

Abstract
It has long been believed that the male-specific region of the human Y chromosome (MSY) is genetically independent from the X chromosome. This idea has been recently dismissed due to the discovery that X–Y gametologous gene conversion may occur. However, the pervasiveness of this molecular process in the evolution of sex chromosomes has yet to be exhaustively analyzed. In this study, we explored how pervasive X–Y gene conversion has been during the evolution of the youngest stratum of the human sex chromosomes. By comparing about 0.5 Mb of human–chimpanzee gametologous sequences, we identified 19 regions in which extensive gene conversion has occurred. From our analysis, two major features of these emerged: 1) Several of them are evolutionarily conserved between the two species and 2) almost all of the 19 hotspots overlap with regions where X–Y crossing-over has been previously reported to be involved in sex reversal. Furthermore, in order to explore the dynamics of X–Y gametologous conversion in recent human evolution, we resequenced these 19 hotspots in 68 widely divergent Y haplogroups and used publicly available single nucleotide polymorphism data for the X chromosome. We found that at least ten hotspots are still active in humans. Hence, the results of the interspecific analysis are consistent with the hypothesis of widespread reticulate evolution within gametologous sequences in the differentiation of hominini sex chromosomes. In turn, intraspecific analysis demonstrates that X–Y gene conversion may modulate human sex-chromosome-sequence evolution to a greater extent than previously thought.
Keywords: X–Y gene conversion, sex chromosome evolution, human Y chromosome, recombination hotspots
Introduction
The human Y chromosome, with its unique genetic features, is a useful tool for investigating many issues regarding a wide range of fields including forensic science (Jobling et al. 1997), human population genetics (Underhill and Kivisild 2007), medical genetics (Krausz et al. 2004), and the analysis of the dynamics of fundamental evolutionary mechanisms of the human genome (Bosch et al. 2004; Repping et al. 2006; Rosser et al. 2009; Trombetta et al. 2010). Therefore, understanding its evolutionary patterns and which molecular mechanisms may affect its genetic variability is a primary topic in human evolutionary genetics.
Human sex chromosomes evolved from a pair of recombining autosomes through the suppression of meiotic recombination (Bachtrog 2013). It has been hypothesized that the suppression of recombination between proto-sex chromosomes did not occur in a single evolutionary event, but in multiple steps (Lahn and Page 1999; Ross et al. 2005). Studies based on sequence divergence between X–Y gametologous (formerly allelic) regions indicate that at least five distinct events of crossing-over suppression have occurred (Ross et al. 2005). The regions of the sex chromosomes that stopped recombining at different time points are called “evolutionary strata.” Average X–Y nucleotide divergence differs significantly among strata and a lower sequence similarity corresponds to a greater elapsed time since the sequences have ceased to recombine (Ross et al. 2005). The oldest stratum was generated in the stem lineage of Theria 166–148 Ma (Bininda-Emonds et al. 2007; Veyrunes et al. 2008; Katsura and Satta 2012), whereas the youngest stratum originated only 30 Ma and it retains a high X–Y sequence similarity (∼95%; Ross et al. 2005; Hughes et al. 2012). On the human X chromosome, the evolutionary strata are arranged in a linearly decreasing order starting from the subtelomeric portion of the short arm. On the Y chromosome, various structural rearrangements have led to the loss of physical continuity between the elements of each stratum (Skaletsky et al. 2003; Ross et al. 2005).
It has long been recognized that meiotic recombination between human sex chromosomes occurs within short telomeric portions, known as pseudoautosomal regions (PARs), which mark the boundaries of a male-specific region (MSY) comprising 95% of the entire chromosome. The MSY is composed of three classes of sequences: X-transposed, X-degenerate, and ampliconic (Skaletsky et al. 2003). The X-transposed regions originated from an X-to-Y transposition 4.7 Ma (Ross et al. 2005). The X-degenerate sequences are remnants of the proto-sex chromosomes and contain all the evolutionary strata. The ampliconic sequences are mainly composed of eight palindromic structures (termed P1–P8), each of which consist in two highly similar intrachromosomal inverted paralogs (or “arms”) separated by a nonduplicated spacer sequence. Palindromic sequences show an arm-to-arm nucleotide identities >99.9%, due to frequent Y–Y gene conversion events (Rozen et al. 2003). Gene conversion (i.e., the unidirectional transfer of genetic information from a “donor” sequence to a highly similar “acceptor”) always initiates by DNA double-strand breaks (DSB) and the genetic transfer can occur between allelic sequences or between highly similar (identity > 90%) nonallelic sequences located on the same or on different chromosomes (Chen et al. 2007).
The view that MSY is a genetically isolated region has been recently dismissed due to the discovery that X-to-Y gene conversion may occur in humans (Rosser et al. 2009; Cruciani et al. 2010; Trombetta et al. 2010). Despite increasing evidence indicating X–Y concerted evolution (Rosser et al. 2009; Iwase et al. 2010; Trombetta et al. 2010; Ellegren 2011; Niederstätter et al. 2013), the study of the dynamics of this molecular mechanism in humans is still in its infancy, and the pervasiveness of X–Y gene conversion in the evolution of human sex chromosomes has yet to be exhaustively explored. A history of gene conversion between paralogous (or gametologous) sequences can be easily detected by examining a four-way alignment of paralogous (or gametologous) and orthologous regions from two closely related species (Osada and Innan 2008; Kijima and Innan 2010). In the absence of conversion, the pattern of nucleotide variation should be dominated by differences between paralogs if duplicated regions originated before speciation. Conversely, if gene conversion was/is active, paralogous sequences should be more similar to each other than to the orthologous ones.
The purpose of this study is to determine how pervasive X–Y gene conversion has been within the youngest evolutionary stratum of human sex chromosomes. To this end, we used an inter- and intraspecies sequence-diversity-based approach. By comparing human X- and Y-specific sequences with their orthologs from chimpanzees, we identified several narrow regions that show a strong signature of historical X–Y concerted evolution. To evaluate whether these regions have also been affected by X-to-Y gene conversion in recent human evolution, we resequenced them in a number of Y chromosome haplogroups that represent an ample range of worldwide human MSY diversity, whereas the role of Y-to-X conversion was investigated using publicly available X chromosome single nucleotide polymorphism (SNP) data. Recent X–Y gene conversion events may be recognized by the switch of GSVs (gametologous sequence variants) from the Y (or X)-specific state to that observed on the gametologous base on the X (or Y) chromosome (Rosser et al. 2009; Trombetta et al. 2010; see Materials and Methods). We identified signals of active X–Y recent gene conversion in at least ten of the regions that show signatures of historical concerted evolution.
Results
Four-Way Sequences Alignment of Humans and Chimpanzees Sex Chromosomes
To identify regions that may have undergone historical gene conversion events within MSY, we first extracted a human X-linked region of approximately 1 Mb (ChrX: 2699520–3689259; GRCh37/hg19) known to be characterized by a high X–Y similarity (evolutionary stratum 5 on the X chromosome in Ross et al. 2005, which corresponds to stratum 9 in Pandey et al. 2013), and searched for gametologous regions on the human Y chromosome and for orthologous regions on the chimpanzee sex chromosomes. We were able to retrieve a total of about 0.5 Mb of human X-linked sequences (456,564 bp; 46.12% of the total extracted sequence), which align with both the human Y chromosome and the chimpanzee sex chromosomes.
To compare the human X- and Y-specific sequences with orthologs from chimpanzees, a four-way sequence alignment was performed. As expected, the overall pattern of nucleotide diversity was dominated by differences between X and Y chromosomes in one species (S-sites) or in both species (N-sites) (fig. 1). Nevertheless, we also identified a total of 266 bivariant sites in which the same nucleotide is shared by the two gametologous sequences of each species (type-C sites; fig. 1). These sites may arise through two possible mechanisms: Two independent mutations at the same gametologous site on the X and Y chromosomes of one species or through gene conversion between sex chromosomes (Osada and Innan 2008; fig. 1).
Fig. 1.

(A) Possible variant sites within a four-way alignment of the orthologous and gametologous sequences from human and chimpanzee sex chromosomes. Different types of sites are shown: C, C-site or conversion site; N, N-site or nonconversion site; S, S-site (or Singleton); M, multiple mutation site; W, other complex site. Invariant sites are not shown. Classification according to Kijima and Innan (2010). (B) Different molecular mechanisms for the formation of variable sites. A type-S site may arise if a mutation occurs in one sex chromosome after human–chimpanzee speciation, whereas an N-site may be generated by a mutation occurring before speciation. If a gene conversion event involves a type-N nucleotide, it generates an S-site, whereas a conversion of an S-site may generate a C-nucleotide or an invariant site depending on the direction of conversion. M- and W-sites may arise only with multiple mutational events.
Following closer and more detailed analysis, C-sites did not appear to be equally distributed, about half of them (126/266) being restricted to 19 C-site-enriched regions (CERs) (≥4 C-sites/kb; hereafter referred to as CER1–19), which cover a total of 13.4 kb (2.9% of the aligned sequences) (table 1). Assuming a uniform distribution for the C-sites, this observation indicates a significant excess (P = 1.0 × 10−7) of the observed number of CERs (supplementary fig. S1, Supplementary Material online) within the alignment.
Table 1.
