-binding domains. Nat Rev Mol Cell Biol. 2008;9:99–111. doi: 10.1038/nrm2328. [DOI] [PubMed] [Google Scholar]
- Leonard TA, Hurley JH. Regulation of protein kinases by lipids. Curr Opin Struct Biol. 2011;21:785–791. doi: 10.1016/j.sbi.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Hou S, Wu X, Nandagopal S, Lin F, Kung S, Marshall AJ. The tandem PH domain-containing protein 2 (TAPP2) regulates chemokine-induced cytoskeletal reorganization and malignant B cell migration. PLOS ONE. 2013;8:e57809. doi: 10.1371/journal.pone.0057809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindhurst MJ, Sapp JC, Teer JK, Johnston JJ, Finn EM, Peters K, Turner J, Cannons JL, Bick D, Blakemore L, Blumhorst C, Brockmann K, Calder P, Cherman N, Deardorff MA, Everman DB, Golas G, Greenstein RM, Kato BM, Keppler-Noreuil KM, Kuznetsov SA, Miyamoto RT, Newman K, Ng D, O’Brien K, Rothenberg S, Schwartzentruber DJ, Singhal V, Tirabosco R, Upton J, Wientroub S, Zackai EH, Hoag K, Whitewood-Neal T, Robey PG, Schwartzberg PL, Darling TN, Tosi LL, Mullikin JC, Biesecker LG. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med. 2011;365:611–619. doi: 10.1056/NEJMoa1104017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo B, Prescott SM, Topham MK. Diacylglycerol kinase zeta regulates phosphatidylinositol 4-phosphate 5-kinase Ialpha by a novel mechanism. Cell Signal. 2004;16:891–897. doi: 10.1016/j.cellsig.2004.01.010. [DOI] [PubMed] [Google Scholar]
- Masters TA, Calleja V, Armoogum DA, Marsh RJ, Applebee CJ, Laguerre M, Bain AJ, Larijani B. Regulation of 3-phosphoinositide-dependent protein kinase 1 activity by homodimerization in live cells. Sci Signal. 2010;3:ra78. doi: 10.1126/scisignal.2000738. [DOI] [PubMed] [Google Scholar]
- McLaughlin S, Wang J, Gambhir A, Murray D. PIP(2) and proteins: interactions, organization, and information flow. Annu Rev Biophys Biomol Struct. 2002;31:151–175. doi: 10.1146/annurev.biophys.31.082901.134259. [DOI] [PubMed] [Google Scholar]
- Mortimer D, Fothergill T, Pujic Z, Richards LJ, Goodhill GJ. Growth cone chemotaxis. Trends Neurosci. 2008;31:90–98. doi: 10.1016/j.tins.2007.11.008. [DOI] [PubMed] [Google Scholar]
- Nalefski EA, Falke JJ. The C2 domain calcium-binding motif: structural and functional diversity. Protein Sci. 1996;5:2375–2390. doi: 10.1002/pro.5560051201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton AC. Lipid activation of protein kinases. J Lipid Res. 2009;50(Suppl):S266–S271. doi: 10.1194/jlr.R800064-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton AC. Protein kinase C: poised to signal. Am J Physiol Endocrinol Metab. 2010;298:E395–E402. doi: 10.1152/ajpendo.00477.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent CA, Weiner OD. The symphony of cell movement: how cells orchestrate diverse signals and forces to control migration. Curr Opin Cell Biol. 2013;25:523–525. doi: 10.1016/j.ceb.2013.07.011. [DOI] [PubMed] [Google Scholar]
- Pilling C, Landgraf KE, Falke JJ. The GRP1 PH domain, like the AKT1 PH domain, possesses a sentry glutamate residue essential for specific targeting to plasma membrane PI(3,4,5)P(3) Biochemistry. 2011;50:9845–9856. doi: 10.1021/bi2011306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer EL, Eddy SR, Bateman A, Finn RD. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer C, Rymarczyk G, Ding L, Kirber MT, Bolotina VM. Role of molecular determinants of store-operated Ca(2+) entry (Orai1, phospholipase A2 group 6, and STIM1) in focal adhesion formation and cell migration. J Biol Chem. 2012;287:40745–40757. doi: 10.1074/jbc.M112.407155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaney KF, Huang CH, Devreotes PN. Eukaryotic chemotaxis: a network of signaling pathways controls motility, directional sensing, and polarity. Annu Rev Biophys. 2010;39:265–289. doi: 10.1146/annurev.biophys.093008.131228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thapa N, Anderson RA. PIP2 signaling, an integrator of cell polarity and vesicle trafficking in directionally migrating cells. Cell Adhes Migr. 2012;6:409–412. doi: 10.4161/cam.21192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian D, Jacobo SM, Billing D, Rozkalne A, Gage SD, Anagnostou T, Pavenstadt H, Hsu HH, Schlondorff J, Ramos A, Greka A. Antagonistic regulation of actin dynamics and cell motility by TRPC5 and TRPC6 channels. Sci Signal. 