

Abstract
The development of selective agents capable of discriminating between protein kinase C (PKC) isoforms and other diacylglycerol (DAG)-responsive C1 domain-containing proteins represents an important challenge. Recent studies have highlighted the role that Ras guanine nucleotide-releasing protein (RasGRP) isoforms play both in immune responses as well as in the development of prostate cancer and melanoma, suggesting that the discovery of selective ligands could have potential therapeutic value. Thus far, the N-methyl-substituted indololactone 1 is the agonist with the highest reported potency and selectivity for RasGRP relative to PKC. Here we present the synthesis, binding studies, cellular assays and biophysical analysis of interactions with model membranes of a family of regioisomers of 1 (compounds 2 to 5) that differ in the position of the linkage between the indole ring and the lactone moiety. These structural variations were studied to explore the interaction of the active complex (C1 domain-ligand) with cellular membranes, which is believed to be an important factor for selectivity in the activation of DAG-responsive C1 domain containing signaling proteins. All compounds were potent and selective activators of RasGRP when compared to PKCα with selectivities ranging from 6 to 65 fold. However, the parent compound 1 was appreciably more selective than any of the other isomers. In intact cells, modest differences in the patterns of translocation of the C1 domain targets were observed. Biophysical studies using giant vesicles as model membranes did show substantial differences in terms of molecular interactions impacting lipid organization, dynamics and membrane insertion. However, these differences did not yield correspondingly large changes in patterns of biological response, at least for the parameters examined.
Keywords: Indolo-lactones, C1 domain, RasGRP, cancer
1. Introduction
The lipid diacylglycerol (DAG) represents one of the central second messengers in cell signaling. Increased levels of DAG in the plasma membrane occur in response to either tyrosine-kinase receptor or G-protein coupled receptor stimulation, primarily through the activation of phospholipase C. This enzyme catalyzes the hydrolysis of phosphatidylinositol 4,5-bisphosphate into a pair of second messengers: inositol triphosphate and DAG. Increased levels of DAG are transduced into cellular signals via various PKC isoforms and six other families of proteins through the interaction with structurally similar C1 domains.1,2,3
C1 domains are zinc finger structures of approximately 50 amino acids in length that were originally discovered as lipid binding modules in PKCs, a family of related kinases that regulate proliferation, differentiation and malignant transformation.4 C1 domains can be sub-classified into two families, those that are responsive to DAG and those that are not. The classic and novel PKC isoforms represent the first recognized and most extensively studied family of effectors for DAG.5,6 The C1 domains of the atypical PKCs provide one paradigm for the so-called “atypical” C1 domains that do not respond to DAG.
There is a large body of evidence supporting the potential of PKC isoforms as therapeutic targets in cancer, immunological, cardiovascular and neurological diseases. Examples of DAG mimetics in clinical trials that bind the C1 domain are the natural products bryostatin 1 and ingenol 3-angelate (PEP005).7 Indeed, the use of ingenol-3-angelate for the topical treatment of actinic keratosis has been approved recently.8,9 After the identification of the PKCs, six other families of proteins with homologous, DAG-responsive C1 domains have been identified. The protein kinase D family is involved in Golgi function, proliferation, metastasis and apoptosis.10 The chimaerins act as inhibitors (GAPs, GTPase activating proteins) for the small GTPase Rac and are candidate tumor suppressors. 11 The Unc-13 family members are responsible for promoting vesicle priming.12 The DAG kinases terminate DAG signaling by phosphorylating DAG.13 MRCK is a downstream effector of cdc42 involved in filopodia formation, contributing to tumor invasion.14 Finally, the RasGRP family members function as activators (GEFs, GTP Exchange factors) for Ras 15 and are prominently expressed in blood cells. RasGRP malfunction likely contributes to autoimmunity and may contribute to blood malignancies. 16 In addition, the role of RasGRP3 in prostate cancer 17 and melanoma 18 has been demonstrated and, very recently, it was shown that RasGRPs are targets of the anticancer drug ingenol-3-angelate.19 Given the important biological roles of RasGRP family members, the discovery of selective agents capable of specifically interacting with their C1 domain could provide exciting lead structures for drug development.
When DAG or phorbol esters bind to the C1 domain, they complete a hydrophobic surface on the top face of it. Likewise, the hydrophobic substituents on the ligand contribute an additional hydrophobic element. Together, these factors drive membrane association of the C1 domain and the accompanying stabilization of the active, membrane-associated conformation of the protein. Because the membrane environment constitutes the third element of the ternary binding complex, the membrane microdomain composition, as well as its variation as a function of localization within the cell, provides a potentially important basis for establishing selectivity of regulation.20
DAG-lactones were developed as a synthetically more tractable alternative to complex natural products such as bryostatin 1 or phorbol esters that are ultrapotent analogues of DAG. DAG-lactones combine a rigid template, responsible for enhanced affinity, with relative chemical simplicity, allowing the synthetic exploration of a wide chemical space through the combined action of a myriad of substituents at the sn-1 and sn-2 positions that we have defined as “chemical zip codes”.21 As reported previously, nanomolar binding affinities for PKC approaching those of natural products have been achieved.20 Furthermore, compounds with marked binding selectivity for RasGRP, as compared to PKC, were developed with the most selective compound incorporating a 1-methyl-1H-indole ring at the sn-2 position of the DAG-lactone (Compound 1, Figure 1).22 Following up on these findings, two other DAG-indololactones were prepared to evaluate the effect on binding affinity and selectivity as a function of the point of attachment on the pyrrole ring of the 1-methyl-1H-indole, as well as the role of the 1-methyl group. While selectivity did not improve, our characterization revealed that the orientation of the 1-methyl-1H-indole ring on the DAG-lactone had substantial effects on the nature of the interaction of the ligand with the membrane. Furthermore, it was found that the removal of the 1-methyl group produced only an insignificant change in binding affinity, suggesting that the free indole NH was not involved in any critical interaction.23 Since the presence of the 1-methyl group greatly facilitates the syntheses, we chose to prepare the complete family of regioisomeric DAG-lactones with an invariant 1-methyl-1H-indole ring to study their PKC and RasGRP binding affinities, biological activity, and translocation characteristics (Figure 1). These isomers differ from one another in terms of the position of the linkage between the benzene ring of the 1-methyl-1H-indole and the DAG-lactone, which changes the orientation of the heteroaromatic ring relative to the lactone template. In addition, we examined the influence of the various 1-methyl-1H-indole ring orientations with model membrane vesicle systems using a range of biophysical techniques. Given that the proposed binding mode for DAG-lactones is with the α-arylidene moiety oriented toward the surface of the C1 domain, adjacent to the lipid interface,24 we postulated that the orientation of the 1-methyl-1H-indole group at this position could influence membrane binding selectivity. Our results show that indeed the orientation of the 1-methyl-1H-indole group plays a critical role in selectivity with the most selective compound being the one linked through position 3 (1). Moreover, the biophysical data suggest a differential mode of membrane interaction depending on the point of attachment. Finally, the successful outcome of this study was critically dependent on the development of a new synthetic strategy for the synthesis of the novel analogues 2-5 that took into account the high reactivity of the 2- and 3-unsubstituted 1-methyl-1H-indole ring.
Figure 1.

Structures of DAG-indololactones.
2. Results
2.1. Chemistry
The synthesis of compound 5 was initially attempted following the same strategy reported for lead compound 1 using bis-silylated lactone 6a as starting material.22 Unfortunately, the selective final monoacylation was not reproducible, giving low yields and varying amounts of diacylated product (Scheme 1). Alternatively, we tried to prepare compound 5 starting from the known lactone 6b, protected with a benzyl and a TBDPS groups as already reported for related derivatives of compound 1.23 In the present case, total decomposition was observed when treating the precursor 7b under typical conditions employed for benzyl group cleavage (BCl3, −78 °C, CH2Cl2) for this type of compounds. In addition, several attempts to selectively remove the benzyl group in the presence of the conjugated double bond by transfer hydrogenation were made. Unfortunately, in our hands, no selectivity could be achieved employing different hydrogen sources and catalysts.25,26 In all cases, we obtained complex mixtures of the desired product together with considerable amounts of the corresponding saturated derivatives.
Scheme 1.
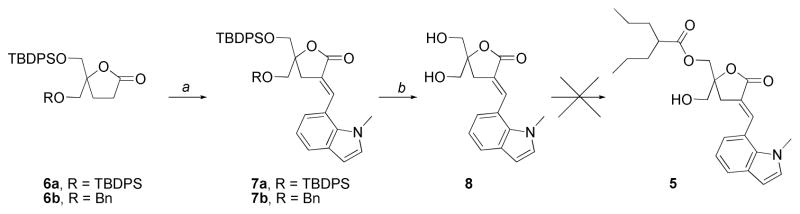
Monoacylation approach towards DAG-indololactones. Reagents and Conditions: (a). i. LiHMDS, indole-7-carboxyaldehyde, THF, −78 °C; ii. Et3N, MsCl, DBU, CH2Cl2, 0°C → rt, 78% (7a), 61% (7b); (b) HF•Et3N, THF, 70 °C, 74%.
In order to overcome these difficulties, we decided to prepare a new starting lactone 11 protected with TBDPS and TBDMS groups. This strategy was based on the possible selective removal, under very mild conditions (PPTS, EtOH),27 of the TBDMS protecting group in the presence of the TBDPS group (Scheme 2). We envisioned that these conditions would be compatible with the presence of highly reactive 2- and 3-unsubstituted indole rings. Therefore, starting from commercially available 1,3-dihydroxyacetone dimer, the monoprotected 1,3-dihydroxyketone was obtained.28 Introduction of the second silyl protective group was performed under standard conditions to yield the fully protected ketone 9. Then, treatment with allylmagnesium chloride produced the homoallylic alcohol 10 in excellent yield and purity. Finally, tandem hydroboration and PCC oxidation yielded the protected lactone 11 with 74% yield.
Scheme 2.
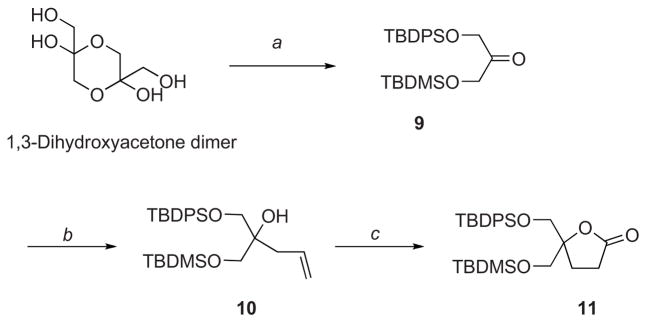
Synthesis of starting lactone 11. Reagents and Conditions: (a) i. TBDPSCl, imidazole, DMF, 0°C, 51%; ii. TBDMSCl, imidazole, CH2Cl2, 0°C, 87%; (b) Allylmagnesium chloride, THF, 0 °C → rt, 89%; (c) i. BH3•SMe2 in THF, CH2Cl2, −78°C → rt; ii. PCC, CH2Cl2, rt, 74% (two steps).
With key intermediate 11 at hand, the desired positional isomers of lead compound 1 were prepared as depicted in Scheme 3. Thus, target compounds 2, 3, 4 and 5 were synthesized by sequential alkylation-elimination followed by selective deprotection of the TBDMS group, acylation of the free primary hydroxyl group, and final deprotection of the second primary hydroxyl group. In all cases, olefination of the aldol products resulting from the reaction with the corresponding 1-methylindole-carboxaldehydes afforded almost exclusively the E-isomers. The E/Z geometry around the double bond was assigned by 1H NMR; the vinyl proton for the E-isomers displayed a characteristic multiplet that was further downfield from that of the corresponding Z-isomers.29 Selective deprotection was achieved by treatment of alkylated products 12a-d in ethanolic solution with PPTS at 60 °C. Under these conditions, the desired alcohols 13a-d were obtained in very good yields. Acylation followed by deprotection afforded the expected DAG-indololactones 2, 3, 4 and 5.
Scheme 3.

Synthesis of regioisomeric DAG-indololactones. Reagents and Conditions: (a) i. LiHMDS, indole-aldehyde, THF, −78 °C; ii. Et3N, MsCl, DBU, CH2Cl2, 0°C → rt; (b) PPTS, EtOH, 60 °C; (c) Et3N, DMAP, (CH2CH2CH3)2CHCOCl, CH2Cl2; (d) HF•Et3N, THF, 70 °C.
2.2. Biological Results
2.2.1. Binding of ligands to PKCα and to RasGRP1/3
To examine possible binding selectivity, the binding affinities of compounds 1-5 to PKCα, to PKCε, to the C1 domain of RasGRP1, and to RasGRP3 were measured by competition for [3H]PDBu binding in the presence of 100 μg/ml phosphatidylserine (Table 1). All compounds were appreciably more selective for the RasGRP isoforms than for PKCα or PKCε, with selectivities ranging from 5 – 84 fold. Relative to both PKC isoforms, the parent compound 1 was the most selective. Compound 3 approached compound 1 in selectivity with respect to PKCα, being about half as selective, and compound 4 was the least selective. Differences in selectivity were somewhat more pronounced when compared to PKCε (up to 64–84-fold) than they were for PKCα (up to 49–65-fold). In terms of absolute potencies for RasGRP1/3, the compounds ranged from slightly more potent than the phorbol ester PDBu to 13-fold less potent. For both PKCα and PKCε, the compounds were all appreciably less potent than PDBu, reflecting their selectivity. Compounds 1 and 3 were the most potent for RasGRP1/3. For RasGRP1/3 and for PKCα, compound 2 was the least potent. For PKCε, compound 1 was a little less potent. RasGRP3 and the C1 domain of RasGRP1 behaved very similarly to one another.
Table 1.