C-Site-Enriched Regions.
| CER ID |
Homo sapiensa |
Pan troglodytesa |
Lb | C-Sites | N-Sites | S-Sitesc |
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ChrX | ChrY | ChrX | ChrY | SHY | SPY | SHX | SPX | ||||
| CER1 | 3679860–3680460 | 7091216–7091814 | 3675948–3676546 | 22898996–22899593 | 601 | 4 | 0 | 10 | 0 | 30 | 0 |
| CER2 | 3671777–3672707 | 7096711–7097641 | 3667630–3668558 | 22893085–22894013 | 931 | 11 | 0 | 6 | 6 | 6 | 3 |
| CER3 | 3603266–3604004 | 7163099–7163862 | 3593349–3594110 | 22826827–22827584 | 776 | 9 | 8 | 15 | 8 | 11 | 4 |
| CER4 | 3600301–3601725 | 7165484–7166941 | 3590386–3591806 | 22823805–22825223 | 1,458 | 7 | 5 | 31 | 8 | 5 | 7 |
| CER5 | 3596033–3596381 | 7168440–7168782 | 3585803–3586149 | 22821952–22822298 | 351 | 4 | 0 | 27 | 1 | 0 | 0 |
| CER6 | 3594322–3594833 | 7169931–7170442 | 3584088–3584597 | 22820296–22820805 | 493 | 5 | 1 | 7 | 4 | 3 | 9 |
| CER7 | 3591725–3593007 | 7171778–7173059 | 3581500–3582777 | 22817686–22818962 | 1,283 | 8 | 5 | 8 | 12 | 22 | 10 |
| CER8 | 3559783–3560184 | 7208985–7209385 | AACZ03178410_random: 258–657 | 22781358–22781750 | 402 | 4 | 0 | 5 | 3 | 0 | 1 |
| CER9 | 3533911–3534399 | 7234931–7235419 | 3537792–3538276 | 22753816–22754299 | 489 | 4 | 0 | 2 | 7 | 4 | 2 |
| CER10 | 3529192–3530503 | 7239647–7240956 | 3532811–3534120 | 22748581–22749881 | 1,321 | 8 | 15 | 28 | 18 | 7 | 8 |
| CER11 | 3525449–3525876 | 7246240–7246670 | 3529302–3529727 | 22742867–22743271 | 441 | 4 | 0 | 5 | 16 | 22 | 6 |
| CER12 | 3455115–3455541 | 7319098–7319526 | 3454452–3454878 | 22663835–22664261 | 429 | 8 | 0 | 3 | 3 | 8 | 2 |
| CER13 | 3436059–3436907 | 7333693–7334559 | 3435069–3435917 | 22643731–22644600 | 884 | 10 | 60 | 8 | 11 | 9 | 2 |
| CER14 | 3362952–3363523 | 7418498–7419244 | 3353968–3354721 | 22552803–22553555 | 757 | 6 | 0 | 2 | 8 | 28 | 2 |
| CER15 | 3278881–3278953 | 14051551–14051623 | GL393313_random: 318610–318682 | 24848003–24848075 | 73 | 4 | 1 | 1 | 0 | 1 | 4 |
| CER16 | 3092315–3092940 | 14295656–14296272 | GL393313_random: 140911–141524 | 22203042–22203656 | 627 | 8 | 2 | 11 | 2 | 30 | 0 |
| CER17 | 2842671–2843504 | 14483248–14484067 | 2842671–2843504 | 17463306–17464121 | 884 | 12 | 0 | 14 | 1 | 20 | 0 |
| CER18 | 2841823–2842477 | 14484255–14484908 | 2833742–2834393 | 17464311–17464962 | 655 | 4 | 22 | 9 | 5 | 1 | 6 |
| CER19 | 2837016–2837544 | 14490130–14490661 | 2830934–2831463 | 17470174–17470703 | 535 | 6 | 0 | 4 | 26 | 8 | 4 |
| Stratum 5d | 3457797–3464821 | 7311695–7318744 | 3485445–3492520 | 22664624–22671623 | 7,498 | 2 | 625 | 44 | 55 | 25 | 27 |
aGenomic position is according to GChR37/hg19 for Homo sapiens and CGSG 2.1.3 for Pan troglodytes.
bNumber of base pairs of the four-way alignment.
cNumber of singleton within the alignment. SHY, S-sites in the human Y chromosome; SPY, S-sites in the chimpanzee Y chromosome; SHX, S-sites in the human X chromosome; SPX, S-sites in the chimpanzee X chromosome.
dNon-CER portion of the four-way alignment representative of the stratum 5.
Pattern of Interspecies Sequence Diversity within CERs
In theory, either a double mutation or X–Y gene conversion could generate a C-site within a four-way alignment of duplicated regions in two closely related species. If X–Y conversion has acted on a CER, we should observe a higher number of type-C nucleotides than what would be expected by chance. To shed light on this issue, we applied the statistical test reported by Osada and Innan (2008) to each of the 19 CERs identified. The null hypothesis is set so that the observed pattern of C-sites could be explained without gene conversion when the effects of multiple independent mutations are taken into account.
For all the 19 CERs, the four-way alignment is shown in supplementary figure S2, Supplementary Material online. For each of these regions the number of type-C and type-N sites was counted, and the expected number of C-sites assuming no gene conversion was calculated (table 2). The number of expected type-C nucleotides was found to be always less than one except in one case (CER14), whereas the observed number ranged from 4 to 12. In each region, the number of observed C-sites is always significantly higher than what would be expected under the hypothesis of C-sites generated by double independent mutations (table 2). These findings strongly support a history of X–Y gene conversion in the evolution of these sequences in at least one of the two species.
Table 2.
Testing for Gene Conversion in CERs.
| CER ID | La | Observed C-Sites | Expected C-Sites | P | Proportion of C-Sites (PC) | Tree Shapeb | Evidence for Conversionc |
Evidence for Conversion by SD Analysisd | |
|---|---|---|---|---|---|---|---|---|---|
| Hsa | Ptr | ||||||||
| CER1 | 601 | 4 | 0.43 | 1 × 10−3 | 1 | ((((Xp,Yp),Yh),Xh),Xo) | No | Yes | Yes |
| CER2 | 931 | 11 | 0.28 | <10−12 | 1 | ((Xp,Yp),(Yh,Xh),Xo) | Yes | Yes | Yes |
| CER3 | 776 | 9 | 0.59 | 1.5 × 10−8 | 0.53 | ((Xp,Yp),(Yh,Xh),Xo) | Yes | Yes | Yes |
| CER4 | 1,458 | 7 | 0.54 | 1.6 × 10−6 | 0.58 | ((((Xp,Yp),Xh),Yh),Xo) | No | Yes | Yes |
| CER5 | 351 | 4 | 0.27 | 1.8 × 10−4 | 1 | (((Xp,Yp),Xh,Yh),Xo) | No | Yes | No |
| CER6 | 493 | 5 | 0.29 | 1.3 × 10−5 | 0.83 | ((((Xh,Yh),Yp),Xp),Xo) | Yes | No | Yes |
| CER7 | 1,283 | 8 | 0.48 | 4.7 × 10−8 | 0.61 | ((((Xp,Yp),Yh),Xh),Xg) | No | Yes | Yes |
| CER8 | 402 | 4 | 0.08 | 1.79 × 10−6 | 1 | ((((Xp,Yp),Xh),Yh),Xo) | No | Yes | Yes |
| CER9 | 489 | 4 | 0.15 | 2 × 10−5 | 1 | ((Xp,Yp),(Yh,Xh),Xg) | Yes | Yes | Yes |
| CER10 | 1,321 | 8 | 0.41 | 1.46 × 10−8 | 0.35 | ((((Yp,Yh),Xh),Xp),Xg) | No | No | Yes |
| CER11 | 441 | 4 | 0.59 | 3.3 × 10−3 | 1 | ((((Yp,Xp),Yh),Xh),Xo) | No | Yes | Yes |
| CER12 | 429 | 8 | 0.32 | 1.8 × 10−9 | 1 | ((Xp,Yp),(Xh,Yh),Xo) | Yes | Yes | Yes |
| CER13 | 884 | 10 | 0.42 | 3.2 × 10−11 | 0.14 | ((Yh,Yp),(Xh,Xp),Xo) | No | No | Yes |
| CER14 | 757 | 6 | 1.25 | 1.8 × 10−3 | 1 | ((((Xp,Yp),Yh),Xh),Xg) | No | Yes | No |
| CER15 | 73 | 4 | 0.451 | 1.2 × 10−3 | 0.8 | ((Xp,Yp),(Xh,Yh),Xo) | Yes | Yes | Yes |
| CER16 | 627 | 8 | 0.88 | 4.13 × 10−6 | 0.8 | ((((Xp,Yp),Yh),Xh),Xo) | No | Yes | Yes |
| CER17 | 884 | 12 | 0.5 | <10−12 | 1 | ((((Xp,Yp),Yh),Xh),Xo) | No | Yes | Yes |
| CER18 | 655 | 4 | 0.16 | 2.7 × 10−5 | 0.15 | ((((Yp,Yh),Xh),Xp),Xg) | No | No | Yes |
| CER19 | 535 | 6 | 0.59 | 3.4 × 10−5 | 1 | ((Xp,Yp),(Xh,Yh),Xo) | Yes | Yes | Yes |
| Stratum 5e | 7,498 | 2 | 0.51 | 9.3 × 10−2 | 3 × 10−2 | ((Yh,Yp),(Xh,Xp),Xg) | No | No | No |
aLength (bp) of the four-way alignment.
bShape of the NJ tree in newick format. Xp and Yp are X and Y chromosomes of Pan troglodytes, respectively; Xh and Yh are X and Y chromosomes of Homo sapiens, respectively; Xo and Xg indicate the X chromosome of Pongo pygmaeus and Gorilla gorilla, respectively.
cEvidence for gene conversion based on the tree shape analysis. Yes or no means the presence or absence of gene conversion in NJ-phylogenetic analyses, respectively.
dEvidence for gene conversion based on the Split Decomposition (SD) analysis. Yes or no means the presence or absence of a reticulation, respectively.
eNon-CER portion of the four-way alignment representative of the stratum 5.