2010;3:ra77. doi: 10.1126/scisignal.2001200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynor D, Milne JL, Insall RH, Kay RR. Ca(2+) signalling is not required for chemotaxis in Dictyostelium. EMBO J. 2000;19:4846–4854. doi: 10.1093/emboj/19.17.4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trovati M, Doronzo G, Barale C, Vaccheris C, Russo I, Cavalot F. Leptin and vascular smooth muscle cells. Curr Pharm Des. 2013 doi: 10.2174/13816128113199990022. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Tsai FC, Meyer T. Ca2+ pulses control local cycles of lamellipodia retraction and adhesion along the front of migrating cells. Curr Biol. 2012;22:837–842. doi: 10.1016/j.cub.2012.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadas O, Burke JE, Zhang X, Berndt A, Williams RL. Structural basis for activation and inhibition of class I phosphoinositide 3-kinases. Sci Signal. 2011;4:re2. doi: 10.1126/scisignal.2002165. [DOI] [PubMed] [Google Scholar]
- von Philipsborn A, Bastmeyer M. Mechanisms of gradient detection: a comparison of axon pathfinding with eukaryotic cell migration. Int Rev Cytol. 2007;263:1–62. doi: 10.1016/S0074-7696(07)63001-0. [DOI] [PubMed] [Google Scholar]
- Waite K, Eickholt BJ. The neurodevelopmental implications of PI3K signaling. Curr Top Microbiol Immunol. 2010;346:245–265. doi: 10.1007/82_2010_82. [DOI] [PubMed] [Google Scholar]
- Wang J, Gambhir A, McLaughlin S, Murray D. A computational model for the electrostatic sequestration of PI(4,5)P2 by membrane-adsorbed basic peptides. Biophys J. 2004;86:1969–1986. doi: 10.1016/S0006-3495(04)74260-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YH, Slochower DR, Janmey PA. Counterion-mediated cluster formation by polyphosphoinositides. Chem Phys Lipids (Special Edition on Phosphoinositides) 2013 doi: 10.1016/j.chemphyslip.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C, Wang X, Chen M, Ouyang K, Song LS, Cheng H. Calcium flickers steer cell migration. Nature. 2009;457:901–905. doi: 10.1038/nature07577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C, Wang X, Zheng M, Cheng H. Calcium gradients underlying cell migration. Curr Opin Cell Biol. 2012;24:254–261. doi: 10.1016/j.ceb.2011.12.002. [DOI] [PubMed] [Google Scholar]
- Weiger MC, Parent CA. Phosphoinositides in chemotaxis. Subcell Biochem. 2012;59:217–254. doi: 10.1007/978-94-007-3015-1_7. [DOI] [PubMed] [Google Scholar]
- Wessels D, Lusche DF, Steimle PA, Scherer A, Kuhl S, Wood K, Hanson B, Egelhoff TT, Soll DR. Myosin heavy chain kinases play essential roles in Ca2+, but not cAMP, chemotaxis and the natural aggregation of Dictyostelium discoideum. J Cell Sci. 2012;125:4934–4944. doi: 10.1242/jcs.112474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Huang XY. Ca2+ influx through L-type Ca2+ channels controls the trailing tail contraction in growth factor-induced fibroblast cell migration. J Biol Chem. 2005;280:27130–27137. doi: 10.1074/jbc.M501625200. [DOI] [PubMed] [Google Scholar]
- Zamburlin P, Ruffinatti FA, Gilardino A, Farcito S, Parrini M, Lovisolo D. Calcium signals and FGF-2 induced neurite growth in cultured parasympathetic neurons: spatial localization and mechanisms of activation. Pflugers Arch. 2013;465:1355–1370. doi: 10.1007/s00424-013-1257-5. [DOI] [PubMed] [Google Scholar]
- Zhu W, Nelson CM. PI3K regulates branch initiation and extension of cultured mammary epithelia via Akt and Rac1 respectively. Dev Biol. 2013;379:235–245. doi: 10.1016/j.ydbio.2013.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziemba BP, Falke JJ. Lateral diffusion of peripheral membrane proteins on supported lipid bilayers is controlled by the additive frictional drags of (1) bound lipids and (2) protein domains penetrating into the bilayer hydrocarbon core. Chem Phys Lipids. 2013:172–173. 67–77. doi: 10.1016/j.chemphyslip.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziemba BP, Knight JD, Falke JJ. Assembly of membrane-bound protein complexes: detection and analysis by single molecule diffusion. Biochemistry. 2012;51:1638–1647. doi: 10.1021/bi201743a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziemba BP, Pilling C, Calleja V, Larijani B, Falke JJ. The PH domain of phosphoinositide-dependent kinase-1 exhibits a novel, phospho-regulated monomer-dimer equilibrium with important implications for kinase domain activation: single-molecule and ensemble studies. Biochemistry. 2013;52:4820–4829. doi: 10.1021/bi400488f. [DOI] [PMC free article] [PubMed] [Google Scholar]