Binding Selectivity of DAG-indololactones for PKCα and PKCε versus RasGRP.
| Compound | PKCαa Ki(nM) | PKCε Ki(nM) | RasGRP3 Ki(nM) | PKCα: RasGRP3 | PKCε: RasGRP3 | RasGRP1-C1 Ki(nM) | PKCα: RasGRP1 | PKCε: RasGRP1 |
|---|---|---|---|---|---|---|---|---|
| 1 | 16.2 ± 1.0 | 21.1 ± 1.9 | 0.33 ± 0.06 | 49 | 64 | 0.25 ± 0.10 | 65 | 84 |
| 2 | 17.8 ± 2.0 | 14.6 ± 2.7 | 1.63 ± 0.18 | 11 | 9.0 | 1.55 ± 0.10 | 10 | 9.4 |
| 3 | 8.25 ± 0.88 | 2.17 ± 0.36 | 0.34 ± 0.01 | 24 | 6.4 | 0.41 ± 0.10 | 24 | 5.3 |
| 4 | 7.5 ± 1.0 | 8.08 ± 0.31 | 1.22 ± 0.21 | 6 | 6.6 | 0.87 ± 0.04 | 9 | 9.3 |
| 5 | 12.9 ± 1.4 | 11.7 ± 0.30 | 1.12 ± 0.05 | 10 | 11 | 0.69 ± 0.14 | 19 | 17 |
| [3H]PDBub | 0.17 ± 0.02 | 0.22 ± 0.05 | 0.42 ± 0.03 | --- | — | 0.12 ± 0.01 | — | — |
Values represent mean ± SEM of triplicate independent experiments
[Kd(nM]
2.2.2. Biological activity of ligands
The biological activities and relative potencies of compounds 1-5 were examined in HEK293 cells and compared with that in HEK293 cells transfected with RasGRP3. PMA was used as a phorbol ester control. Erk1/2 phosphorylation is a reporter for activation of the Ras pathway and thus should reflect RasGRP3 activation. Phosphorylation of PKCδ at S299 provides a measure of PKCδ activation,30 while phosphorylation of RasGRP3 at T133 is thought to be needed for activation,31,32 along with occupancy of its C1 domain by DAG, phorbol ester, or related ligands,33 and is brought about by various PKC isoforms.34,35 All five compounds behaved similarly, within the range of resolution of the experiments. As illustrated for compound 1 (Figure 2A), Erk1/2 phosphorylation was greatly enhanced in the HEK cells overexpressing RasGRP3. Phosphorylation began to be seen at 1 μM and was intense by 10 μM. RasGRP3 T133 phosphorylation increased appreciably less with increasing ligand concentration than did Erk1/2 phosphorylation, consistent with the important role of C1 domain occupancy by the ligand for RasGRP activity. The dose response curve for PKCδ S299 phosphorylation in response to the compounds was shifted to somewhat higher concentrations relative to that of RasGRP3 T133 phosphorylation, suggesting that other PKC isoforms may be more important than PKCδ for RasGRP3 phosphorylation. A generally similar pattern of responses was observed with PMA, although the potency of PMA was considerably greater (Figure 2B). Within the resolution of the experiments, results for compounds 2 – 5 were similar (Supplementary Figure 1).
Figure 2.
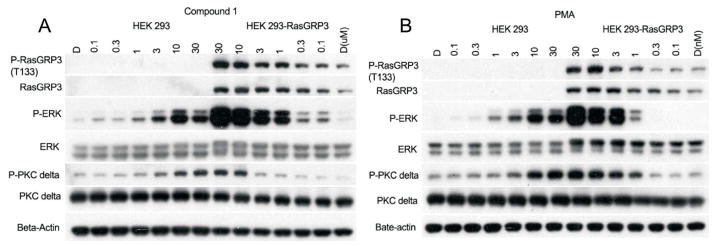
Response of HEK293 and HEK293 cells overexpressing RasGRP3 to treatment with compound 1 (A) or with PMA (B). Cells were treated for 30 min with the indicated concentrations of compound, after which the cells were lysed and the lysates subjected to electrophoresis and immunoblotting with the antibodies as indicated. Results are representative of the triplicate experiments performed.
Biological activity was also examined in Ramos cells, which endogenously express RasGRP3 to a high level. Once again all five compounds showed approximately similar potencies for Erk1/2 activation and for PKCδ S299 phosphorylation, as well as for phosphorylation of PKD, a downstream substrate of PKC (Supplementary Figure 2).
2.2.3. Translocation of PKCs and RasGRP3 in response to ligands
Translocation of PKC isoforms in response to phorbol esters or other DAG analogs is a sensitive reporter of the PKC – ligand – membrane interactions.36,37 We visualized cellular translocation of exogenously expressed GFP-PKCα, YFP-PKCε, GFP-PKCδ, and GFP-RasGRP3 as a function of time of treatment with 10 μM ligand. The GFP-PKCε, GFP-PKCδ and GFP-RasGRP3 were expressed in LNCaP cells, which we have extensively used for visualization of PKC translocation, as well as for biological analysis of PKC ligands.38– 40 GFP-PKCα was expressed in CHO cells, since PKCα shows only limited translocation in response to phorbol esters in LNCaP cells, presumably reflecting a low basal level of internal calcium, which functions as a co-activator of classic PKC isoforms.38 Representative images are presented for PKCα, for PKCε, for PKCδ, and for RasGRP3 (Figure 3A, 3C, 3E, and 2F, respectively). For PKCα and PKCε, which showed substantial differences among ligands, the results were also quantitated as the ratio of membrane/cytoplasmic signal (Figure 3B, 3D, respectively). Quantitation of images indicated that, for PKCα, compounds 3, 4, and 5 induced more translocation that did 1 and 2. It was also noteworthy that the rates of translocation varied, with response to 4 reaching a maximum after 2–5 min; those for 1 and PMA were appreciably slower. For PKCε, compounds 2 and 3 induced more rapid translocation than did 1, 4, and 5. For PKCδ, little difference was seen among compounds, all of which induced less translocation than did PMA. For RasGRP3, all of the compounds induced greater clustering at internal membranes, as evidenced by increased patchiness of its distribution, but without increased association with the plasma membrane. For further comparison, we also examined translocation in response to PDBu (Supplementary Figure 3). PDBu, being less lipophilic than PMA, induces localization of PKCδ predominantly to internal membranes rather than the plasma membrane. Its effects on PKCα, PKCε, and RasGRP3 are similar to those of PMA but somewhat less complete, reflecting its lower absolute affinity.
Figure 3.

Translocation of different PKC isoforms in living cells. CHO-K1 cells were transiently transfected with GFP-PKCα (A, B) and LNCaP cells were transiently transfected with YFP-PKCε (C, D), GFP- PKCδ (E), or GFP-RasGRP3 (F). The translocation pattern was examined by confocal microscopy as a function of time after treatment with the indicated drugs (10,000 nM) or PMA (1000 nM). Each panel represents images typical of the experiments performed (n = 3–6). Bars are 10 μm. Quantitative analysis of PKCα (B) and PKCε (D) translocation. Average ± SEM of calculated values are presented.
2.3. Biophysical Studies
To assess the modes of membrane interactions of DAG-indololactones 1-5 and to determine both the effects of the compounds upon lipid organization and dynamics and their relative insertion into membrane bilayers, we applied several fluorescence spectroscopy techniques. Figure 4 depicts the results of fluorescence anisotropy measurements utilizing giant PC/PG vesicles, which also contained the fluorescent dye diphenylhexatriene-trimethylammonium (DPH-TMA). DPH-TMA is embedded in the hydrophobic environment of the lipid bilayer, and changes to its fluorescence anisotropy generally provide a mean for evaluating modulation of bilayer fluidity induced by membrane-active compounds.41,42
Figure 4.

DAG-indololactones modulation of fluorescense anisotropy of DHP-TMA embedded in giant PC/PG vesicles. DHP-TMA was present in 1.25 μM concentration. Anisotropy was recorded in control vesicles without compound addition (left column), and in the presence of two concentrations of DAG-indololactones 1 to 5: 2.42 μM (middle column) and 6.05 μM (right column).
The fluorescence anisotropy data in Figure 4 point to distinct bilayer modulation effects induced by the DAG-indololactones studied. All five compounds appear to modulate the fluorescence anisotropy, albeit to different degrees. Specifically, the data presented in Figure 4 point to roughly two main groupings. Compounds 2 and 3 exhibited only minor changes in anisotropy, indicating minimal effect upon bilayer fluidity. Compounds 1, 4, and 5, however, gave rise to a much more significant increase in fluorescence anisotropy, corresponding to lowered bilayer fluidity upon binding of these indololactones.
While Figure 4 highlights the impact of the DAG-indololactones upon the bilayer properties, we carried out fluorescence spectroscopy experiments utilizing the intrinsic 1-methyl-1H-indole fluorescence emission43 to probe the effect of vesicle interactions upon the DAG-indololactones’ environment (Figure 5). Figure 5A indicates that the intrinsic fluorescence emission of 1, 3 and 5 is low. However, the fluorescence signals of 2 and 4 undergo dramatic modulations ascribed to bilayer internalization (Figure 5B–C). Specifically, the fluorescence emission peaks of both 2 and 4 clearly increase in intensity and shift to lower wavelengths following increasing vesicle concentrations in the aqueous solutions. These effects are indicative of more hydrophobic environments of the 1-methyl-1H-indole moieties,44 most likely brought about by insertion of these two DAG-indololactones into the bilayer. Importantly, the fluorescence shift appears most significant in the case of DAG-indololactone 4, which is consistent with the anisotropy data in Figure 4 pointing to pronounced interaction of this compound with the lipid bilayer. Additionally, the fluorescence excitation and emission spectra of indololactones 1 to 5 in solvents of varying polarities were measured (Supplementary Figure 4). In accordance with the effect of lipid addition, the results showed a strong dependence of fluorescence emission with solvent polarity. A shift towards lower wavelengths was observed with decreasing solvent polarity.
Figure 5.

Fluorescence emission spectra of DAG-indololactones 1 to 5 in the absence and in the presence of increasing concentration of giant vesicles. (a) Compounds 1 (- - -), 3 (····) and 5 (solid line); samples (6.05μM) in buffer in the presence DMPG/egg-PC giant vesicle solutions (30μL) in the scale of compound 4. Excitation wavelength = 358 nm, 343nm, 364 nm respectively; (b) Compound 2 (6.05μM), control sample (- - -) titrated with an increasing volume of DMPG/egg-PC giant vesicle solutions (5, 10, 20, 30μL). Excitation wavelength = 373 nm; (c) Compound 4 (6.05μM), control sample (- - -) titrated with an increasing volume of DMPG/egg-PC giant vesicle solutions (5, 10, 20, 30μL). Excitation wavelength = 349 nm.
To further probe membrane interactions of the DAG-indololactones we carried out fluorescence energy transfer experiments utilizing giant egg-PC/PG vesicles containing the fluorescent dye NBD-PE. Figure 6 depicts the results of fluorescence resonance energy transfer (FRET) experiments in which the fluorescence emission spectra of NBD-PE were recorded following excitation at 330–370 nm (the intrinsic excitation wavelength of the DAG-indololactones). In addition, one measurement for each sample was done also in the presence of 20% Triton X-100 that disrupts vesicles and, consequently, eliminates the FRET.
Figure 6.

Fluorescence energy transfer induced by DAG-indololactones in giant egg-PC/PG vesicles containing NDB-PE. (a) Compound 4, 6.05μM (excitation wavelength = 349 nm). NBD-PE (····), NBD-PE+4 (solid line), and NBD-PE+4+Triton (- - -); (b) compound 5, 6.05μM (excitation wavelength = 364 nm). NBD-PE (····), NBD-PE+5 (solid line) and NBD-PE+5+Triton (- - -); (c) compound 3, 6.05μM (excitation wavelength = 343 nm). NBD-PE (····), NBD-PE+3 (solid line) and NBD-PE+3+Triton (- - -); (d) compound 1, 6.05μM (excitation wavelength = 358 nm). NBD-PE (····), NBD-PE+1 (solid line) and NBD-PE+1+Triton (- - -).
The FRET data in Figure 6 provide further insight into membrane interactions of the DAG-indololactones and the differences among the compounds. Consistent with the fluorescence anisotropy data (Figure 4) and fluorescence emission analysis (Figure 5), 4 appears to enmesh most efficiently within the bilayer, giving rise to significant FRET to the bilayer-embedded NBD-PE (Figure 6A). The FRET results also underscore highly effective FRET in the case of 5 (Figure 6B), likewise consistent with the anisotropy results in Figure 3. Intriguingly, DAG-indololactone 3 also appears to facilitate FRET, giving rise to the notable increase in the fluorescence emission of the co-encapsulated NBD-PE (Figure 6C). This result is somewhat surprising in light of the relatively smaller effect of 3 upon the DPH-TMA anisotropy (Figure 4) and suggests that this DAG-indololactone undergoes distinct bilayer interactions in comparison with the other compounds studied. In this context, it should be emphasized that an insignificant FRET effect was recorded in the case of 1 (Figure 6D), supporting the proposal that 1 experiences a different mode of bilayer insertion. By addition of Triton X-100, the emission spectra of each DAG-indololactone were obtained after irradiation at its corresponding intrinsic excitation wavelength, as a consequence of vesicle disruption and FRET elimination. [No FRET could be measured in the case of 2 since there is a significant mismatch between its emission wavelength and the excitation wavelength of NBD-PE].
2.4. Modeling Studies
In order to explain and reconcile the biological and biophysical results, we decided to investigate the interaction of the DAG-indololactone compounds through molecular dynamics simulations with a modeled bilayer that approximates the solvation effects of a lipid membrane, including the heterogeneous dielectric environment of the membrane interfacial region where water interacts with the lipid headgroups and glycerol backbones.45 The results show that all the compounds reach equilibrium at approximately the same depth in the bilayer (Figure 7A), with both the indole ring and the branched alkyl sidechain located in the interfacial region. This is consistent with experimental NMR46,47,48 and all-atom molecular dynamics results49,50 for the bilayer localization of indole and tryptophan. Shown in this figure also are the approximate experimentally determined locations of DPH-TMA (the central double bond)51 and NBD-PE.52
Figure 7.


(a) Histogram of the density of indole ring atoms (solid lines) and branched alkyl sidechain atoms (dashed lines) as a function of distance from the bilayer center. (b) Histogram of the occupancy of the angle between the indole emission dipole moment and the bilayer normal.