To evaluate the extent of gene conversion in these regions, we computed the proportion of C-sites compared with N-sites (PC values; Kijima and Innan 2010). With few exceptions, the number of C-sites largely exceeds the number of N-sites, and, overall, PC values are high (table 2). This finding highlights the pervasive role of gene conversion in shaping sequence diversity of these regions.
Interspecies Phylogenetic Analysis of CERs
Suppression of recombination within the youngest X–Y stratum vastly predates the human–chimpanzee split. Thus, in absence of X–Y homogenization, a phylogeny comprising both gametologous and orthologous sequences is expected to be dominated by a clustering of orthologs. On the other hand, if gene conversion has been active, we would expect to see clustering of gametologs. In order to examine the phylogenetic history of CERs, a neighbor-joining (NJ) analysis (Saitou and Nei 1987) of these regions was performed. We also constructed a phylogenetic tree (in newick format, table 2) for a non-CER fragment of about 7 kb of the alignment, which likely represents the evolutionary history of the entire evolutionary stratum in absence of gene conversion. The inspection of the tree shape for each CER (table 2 and supplementary fig. S3, Supplementary Material online) showed a clustering of paralogous sequences in all but three cases (CER10, CER13, and CER18). Six blocks (CER2, CER3, CER9, CER12, CER15, and CER19) showed a clustering of gametologous sequences in humans and chimpanzees, which strongly suggests that gene conversion has occurred in both species. Ten CERs showed clustering of gametologous sequences for either of the two species, only one of which (CER6) showed evidence of past X–Y conversion in humans (table 2 and supplementary fig. S3, Supplementary Material online). The three blocks that did not show phylogenetic signals of X–Y homogenization in the NJ tree are also the CERs with the lowest PC values (table 2).
To further explore the molecular relationships among gametologous regions, we performed a split decomposition analysis (Huson and Bryant 2006), which displays phylogenetic conflicting signals as a reticulated structure. A reticulation may be generated by various molecular mechanisms, such as recurrent mutations or gametologous gene conversion. The presence of a reticulation at the center of the network was observed for almost all the CERs (supplementary fig. S3, Supplementary Material online). No reticulation was observed in CER5 and CER14, which, on the other hand, showed tight clustering for the chimpanzee sequences within the phylogeny. This could be the consequence of strong X–Y gene conversion, which may have completely eradicated conflicting signals of gametologous/orthologous relationships.
Distribution of CERs within Human Sex Chromosomes
The CERs seem to be unevenly distributed on the human sex chromosomes. Out of the 19 blocks, 12 lie within the two active genes PRKX and ARSD on the X chromosome and their gametologous pseudogenes (PRKY and ARSDP) on the Y chromosome (figs. 2 and 3, supplementary table S1, Supplementary Material online). The remaining seven CERs were found in intergenic regions, with CER1 and CER2 adjacent (∼40 kb) to the 5′-end of the PRKX/PRKY genes (figs. 2 and 3). We next investigated whether there is a tendency of CERs to cover exonic sequences on the X chromosome. Four of the nine hotspots within the PRKX gene totally cover four different exons and one CER partially covers the 3′-UTR of the gene (fig. 2 and supplementary table S1, Supplementary Material online). This results in a significant excess (P < 10−4, chi-square test) of exonic sequences covered by CERs. This finding suggests that functional differentiation between gametologous genes involved in gene conversion can be erased or functionality could be destroyed. The evolutionary cost of this may be counterbalanced by the beneficial effects of gene-conversion-mediated repair of DSBs (see Discussion).
Fig. 2.

Distribution of CERs on the human X chromosome: (A) Schematic representation of the X chromosome. (B) The different tracks report the following features (from top to bottom): Scale bar; genomic coordinates from the GRCh37 human genome reference sequence; regions of the X chromosomes used for the four-way alignment position of CERs (red: CERs involved in recent X-to-Y gene conversion); previously known X–Y gene conversion hotspots (in green, ARSDP and HSA hotspots); UCSC genes.
Fig. 3.

Distribution of CERs on the human Y chromosome: (A) Schematic representation of the Y chromosome. (B) The different tracks report the following features (from top to bottom): Scale bar; genomic coordinates from the GRCh37 human genome reference sequence; regions of the X chromosomes used for the four-way alignment position of CERs (red: CERs involved in recent X-to-Y gene conversion); previously known X–Y gene conversion hotspots (in green, ARSDP and HSA hotspots); UCSC genes (the ARSDP position is inferred by BLAT analysis).
Pattern of Intraspecies Genetic Diversity within CERs of the Human MSY
To determine whether the gametologous sequences have also been engaged in X-to-Y gene conversion during recent human evolution, we studied the genetic diversity of the human Y chromosome for 17 of the gene conversion hotspots identified through the interspecies comparison. CER2 was not resequenced because its genetic diversity has been previously described (Rosser et al. 2009; Cruciani et al. 2010). CER4 has not been sequenced due to the failure of primer activity in polymerase chain reaction (PCR) amplification.
Overall, 16 kb of the MSY was resequenced, including CERs and surrounding regions (supplementary table S2, Supplementary Material online), in 68 chromosomes representing different haplogroups of the Y phylogeny (fig. 4). We found a total of 52 SNPs and 2 indel polymorphisms (V337 and V327) (table 3). Seven of these SNPs (V270, V274, V275, V300, V329, V332, and V335) correspond to SNPs, which are present in the build 137 of the Single Nucleotide Polymorphism database (dbSNP). The nucleotide diversity for the entire region was estimated as π = 2.3 ± 0.2 × 10−4, a slightly higher value than previously observed for the MSY (Shen et al. 2000; Trombetta et al. 2010).
Fig. 4.

SNPs identified in the sequenced regions. To the left, a simplified version of the Y-chromosome tree showing the phylogenetic relationships of the chromosomes analyzed (Karafet et al. 2008; Scozzari et al. 2012). SNP names and CER numbers are given at the top and at the bottom, respectively. Square colors represent the allelic state for each SNP: White, ancestral allele; red, the SNP has arisen in an X–Y GSV and the derived state is equal to the gametologous base on the X; and green, the SNP has arisen in an identical site between X and Y; ×-marked squares, missing data SNPs V288 and V289 are in regions that do not align with any region of the X chromosome (see table 3).
Table 3.
List of Y-Polymorphisms Identified in This Study.
| SNP | Y-Positiona | Mutation | ChrXb | Haplogroupc | CERs | dbSNP Build 137 |
|---|---|---|---|---|---|---|
| V290 | 7091768 | A to T | A | A3b-M114 | 1 | — |
| V335 | 7163481 | C to T | C | E1b1a1-M2 | 3 | rs9785813 |
| V336 | 7163514 | A to T | A | A3a-M28 | — | |
| V337 | 7163703 | Del TAG | Ins | E1b1c-M329 | — | |
| V301 | 7169816 | C to T | C | A2-M6 | 6 | — |
| V302 | 7169899 | C to T | C | DE | — | |
| V318.1 | 7170441 | A to C | C | E1b1a1g1a*-M290* | — | |
| V318.2 | 7170441 | A to C | C | E2b*-M98* | — | |
| V327 | 7171673 | Ins AC | del | E1b1b1a*-V68* | 7 | — |
| V328 | 7171834 | G to T | G | J1d-P56 | — | |
| V329 | 7171957 | G to A | G | H1-M52 | rs35147341 | |
| V330 | 7172251 | C to G | C | B2b4b-M211 | — | |
| V331 | 7172768 | G to A | G | B1*-M236* | — | |
| V332.1 | 7173143 | G to A | A | E1b1a1a-M58 | rs9786714 | |
| V332.2 | 7173143 | G to A | A | F2-IJ-K | rs9786714 | |
| V283 | 7209376 | G to A | G | E1b1a1*-M2* | 8 | — |
| V284 | 7209401 | G to T | G | E1a*-M33* | — | |
| V334 | 7209078 | C to G | C | O2b-P49 | — | |
| V297 | 7234782 | C to T | C | J2-M172 | 9 | — |
| V299 | 7235118 | C to A | C | B2b*-M112* | — | |
| V300 | 7235379 | G to A | G | A3b-M144 | rs35195174 | |
| V319 | 7239870 | C to T | (C/T) rs145173948 | B2a1*-M218* | 10 | — |
| V320 | 7240836 | A to C | A | O-M175 | — | |
| V339 | 7246397 | A to G | A | A1b-V148 | 11 | — |
| V279 | 7319200 | T to C | T | E1b1b1b1-M34 | 12 | — |
| V280 | 7319490 | C to T | C | A3b-M144 | — | |
| V281 | 7319576 | A to C | A | A2-V218 | — | |
| V282 | 7319580 | T to C | T | E1b1b1-M35 | — | |
| V340 | 7333516 | A to G | A | R1b1a*-V88* | 13 | — |
| V291 | 7333543 | C to T | C | J2b-M12 | — | |
| V292 | 7334323 | T to C | T | O1a2-M50 | — | |
| V293 | 7418611 | G to A | (C/G) rs187021908 | F*-M89* | 14 | — |
| V294 | 7419117 | C to T | C | B2b3-M30 | — | |
| V295 | 7419266 | G to A | G | A3a-M28 | — | |
| V296 | 7419349 | G to C | G | A2-M6 | — | |
| V321 | 14051677 | C to T | C | A2-M114 | 15 | — |
| V322 | 14051686 | A to G | G | E*-M96* | — | |
| V323 | 14051820 | C to T | C | A3a-M28 | — | |
| V324 | 14051832 | C to A | C | E1b1b1a1d-V65 | — | |
| V325 | 14051863 | C to T | T | B2b4b-M211 | — | |
| V326 | 14051913 | G to A | G | A3b1b-V11 | — | |
| V285 | 14295658 | C to T | C | O-M175 | 16 | — |
| V286 | 14295751 | C to G | C | C1-M8 | — | |
| V287 | 14295977 | G to A | G | D2a1-M125 | — | |
| V288 | 14296291 | G to A | NA | A3a-M28 | — | |
| V289 | 14296306 | T to G | NA | B2a1a2a2a*-P50* | — | |
| V270 | 14482977 | G to C | G | A3-M32 | 17 | rs34555243 |
| V271 | 14483622 | T to C | C | C3c-M48 | — | |
| V272 | 14483887 | A to C | A | B2b4b-M211 | — | |
| V333 | 14483992 | G to C | G | O1a2-M50 | — | |
| V273 | 14484106 | G to T | G | B1*-M236* | — | |
| V274 | 14484379 | A to C | C | I-M170 | 18 | rs113822196 |
| V275 | 14484394 | C to T | T | I*-M170* | rs113686211 | |
| V257 | 14484596 | C to T | C | E1b1b1b-V257 | — | |
| V276 | 14484897 | G to A | G | A2-T | — | |
| V277 | 14490108 | G to A | G | D2a1-M125 | 19 | — |
aPosition according to the February 2009 Y-chromosome reference sequence of the UCSC genome browser.