The measured changes in DPH-TMA fluorescence anisotropy sense the perturbation induced by the insertion of the indololactones in the bilayer and, as such, they depend on several factors that influence the fluidity and ordering in the acyl chain region of the membrane. This is not directly observable in our simulations, since we are not explicitly including any lipid atoms, and since the DAG-indololactones do not penetrate deeply into this region of the bilayer (Figure 7A), but may be related to changes in lipid headgroup spacing and hydration53 induced to different degrees by the different DAG-indololactones.
On the other hand, FRET measurements are directly related to the distance and the angle between the dye (NBD) and the indole ring of the DAG-indololactones. Although all the indole rings occupy approximately the same depth in the bilayer, the magnitude and efficiency of energy transfer in FRET is also a function of the relative angles of the donor emission dipole of the indole and the acceptor absorption dipole of NBD. Indole has two excited states, La and Lb, whose energies and dipole moment directions depend both on the polarity of the solvent environment and on the nature and position of any ring substituents.54,55 Here for simplicity we assumed that in the bilayer environment the emitting state will be Lb, and defined an approximate dipole moment as a vector from the 3-position to the 7-position on the ring. In Figure 7B the occupancy of the angle between this vector and the bilayer normal is calculated for each compound. It can be observed that compound 1 is an outlier with a significantly more restricted range of motion, differing from the other compounds that exhibit a greater freedom of rotation. The fluorescence resonance energy transfer (FRET) depicted in Figure 6 shows that for compounds 3, 4, and 5 there is a significant FRET to the bilayer-embedded NBD agreeing with the proximity observed in our simulations between it and the indole moieties as well as the orientational heterogeneity of the indole ring. In the case of compound 1, however, the simulations suggest that the insignificant FRET observed may be a function of the restricted rotation of the indole ring in the bilayer environment. This may also be an explanation for the low intrinsic fluorescence emission of this compound, as shown in Figure 5.
The conformation that all DAG-lactones must adopt in order to bind the C1 domain has been well-established by our previous modeling studies.56,57,58,59,60 Figure 8 shows a representation of compound 1 relative to the binding site loops of the C1 domain and the bilayer dielectric environment. The carbon atoms of the glycerol backbone (or what would be the glycerol backbone if these compounds were standard open-chain diacylglycerols) are numbered.61 For effective C1 domain binding, the θ4 dihedral angle (around the bond between carbon atoms 1 and 2) must be the +sc rotamer (~60°), and the plane of the glycerol backbone should be approximately 45° relative to the plane of the bilayer. The occupancy of these angular conformations, as well as the average depth of the chiral carbon in the lactone ring (atom 2 of the glycerol backbone), which falls near the center of the binding site, are summarized for our simulations of each DAG-indololactone in Table 2.
Figure 8.
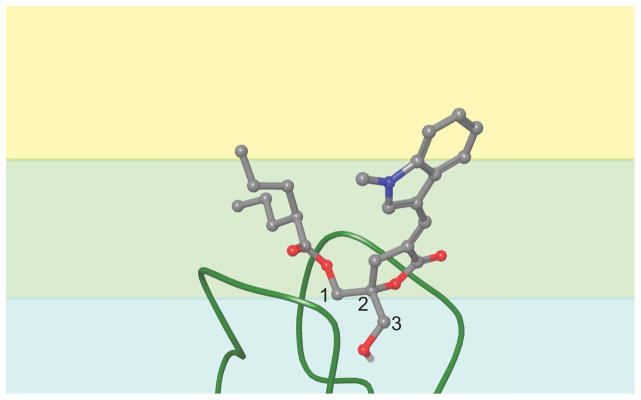
Cartoon representation of the C1 domain binding conformation of a DAG-indololactone. The carbon atoms in the glycerol backbone are numbered. The colored background represents the varying dielectric environments in the modeled bilayer: blue = lipid headgroups and water; green = ester groups and glycerol backbone; yellow = hydrophobic acyl chains.
Table 2.
Binding Conformation of DAG-indololactones
| Compound | ring deptha | backbone angleb | +sc rotamerc |
|---|---|---|---|
|
| |||
| 1 | 17.38 | 44.31 | 0.63 |
| 2 | 17.34 | 43.10 | 0.51 |
| 3 | 17.54 | 48.31 | 0.50 |
| 4 | 17.58 | 59.04 | 0.50 |
| 5 | 17.34 | 49.18 | 0.44 |
Average distance of ring chiral carbon from bilayer center
Average value of angle between plane of glycerol backbone and plane of bilayer
Occupancy of 60±30° range for θ4 dihedral angle
All of the DAG-indololactones reach equilibrium at essentially identical membrane depths for the lactone ring, and also adopt an average angle relative to the bilayer plane that is close to 45°. This suggests that the depth and angle of membrane penetration by the C1 domain for ligand binding will be nearly the same for all compounds. Additionally, the binding conformation of the θ4 dihedral angle, which controls the position of the sn-1 branched alkyl sidechain, is energetically accessible and occupied a significant fraction of the time in our simulations for each compound. These results shed light on the relatively minor differences in binding affinity, for a given C1 domain, between the DAG-indololactones (Table 1).
3. Discussion
The synthesis of DAG-indololactones with an unsubstituted 1-methyl-1H-indole ring at the 3- and/or 2-positions required the preparation of the lactone precursor 11 where the two hydroxyl moieties were distinctively protected either as TBDPS or TBDMS ethers. This material was prepared from economical, commercially available 1,3-dihydroxyacetone dimer in 5 steps with an overall yield of 29%. This result constitutes a valuable synthetic achievement when compared to known synthetic routes towards DAG-lactones that employ expensive glycidyl 4-methoxyphenyl ether as starting material.62 With 11 at hand, the development of a synthetic approach based on the selective removal of the TBDMS group under mild conditions allowed the preparation of the desired indololactones 2-5 that we have failed to synthesize employing already developed conditions. In the case of the monoacylation approach reported for the synthesis of compound 1, no selectivity could be achieved and low and variable yields of the desired indololactone 5 were obtained. When trying the use of 6b, differentially protected as benzyl and a TBDPS ethers, the benzyl group could not be removed either because of poor stability of the molecule in the presence of a Lewis acid (position 2 and 3 of the indole ring are very reactive) or because of lack of selectivity in transfer hydrogenation conditions.22,23
It has become increasingly clear that physical perturbations of lipid membranes such as fluidity, curvature, hydrocarbon volume and headgroup separation contribute to the modulation of PKC activity.63 It is also accepted that lipid-protein interactions can provide the necessary specificity and affinity to achieve a particular subcellular localization of signaling proteins.64 Thus, membrane lipids may help compartmentalize signaling complexes and regulate the spatiotemporal dynamics of PKC activations through interacting membrane microdomains, some of which can be formed transiently during signaling after release of DAG. Accordingly, we surmise that the specific cellular localization of the C1 domain containing protein ought to be determined in part by the different lipid composition of the membranes, the targeting information intrinsic to the individual C1 domain bound to DAG, and the binding to scaffolding or signaling proteins.
The most useful pharmacological probes for C1 domains —the families of phorbol esters— are not known to distinguish between DAG-responsive C1 domain containing proteins. Therefore, our objective has been to design DAG-lactones that could selectively direct the flow of information of DAG signaling pathways through their multiple signal transducers in order to achieve a specific cellular response with a potential therapeutic value. Previously, a combinatorial approach allowed the identification of DAG-lactones containing substituted aromatic rings at sn-1 and sn-2 positions that showed important selectivity for RasGRP3.21,65 The discovery of these compounds provided additional support for the feasibility of designing compounds selective for specific members of the families of signaling proteins with DAG-responsive C1 domains, through the combination of a “core” DAG-lactone with an array of sn-1 and sn-2 substituents acting as a “chemical zip codes” as we have described before.21
With this in mind, further variation of the aromatic moieties identified an DAG-indololactone 1 that was the most selective and potent compound known for the activation of RasGRP3.22 On the basis of this finding we have synthesized compounds that differ with respect to the point of attachment of the indole ring to the DAG-lactone template. This variation influences the orientation of the indole relative to the lactone ring and, consequently, could affect membrane interactions and selectivity. All isomers tested were selective and potent activators when comparing binding affinities towards RasGRP1/3 vs. PKCα. However, at the cellular level, RasGRP3 activation differences amongst the compounds in HEK293 cells were modest, suggesting that at this level of analysis these structural changes were not sensed by the lipid environment of the cell membrane. On the other hand, translocation experiments in living cells did show measurable differences between PKC isoforms although not with RasGRP1/3. Significantly, at the molecular level, biophysical experiments were able to detect important differences in the way these molecules interact with model membranes.
A global structure-activity relationship (SAR) for these compounds based on the results from all the biological assays used in this study is difficult to achieve. Each individual assay provides strong evidence that the compounds behave differently, showing unique properties depending on the specific assay and the conditions of the test. For example, individual affinities for PKCα, PKCε and RasGRP1/3, which were determined in a milieu with a high concentration of phosphatidyl serine (100 μg/ml), show modest variations relative to the point of attachment of the indole ring. This result was supported by the modeling, which showed little difference in average membrane depth and orientation across the DAG-indololactone compound series, suggesting that differences in the indole attachment point, and consequent strong differences in indole-bilayer interactions, may not necessarily be reflected in differences in C1 domain binding patterns.
Previously, it was determined that between DAG-lactones linked at the 2- and 3-positions of the indole, the one linked through the 3-position (compound 1) had greater discriminating capacity between PKC and RasGRP.23 In the present study, the point of attachment through the four remaining options available on the benzene ring (compounds 2-5) produced somewhat smaller changes in binding affinity with greater discriminating selectivity (24-fold) for the compound linked through the 5-position (compound 3). Overall, amongst all of the indole-substituted DAG-lactones, compound 1 still shows the greatest selectivity between PKC and RasGRP with ratios ranging from 49-fold to 84-fold (Table 1).
The response to compounds 1-5 in HEK293 and HEK293 cells overexpressing RasGRP3 showed that in this type of assay all five compounds behaved similarly within the range of resolution of the experiments. A similar pattern of activity was also evidenced in Ramos cells, which endogenously express high levels of RasGRP3. The response with PMA was analogous although in all cases the potency of PMA was considerably greater. Here again the conditions of the assay are widely different from the binding affinity experiments, as the ligand-protein complexes have to be translocated to the cellular membrane. These interactions therefore occur in a different lipid milieu.
Another way of examining protein-ligand-membrane interactions for compounds 1-5 was to observe the extent of translocation of various PKC isoforms and RasGRP3. These were visualized with exogenously expressed GFP-PKCα, YFP-PKCε, GFP-PKCδ, and GFP-RasGRP3 as a function of time with 10 μM ligand using CHO cells for GFP-PKCα and LNCaP cells for the rest of the enzymes. In these assays substantial differences among ligands were observed, for example between PKCα and PKCε, but little differences if any were detected for GFP-PKCδ and GFP-RasGRP3. For PKCα, compounds 3, 4, and 5 induced more translocation than did 1 and 2, whereas for PKCε the difference was not with the extent of translocation but rather with its rate, where compounds 2 and 3 induced a more rapid translocation than did 1, 4, and 5. These differences obviously reflect different modes of PKC-ligand-membrane interactions, which are probably a function of the different lipid composition of the membranes. Finally, the biophysical studies were conducted employing giant egg-PC/PG vesicles, which obviously do not correspond to a typical cellular membrane composition but which are useful to explore more specific molecular interactions impacting lipid organization and dynamics and the extent of insertion into membrane bilayers. Overall, the biophysical experiments confirmed that all five DAG-indololactones experience significant interactions with membranes and all associate with the membrane bilayer to a varied degree. The experiments pointed, however, to distinct mechanisms of bilayer binding and insertion. In particular, 4 appears to exhibit the most pronounced bilayer interactions and insertion, thereby both affecting most significantly the lipid organization (Figure 4) and, conversely, experiencing the most hydrophobic environment following bilayer association (Figures 5 and 6). Interestingly, compound 1 showed significantly reduced fluorescent energy transfer to the NBD dye and restricted rotation of the indole ring in the interfacial region of the bilayer. This could be related perhaps with its higher selectivity towards RasGRP, but this may be confirmed with further experimentation.
Our modeling results are consistent with physicochemical studies with membrane proteins that demonstrate that tryptophan has a penchant for membrane surfaces due to distinct interfacial interactions.46 Such interactions may involve the formation of hydrogen bonds between the indole’s NH and lipid acyl carbonyls66 as well as tryptophan-lipid cation-π interactions.47 In the specific case of the DAG-indololactones we found no differences between compound 1 and the analogue with the free indole NH in terms of PKC binding affinity23 suggesting that the primary force responsible for tryptophan’s interfacial preference in these compounds is the aromaticity of the indole ring, its pi electron cloud, and the associated quadrupole moment. This is confirmed by our molecular dynamics simulations where the bilayer solvent dielectric field (in the absence of any hydrogen bonds or other specific atomic interactions) is enough to localize and orient the indole group.
4. Conclusions
In summary, we believe that the structural differences between these DAG-lactones reflect changes limited to the membrane environment. Contrary to the contacts of the critical pharmacophores on the DAG-lactone with the protein environment, which involve precise hydrogen bonding interactions with the C1 domain, interactions with the lipid environment are less sensitive to relatively small structural changes amongst isosteres. The biophysical differences detected are real but perhaps of lesser magnitude in the protein-ligand-membrane interactions equation.
5. Experimental section
5.1. General Procedures
All chemical reagents were commercially available. [20-3H]phorbol 12,13-dibutyrate (PDBu) was obtained from Perkin-Elmer, Waltham, MA. PMA was from LC Laboratories, Woburn, MA, USA. Melting points were determined on an Electrothermal IA9000series digital melting point apparatus and are uncorrected. Column chromatography was performed on a Teledyne Isco CombiFlash Companion instrument under gradient elution conditions with RediSep disposable flash columns. Analytical TLC was performed on Merck silica gel 254F plates. 1H and 13C NMR spectra were recorded on a Bruker Avance DPX 400 instrument at 400 and 100 MHz, respectively. Spectra are referenced to the solvent in which they were run (7.26 ppm for CDCl3). Both low resolution and high resolution positive ion electrospray ionization (ESI) mass spectra were obtained for all compounds. Low resolution ESI analysis was carried out on an Agilent LC/MSD single quadrupole system, equipped with an in-line diode-array UV detector, to assess compound identity and homogeneity. Initial analyses were carried out in flow-injection analysis (FIA) mode with the sample injected directly into the LC/MSD using 1:1 CH3OH/H2O containing 0.1% CH3COOH at a flow rate of 300 μl/min. Final DAG-lactone products were additionally analyzed by LC/MS using a narrow-bore (100 × 2.1 mm), small-particle (3.5-μm), Zorbax Rapid-Resolution reversed-phase C18 column coupled with a C18 guard column (12.5 × 2.1 mm) eluted with a 5–90% gradient of CH3OH/H2O containing 0.1% CH3COOH at a flow rate of 300 μl/min for separations. Both the total-ion chromatogram (TIC) and the UV-chromatogram were used to confirm compound purity. The full scan (210 – 400 nm) diode-array UV spectra for both FIA and LC/MS analyses of the final DAG-lactone products were also generated and compared to assess similarity. UV spectra in all cases were equivalent and are reported for the individual products. High resolution MS analysis was conducted on a Thermo-Fisher LTQ-XL Orbitrap hybrid mass spectrometer operated at a resolution of 30,0000 (FWHM) using either FIA or LC/MS sample introduction, depending on which mode was the most suitable based on previous low resolution analyses. For LC/MS analyses on the Orbitrap, a narrow-bore (50 × 2.1 mm) Zorbax Rapid-Resolution reversed-phase C18 column coupled with a C18 guard column was eluted with a 5–90% gradient of CH3CN/H2O containing 0.1% HCOOH at 250 μl/min. The resulting accurate mass measurement of a molecular species ([M+H]+, [M+Na]+ or M+NH4]+) was then used to determine a unique elemental composition for each particular compound. Where appropriate, 1H and 13C NMR data were used to set elemental constraints for this calculation. Elemental analyses were performed by Atlantic Microlab, Inc., Norcross, GA and by UMYMFOR-CONICET, Argentina. 1,2-Dimyristoyl-sn-glycerophosphocholine (DMPC), 1-(3 sn-phosphatidyl)-rac-glycerol sodium salt (PG), phosphatidylserine, and the fluorescent dye 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl])(ammonium salt) (NBD-PE) were purchased from Avanti (Alabaster, AL, USA). Sodium dithionite (Na2O4S2) and Tris(hydroxymethyl)aminomethane (TRIZMA base buffer, C4H11NO3) KCl, and sucrose were purchased from Sigma–Aldrich. 1-(4-Trimethylammoniumphenyl)-6-phenyl-1,3,5-hexatriene (TMA-DPH) was obtained from Molecular Probes, Inc. (Eugene, Oregon, USA).
5.2. Chemistry
(E)-5,5-bis((tert-butyldiphenylsilyloxy)methyl)-3-((1-methyl-1H-indol-7-yl)methylene)dihydrofuran-2(3H)-one (7a)
A solution of 6a (549 mg, 0.88 mmol) in anhydrous tetrahydrofuran (8 mL) at −78°C was treated dropwise with LiHMDS (1.3 mL, 1.30 mmol) and stirred at the same temperature for 0.3 h. A solution of indole-7-carboxyaldehyde (293 mg, 1.83 mmol) in anhydrous tetrahydrofuran (1 mL) was added dropwise, and the mixture was stirred at −78 °C for 2 h. A saturated aqueous solution of NH4Cl (15 mL) was added at room temperature and the aqueous phase was extracted with ethyl ether (3 × 20 mL). The organic phase was dried (Na2SO4) and concentrated to give a mixture of diastereomers, which was used directly in the next step. Thus, the residue was dissolved in anhydrous dichloromethane (6 mL) and treated with triethylamine (0.54 mL, 3.52 mmol) and MsCl (0.14 mL, 1.76 mmol) at 0°C for 2 h. DBU (0.66 mL, 4.4 mmol) was added and the mixture was allowed to reach room temperature for 2 h. The reaction was quenched by the addition of NH4Cl (ss, 10 mL) and the aqueous phase was extracted with CH2Cl2 (3 × 20 mL). The organic phase was dried (Na2SO4) and concentrated. The residue was purified by silica gel flash column chromatography (gradient 0–20% EtOAc/hexanes) to give 528 mg (78% yield) of 7a as a colorless syrup. 1H NMR (400 MHz, CDCl3) δ 8.25 (m, 1 H, –CH=C–), 7.65 (d, J = 7.6 Hz, 1 H, H-4′), 7.57 (d, J = 7.8 Hz, 8 H, Ph), 7.38 – 7.16 (m, 13 H, Ph, H-6′), 7.10 (t, J = 7.6 Hz, 1 H, H-5′), 6.98 (d, J = 3.0 Hz, 1 H, H-2′), 6.52 (d, J = 3.0 Hz, 1 H, H-3′), 3.89 (s, 3 H, CH3N–), 3.82 (d, J = 10.6 Hz, 2 H, – CHaHOTBDPS), 3.74 (d, J = 10.6 Hz, 2 H, –CHHbOTBDPS), 3.07 (mAB, 2 H, H-4), 0.99 (s, 18 H, –SiC(CH3)3); 13C NMR (50 MHz, CDCl3) δ 171.44, 135.58, 135.51, 134.42, 132.83, 132.59, 132.38, 131.26, 130.38, 129.78, 127.73, 126.61, 122.85, 122.50, 119.84, 119.21, 101.38, 85.55, 66.15, 53.39, 37.43, 32.00, 26.71, 19.23; IR (neat) 1751 cm−1; ESI-MS m/z 781 [M+NH4]+, 764 [M+H+]; HRMS (ESI) m/z [M + H]+ calcd for C48H53NO4Si2: 764.3586, found: 764.3553.
(E)-5-(Benzyloxymethyl)-5-(tert-butyldiphenylsilyloxy)methyl)-3-((1-methyl-1H-indol-7-yl)methylene)dihydrofuran-2(3H)-one (7b)
A solution of 6b (302 mg, 0.63 mmol) in anhydrous tetrahydrofuran (6 mL) at −78°C was treated dropwise with LiHMDS (0.95 mL, 0.945 mmol) and stirred at the same temperature for 0.3 h. A solution of indole-7-carboxyaldehyde (200 mg, 1.26 mmol) in anhydrous tetrahydrofuran (1 mL) was added dropwise, and the mixture was stirred at −78 °C for 2 h. A saturated aqueous solution of NH4Cl (15 mL) was added at room temperature and the aqueous phase was extracted with ethyl ether (3 × 20 mL). The organic phase was dried (Na2SO4) and concentrated to give a mixture of diastereomers, which was used directly in the next step. Thus, the residue was dissolved in anhydrous dichloromethane (5 mL) and treated with triethylamine (0.39 mL, 2.58 mmol) and MsCl (0.10 mL, 1.29 mmol) at 0°C for 2 h. DBU (0.49 mL, 3.23 mmol) was added and the mixture was allowed to reach room temperature for 2 h. The reaction was quenched by the addition of NH4Cl (ss, 10 mL) and the aqueous phase was extracted with CH2Cl2 (3 × 20 mL). The organic phase was dried (Na2SO4) and concentrated. The residue was purified by silica gel flash column chromatography (gradient 0–20% EtOAc/hexanes) to give 240 mg (61% yield) of 7b as a colorless syrup. 1H NMR (400 MHz, CDCl3) δ 8.25 (m, 1 H, –CH=C–), 7.64 (d, J = 7.6 Hz, 1 H, H-4′), 7.59 (d, J = 7.8 Hz, 4 H, Ph), 7.38 – 7.18 (m, 12 H, Ph, H-6′), 7.11 (t, J = 7.6 Hz, 1 H, H-5′), 7.03 (d, J = 3.0 Hz, 1 H, H-2′), 6.50 (d, J = 3.0 Hz, 1 H, H-3′), 4.53 (mAB, 2 H, –OCH2Ph), 3.87 (s, 3 H, CH3N–), 3.82 (d, J = 10.6 Hz, 1 H, –CHaHOTBDPS), 3.75 (d, J = 10.6 Hz, 1 H, –CHHbOTBDPS), 3.62 (mAB, 2 H, –CH2OCH2Ph), 3.10 (mAB, 2 H, H-4), 1.00 (s, 9 H, –SiC(CH3)3); 13C NMR (50 MHz, CDCl3) δ 171.33, 137.58, 135.56, 134.36, 132.82, 132.73, 132.57, 131.27, 130.32, 129.76, 128.36, 127.68, 126.37, 122.91, 122.84, 122.58, 122.51, 119.78, 119.23, 101.41, 84.79, 73.70, 71.91, 66.37, 37.45, 32.40, 26.72, 19.34; IR (neat) 1749 cm−1; ESI-MS m/z 616 [M+H+]; HRMS (ESI) m/z [M + H]+ calcd for C39H41NO4Si: 616.2883, found: 616.2892.
(E)-5,5-bis(hydroxymethyl)-3-((1-methyl-1H-indol-7-yl)methylene)dihydrofuran-2(3H)-one (8)
To a solution of 7 (164 mg, 0.21 mmol) in anhydrous THF (8 ml) was added triethylamine trihydrofluoride (0.34 mL, 2.13 mmol). The reaction mixture was stirred at 70°C for 6 h. The crude solution was then concentrated in vacuo and purified by silica gel flash column chromatography (gradient 50–100% EtOAc/hexanes) to give 46 mg (75 % yield) of 8 as a yellow syrup: 1H NMR (400 MHz, CDCl3, CD3OD) δ 8.32 (m, 1 H, –CH=C–), 7.65 (d, J = 7.6 Hz, 1 H, H-4′), 7.29 (d, J = 7.3 Hz, 1 H, H-6′), 7.11 (t, J = 7.6 Hz, 1 H, H-5′), 7.03 (d, J = 3.0 Hz, 1 H, H-2′), 6.51 (d, J = 3.0 Hz, 1 H, H-3′), 4.04 (s, 3 H, CH3N–), 3.75 (d, J = 12.1 Hz, 2 H, –CHaHOH), 3.68 (d, J = 12.1 Hz, 2 H, –CHHbOH), 3.10 (d, J = 2.8 Hz, 2 H, H-4); 13C NMR (100 MHz, CDCl3) δ 172.47, 134.25, 133.96, 131.29, 130.29, 125.56, 123.13, 122.61, 119.31, 119.12, 101.31, 86.32, 64.14, 37.28, 31.43; IR (neat) 3493, 2925, 1690, 1627, 722 cm−1, ESI-MS m/z 310 [M+Na]+, 288 [M+H+]; HRMS (ESI) m/z [M + H]+ calcd for C16H17NO4: 288.1230, found: 288.1229.
1-(tert-Butyl-dimethyl-silanyloxy)-3-(tert-butyl-diphenyl-silanyloxy)-propan-2-one (9)
A solution of 1,3-dihydroxyacetone dimer (11.0 g, 60.79 mmol) and imidazole (1.84 g, 26.88 mmol) in anhydrous DMF (40 mL) at 0 °C was treated dropwise with a solution of TBDPSCl (9.6 mL, 37.68 mmol) in anhydrous DMF (9 mL). The reaction mixture was stirred at this temperature for 4 h. Water was added (40 mL) and the mixture was extracted with dichloromethane (3 × 40 mL). The combined organic phases were dried (Na2SO4) and concentrated. The residue was purified by silica gel flash column chromatography (gradient 0–20% EtOAc)/hexanes) to give 6.31 g of slightly impure monoprotected product 1-(tert-butyldiphenylsilyloxy)-3-hydroxypropan-2-one28 that was used as such for the next step. Thus, to a solution of 1-(tert-butyldiphenylsilyloxy)- 3-hydroxypropan-2-one (6.31 g, 19.20 mmol) in anhydrous dichloromethane (80 ml) were added imidazole (3.0 g, 44.00 mmol) and a solution of TBDMSCl (3.43 g, 22.66 mmol) in anhydrous dichloromethane (40 ml). The reaction mixture was stirred at 0 °C for 2 h. Water (80 mL) was added and the aqueous phase was extracted with dichloromethane (3 × 80 mL). The organic phase was dried (Na2SO4) and concentrated. The residue was purified by silica gel flash column chromatography (gradient 0–5% EtOAc/hexanes) to give 7.42 g (44 % two steps yield) of 9 as a white solid: mp 56 °C; 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 6.7 Hz, 4 H, Ph), 7.46 – 7.37 (m, 6 H, Ph), 4.42 (s, 2H, –CH2OTBDMS)*, 4.40 (s, 2H, –CH2OTBDPS)*, 1.10 (s, 9 H, – SiC(CH3)3)(CH3)2)*, 0.85 (s, 9 H, –SiPh2C(CH3)3)*, 0.02 (s, 6 H, –Si(t-Bu)(CH3)2); 13C NMR (100 MHz, CDCl3) δ 208.58, 136.17, 133.29, 130.66, 128.54, 69.14, 68.68, 27.43, 26.41, 19.92, 18.98, −4.91; IR (neat) 1740, 1097, 837, 825, 777, 741, 702 cm−1; ESI-MS m/z 465 [M+Na]+. Elemental Analysis Calcd. for C25H38O3Si2: %C,67.82; %H, 8.65. Found: %C, 67.91; %H, 8.71.
*Signals could be interchanged.
2-(tert-Butyl-dimethyl-silanyloxymethyl)-1-(tert-butyl-diphenyl-silanyloxy)-pent-4-en-2-ol (10)
To a solution of 9 (7.38 g, 16,67 mmol) in anhydrous tetrahydrofuran (30 mL) was added allylmagnesium chloride (2 M, 20 mL, 40.00 mmol). The reaction mixture was stirred at room temperature for 2 h. A saturated aqueous solution of NH4Cl (50 mL) was added and the aqueous phase was extracted with EtOAc (3 × 100 mL). The organic phase was dried (Na2SO4) and concentrated. The residue was purified by silica gel flash column chromatography (gradient 0–5% EtOAc/hexanes) to give 7.18 g (89 % yield) of 10 as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.65 (d, J = 6.7 Hz, 4 H, Ph), 7.44 – 7.35 (m, 6 H, Ph), 5.92 – 5.81 (m, 1H, –CH2CH=CH2), 5.07 (m, 2 H, –CH2CH=CH2), 3.60 (d, 1 H, J = 9.0 Hz, –CHaHOTBDPS)*, 3.58 (d, 1 H, J = 9.0 Hz, – CHaHOTBMPS)*, 3.52 (d, 2 H, J = 9.0 Hz, –CHHbOTBDPS, –CHHbOTBDMS), 2.53 (s, 1 H, -OH), 2.30 (d, J = 7.2 Hz, –CH2CH=CH2), 1.07 (s, 9 H, -SiC(CH3)3), 0.87 (s, 9 H, - SiC(CH3)3), 0.05 (s, 6 H, -Si(t-Bu)(CH3)2); 13C NMR (100 MHz, CDCl3) δ 136.30, 136.26, 134.09, 133.87, 133.82, 130.39, 128.36, 118.53, 74.73, 66.53, 65.80, 39.03, 27.57, 26.54, 19.96, 18.86, −4.80, −4.84; IR (neat) 3072, 2954, 2929, 2857, 1471, 1428, 1252, 1080, 835, 776, 739, 700 cm−1 (OH); ESI-MS m/z 507 [M+Na]+. Elemental Analysis Calcd. for C28H44O3Si2: %C, 69.37; %H, 9.15. Found: %C, 69.61; %H, 9.29.