bGametologous base on the X chromosome. NA, no X–Y alignment. The two X-SNPs have a MAF < 1%.
cHaplogroup nomenclature by lineage and last mutation. Nomenclature according to Karafet et al. (2008) and Scozzari et al. (2012).
If gene conversion is operating between X–Y human sequences, we would expect 1) that the number of Y SNPs found in X–Y GSV sites is higher than expected by chance and 2) that the Y-linked derived allele (as inferred from the Y phylogeny) is the same as the gametologous site on the X. The observation of recurrent mutation in the Y phylogeny and shared X–Y polymorphisms are additional common outcomes of gametologous gene conversion. Overall, within the sequenced regions (16,014 bp), we counted a total of 658 GSVs and we found a significant excess of SNPs in GSVs (observed = 7, expected number = 2, P = 5.7 × 10−3) with the Y-linked derived allele corresponding to the gametologous base on the X chromosome. Two of the seven polymorphisms in X–Y GSVs are recurrent in the Y phylogeny (table 3 and fig. 4) and a single polymorphism (V319 in CER10; table 3) is shared between X and Y chromosomes. Overall, these findings suggest the involvement of X-to-Y gene conversion events in generating the observed diversity.
More specifically, signals of X-to-Y gene conversion were detected in five CERs (CER6, CER7, CER15, CER17, and CER18; table 3 and fig. 4). Within CER6, three SNPs were found, one of which (V318) occur in an X–Y GSV with the derived allele equal to the gametologous base on the X chromosome. Moreover, this mutation independently arose in E1b1a1g1*-U209* and E2b*-M98* haplogroups (fig. 4). The presence of this single recurrent SNP results in an excess of mutations in GSV for this region (expected mutations in GSV = 0.1; P = 4.5 × 10−3). In CER7, we identified two independent mutations at a single X–Y GSV (V332) (expected mutations in GSV = 0.3; P = 2.8 × 10−2), which are recurrent in various phylogenetic contexts (fig. 4 and table 3). Out of six mutations identified in CER15, two (V322 and V325) overwrite the pre-existing differences between the gametologous regions (expected mutations in GSV = 0.2; P = 1.4 × 10−2). Within the CER17–CER18 contiguous regions, we found a total of nine SNPs (table 3), three of which (V271, V274, and V275) resulted in the homogenization of X–Y sequence differences (expected mutations in GSV = 0.35; P = 4.2 × 10−3).
Dynamics of X-to-Y Gene Conversion in Humans
Following the criteria reported in Trombetta et al. (2010), we counted a minimum of nine X-to-Y independent gene conversion events (considering that the recurrence of SNPs V318 and V332 was due to two independent conversions each) and calculated the maximum and minimum lengths of the observed conversion tracts in each hotspot region (supplementary table S3, Supplementary Material online). The minimum observed tract length is always 1 bp as we observed no co-converted GSVs. The maximum gene-conversion tract, measured as the distance between the two nearest nonconverted GSVs flanking the converted site, ranges from 9 to 163 bp (supplementary table S3, Supplementary Material online) with an average length of 47 bp. In principle, the mutational patterns observed within the Y-linked CERs could also be explained by X–Y double crossover. However, the most likely explanation remains X-to-Y gene conversion, due to the short length of the observed sequence changes (Chen et al. 2007).
We used the equation reported in Cruciani et al. (2010) to calculate the X-to-Y gene conversion rate for each of the active hotspots described earlier (supplementary table S4, Supplementary Material online). We obtained an average rate of X-to-Y gene conversion that ranges from a minimum of 1.8 × 10−8 to a maximum of 1.1 × 10−6 per base per generation (assuming a 25-year generation time) for the CERs involved in X-to-Y gene conversion (supplementary table S4 and fig. S4, Supplementary Material online). This is a clear underestimate of the real rate because X-to-Y conversion involving identical sites cannot be detected using this approach.
Our estimates (supplementary table S4, Supplementary Material online) are lower than previously reported for other hotspots (supplementary fig. S4, Supplementary Material online) (Cruciani et al. 2010). The lower value of the conversion rate is comparable to the MSY mutation rate (Francalacci et al. 2013; Mendez et al. 2013; Poznik et al. 2013; Scozzari et al. 2014), whereas the upper value is 1–2 orders of magnitude higher (supplementary fig S4, Supplementary Material online).
Pattern of Intraspecies Genetic Diversity within CERs of the Human X Chromosome
To understand the role of human Y-to-X gene conversion in X chromosome sequence evolution, we used publicly available SNPs data for the 19 X-linked CERs. Mining the information contained in the build 137 of the dbSNP, we identified a total of 69 SNPs and 3 indel polymorphisms (supplementary table S5, Supplementary Material online) out of a total of 13,027 bp of the human X chromosome.
Similarly to X-to-Y gene conversion detection, if Y-to-X gene conversion is operating, we would expect 1) that the number of X-linked SNPs found in X–Y GSV sites is higher than expected by chance and 2) that the X-linked derived allele (as inferred using orthologous sequences) is the same as the gametologous site on the Y chromosome.
In total, within the X-linked sequences (which include CER2 and CER4 that were not analyzed for Y chromosome), we counted 432 GSVs and observed a significantly high number of SNPs in GSVs (observed = 16; expected number = 2.3; P < 10−6) with the X-linked derived allele corresponding to the gametologous base on the Y chromosome. This would seem to indicate that Y-to-X gene conversion events are involved in the generation of the observed diversity.
More specifically, mutations in X–Y GSVs were detected in ten CERs (CER1–4, CER6, CER7, CER10, CER11, CER13, and CER19; supplementary table S5, Supplementary Material online), with a significant excess of SNPs in X–Y GSVs in five of them (CER3, CER4, CER6, CER10, and CER11; supplementary table S5, Supplementary Material online).
Discussion
The major evolutionary feature of chromosomal sex-determination systems is the suppression of recombination between sex chromosomes in heterogametic sex, followed by the structural decay of the sex-specific chromosome (Ellegren 2011). This is well illustrated by the evolutionary path of human sex chromosomes, which have evolved from a single pair of recombining autosomes that began to differentiate due to the arrest of meiotic recombination. It has been hypothesized that the process of sequence differentiation between sex chromosomes involved at least five successive events of crossing-over suppression, which generated discrete clusters (termed “evolutionary strata”) with specific X–Y nucleotide differences depending on the time of recombination arrest (Lahn and Page 1999; Ross et al. 2005). Recently, Pandey et al. (2013) developed an algorithm to identify different evolutionary strata on the X chromosome also for the regions without a Y chromosome counterpart. They identified a total of nine evolutionary strata on the X chromosome (four of them without a Y chromosome gametolog region) corresponding to as many crossing-over suppressions. A near perfect match was observed between stratum 5 (identified through XY sequence comparison by Ross et al. 2005) and stratum 9 (identified by Pandey et al. 2013). Hereafter we will refer to this region as stratum 5, because no more than five strata may be recognized on the human Y chromosome.
For a long time, it was believed that recombination between human sex chromosomes was limited to PARs. In recent years, however, the idea that human MSY did not have an independent evolutionary history begun to emerge with the discovery that recombination, in the form of X-to-Y gene conversion, is active in some portions of this genomic region. To date, only three narrow (20–700 bp) regions of MSY have been found to be active X-to-Y gene conversion hotspots in humans (Rosser et al. 2009; Cruciani et al. 2010; Trombetta et al. 2010). One hotspot lies within the VCY genes, in the P8 palindrome, whereas the other two hotspots are located in the evolutionary stratum 5: One in the ARSDP pseudogene (Trombetta et al. 2010; Niederstätter et al. 2013) and the other (termed HSA) near the 5′-end of the PRKY (Schiebel et al. 1997; Rosser et al. 2009; Cruciani et al. 2010). Moreover, using a phylogenetic approach, ancient episodes of X–Y conversion have been inferred in different primates within a 10-kb region of stratum 4 (Iwase et al. 2010) and in Faelidae within ZFX/ZFY genes (Slattery et al. 2000).
In this study, we demonstrate the existence of several X–Y gene conversion hotspots, highlighting that this molecular process can be effective in modulating the sequence landscape of sex chromosomes. More specifically, using human–chimpanzee sequence comparisons of the stratum 5, we identified 19 regions in which X–Y gene conversion has historically occurred. Furthermore, through intraspecific phylogenetic investigation, we found that at least six of these regions have undergone X-to-Y gene conversion during recent human evolution. We also found footprints of recent Y-to-X gene conversion in at least five CERs.
Our interspecies analyses were based on a four-way alignment of human and chimpanzee sex chromosomes. Within the alignment, based on the configuration of nucleotides, variable sites can be classified into several categories (fig. 1A). As the separation of stratum 5 vastly predates human–chimpanzee speciation (Ross et al. 2005), one would expect, assuming no gene conversion, to observe a higher number of N-sites than S-sites and a similar number of singletons between orthologous chromosomes. An overrepresentation of S-sites on the Y chromosome relative to X chromosome is also expected given the higher mutation rate on the Y versus the X chromosome. Furthermore, we would expect to observe a low number of (or zero) C-sites and consequently very low PC values (Osada and Innan 2008). This situation was observed for almost all the alignment. In table 1, there is an example of an uninterrupted 7-kb fragment of alignment, which is representative of stratum 5 (tables 1 and 2) where 625 N-sites, 151 S-sites, and 2 C-sites were counted. The opposite can be seen for 19 narrow regions (CERs1–19, table 1) where the number of observed type-C nucleotide is always significantly higher than expected and PC values are overall very high (table 2). The most parsimonious explanation for this observation is that past gene conversion has played a role in shaping the genetic diversity of these regions in one or in both species. Gene conversion, together with the primary effect of increasing the number of C-sites and decreasing the number of N-sites, may either create or eliminate S-sites within the alignment (fig. 1B). The pattern of variation of the S-sites within the four-way alignment (table 1) can be used to make some inferences about the species (chimpanzee or human) and the direction (X-to-Y or Y-to-X) in which prevalent (but not necessarily exclusive) historical conversion events occurred (fig. 1). For example, a high number of singletons in the X chromosome of Homo sapiens (SHX, see table 1) may indicate prevalent Y-to-X gene conversion in the chimpanzee. This is well illustrated by the situation observed in CER1, in which no singletons were observed in the chimpanzee sex chromosomes, whereas, in humans, several singletons were counted with a three-time excess on the X-chromosome (table 1). In general, a sex chromosome of one species mostly acting as an “acceptor” sequence of gene conversion may result in an excess of singletons in the orthologous chromosome. We analyzed the pattern of variation of S-sites within CERs, and no clear evidence for preferential direction of conversion was observed for 11 blocks (CER2, CER6–13, CER15, and CER18). A preferential X-to-Y gene conversion may be hypothesized in CER4–5 (in chimpanzees) and CER19 (in humans), whereas preferential events in the opposite direction may be inferred in three regions (CER1, CER14, and CER16) in chimpanzees. No particular direction, but a prevalent gene conversion activity in chimpanzees, may be inferred for CER3 and CER17.
Two X–Y gene conversion hotspots (HSA and ARSDP) have been previously described within stratum 5 (Rosser et al. 2009; Trombetta et al. 2010), but only one of them has been detected through the interspecific alignment analysis performed here. In fact, HSA overlaps CER2 in which nine C-sites were counted. Conversely, no C-sites were observed within the four-way alignment of the ARSDP hotspot previously reported. This fact highlights the possibility that some active hotspots may not be recognizable by analyzing the interspecies alignments. The lack of C-sites within the ARSDP hotspot may be probably due to a recent activation of X–Y conversion, which has not had enough time to accumulate an excess of C-sites.
By using an interspecific phylogenetic approach for each CER, we observed a tight clustering of gametologous sequences and/or conflicting evolutionary signals which would seem to indicate gametologous gene conversion. In particular, at least six CERs have had a history of X–Y exchange in both humans and chimpanzees (table 2 and supplementary fig. S3, Supplementary Material online). The existence of shared hotspots across the two species may suggest that their origin predates human–chimpanzee speciation, as already pointed out for the hotspot in the VCY genes (Trombetta et al. 2010). This finding adds to the previous body of evidence in favor of a longer lifespan of nonallelic homologous recombination (NAHR) hotspots compared with the allelic homologous recombination (AHR) ones. In fact, most AHR hotspots that have been detected in the human genome have been shown to not exist in the chimpanzee genome, which indicates a rapid turnover for them (Ptak et al. 2004; Winckler et al. 2005; Auton et al. 2012; Fawcett and Innan 2013), whereas recent studies of NAHR in different primates have shown that these are often shared across evolutionarily distant primate species (Rozen et al. 2003; Bosch et al. 2004; Hurles et al. 2004; Lee et al. 2008; Perry et al. 2008; Iwase et al 2010; Fawcett and Innan 2013).
Gene conversion is a common outcome in recombination-mediated DSB repair processes. This molecular mechanism leads to the formation of a “Holliday junction” that can be resolved by either gene conversion or crossing-over. Therefore, the propensity to resolve DSB with X–Y gene conversion might result in a similar propensity for ectopic X–Y crossing-over. Comparing the positions of the CERs identified here to sites in which an illegitimate X–Y crossing-over has been previously described (Vollrath et al. 1992; Schiebel et al. 1997) made it possible to investigate the potential overlap between gene conversion hotspots and translocation hotspots. Through deletion mapping analysis, it was previously observed that abnormal X–Y interchange happens particularly frequently between X–Y gametologous regions harboring the PRKX/PRKY genomic loci (Schiebel et al. 1997), the same region in which we found more than half of the CERs. In particular, gene conversion hotspots CER2 and CER11 overlap two well-characterized PRKX/PRKY translocation hotspots (HSA and HSB, respectively), in which a high frequency of crossing-over (causing a human genomic disorder known as “sex reversal” [46, XX males and 46, XY females]) has been previously observed (Schiebel et al. 1997). Furthermore, almost all of the CERs identified in this study fall within regions in which at least one pathological illegitimate crossing-over between sex chromosomes has been reported (supplementary table S6, Supplementary Material online). Although it is not possible to mark the exact point of X–Y translocation due to the low resolution of these studies (Vollrath et al. 1992; Schiebel et al. 1997), it is tempting to speculate that CERs well correlate with the chromosome breakpoints in which abnormal exchange occurred. This situation is similar to that observed by Lange et al. (2009) within ampliconic sequences, where both crossover and noncrossover (gene conversion) pathways are active between Y chromosome palindrome arms. Y–Y crossing-over often results in isodicentric Y chromosomes indicating that interchromatid exchange has occurred (Lange et al. 2009). A model has been proposed in which Y–Y gene conversion may be useful in protecting Y chromosome against its evolutionary degradation (Charlesworth 2003; Rozen et al. 2003; Connallon and Clark 2010; Marais et al. 2010; Betrán et al. 2012; Hallast et al. 2013), by facilitating the efficient removal of Y-linked deleterious alleles from the population. There is little obvious fitness cost for this process, as long as crossover events are rare (Lange et al. 2009). This model can be difficult to apply to X–Y gene conversion. In fact, most theories regarding the evolutionary differentiation of X and Y chromosomes posit that recombination between them will be costly because of functional differences between the X-linked and Y-linked gene copies. Sexually antagonistic alleles potentially drive recombination suppression, and these readily accumulate near male-specific regions or male-determining loci (Jordan and Charlesworth 2012; Charlesworth et al. 2014). For those genes where the optimal sequence is the same on the X and Y, gene conversion might be beneficial by aiding the removal of Y-linked deleterious mutations, yet many other genes might not fit this constraint, and for these, gene conversion would be costly, due to the introduction of deleterious variation from the X to the Y, or vice versa. However, it could be argued that, before DNA replication, no homologous sequences can be used by the “haploid” MSY and its gametologous counterpart to repair DSBs in males. So X–Y gene conversion may be the “extrema ratio” for the haploid male sequences to maintain their integrity. In this view, we can hypothesize that CERs may be regions of genomic instability and that gene conversion is the molecular pathway which repairs these regions.
By analyzing the intraspecific MSY diversity, we find here footprints of recent X-to-Y gene conversion in five human CERs (CER6, CER7, CER15, CER17, and CER18). Previous studies on the intraspecific diversity of HSA (overlapping CER2) have shown that this region is also involved in X–Y gene conversion (Rosser et al. 2009; Cruciani et al. 2010), increasing to at least 6 the number of CERs involved in gene conversion in recent human evolution.