5-((tert-butyldimethylsilyloxy)methyl)-5-((tertbutyldiphenylsilyloxy) methyl)dihydrofuran-2(3H)-one (11)
A THF solution of BH3·SMe2 (2.0 M, 15.5 mL, 30.95 mmol) was added slowly to a stirred solution of 10 (6.0 g, 12.37 mmol) in anhydrous THF (50 mL) at −78°C. The reaction mixture was stirred at −78°C for 0.25 h and then at room temperature for 4 h. Methanol (30 mL) was added slowly and the resulting solution was concentrated to yield a colorless oil that was used as such in the next step. The obtained oil (6.92 g) was dissolved in anhydrous dichloromethane (160 mL) and PCC (20.02 g, 92.99 mmol) and 4Å molecular sieves (7.02 g) were added. The reaction mixture was stirred at room temperature for 72 h. Ethyl ether (150 mL) was added and the resulting solution was stirred at room temperature for 1 h. The reaction mixture was then filtrated through a column of silica gel and concentrated. The residue was purified by silica gel flash column chromatography (gradient 0–10% EtOAc/hexanes) to give 4.55 g (74 % yield) of 11 as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.65 (d, J = 6.8 Hz, 4 H, Ph), 7.44 – 7.37 (m, 6 H, Ph), 3.72 (d, 1 H, J = 10.9 Hz, –CHaHOTBDPS)*, 3.71 (d, 1 H, J = 10.6 Hz, –CHaHOTBMPS)*, 3.65 (d, 1 H, J = 9.0 Hz, –CHHbOTBDMS)≠, 3.64 (d, 1 H, J = 9.0 Hz, –CHHbOTBDPS)≠, 2.60 (t, J =8.5 Hz, 2 H, H-3), 2.16 (t, J = 8.2 Hz, 2 H, H-4), 1.06 (s, 9 H, -C(CH3)3), 0.85 (s, 9 H, -C(CH3)3), 0.03 (s, 6 H, -Si(CH3)2); 13C NMR (50 MHz, CDCl3) δ 177.18, 135.61, 135.57, 132.83, 132.62, 129.88, 127.82, 88.19, 66.47, 65.92, 29.45, 26.78, 25.75, 25.54, 19.22, 18.17, −5.55, −5.61; IR (neat) 2954, 2929, 2857, 1778, 1112, 836, 777, 738, 700 cm−1; ESI-MS m/z 521 [M+Na]+, 499 [M+H]+, 421 [M+H-C6H6]+; HRMS (ESI) m/z [M + H]+ calcd for C28H42O4Si2: 499.2694, found: 499.2693. Elemental Analysis Calcd. For C28H42O4Si2: %C, 67.42; %H, 8.49. Found: %C, 67.48; %H, 8.30.
*, ≠ Signals could be interchanged.
General Procedure A. Aldol condensation followed by olefination. 67, 62
Under argon, a solution of 11 (1 equiv) in THF (5 mL/mmol) at −78 °C was treated dropwise with [(CH3)3Si]2N-Li (LiHMDS, 1M in THF, 1.5 equiv) and stirred at the same temperature for 0.5 h. A solution of indole aldehyde (1.5 equiv dissolved in THF) was then added dropwise, and the reaction was stirred at −78 °C for 2 h. The reaction mixture was then quenched with a saturated aqueous solution of NH4Cl and allowed to warm to room temperature. The layers were separated, and the aqueous layer was extracted with Et2O (3 ×). The combined organics were washed with H2O (2 ×) and brine (1×), dried (Na2SO4), and concentrated in vacuo. The obtained residue was used directly without purification. Thus, it was dissolved in CH2Cl2 (10 mL/mmol) and Et3N (4 equiv) was added. The resulting solution was cooled to 0 °C, treated dropwise with CH3SO2Cl (MsCl, 2 equiv), and then stirred at room temperature for 1–2 h. The reaction mixture was then cooled again to 0 °C and treated dropwise with 1,8-diazabicyclo[5.4.0]non-5-ene (DBU, 5 equiv). When the addition of DBU was completed, the reaction mixture was allowed to reach room temperature overnight. The residue was treated with EtOAc (10 mL/mmol) and saturated solution of NH4Cl (10 mL/mmol). The layers were separated, and the aqueous layer was extracted with EtOAc (1 ×). The combined organics were washed with H2O (2 ×) and brine (1 ×), dried (Na2SO4), and concentrated in vacuo. Purification by silica gel flash column chromatography (gradient EtOAc/hexanes) gave 12 (a–d).
(E)-5-((tert-butyldimethylsilyloxy)methyl)-5-((tert-butyldiphenylsilyloxy)methyl)-3-((1-methyl-1H-indol-4-yl)methylene)dihydrofuran-2(3H)-one (12a)
Starting from 11 (1.30 g, 2.60 mmol)and following general procedure A, 12a was isolated as a greenish oil (997 mg, 60% yield); 1H NMR (400 MHz, CDCl3) δ 8.01 (m, 1 H, –CH=C–), 7.62 (t, J = 6.6 Hz, 4H, Ph), 7.40 – 7.28 (m, 9 H, Ph, H-5, H-6, H-7), 7.15 (d, J = 2.8 Hz, 1 H, H-2′), 6.73 (d, J = 2.8 Hz, 1 H, H-3′), 3.84 (s, 3 H, CH3N), 3.77 (m, 4 H, –CH2OTBDPS, –CH2OTBDMS), 3.14 (mAB, 2 H, H-4), 0.99 (s, 9 H, –C(CH3)3), 0.81 (s, 9 H, -C(CH3)3), 0.02 (d, J = 3.5 Hz, 6 H, –Si(CH3)2):13C NMR (50 MHz, CDCl3) δ 172.05, 136.85, 135.63, 135.60, 132.92, 132.81, 132.76, 129.76, 129.73, 129.49, 127.74, 127.02, 125.50, 121.37, 119.38, 110.68, 99.60, 85.34, 66.19, 65.39, 32.99, 32.55, 26.66, 25.71, 19.20, 18.14, −5.54; IR (neat) 2928, 2856, 1749, 1110, 836, 777, 700 cm−1; ESI-MS m/z 662 [M+Na]+. Elem. Anal. Calcd. for C38H49NO4Si2: C, 71.32; H, 7.72; N, 2.19. Found: C, 70.87; H, 7.84; N, 1.91.
(E)-5-((tert-butyldimethylsilyloxy)methyl)-5-((tert-butyldiphenylsilyloxy)methyl)-3-((1-methyl-1H-indol-5-yl)methylene)dihydrofuran-2(3H)-one (12b)
Starting from 11 (382 mg, 0.76 mmol) and following general procedure A, 12b was isolated as a colorless oil (250 mg, 51% yield); 1H NMR (400 MHz, CDCl3) δ 7.78 (sa, 1 H, H–4′), 7.66 – 7.62 (m, 5 H, CH=C, Ph), 7.40 – 7.30 (m, 8 H, Ph, H–6′, H–7′), 7.10 (d, J = 3.0 Hz, 1 H, H–2′), 6.55 (d, J = 3.0 Hz, 1 H, H–3′), 3.82 (s, 3 H, CH3N), 3.77 (m, 4 H, –CH2OTBDPS, –CH2OTBDMS), 3.13 (mAB, 2 H, H–4), 1.01 (s, 9 H, – C(CH3)3), 0.82 (s, 9 H, –C(CH3)3), 0.03 (s, 3 H, –Si(CH3)2), 0.02 (s, 3 H, –Si(CH3)2);13C NMR (50 MHz, CDCl3) δ 172.45, 137.52, 137.14, 135.63, 132.92, 132.77, 130.07, 129.75, 128.70, 127.74, 126.57, 123.95, 123.55, 122.06, 109.54, 102.00, 85.15, 66.17, 65.39, 32.96, 32.26, 26.64, 25.71, 19.19, 18.14, −5.52; IR (neat) 2928, 2856, 1749, 1110, 836, 777, 700 cm−1; ESI-MS m/z 662 [M+Na]+, 640 [M+H]+, 562 [M+H-C6H6]+; HRMS (ESI) m/z [M + H]+ calcd for C38H49NO4Si2: 640.3273, found: 640.3271.
(E)-5-((tert-butyldimethylsilyloxy)methyl)-5-((tert-butyldiphenylsilyloxy)methyl)-3-((1-methyl-1H-indol-6-yl)methylene)dihydrofuran-2(3H)-one (12c)
Starting from 11 (565 mg, 1.13 mmol) and following general procedure A, 12c was obtained as a yellowish oil (457 mg, 63% yield); 1H NMR (400 MHz, CDCl3): δ 7.68 – 7.61 (m, 6 H, CH=C, Ph, H-4′), 7.43 – 7.28 (m, 8 H, Ph, H-5′, H-7′), 7.17 (d, J = 3.0 Hz, 1 H, H-2′), 6.52 (d, J = 2.8 Hz, 1 H, H-3′), 3.82 (s, 3 H, CH3N–), 3.76 (m, 4 H, –CH2OTBDPS, –CH2OTBDMS), 3.14 (m, 2 H, H-4), 1.01 (s, 9 H, –C(CH3)3), 0.82 (s, 9 H, –C(CH3)3), 0.03 (s, 3 H, –Si(CH3)2), 0.02 (s, 3 H, –Si(CH3)2);13C NMR (50 MHz, CDCl3): δ 172.27, 137.43, 136.70, 135.60, 132.77, 131.20, 129.77, 129.63, 128.61, 127.74, 122.96, 121.13, 121.07, 111.79, 101.36, 85.18, 66.20, 65.36, 32.87, 32.35, 26.67, 25.71, 19.22, 18.14, −5.50; IR (neat): 2928, 2856, 1747, 1111, 836, 777, 700 cm−1 (CO); ESI-MS m/z 662 [M+Na]+, 640 [M+H]+; HRMS (EI) m/z [M + H]+ calcd for C38H49NO4Si2: 640.3273, found: 640.3271.
(E)-5-((tert-butyldimethylsilyloxy)methyl)-5-((tert-butyldiphenylsilyloxy)methyl)-3-((1-methyl-1H-indol-7-yl)methylene)dihydrofuran-2(3H)-one (12d)
Starting from 11 (710 mg, 1.42 mmol) and following general procedure A, 12d was obtained as a yellowish oil (674 mg, 74% yield); 1H NMR (400 MHz, CDCl3) δ 8.25 (m, 1 H, –CH=C–), 7.65 (d, J = 7.8 Hz, 1 H, H-4′), 7.60 (d, J = 7.1 Hz, 4 H, Ph), 7.38-7.22 (m, 7 H, Ph, H-6′), 7.11 (t, J = 7.6 Hz, 1 H, H-5′), 6.99 (d, J = 2.5 Hz, 1 H, H-2′), 6.51 (d, J = 2.5 Hz, 1 H, H-3′), 3.96 (s, 3 H, CH3N–), 3.79 (d, 1 H, J = 10.6 Hz, –CHaHOTBDPS)*, 3.77 (d, 1 H, J = 10.6 Hz, –CHaHOTBMPS)*, 3.71 (d, 2 H, J = 10.6 Hz, –CHHbOTBDMS, –CHHbOTBDPS), 3.08 (mAB, 2 H, J = 7.1 Hz, H-4), 1.01 (s, 9 H, –C(CH3)3), 0.83 (s, 9 H, –C(CH3)3), 0.03 (s, 6 H, –Si(CH3)2); 13C NMR (50 MHz, CDCl3) δ 171.43, 135.60, 135.51, 134.40, 132.89, 132.63, 132.31, 131.26, 130.36, 129.77, 127.71, 126.81, 122.79, 122.47, 119.88, 119.21, 101.36, 85.53, 66.17, 65.39, 37.44, 31.68, 26.70, 25.71, 19.22, 18.14, −5.52; IR (neat) 2952, 2928, 2856, 1750, 1111, 836, 778, 700 cm−1; ESI-MS m/z 662 [M+Na]+, 640 [M+H]+, 562 [M+H-C6H6]+; HRMS (ESI) m/z [M + H]+ calcd for C38H49NO4Si2: 640.3273, found: 640.3276.
*Signals could be interchanged.
General Procedure B. TBDMS deprotection
Pyridinium p-toluene sulfonate (0.80 eq) was added to a solution of fully protected DAG-indololactone (12a–d) in absolute ethanol (10 mL/mmol). The reaction mixture was stirred at 60 °C till disappearance of starting material by TLC. Brine solution was added and the mixture was extracted with AcOEt (3 ×). The combined organic layers were dried (Na2SO4) and the volatiles were removed. The residue was purified by silica gel flash column chromatography (gradient EtOAc/hexanes) to yield compounds 13a–d.