X-to-Y gene conversion rate estimates for the five CERs showing X–Y GSV homogenization range from a minimum mean value of 1.8 × 10−8 to a maximum of 1.1 × 10−6 conversion/base/generations. This rate is similar to that reported by Cruciani et al. (2010) for HSA/CER2 and significantly lower than the value calculated for VCY (supplementary fig. S4, Supplementary Material online) (Trombetta et al. 2010). In line with previous considerations, the higher rate estimated for VCY does not necessarily reflect a more intense gene conversion activity, but can be the consequence of multiple VCX sequences acting as donors. The four divergent donor sequences involved in VCX-to-VCY gene conversion (Trombetta et al. 2010) could be a continuous source of X–Y GSV, which would mean that X–Y differences will probably never be zero. Conversely, with only one donor sequence, conversion events can only decrease the number of GSVs.
Present estimates of X-to-Y gene conversion rate are considerably lower than that reported for Y–Y gene conversion (Rozen et al. 2003; Hallast et al. 2013), but similar or even much higher than recent estimates of MSY mutation rate (Francalacci et al. 2013; Mendez et al. 2013; Poznik et al. 2013; Scozzari et al. 2014). Thus, although the present estimates are based on relatively small data sets, it is clear that X-to-Y gene conversion can be highly effective in increasing the level of diversity among human Y chromosomes, although this effect is restricted to the X–Y GSV sites.
The gene-conversion-tract lengths here observed (mean maximum tract length across sites: 47 bp) are comparable with those previously obtained for other X-to-Y gene conversion hotspots: 118 bp at HSA (Cruciani et al. 2010), 95 bp at VCX/VCY (Trombetta et al. 2010), and 64 bp in the ARSDP pseudogene (Niederstätter et al. 2013). These figures are in the range of ectopic gene-conversion-tract lengths for autosomal hotspots (see Chen et al. 2007 for a review). The tract length similarity observed for autosomic and nonautosomic ectopic gene conversion may indicate a similar molecular mechanism regardless of the chromosomal context in which it occurs. The existence of such pervasive X-to-Y gene conversion raises serious questions about the use of SNPs as stable markers in the construction of the phylogenetic tree of the Y chromosome and their use in forensic applications. Events of gene conversion can produce phylogenetically incoherent SNPs creating the same derived polymorphism in several branches of the MSY phylogeny (as observed in fig. 4), or changing the derived state of a SNP in its ancestral state, as already suggested (Adams et al. 2006; Trombetta et al. 2010; Niederstätter et al. 2013). The use of mutations occurring in gene-conversion prone regions may lead to an altered structure of the tree, obscuring signals from other phylogenetic markers. In this regard, it would be interesting to evaluate whether the high proportion (2.9%) of recurrent mutations reported in a recent high-coverage MSY resequencing study (Wei et al. 2013) may be due to gene conversion events. This emphasizes the importance of identifying and characterizing new ectopic gene conversion hotspots within MSY.
Mining the information contained in the dbSNP we also identified signals of Y-to-X gene conversion. In particular, at least five X-linked CERs seem to be active as X–Y gene conversion acceptor sequences. The absence of an unambiguous phylogeny for the X chromosome and the occurrence of recombination during female meiosis make it difficult to understand the evolutionary dynamics (tract lengths and conversion rate) for Y-to-X gene conversion events. Interestingly, with the exception of CER6, no correspondence between X- and Y-linked CERs involved in recent gene conversion was observed, indicating a possible difference between sex chromosomes in the occurrence of DSBs.
Using our approach, the lack of footprints of recent exchange within some of the CERs does not necessarily imply that gene conversion is not ongoing in these sequences. One of the effects of X–Y conversion is that nucleotide differences between paralogous sequences disappear, making it difficult to recognize gene conversion because we rely on the presence of X–Y GSVs to detect it. Therefore, it is possible that, with intraspecific diversity analysis, some X–Y gene conversion events (or entire hotspots) have gone undetected.
To sum up, our results revise and expand previous research on X–Y gene conversion (Rosser et al. 2009; Cruciani et al. 2010; Trombetta et al. 2010) clearly indicating that productive recombination is still active on different portions of stratum 5 of the sex chromosomes, supporting the idea that the sequence landscape of MSY could be modulated by the transfer of genetic information from the X chromosome and vice versa.
Materials and Methods
Interspecies Data Analysis
An approximately 1-Mb human X-linked region (ChrX: 2699520–3689259), which corresponds to the youngest evolutionary stratum (stratum 5 sensu Ross et al. [2005], which corresponds to stratum 9 following the criteria of Pandey et al. [2013]), was extracted from the X chromosome sequence reported in the February 2009 assembly of the UCSC Genome Browser (http://genome.ucsc.edu/, last accessed May 20, 2014). Afterwards, the whole region was divided into 5-kb windows (50% overlapping) to search for paralogous and orthologous regions in humans (GRCh37/hg19) and chimpanzees (CGSG 2.1.3), using the BLAT function of the UCSC genome browser.
The four-way alignment (X and Y chromosomes, both from humans and chimpanzees) was performed using the ClustalW2 software (http://www.ebi.ac.uk/Tools/clustalw2/, last accessed May 20, 2014; Larkin et al. 2007). A portion of the alignment was eliminated from further analysis if 1) one species showed a nonspecified sequence (i.e., a stretch of N), and/or 2) one species showed a larger gap than 100 bp (due to insertion/deletions difference among the four sequences). Variable sites within the alignment have been identified using the DNAsp ver. 4.50.1 software (Rozas et al. 2003). A CER was defined as a region of the alignment with four or more C-sites per kb. The start and end of each CER were considered to be the nucleotide positions of the two outermost C-sites observed in that CER. For S-site and N-site counting, we arbitrarily chose to consider a deletion of more than 10 bp as a single site, which is generated by a single event. An approximately 150-bp deletion in the human X chromosome was considered for the alignment of CER14, due to the large number of C-sites within the alignment. The P value for the observed number of CERs was obtained using a random permutation test. More specifically, we randomized the distribution for the 266 observed C-sites within 456,564 bp of alignment (using the “randbetween” function of Microsoft Excel software), and counted the region with four or more C-sites/kb. This process was replicated 1,000 times, which produced the null distribution of CERs (supplementary fig. S1, Supplementary Material online). We determined the P value of observing ≥19 CERs assuming that their null distribution follows a Poisson distribution.
The expected number of C-sites is given by the formula:
where L is the length of the alignment (in bp) and P0 is the number of nucleotide substitutions per site between the orthologous sequences as indicated in Osada and Innan (2008). The P values resulting from the test for gene conversion (table 2) were calculated according to Osada and Innan (2008), using the formula:
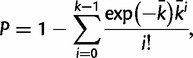 |
in which K is the observed number of C-sites. The PC value (Kijima and Innan 2010) was calculated using the following equation: PC = nC/(nC + nN), where nC and nN are the numbers of type-C and type-N sites, respectively. Interspecific NJ trees and Split Decomposition networks were constructed using the SplitsTree program (Huson and Bryant 2006).
Identification of SNPs on the Human Y Chromosome
We analyzed a total of 68 samples chosen from the collections of the authors. Human Y chromosomes to be sequenced were selected on the basis of their haplogroups, which had been determined in previous studies (Cruciani et al. 2004, 2007, 2011; Trombetta et al. 2011; Scozzari et al. 2012). Haplogroups to be analyzed were chosen in order to maximize the coverage of the Y phylogeny (fig. 4). In most cases, DNA was prepared from fresh venous blood. The study was approved by the ethical committee of the “Policlinico Umberto I, Sapienza Università di Roma.”
Overall, 16 kb from the stratum 5 of the human Y chromosome was sequenced. We designed PCR and sequence primers (available on request) for each CER on the basis of the Y-chromosome sequence reported in the February 2009 assembly of the UCSC Genome Browser using Primer3 software (http://genome.ucsc.edu/; http://frodo.wi.mit.edu/primer3/, last accessed May 20, 2014). Sequencing templates were obtained through PCR in a 50 µl reaction containing 50 ng of genomic DNA, 200 µM of each deoxyribonucleotide, 2.5 mM MgCl2, 1 unit of Taq polymerase, and 10 pmol of each primer. A touch-down PCR program was used with an annealing temperature decreasing from 62 to 55 °C over 14 cycles, followed by 30 cycles with an annealing temperature of 55 °C. Y-specificity of the PCR products was confirmed by using female genomic DNAs as a negative control. Following DNA amplification, PCR products were purified using the 715QIAquick PCR purification kit (Qiagen, Hilden, Germany). Cycle sequencing was performed using the BigDye Terminator Cycle Sequencing Kit with Amplitaq DNA polymerase (Applied Biosystems, Foster City, CA) and an internal or PCR primer. Cycle sequencing products were purified by ethanol precipitation and run on an ABI Prism 3730XL DNA sequencer (Applied Biosystems). Chromatograms were aligned and analyzed for mutations using Sequencher 4.8 (Gene Codes Corporation, Ann Arbor, MI).
Identification of SNPs on the Human X Chromosome
Human X-linked polymorphisms located within the CERs were extracted from the build 137 of dbSNP (Supplementary table S5, Supplementary Material online) using the table function of the UCSC genome browser. We only used polymorphisms with a minor allele frequency >1%.
Intraspecies Data Analysis
Gene conversion between gametologs can have two effects: On the one hand, it can increase overall sequence similarity between them; on the other hand, when it involves GSVs, it can generate an excess of genetic diversity among allelic copies (Trombetta et al. 2010). This is because the gametologous base on the donor sequence will change the base on the acceptor chromosome (Rosser et al. 2009; Trombetta et al. 2010) and a new Y (or X) chromosome SNP will be observed in the population. When gene conversion events between sex chromosomes have contributed to their variation, we expect to find an excess of SNPs at X–Y GSV sites, where the Y-linked derived allele is the same as the gametologous sequence on the X or vice versa. Critical points for the success of this analysis regard the correct inference about the direction of the mutation and the identification of an actual ancestral X or Y donor sequence.