(E)-5-((tert-butyldiphenylsilyloxy)methyl)-5-(hydroxymethyl)-3-((1-methyl-1H-indol-4-yl)methylene)dihydrofuran-2(3H)-one (13a)
Starting from 12a (997 mg, 1.55 mmol) and following general procedure B, 13a was obtained as a yellow oil (550 mg, 68% yield); 1H NMR (400 MHz, CDCl3) δ 8.05 (m, 1 H, CH=C), 7.60 (t, J = 6.6 Hz, 4 H, Ph), 7.41-7.28 (m, 9 H, Ph, H-5′, H-6′, H-7′), 7.16 (d, J = 2.8 Hz, 1 H, H-2′), 6.73 (d, J = 2.5 Hz, 1 H, H-3′), 3.84 (s, 3 H, CH3N–), 3.74 (m, 4 H, –CH2OTBDPS, –CH2OH), 3.21 (mAB, 2 H, 17.6 Hz, H-4a,b), 1.89 (t, J = 6.8 Hz, 1 H, OH), 0.99 (s, 9 H, –C(CH3)3), 13C NMR (50 MHz, CDCl3) δ 171.92, 136.85, 135.63, 135.57, 133.85, 132.69, 132.48, 129.89, 129.57, 127.80, 126.66, 124.56, 121.39, 119.59, 111.00, 99.50, 85.27, 66.20, 65.24, 33.02, 32.49, 26.64, 19.16; IR (neat) 3401, 2929, 2856, 1732, 1111, 730, 700 cm−1; ESI-MS m/z 548 [M+Na]+, 526 [M+H]+, 448 [M+H-C6H6]+; HRMS (ESI) m/z [M + H]+ calcd for C32H35NO4Si: 526.2408, found: 526.2400.
(E)-5-((tert-butyldiphenylsilyloxy)methyl)-5-(hydroxymethyl)-3-((1-methyl-1H-indol-5-yl)methylene)dihydrofuran-2(3H)-one (13b)
Starting from 12b (250 mg, 0.39 mmol) and following general procedure B, 13b was obtained as a white solid (158 mg, 77% yield); mp 185–186 °C; 1H NMR (400 MHz, CDCl3) δ 7.79 (s, 1 H, H-4′), 7.69 (t, J = 2.5 Hz, 1 H, CH=C), 7.61 (m, 4 H, Ph), 7.41 – 7.30 (m, 8 H, Ph, H-6′, H-7′), 7.11 (d, J = 3.0 Hz, 1 H, H-2′), 6.55 (d, J = 3.0 Hz, 1 H, H-3′), 3.83 (s, 3 H, CH3N), 3.79 (m, 4 H, –CH2OTBDPS, –CH2OH), 3.18 (mAB, 2 H, H-4), 1.00 (s, 9 H, (–C(CH3)3); 13C NMR (50 MHz, CDCl3) δ 172.22, 138.62, 137.32, 135.62, 135.60, 132.75, 132.58, 130.19, 129.88, 128.78, 127.79, 126.33, 124.13, 123.78, 121.21, 109.60, 102.13, 85.00, 66.27, 65.37, 33.00, 32.27, 26.69, 19.20; IR (neat) 3372, 2923, 2858, 1708, 1113, 805, 699 cm−1; ESI-MS m/z 548 [M+Na]+, 526 [M+H]+, 448 [M+H-C6H6]+; HRMS (ESI) m/z [M + H]+ calcd for C32H35NO4Si: 526.2408, found: 526.2401.
(E)-5-((tert-butyldiphenylsilyloxy)methyl)-5-(hydroxymethyl)-3-((1-methyl-1H-indol-6-yl)methylene)dihydrofuran-2(3H)-one (13c)
Starting from 12c (457 mg, 0.71 mmol) and following general procedure B, 13c was obtained as a white solid (167 mg, 45% yield); mp 171°C, 1H NMR (400 MHz, CDCl3) δ 7.70 (t, J = 2.6 Hz, 1 H, CH=C), 7.66 (d, J = 8.3 Hz, 1 H, H-4′), 7.60 (m, 4 H, Ph), 7.42 – 7.25 (m, 8 H, Ph, H-5′, H-7′), 7.17 (d, J = 3.0 Hz, 1 H, H-2′), 6.52 (d, J = 3.0 Hz, 1 H, H-3′), 3.81 (s, 3 H, –NCH3), 3.79 (m, 4 H, –CH2OTBDPS, –CH2OH), 3.19 (mAB, 2 H, H-4), 1.00 (s, 9 H, (–C(CH3)3); 13C NMR (50 MHz, CDCl3) δ 172.24, 138.42, 136.64, 135.60, 132.69, 132.48, 131.40, 129.86, 128.26, 127.77, 122.15, 121.22, 121.10, 111.96, 101.39, 85.24, 66.20, 65.15, 32.90, 32.17, 26.64, 19.19; IR (neat) 3424, 2929, 2857, 1708, 1111, 1056, 802, 698 cm−1; ESI-MS m/z 548 [M+Na]+, 526 [M+H]+, 448 [M+H-C6H6]+; HRMS (ESI) m/z [M + H]+ calcd for C32H35NO4Si: 526.2408, found: 526.2409.
(E)-5-((tert-butyldiphenylsilyloxy)methyl)-5-(hydroxymethyl)-3-((1-methyl-1H-indol-7-yl)methylene)dihydrofuran-2(3H)-one (13d)
Starting from 12d (316 mg, 0.63 mmol) and following general procedure B, 13d was obtained as a white solid (180 mg, 54% yield); mp 165 – 167°C; 1H NMR (400 MHz, CDCl3) δ 8.30 (m, 1 H, CH=C), 7.67 (d, J = 7.6 Hz, 1 H, H-4′), 7.57 (d, J = 7.6 Hz, 4 H, Ph), 7.36 (m, 2 H, Ph), 7.26 (m, 5 H, H-6′, Ph), 7.12 (t, J = 7.6 Hz, 1 H, H-5′), 7.00 (d, J = 3.0 Hz, 1 H, H-2′), 6.52 (d, J = 3.0 Hz, 1 H, H-3′), 3.94 (s, 3 H, –NCH3), 3.85-3.69 (m, 4 H, –CH2OTBDPS, –CH2OH), 3.12 (mAB, 2 H, H-4), 1.88 (t, J = 6.8 Hz, 1 H, OH), 1.00 (s, 9 H, –C(CH3)3); 13C NMR (50 MHz, CDCl3) δ 171.49, 135.57, 135.48, 134.37, 133.27, 132.66, 132.37, 131.35, 130.39, 129.86, 127.74, 125.87, 123.14, 122.64, 119.56, 119.24, 101.42, 85.59, 66.11, 65.12, 37.50, 31.74, 26.67, 19.22; IR (neat) 3391, 2929, 2857, 1708, 1296, 1112, 796, 700 cm−1; ESI-MS m/z 548 [M+Na]+, 526 [M+H]+, 448 [M+H-C6H6]+; HRMS (ESI) m/z [M + H]+ calcd for C32H35NO4Si: 526.2408, found: 526.2407.
General Procedure C. Acylation
A solution of 13 (a–d) (1 equiv) in dichloromethane (12 ml/mmol) was treated with Et3N (3 equiv), valproic acid chloride (1.3 equiv) and a catalytic amount of DMAP (0.1 equiv). The reaction was stirred at room temperature and monitored by TLC, and upon completion it was concentrated in vacuo. Purification by silica gel flash column chromatography gave 14 (a–d).
(E)-(2-((tert-butyldiphenylsilyloxy)methyl)-4-((1-methyl-1H-indol-4-yl)methylene)-5-oxotetrahydrofuran-2-yl)methyl 2-propylpentanoate (14a)
Starting from 13a (549 mg, 1.04 mmol) and following general procedure C, 14a was obtained as an amber oil (623 mg, 92% yield); 1H NMR (400 MHz, CDCl3) δ 8.06 (m, 1 H, CH=C), 7.61 (m, 4 H, Ph), 7.42- 7.22 (m, 9 H, Ph, H-5′, H-6′, H-7′), 7.16 (d, J = 3.0 Hz, 1 H, H-2′), 6.72 (d, J = 2.5 Hz, 1 H, H-3′), 4.30 (mAB, 2 H, –CH2CO2–), 3.85 (s, 3 H, –NCH3), 3.81 (d, 1 H, J = 10.6 Hz, –CHaHOTBDPS), 3.73 (d, 1 H, J = 10.6 Hz, –CHHbOTBDPS), 3.22 (dd, 1 H, J = 17.7, 2.5 Hz, C-H4a), 3.06 (dd, 1 H, J = 17.7, 2.8 Hz, C-H4b), 2.34 (m, 1 H, (CH3CH2CH2)2CHCO2CH2–), 1.15 – 1.56 (m, 8 H, (CH3CH2CH2)2CHCO2CH2–), 1.00 (s, 9 H, –C(CH3)3), 0.78 (t, J = 7.32 Hz, 3 H, (CH3CH2CH2)2CHCO2CH2), 0.74 (t, J = 7.07 Hz, 3 H, (CH3CH2CH2)2CHCO2CH2);13C NMR (50 MHz, CDCl3) δ 175.97, 171.49, 136.85, 135.60, 135.54, 133.65, 132.54, 132.37, 129.89, 129.57, 127.80, 126.54, 124.04, 121.36, 119.44, 111.03, 99.44, 83.17, 66.32, 65.33, 45.13, 34.38, 33.02, 26.61, 20.56, 19.16, 13.86; IR (neat): 2956, 2931, 2858, 1751, 1736, 1105, 742, 701 cm−1; ESI-MS m/z 674 [M+Na]+, 652 [M+H]+; HRMS (ESI) m/z [M + H]+ calcd for C40H49NO5Si: 652.3453, found: 652.3453.
(E)-(2-((tert-butyldiphenylsilyloxy)methyl)-4-((1-methyl-1H-indol-6-yl)methylene)-5-oxotetrahydrofuran-2-yl)methyl 2-propylpentanoate (14c)
Starting from 13c (167 mg, 0.32 mmol) and following general procedure C, 14c was obtained as an amber oil (198 mg, 96% yield); 1H NMR (400 MHz, CDCl3) δ 7.71 (m, 1 H, CH=C), 7.66 (d, J = 8.3 Hz, 1 H, H-4′), 7.61 (m, 4 H, Ph), 7.41 – 7.24 (m, 8 H, Ph, H-5′, H-7′), 7.18 (d, J = 2.8 Hz, 1 H, H-2′), 6.53 (d, J = 3.0 Hz, 1 H, H-3′), 4.33 (mAB, 2 H, –CH2OCO–), 3.82 (s, 3 H, –NCH3), 3.81 (d, 1 H, J = 10.6 Hz, –CHaHOTBDPS), 3.76 (d, 1 H, J = 10.6 Hz, –CHHbOTBDPS), 3.23 (dd, 1 H, J = 17.2, 2.5 Hz, C-4a), 3.06 (dd, 1 H, J = 17.2, 2.5 Hz, C-4b), 2.35 (m, 1 H, (CH3CH2CH2)2CHCO2CH2–), 1.48 (m, 2 H, (CH3CH2CH2)2CHCO2CH2–), 1.35 (m, 2 H, (CH3CH2CH2)2CHCO2CH2–), 1.23 (m, 4 H, (CH3CH2CH2)2CHCO2CH2–), 1.01 (s, 9 H, –C(CH3)3), 0.79 (t, 3 H, J = 7.3 Hz, (CH3CH2CH2)2CHCO2CH2–), 0.75 (t, 3 H, J = 7.3 Hz, (CH3CH2CH2)2CHCO2CH2–); 13C NMR (50 MHz, CDCl3) δ 176.00, 171.67, 138.29, 136.72, 135.62, 135,57, 132.61, 132.42, 131.43, 129.91, 128.23, 127.81, 121.59, 121.16, 120.95, 112.09, 103.74, 101.47, 83.00, 66.35, 65.37, 45.17, 34.41, 32.92, 32.84, 26.68, 20.59, 19.23, 13.87; IR (neat) 2956, 2931, 2858, 1737, 1105, 810, 701 cm−1; ESI-MS m/z 674 [M+Na]+, 652 [M+H]+; HRMS (ESI) m/z [M + H]+ calcd for C40H49NO5Si: 652.3453, found: 652.3449.
(E)-(2-((tert-butyldiphenylsilyloxy)methyl)-4-((1-methyl-1H-indol-7-yl)methylene)-5-oxotetrahydrofuran-2-yl)methyl 2-propylpentanoate (14d)
Starting from 13d (180 mg, 0.34 mmol) and following general procedure C, 14d was obtained as an amber oil (191 mg, 86% yield); 1H NMR (400 MHz, CDCl3) δ 8.31 (m, 1 H, CH=C), 7.67 (d, J = 7.6 Hz, 1 H, H-4′), 7.58 (d, J = 6.9 Hz, 4 H, Ph), 7.40-7.17 (m, 7 H, Ph, H-6′), 7.11 (t, J = 7.6 Hz, 1 H, H-5′), 7.00 (d, J = 2.8 Hz, 1 H, H-2′), 6.52 (d, J = 3.0 Hz, 1 H, H-3′), 4.29 (qAB, 2 H, J = 11.9 Hz, –CH2OCO–), 3.96 (s, 3 H, –NCH3), 3.76 (qAB, 2 H, J = 10.6 Hz, –CH2OTBDPS), 3.16 (dd, 1 H, J = 17.6, 2.1 Hz, H-4a), 3.01 (dd, 1 H, J = 17.6, 2.1 Hz, H-4b), 2.36 (m, 1 H, (CH3CH2CH2)2CHCO2CH2–), 1.52 (m, 2 H, (CH3CH2CH2)2CHCO2CH2–), 1.37 (m, 2 H, (CH3CH2CH2)2CHCO2CH2–), 1.24 (m, 4 H, (CH3CH2CH2)2CHCO2CH2–), 1.01 (s, 9 H, –C(CH3)3), 0.81 (t, 3 H, J = 7.1 Hz, (CH3CH2CH2)2CHCO2CH2–), 0.79 (t, 3 H, J = 7.1 Hz, (CH3CH2CH2)2CHCO2CH2–); 13C NMR (50 MHz, CDCl3) δ 175.88, 170.91, 135.57, 135.48, 134.43, 133.27, 132.60, 132.31, 131.38, 130.50, 129.92, 127.80, 125.23, 123.23, 122.50, 119.50, 119.27, 101.51, 83.29, 66.17, 65.18, 45.15, 37.53, 34.41, 32.32, 26.70, 20.62, 20.56, 19.22, 13.86; IR (neat) 2957, 2931, 2858, 1736, 1111, 701 cm−1; ESI-MS m/z 674 [M+Na]+; HRMS (ESI) m/z [M + H]+ calcd for C40H49NO5Si: 652.3453, found: 652.3456.