The direction of the mutation for each Y-linked polymorphism was unambiguously determined by placing it in the context of the well-known intraspecific human Y chromosome phylogeny (Karafet et al. 2008, Scozzari et al. 2012, 2014). In absence of an unambiguous human X chromosome phylogeny, the direction for mutations on this chromosome was inferred by using the orthologous position on the chimpanzee X chromosome. It should be noted that this inference may be affected by mutation occurring in the chimpanzee lineage, which may lead to the erroneous assignment of mutation direction (Chimpanzee Sequencing and Analysis Consortium 2005).
The ancestral sequence for the human Y chromosome was determined by using Y chromosome phylogenetic information obtained in this study (fig. 4), whereas the actual ancestral sequence for the X chromosome was obtained from the reference sequence (GRCh37/hg19) taking into account outgroup information in correspondence to polymorphic sites.
Comparing the ancestral X and Y sequences, a site was considered to be a GSV whenever a difference was found between them. We arbitrarily chose not to consider sequences of more than five nonaligning contiguous bases as an X–Y GSV. For each CER, the expected number of mutations falling in X–Y GSVs was calculated as the product of the total number of SNPs detected in this study and the proportion of GSVs observed in the same region. To evaluate whether the Y- or the X-linked SNPs were randomly distributed compared with X–Y GSV and non-GSV, we used a Fisher exact test on 2 × 2 contingency tables (variant sites/invariant sites vs. GSVs/non-GSVs). The null hypothesis is set assuming a random occurrence of mutations within the sequence relative to GSV sites. The test was also applied to each of the CERs showing SNPs occurring at GSV sites.
The rate of X-to-Y gene conversion was calculated using the equation of Cruciani et al. (2010):
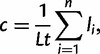 |
where c is the estimated rate of gene conversion per base per generation, n is the number of observed gene conversion events, li is the length in bp of the ith gene conversion events, L is the length in bp of the CER under study, and t is the number of generations in the tree. To estimate the total time spanned by all branches in the tree (the [t] parameter in the previous equation), we used the equation of Repping et al. (2006):
with the following parameters: We considered 47 as the total number of SNPs identified in the analyzed regions (considering only SNPs that were not due to X-to-Y gene conversion [Stot]), 2.5 as the average number of mutations on path from the root (Sh), and 141,500 years as the time to the most recent common ancestor (th) (Cruciani et al. 2011). The maximum and minimum gene conversion tracts were used to calculate the range of gene conversion rate. The number of generations was calculated using a generation time of 25 years. It is worth noting that the lack of an unambiguous phylogeny for the X chromosome implies that a conversion rate for the X chromosome cannot be estimated in the same way as for the Y.
Nucleotide diversity (π) and its standard deviation were calculated according to Nei (1987).
Supplementary Material
Supplementary tables S1–S6 and figures S1–S4 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
This work was supported by Sapienza Università di Roma, Ricerche Universitarie grant number C26A13S9AR to F.C.; and Istituto Pasteur—Fondazione Cenci Bolognetti, Programmi di Ricerca 2013–2014 to F.C.
References
- Adams SM, King TE, Bosch E, Jobling MA. The case of the unreliable SNP: recurrent back-mutation of Y-chromosomal marker P25 through gene conversion. Forensic Sci Int. 2006;159:14–20. doi: 10.1016/j.forsciint.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Auton A, Fledel-Alon A, Pfeifer S, Venn O, Ségurel L, Street T, Leffler EM, Bowden R, Aneas I, Broxholme J, et al. A fine-scale chimpanzee genetic map from population sequencing. Science. 2012;336:193–198. doi: 10.1126/science.1216872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachtrog D. Y-chromosome evolution: emerging insights into processes of Y-chromosome degeneration. Nat Rev Genet. 2013;14:113–124. doi: 10.1038/nrg3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betrán E, Demuth JP, Williford A. Why chromosome palindromes? Int J Evol Biol. 2012;2012:207958. doi: 10.1155/2012/207958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bininda-Emonds OR, Cardillo M, Jones KE, MacPhee RD, Beck RM, Grenyer R, Price SA, Vos RA, Gittleman JL, Purvis A. The delayed rise of present-day mammals. Nature. 2007;446:507–512. doi: 10.1038/nature05634. [DOI] [PubMed] [Google Scholar]
- Bosch E, Hurles ME, Navarro A, Jobling MA. Dynamics of a human interparalog gene conversion hotspot. Genome Res. 2004;14:835–844. doi: 10.1101/gr.2177404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B. The organization and evolution of the human Y chromosome. Genome Biol. 2003;4:226. doi: 10.1186/gb-2003-4-9-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Jordan CY, Charlesworth D. The evolutionary dynamics of sexually antagonistic mutations in pseudoautosomal regions of sex chromosomes. Evolution. 2014;68:1339–1350. doi: 10.1111/evo.12364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J-M, Cooper DN, Chuzhanova N, Férec C, Patrinos GP. Gene conversion: mechanisms, evolution and human disease. Nat Rev Genet. 2007;8:762–775. doi: 10.1038/nrg2193. [DOI] [PubMed] [Google Scholar]
- Chimpanzee Sequencing and Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437:69–87. doi: 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- Connallon T, Clark AG. Gene duplication, gene conversion and the evolution of the Y chromosome. Genetics. 2010;186:277–286. doi: 10.1534/genetics.110.116756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruciani F, La Fratta R, Santolamazza P, Sellitto D, Pascone R, Moral P, Watson E, Guida V, Colomb EB, Zaharova B, et al. Phylogeographic analysis of haplogroup E3b (E-M215) Y chromosomes reveals multiple migratory events within and out of Africa. Am J Hum Genet. 2004;74:1014–1022. doi: 10.1086/386294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruciani F, La Fratta R, Trombetta B, Santolamazza P, Sellitto D, Colomb EB, Dugoujon J-M, Crivellaro F, Benincasa T, Pascone R, et al. Tracing past human male movements in northern/eastern Africa and western Eurasia: new clues from Y-chromosomal haplogroups E-M78 and J-M12. Mol Biol Evol. 2007;24:1300–1311. doi: 10.1093/molbev/msm049. [DOI] [PubMed] [Google Scholar]
- Cruciani F, Trombetta B, Macaulay V, Scozzari R. About the X-to-Y gene conversion rate. Am J Hum Genet. 2010;86:495–497. doi: 10.1016/j.ajhg.2010.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruciani F, Trombetta B, Massaia A, Destro-Bisol G, Sellitto D, Scozzari R. A revised root for the human Y chromosomal phylogenetic tree: the origin of patrilineal diversity in Africa. Am J Hum Genet. 2011;88:814–818. doi: 10.1016/j.ajhg.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren H. Sex-chromosome evolution: recent progress and the influence of male and female heterogamety. Nat Rev Genet. 2011;12:157–166. doi: 10.1038/nrg2948. [DOI] [PubMed] [Google Scholar]
- Fawcett JA, Innan H. The role of gene conversion in preserving rearrangement hotspots in the human genome. Trends Genet. 2013;29:561–568. doi: 10.1016/j.tig.2013.07.002. [DOI] [PubMed] [Google Scholar]
- Francalacci P, Morelli L, Angius A, Berutti R, Reinier F, Atzeni R, Pilu R, Busonero F, Maschio A, Zara I, et al. Low-pass DNA sequencing of 1200 Sardinians reconstructs European Y-chromosome phylogeny. Science. 2013;341:565–569. doi: 10.1126/science.1237947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallast P, Balaresque P, Bowden GR, Ballereau S, Jobling MA. Recombination dynamics of a human Y-chromosomal palindrome: rapid GC-biased gene conversion, multi-kilobase conversion tracts, and rare inversions. PLoS Genet. 2013;9:e1003666. doi: 10.1371/journal.pgen.1003666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JF, Skaletsky H, Brown LG, Brown LG, Pyntikova T, Graves T, Fulton RS, Dugan S, Ding Y, Buhay CJ, et al. Strict evolutionary conservation followed rapid gene loss on human and rhesus Y chromosomes. Nature. 2012;483:82–86. doi: 10.1038/nature10843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurles ME, Willey D, Matthews L, Hussain SS. Origins of chromosomal rearrangement hotspots in the human genome: evidence from the AZFa deletion hotspots. Genome Biol. 2004;5:R55. doi: 10.1186/gb-2004-5-8-r55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23:254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- Iwase M, Satta Y, Hirai H, Hirai Y, Takahata N. Frequent gene conversion events between the X and Y homologous chromosomal regions in primates. BMC Evol Biol. 2010;10:225. doi: 10.1186/1471-2148-10-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobling MA, Pandya A, Tyler-Smith C. The Y chromosome in forensic analysis and paternity testing. Int J Legal Med. 1997;110:118–124. doi: 10.1007/s004140050050. [DOI] [PubMed] [Google Scholar]
- Jordan CY, Charlesworth D. The potential for sexually antagonistic polymorphism in different genome regions. Evolution. 2012;66:505–516. doi: 10.1111/j.1558-5646.2011.01448.x. [DOI] [PubMed] [Google Scholar]
- Karafet TM, Mendez FL, Meilerman MB, Underhill PA, Zegura SL, Hammer MF. New binary polymorphisms reshape and increase resolution of the human Y chromosomal haplogroup tree. Genome Res. 2008;18:830–838. doi: 10.1101/gr.7172008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsura Y, Satta Y. No evidence for a second evolutionary stratum during the early evolution of mammalian sex chromosomes. PLoS One. 2012;7:e45488. doi: 10.1371/journal.pone.0045488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kijima TE, Innan H. On the estimation of the insertion time of LTR retrotransposable elements. Mol Biol Evol. 2010;27:896–904. doi: 10.1093/molbev/msp295. [DOI] [PubMed] [Google Scholar]
- Krausz C, Quintana-Murci L, Forti G. Y chromosome polymorphisms in medicine. Ann Med. 2004;36:573–583. doi: 10.1080/07853890410018853. [DOI] [PubMed] [Google Scholar]
- Lahn BT, Page DC. Four evolutionary strata on the human X chromosome. Science. 1999;286:964–967. doi: 10.1126/science.286.5441.964. [DOI] [PubMed] [Google Scholar]
- Lange J, Skaletsky H, van Daalen SK, Embry SL, Korver CM, Brown LG, Oates RD, Silber S, Repping S, Page DC. Isodicentric Y chromosomes and sex disorders as byproducts of homologous recombination that maintains palindromes. Cell. 2009;138:855–869. doi: 10.1016/j.cell.2009.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. ClustalW and ClustalX version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Lee AS, Gutiérrez-Arcelus M, Perry GH, Vallender EJ, Johnson WE, Miller GM, Korbel JO, Lee C. Analysis of copy number variation in the rhesus macaque genome identifies candidate loci for evolutionary and human disease studies. Hum Mol Genet. 2008;17:1127–1136. doi: 10.1093/hmg/ddn002. [DOI] [PubMed] [Google Scholar]
- Marais GA, Campos PR, Gordo I. Can intra-Y gene conversion oppose the degeneration of the human Y chromosome? A simulation study. Genome Biol Evol. 2010;2:347–357. doi: 10.1093/gbe/evq026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez FL, Krahn T, Schrack B, Krahn AM, Veeramah KR, Woerner AE, Fomine FL, Bradman N, Thomas MG, Karafet TM, et al. An African American paternal lineage adds an extremely ancient root to the human Y chromosome phylogenetic tree. Am J Hum Genet. 2013;92:454–459. doi: 10.1016/j.ajhg.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M. Molecular evolutionary genetics. New York: Columbia University Press; 1987. [Google Scholar]
- Niederstätter H, Berger B, Erhart D, Willuweit S, Geppert M, Gassner C, Schennach H, Parson W, Roewer L. Multiple recurrent mutations at four human Y-chromosomal single nucleotide polymorphism sites in a 37bp sequence tract on the ARSDP1 pseudogene. Forensic Sci Int Genet. 2013;7:593–600. doi: 10.1016/j.fsigen.2013.05.010. [DOI] [PubMed] [Google Scholar]
- Osada N, Innan H. Duplication and gene conversion in the Drosophila melanogaster genome. PLoS Genet. 2008;4:e1000305. doi: 10.1371/journal.pgen.1000305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey RS, Wilson Sayres MA, Azad RK. Detecting evolutionary strata on the human X chromosome in the absence of gametologous Y-linked sequences. Genome Biol Evol. 2013;5:1863–1871. doi: 10.1093/gbe/evt139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry GH, Yang F, Marques-Bonet T, Murphy C, Fitzgerald T, Lee AS, Hyland C, Stone AC, Hurles ME, Tyler-Smith C, et al. Copy number variation and evolution in humans and chimpanzees. Genome Res. 2008;18:1698–1710. doi: 10.1101/gr.082016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poznik GD, Henn BM, Yee MC, Sliwerska E, Euskirchen GM, Lin AA, Snyder M, Quintana-Murci L, Kidd JM, Underhill PA, et al. Sequencing Y chromosomes resolves discrepancy in time to common ancestor of males versus females. Science. 2013;341:562–565. doi: 10.1126/science.1237619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptak SE, Roeder AD, Stephens M, Gilad Y, Paäbo S, Przeworski M. Absence of the TAP2 human recombination hotspot in chimpanzees. PLoS Biol. 2004;2:849–855. doi: 10.1371/journal.pbio.0020155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repping S, van Daalen SKM, Brown LG, Korver CM, Lange J, Marszalek JD, Pyntikova T, van der Veen F, Skaletsky H, Page DC, et al. High mutation rates have driven extensive structural polymorphism among human Y chromosomes. Nat Genet. 2006;38:463–467. doi: 10.1038/ng1754. [DOI] [PubMed] [Google Scholar]
- Ross MT, Grafham DV, Coffey AJ, Scherer S, McLay K, Muzny D, Platzer M, Howell GR, Burrows C, Bird CP, et al. The DNA sequence of the human X chromosome. Nature. 2005;434:325–337. doi: 10.1038/nature03440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosser ZH, Balaresque P, Jobling MA. Gene conversion between the X chromosome and the male-specific region of the Y chromosome at a translocation hotspot. Am J Hum Genet. 2009;85:130–134. doi: 10.1016/j.ajhg.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas J, Sánchez-DelBarrio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- Rozen S, Skaletsky H, Marszalek JD, Minx PJ, Cordum HS, Waterston RH, Wilson RK, Page DC. Abundant gene conversion between arms of palindromes in human and ape Y chromosomes. Nature. 2003;423:873–876. doi: 10.1038/nature01723. [DOI] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–4425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Schiebel K, Winkelmann M, Mertz A, Xu X, Page DC, Weil D, Petit C, Rappold GA. Abnormal X-Y interchange between a novel isolated protein kinase gene, PRKY, and its homologue, PRKX, accounts for one third of all (Y+)XX males and (Y-)X-Y females. Hum Mol Genet. 1997;6:1985–9198. doi: 10.1093/hmg/6.11.1985. [DOI] [PubMed] [Google Scholar]
- Scozzari R, Massaia A, D’Atanasio E, Myres NM, Perego UA, Trombetta B, Cruciani F. Molecular dissection of the basal clades in the human Y chromosome phylogenetic tree. PLoS One. 2012;7:e49170. doi: 10.1371/journal.pone.0049170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scozzari R, Massaia A, Trombetta B, Bellusci G, Myres NM, Novelletto A, Cruciani F. An unbiased resource of novel SNP markers provides a new chronology for the human Y chromosome and reveals a deep phylogenetic structure in Africa. Genome Res. 2014;24:535–544. doi: 10.1101/gr.160788.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen P, Wang F, Underhill PA, Franco C, Yang W-H, Roxas A, Sung R, Lin AA, Hyman RW, Vollrath D, et al. Population genetic implications from sequence variation in four Y chromosome genes. Proc Natl Acad Sci U S A. 2000;97:7354–7359. doi: 10.1073/pnas.97.13.7354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaletsky H, Kuroda-Kawaguchi T, Minx PJ, Cordum HS, Hillier L, Brown LG, Repping S, Pyntikova T, Ali J, Bieri T, et al. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature. 2003;423:825–837. doi: 10.1038/nature01722. [DOI] [PubMed] [Google Scholar]
- Slattery JP, Sanner-Wachter L, O’Brien SJ. Novel gene conversion between X–Y homologues located in the non-recombining region of the Y chromosome in Felidae (Mammalia) Proc Natl Acad Sci U S A. 2000;97:5307–5312. doi: 10.1073/pnas.97.10.5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trombetta B, Cruciani F, Sellitto D, Scozzari R. A new topology of the human Y chromosome haplogroup E1b1 (E-P2) revealed through the use of newly characterized binary polymorphisms. PLoS One. 2011;6:e16073. doi: 10.1371/journal.pone.0016073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trombetta B, Cruciani F, Underhill PA, Sellitto D, Scozzari R. Footprints of X-to-Y gene conversion in recent human evolution. Mol Biol Evol. 2010;27:714–725. doi: 10.1093/molbev/msp231. [DOI] [PubMed] [Google Scholar]
- Underhill PA, Kivisild T. Use of y chromosome and mitochondrial DNA population structure in tracing human migrations. Annu Rev Genet. 2007;41:539–564. doi: 10.1146/annurev.genet.41.110306.130407. [DOI] [PubMed] [Google Scholar]
- Veyrunes F, Waters PD, Miethke P, Rens W, McMillan D, Alsop AE, Grützner F, Deakin JE, Whittington CM, Schatzkamer K, et al. Bird-like sex chromosomes of platypus imply recent origin of mammal sex chromosomes. Genome Res. 2008;18:965–973. doi: 10.1101/gr.7101908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollrath D, Foote S, Hilton A, Brown LG, Beer-Romero P, Bogan JS, Page DC. The human Y chromosome: a 43-interval map based on naturally occurring deletions. Science. 1992;258:52–59. doi: 10.1126/science.1439769. [DOI] [PubMed] [Google Scholar]
- Wei W, Ayub Q, Chen Y, McCarthy S, Hou Y, Carbone I, Xue Y, Tyler-Smith C. A calibrated human Y-chromosomal phylogeny based on resequencing. Genome Res. 2013;23:388–395. doi: 10.1101/gr.143198.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winckler W, Myers SR, Richter DJ, Onofrio RC, McDonald GJ, Bontrop RE, McVean GA, Gabriel SB, Reich D, Donnelly P, et al. Comparison of fine-scale recombination rates in humans and chimpanzees. Science. 2005;308:107–111. doi: 10.1126/science.1105322. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.