General Procedure D. TBDPS Deprotection
To a solution of compounds 14a–d in anhydrous THF (20 mL/mmol) was added triethylamine hydrofluoride (10 eq). The reaction mixture was stirred at 70°C until the starting material disappeared by TLC analysis. Volatiles were removed and the residue was purified by silica gel flash column chromatography to yield pure final DAG-indololactones 2, 3, 4 and 5.
(E)-(2-(hydroxymethyl)-4-((1-methyl-1H-indol-4-yl)methylene)-5-oxotetrahydrofuran-2-yl)methyl 2-propylpentanoate (2)
Starting from 14a (547 mg, 0.84 mmol) and following general procedure D, 2 was obtained as a yellow crystalline solid (287 mg, 83% yield); mp 100 – 102°C; 1H NMR (400 MHz, CDCl3) δ 8.10 (t, J = 2.8 Hz, 1 H, CH=C), 7.40 (dd, J = 6.4, 2.0 Hz, 1 H, H-7′), 7.27 (m, 2 H, H-5′, H-6′), 7.17 (d, J = 3.0 Hz, 1 H, H-2′), 6.73 (d, J = 3.0 Hz, 1 H, H-3′), 4.31 (qAB, 2 H, J = 12.1 Hz, –CH2OCO–), 3.84 (s, 3 H, –NCH3), 3.76 (qAB, J = 12.1 Hz, 2 H, –CH2OH), 3.26 (dd, 1 H, J = 17.7, 2.8 Hz, H-4a), 3.09 (dd, 1 H, J = 17.7, 2.8 Hz, H-4b), 2.38 (m, 1 H, (CH3CH2CH2)2CHCO2CH2–), 2.15 (m, 1 H, OH), 1.53 (m, 2 H, (CH3CH2CH2)2CHCO2CH2–), 1.37 (m, 2 H, (CH3CH2CH2)2CHCO2CH2–), 1.21 (m, 4 H, (CH3CH2CH2)2CHCO2CH2–), 0.81 (t, 3 H, J = 7.3 Hz, (CH3CH2CH2)2CHCO2CH2–), 0.79 (t, 3 H, J = 7.3 Hz, (CH3CH2CH2)2CHCO2CH2–); 13C NMR (50 MHz, CDCl3) δ 176.37, 171.36, 136.91, 134.72, 130.08, 129.65, 126.32, 123.17, 121.41, 119.64, 111.33, 99.36, 83.16, 65.22, 64.95, 45.19, 34.38, 33.02, 32.75, 20.57, 13.86, 13.83; IR (neat) 3465, 2932, 1724, 1225, 1042, 731, 746 cm−1; UV (CH3OH/H2O [1:1] with 0.1% CH3CO2H) λmax @ 256 nm, 363 nm (conjugated 1-methyl indole); HPLC (gradient elution) pure (>98%); ESI-MS m/z 436 [M+Na]+, 414 [M+H]+; HRMS (ESI) m/z [M + H]+ calcd for C24H31NO5: 414.2275, found: 414.2269.
(E)-(2-(hydroxymethyl)-4-((1-methyl-1H-indol-5-yl)methylene)-5-oxotetrahydrofuran-2-yl)methyl 2-propylpentanoate (3)
Starting from 13b (137 mg, 0.26 mmol) and following general procedures C and D, 3 was obtained as a white solid (48 mg, 44% yield); mp 107 – 109°C; 1H NMR (400 MHz, CDCl3) δ 7.80 (s, 1 H, H-4′), 7.73 (sa, 1 H, CH=C), 7.37 (m, 2 H, H-6′, H-7′), 7.10 (d, J = 3.0 Hz, 1 H, H-2′), 6.54 (d, J = 3.0 Hz, 1 H, H-3′), 4.35 (qAB, 2 H, J = 11.8 Hz, –CH2OCO–), 3.82 (s, 3 H, –NCH3), 3.80 (dd, 1 H, J = 12.2, 6.6 Hz, –CHaHOH), 3.73 (dd, 1 H, J = 12.2, 5.8 Hz, –CHHbOH), 3.26 (dd, 1 H, J = 17.4, 2.8 Hz, H-4a), 3.08 (dd, 1 H, J = 17.4, 2.8 Hz, H-4b), 2.45 (t, 1 H, J = 6.3 Hz, OH), 2.39 (m, 1 H, (CH3CH2CH2)2CHCO2CH2–), 1.54 (m, 2 H, (CH3CH2CH2)2CHCO2CH2–), 1.37 (m, 2 H, (CH3CH2CH2)2CHCO2CH2–), 1.23 (m, 4 H, (CH3CH2CH2)2CHCO2CH2–), 0.82 (t, 3 H, J = 7.3 Hz, (CH3CH2CH2)2CHCO2CH2–), 0.78 (t, 3 H, J = 7.3 Hz, (CH3CH2CH2)2CHCO2CH2–); 13C NMR (50 MHz, CDCl3), δ 176.42, 171.79, 139.45, 137.40, 130.30, 128.78, 125.95, 124.09, 123.93, 119.78, 109.66, 102.14, 83.01, 65.28, 64.97, 45.17, 34.39, 34.38, 32.98, 32.49, 20.58, 20.57, 13.88, 13.84; IR (neat) 3363, 2952, 2932, 2872, 1741, 1717, 1147, 811, 714 cm−1; UV (CH3OH/H2O [1:1] with 0.1% CH3CO2H) λmax @ 280 nm, 329 nm (conjugated 1-methyl indole); HPLC (gradient elution) pure (>98%); ESI-MS m/z 849 [M2+Na]+, 436 [M+Na]+, 414 [M+H]+; HRMS (ESI) m/z [M + H]+ calcd for C24H31NO5: 414.2275, found: 414.2259.
(E)-(2-(hydroxymethyl)-4-((1-methyl-1H-indol-6-yl)methylene)-5-oxotetrahydrofuran-2-yl)methyl 2-propylpentanoate (4)
Starting from 14c (146 mg, 0.22 mmol) and following general procedure D, 4 was obtained as a pale yellow oil (59 mg, 63% yield); 1H NMR (400 MHz, CDCl3) δ 7.73 (m, 1 H, CH=C), 7.65 (d, J = 8.3 Hz, 1 H, H-4′), 7.43 (s, 1 H, H-7′), 7.27 (dd, J = 9.1, 2.8 Hz, 1 H, H-5′), 7.18 (d, J = 3.0 Hz, 1 H, H-2′), 6.52 (d, J = 2.8 Hz, 1 H, H-3′), 4.31 (qAB, 2 H, J = 11.9 Hz, –CH2OCO–), 3.83 (s, 3 H, –NCH3), 3.81 (d, J = 12.1 Hz, 1 H, –CHaHOH), 3.73 (d, J = 12.1 Hz, 1 H, –CHHbOH), 3.28 (dd, 1 H, J = 17.3, 2.5 Hz, H-4a), 3.08 (dd, 1 H, J = 17.3, 2.5 Hz, C-4b), 2.37 (m, 1 H, (CH3CH2CH2)2CHCO2CH2–), 1.54 (m, 2 H, (CH3CH2CH2)2CHCO2CH2–), 1.37 (m, 2 H, (CH3CH2CH2)2CHCO2CH2–), 1.23 (m, 4 H, (CH3CH2CH2)2CHCO2CH2–), 0.82 (t, J = 7.3 Hz, 3 H, (CH3CH2CH2)2CHCO2CH2–), 0.77 (t, J = 7.3 Hz, 3 H, (CH3CH2CH2)2CHCO2CH2–); 13C NMR (100 MHz, CDCl3) δ 176.34, 171.67, 139.31, 136.66, 131.59, 130.07, 127.91, 121.19, 121.00, 120.71, 112.28, 101.47, 83.11, 65.29, 64.91, 45.17, 34.39, 32.92, 32.44, 20.56, 13.87, 13.81; IR (neat) 3435, 2956, 2932, 2872, 1732, 1644, 1164, 1043, 731, 625 cm−1; UV (CH3OH/H2O [1:1] with 0.1% CH3CO2H) λmax @ 271 nm, 325 nm (conjugated 1-methyl indole); HPLC (gradient elution) pure (>98%); ESI-MS m/z 436 [M+Na]+, 414 [M+H]+; HRMS (ESI) m/z [M + H]+ calcd for C24H31NO5: 414.2275, found: 414.2274; Elem. Anal. Calcd. for C24H31NO5: %C, 69.71; %H, 7.56; %N, 3.39. Found: %C, 69.70; %H, 7.49, %N, 3.40.
(E)-(2-(hydroxymethyl)-4-((1-methyl-1H-indol-7-yl)methylene)-5-oxotetrahydrofuran-2-yl)methyl 2-propylpentanoate (5)
Starting from 14d (94 mg, 0.14 mmol) and following general procedure D, 5 was obtained as a pale yellow oil (40 mg, 67% yield); 1H NMR (400 MHz, CDCl3) δ 8.38 (m, 1 H, CH=C), 7.66 (d, J = 7.6 Hz, 1 H, H-4′), 7.24 (d, J = 7.6 Hz, 1 H, H-6′), 7.11 (t, J = 7.6 Hz, 1 H, H-5′), 7.01 (d, J = 3.1 Hz, 1 H, H-2′), 6.51 (d, J = 3.1 Hz, 1 H, H-3′), 4.34 (qAB, 2 H, J = 11.9 Hz, –CH2OCO–), 4.05 (s, 3 H, –NCH3), 3.79 (dd, J = 6.6, 12.1 Hz, –CHaOH), 3.70 (dd, J = 6.6, 12.1 Hz, –CHHbOH), 3.21 (dd, 1 H, J = 17.7, 2.8 Hz, H-4a), 3.03 (dd, 1 H, J = 17.7, 2.8 Hz, H-4b), 2.39 (m, 1 H, (CH3CH2CH2)2CHCO2CH2–), 2.13 (t, 1 H, J = 6.8 Hz, –OH), 1.55 (m, 2 H, (CH3CH2CH2)2CHCO2CH2–), 1.40 (m, 2 H, (CH3CH2CH2)2CHCO2CH2–), 1.24 (m, 4 H, (CH3CH2CH2)2CHCO2CH2–), 0.83 (t, J = 7.3 Hz, 3 H, (CH3CH2CH2)2CHCO2CH2–), 0.81 (t, J = 7.3 Hz, 3 H, (CH3CH2CH2)2CHCO2CH2–); 13C NMR (50 MHz, CDCl3) δ 176.25, 170.84, 134.41, 131.48, 130.57, 124.51, 123.53, 122.65, 119.31, 101.62, 83.34, 65.09, 64.88, 45.21, 37.63, 34.43, 32.14, 20.61, 13.86; IR (neat) 3430, 2957, 2931, 2872, 1732, 1292, 1207, 1015, 719 cm−1;); UV (CH3OH/H2O [1:1] with 0.1% CH3CO2H) λmax @ 261 nm, 355 nm (conjugated 1-methyl indole); HPLC (gradient elution) pure (>98%); ESI-MS m/z 436 [M+Na]+, 414 [M+H]+; HRMS (ESI) m/z [M+H]+ calcd for C24H31NO5: 414.2275, found: 414.2275; Elem. Anal. Calcd. For: C24H31NO5: %C, 69.71; %H, 7.56; %N, 3.39. Found: %C, 69.44; %H, 7.51, %N, 3.43.
5.3. Biological studies
Cell culture
LNCaP cells (ATCC, Manassas, VA) were cultured in RPMI-1640 medium (ATCC) containing 4 mM L-glutamine supplemented with 10% FBS (ATCC). CHO-K1 cells (ATCC) were grown in F-12 medium (Invitrogen) supplemented with 10% FBS (ATCC). HEK-293-RasGRP3 cells were grown in DMEM supplemented with 10% FBS (ATCC).
Molecular Constructs
The constructs used for confocal imaging (human GFP-PKCα, human YFP-PKCε, and human GFP-PKCδ) were described earlier.38 Human RasGRP3 was PCR amplified and cloned into the pQBI25fN1 vector using NheI restriction enzyme. Sequence analysis of the construct was conducted by the DNA Minicore (Center for Cancer Research, NCI, National Institutes of Health).
Ligand binding
Binding of [3H]PDBu to PKCα, to RasGRP3, and to RasGRP1-C1 and competition for binding by DAG-indololactones were measured in the presence of 100 μg/ml phosphatidylserine as described previously.23 To optimize stability, binding to PKCα and RasGRP1-C1 was performed at 37°C; binding to RasGRP3 was performed at 18°C.
Biological activity of ligands
HEK-293 cells and HEK-293 with overexpressed RasGRP3 (LEGFP-HEK-N-RasGRP3) cells were seeded at 2 × 106/dish, cultured for 24 h, then treated with different concentrations of PMA or indololactones 1–5 for 30 min. The cells were then collected, lysed, and western blotting was performed. Briefly, equal amounts of protein were separated by SDS gel electrophoresis and then transferred to immobilon-P membranes (Millipore Corp.). The membranes were blocked and labeled with primary antibodies and secondary antibodies. The signals were developed by the ECL western blotting detection system and imaged on BioMax XAR film. The following antibodies were used: anti-RasGRP3(1:1000), ERK (1:2000) and p-ERK (1:1000) antibodies (Cell Signaling Technology Inc.); anti-p-RasGRP3(pT133) (1:2000), p-PKCδ (pS299)(1:3000) antibodies (Epitomics Company); anti-PKCδ (1:2000) and β-actin (1:2000) antibodies (Santa Cruz Biotechnology Inc). Results represent triplicate independent experiments for all compounds.
Ramos cells grown in RPMI-1640 (ATCC) containing 10% fetal bovine serum and 1% L-glutamine were plated in 6 cm dishes at a concentration of 300,000 cells/ml. After 24 hours cells were exposed to compounds at 0.01, 0.1, 1, and 10 μM concentrations for 30 minutes. Cells were then pelleted and cell lysates were prepared in PBS containing 0.5% Triton X-100 with protease and phosphatase inhibitors (Roche). Cells were sonicated 3 times for 6 seconds each and 7.5 μg of protein lysate (concentration calculated using BIO-RAD DC protein assay) in SDS containing beta-mercaptoethanol was separated on 10% Tris-glycine gels (Novex) and transferred to nitrocellulose membranes (Whatman). The membranes were blocked with 5% nonfat dry milk (BIO-RAD) and incubated overnight in the primary antibody, washed with PBS containing 0.05% Triton X-100 and incubated 1 hour in the secondary antibody and washed in PBS containing 0.05% Triton X-100. The blots were developed by ECL (Amersham) and signals detected on chemiluminescence film (Amersham). The primary antibodies used were: RasGRP3 (C33A3, Cell Signaling), pRasGRP3 (pT133, Epitomics), PKCδ (Epitomics), pPKCδ (S299, Epitomics), pPKD1 (S744/748, Cell Signaling), ERK (Cell Signaling), pERK (T202/Y204, Cell Signaling), PKCα (c-terminus, Epitomics), and PKCε (C-15, Santa Cruz). The secondary antibody was a horseradish peroxidase conjugated anti-rabbit (BIO-RAD) antibody.
Visualization of translocation of PKC isoforms and RasGRP3 in living cells in response to ligands
CHO-K1 cells (40,000 cells/ml) or LNCaP cells (80,000 cells/ml) were plated onto ibidi dishes (ibidi LLC, Verona, WI). After 24 or 48 hours in culture, respectively, cells were transfected with recombinant constructs using Lipofectamine reagent and Plus reagent (both from Invitrogen) according to the manufacturer’s protocol. Cellular expression and translocation of fluorescent fusion proteins after drug treatment were examined 24 h after transfection using a Zeiss LSM 510 NLO confocal microscopy system (Carl Zeiss, Inc., Thornwood, NY) with excitation from a 30-milliwatt argon laser tuned to 488 nm and emission collected with a BP 500–530 filter. Images were acquired every 30 s for 30 min at varying zoom settings (1.4–2) using Zeiss AIM software and a 63 × 1.4 NA Zeiss Plan-Apochromat oil immersion objective. Quantitation of confocal images was performed as described earlier.57 In each cell, regions of 4 μm2 each were selected in the cytoplasm and the cell membrane. Mean intensities in the selected regions were calculated using the Zeiss AIM software at the indicated time points. The ratio of the intensities for membrane and cytoplasm was then calculated and normalized to the time 0 values. The increase in the membrane/cytoplasm ratio indicates translocation.
5.4. Biophysical studies
Giant vesicle (GV) preparation
GVs were prepared through the rapid evaporation method.68 GVs comprising combinations of lipids were prepared in the molar ratio of 9:1, DMPC/DMPG. The lipid constituents were dissolved in chloroform/ethanol (1:1, v/v) and subsequently added to a round-bottom flask (250 mL) containing chloroform (1 mL). The aqueous phase (5 mL of 0.1 M sucrose, 0.1 M KCl, 2 mM Tris solution, pH 7.4) was then carefully added along the flask walls. The organic solvent was removed in a rotary evaporator under reduced pressure (final pressure 40 mbar) at 25°C and 40 rpm. After evaporation for 4–5 min, an opalescent fluid was obtained with a volume of approximately 5 mL.
Fluorescence measurements
Changes in the compound’s intrinsic emission were measured for 2.42 μM and 6.05 μM solutions titrated with DMPC/DMPG giant vesicles. Fluorescence emission spectra were acquired at 27°C on a FL920 spectrofluorimeter (Edinburgh, Co., Edinburgh, UK) by using the compound’s excitation wavelength. Total sample volumes were 1 mL, and the solutions were placed in a quartz cell with a 0.5 cm optical path-length. Light scattering from the vesicles was confirmed to account for less than 5% of the emission intensity.
Fluorescence anisotropy
The fluorescence probe DHP-TMA was incorporated into the GVs, that were prepared according to the procedure described above, by adding the dye dissolved in THF (1 mM) to the vesicles up to a final concentration of 1.25 μM and incubating at room temperature for 30 min in order to allow the incorporation of the probe into the vesicles. The required amount of solutions of compounds 1 to 5 (DMSO, 1 mg/mL) were added to the vesicles containing TMA-DPH probe (30 μL) and the volume was adjusted to 1 mL with buffer (0.1 M sucrose, 0.1 M KCl, 2 mM Tris solution, pH 7.4). The samples were placed in a quartz cell with a 0.5 cm optical path-length. TMA-DPH fluorescence anisotropy was measured at 430 nm (excitation 355 nm) on a FL920 spectrofluorimeter. Anisotropy values were automatically calculated by the spectrofluorimeter software by using conventional methodology. The concentrations of compounds were 2.42 μM and 6.05 μM.
Förster resonance energy transfer (FRET) measurements
Vesicles prepared according to the procedure described above were additionally supplemented with NBD-PE at a 100:1 (phospholipid:fluorophore) molar ratio. Indololactones 1 to 5 were dissolved in DMSO (1 mg/mL) and the required amount of each solution was placed in a quartz cell with a 0.5 cm optical path-length containing 30 μl of vesicles and the volume was adjusted to 1 mL with buffer (0.1 M sucrose, 0.1 M KCl, 2 mM Tris solution, pH 7.4). Emission spectra were acquired at 530 nm (NBD-PE) with an excitation wavelength of the indololactone at 25°C on a FL920 spectrofluorimeter. Energy transfer was determined by three types of measurement: 1. only vesicles, 2. vesicles with addition of indololactone and 3. vesicles with addition of indololactone and addition of 20% Triton X-100.
5.5 Molecular modeling
Molecular dynamics simulations with an implicit solvent bilayer were run in CHARMM69 version c35b5. Forcefield parameters for the lactone ring and alkyl sidechain of the DAG-indololactones were developed by analogy with existing parameters for lipids70 and furanose sugars, and checked against ab-initio geometry-optimized structures calculated at the RHF/6-31G* theory level. Parameters for the N-methylindole sidechain were based on tryptophan, and the indole-lactone linker was derived from styrene in the CHARMM General Force Field.71
The implicit solvent bilayer was simulated using the HDGB (heterogeneous dielectric generalized Born) method [ref 44] in CHARMM. In this method the bilayer is modeled as a series of slabs, set up perpendicular to the z-axis, with varying dielectric constants. The inner slab, centered at z = 0 with a width of 20 Å, has a dielectric constant of 2.0 as a representation of the hydrophobic core of the bilayer, a middle slab with a thickness of 5 Å has a dielectric constant of 7.0 as a representation of the glycerol backbone region, and the outer slab has a dielectric constant of 80.0 for bulk water. An apparent dielectric constant as a function of membrane depth z was calculated by solving the Poisson equation for a probe ion moving across the membrane. This apparent dielectric is then used to calculate the electrostatic component of the solvation energy for each atom by solving the Born equation. The surface tension coefficient for the nonpolar portion of the solvation energy was 0.015 kcal/mol·Å2.
Four molecular dynamics simulations were run for each indololactone, each starting from a different orientation relative to the bilayer, with two inside the membrane core at z = 0 and two outside the membrane entirely at z = 25. Each simulation underwent 200 steps of adopted-basis Newton Raphson miminization, was heated in 10-degree increments from a starting temperature of 50K to 300K over the course of 10 ps, and run at constant temperature for 100 ns, with coordinate snapshots saved every 5 ps. Any rotational and translational movement of the molecules was stopped after every picosecond. After 20 ns of simulation time, each of the indololactones had reached its equilibrium position in the membrane, and the remaining 80 ns were used for analysis. The absolute value of the z-position of each heavy atom in the indole ring and branched alkyl group was extracted from each saved coordinate snapshot, binned in 0.25 Å increments from z = 0 to z =30, and averaged over the four simulations, to give the density of each type of atom at each z-position. The transition dipole moment of the indole ring was defined as a vector from atom CG to atom CZ2. This vector was then normalized, and its angle to the bilayer normal (which is the z-axis) was calculated by taking the arccosine of its z-component. This angle was extracted from each saved coordinate snapshot, binned in 5 degree increments from −180 to 180, and averaged over the four simulations, to give the occupancy of each angle.
Supplementary Material
Acknowledgments
The research was supported in part by The National Institute of Industrial Technology (INTI) and Consejo Nacional de Investigación Científica y Tecnológica (CONICET); in part by the Intramural Research Program of the National Institutes of Health, Center for Cancer Research, National Cancer Institute (Z1A BC 005720) and in part with Federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract HHSN261200800001E.
Abbreviations
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- DMAP
4-dimethylaminopyridine
- DMPC/DMPG
1,2-dimyristoyl-sn-glycero-3-phospocholine/1,2-dimyristoyl-sn-glycero-3-phosphoglycerol
- Erk1/2
extracellular signal-regulated kinases 1 and 2
- LiHMDS
lithium hexamethyldisilazide
- MsCl
methanesulfonyl chloride
- PCC
pyridinium chlorochromate
- NBD-PE
[1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl)] (ammonium salt)
- PDBu
[20-3H]phorbol 12,13-dibutyrate
- PKC
protein kinase C
- PC
phosphatidyl choline
- PG
1-(3-sn-phosphatidyl)-rac-glycerol sodium salt
- PKD
protein kinase D
- PMA
phorbol 12-myristate 13-acetate
- PPTS
pyridinium p-toluene sulfonate
- RasGRP
Ras guanine nucleotide-releasing protein
- TBDMSCl
tert-butyldimethylsilyl chloride
- TBDPSCl
tert-butyldiphenylsilyl chloride
Footnotes
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nishizuka Y. Science (80-) 1992;258:607–614. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- 2.Rhee SG. Annu Rev Biochem. 2001;70:281–312. doi: 10.1146/annurev.biochem.70.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oude Weernink PA, Han L, Jakobs KH, Schmidt M. Biochim Biophys Acta - Biomembr. 2007;1768:888–900. doi: 10.1016/j.bbamem.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 4.Griner EM, Kazanietz MG. Nat Rev Cancer. 2007;7:281–294. doi: 10.1038/nrc2110. [DOI] [PubMed] [Google Scholar]
- 5.Newton AC. Chem Rev. 2001;101:2353–2364. doi: 10.1021/cr0002801. [DOI] [PubMed] [Google Scholar]
- 6.Newton AC, Johnson JE. Biochim Biophys Acta - Rev Biomembr. 1998;1376:155–172. doi: 10.1016/s0304-4157(98)00003-3. [DOI] [PubMed] [Google Scholar]
- 7.Boije af Gennäs G, Talman V, Yli-Kauhaluoma J, Tuominen RK, Ekokoski E. Curr Top Med Chem. 2011;11:1370–1392. doi: 10.2174/156802611795589584. [DOI] [PubMed] [Google Scholar]
- 8.http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.DrugDetails.
- 9.http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002275/human_med_001600.jsp&mid=WC0b01ac058001d124.
- 10.Wang QJ. Trends Pharmacol Sci. 2006;27:317–323. doi: 10.1016/j.tips.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 11.Bruinsma SP, Baranski TJ. Cell Cycle. 2007;6:2440–2444. doi: 10.4161/cc.6.20.4786. [DOI] [PubMed] [Google Scholar]
- 12.Silinsky EM, Searl TJ. Br J Pharmacol. 2003;138:1191–1201. doi: 10.1038/sj.bjp.0705213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Topham MK. J Cell Biochem. 2006;97:474–484. doi: 10.1002/jcb.20704. [DOI] [PubMed] [Google Scholar]
- 14.Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, Sahai E. Nat Cell Biol. 2007;9:1392–1400. doi: 10.1038/ncb1658. [DOI] [PubMed] [Google Scholar]
- 15.Stone JC. Biochem Soc Trans. 2006;34:858–861. doi: 10.1042/BST0340858. [DOI] [PubMed] [Google Scholar]
- 16.Stone JC. Genes and Cancer. 2011;2:320–334. doi: 10.1177/1947601911408082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang D, Kedei N, Li L, Tao J, Velasquez JF, Michalowski AM, Tóth BI, Marincsák R, Varga A, Bíró T, Yuspa SH, Blumberg PM. Cancer Res. 2010;70:7905–7917. doi: 10.1158/0008-5472.CAN-09-4729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang D, Tao J, Li L, Kedei N, Tóth ZE, Czap A, Velasquez JF, Mihova D, Michalowski AM, Yuspa SH, Blumberg PM. Oncogene. 2011;30:4590–4600. doi: 10.1038/onc.2011.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song X, Lopez-Campistrous A, Sun L, Dower Na, Kedei N, Yang J, Kelsey JS, Lewin NE, Esch TE, Blumberg PM, Stone JC. PLoS One. 2013;8:e72331. doi: 10.1371/journal.pone.0072331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marquez VE, Blumberg PM. Acc Chem Res. 2003;36:434–443. doi: 10.1021/ar020124b. [DOI] [PubMed] [Google Scholar]
- 21.Duan D, Sigano DM, Kelley JA, Lai CC, Lewin NE, Kedei N, Peach ML, Lee J, Abeyweera TP, Rotenberg SA, Kim H, Kim YH, El Kazzouli S, Chung J-U, Young HA, Young MR, Baker A, Colburn NH, Haimovitz-Friedman A, Truman J-P, Parrish DA, Deschamps JR, Perry NA, Surawski RJ, Blumberg PM, Marquez VE. J Med Chem. 2008;51:5198–5220. doi: 10.1021/jm8001907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.El Kazzouli S, Lewin NE, Blumberg PM, Marquez VE. J Med Chem. 2008;51:5371–5386. doi: 10.1021/jm800380b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gal N, Kolusheva S, Kedei N, Telek A, Naeem TA, Lewin NE, Lim L, Mannan P, Garfield SH, El Kazzouli S, Sigano DM, Marquez VE, Blumberg PM, Jelinek R. Chem Bio Chem. 2011;12:2331–2340. doi: 10.1002/cbic.201100246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kang JH, Benzaria S, Sigano DM, Lewin NE, Pu Y, Peach ML, Blumberg PM, Marquez VE. J Med Chem. 2006;49:5371–5386. doi: 10.1021/jm060011o. [DOI] [PubMed] [Google Scholar]
- 25.Jung ME, Usui Y, Vu CT. Tetrahedron Lett. 1987;28:5977–5980. [Google Scholar]
- 26.Choquet-farnier C, Stasik I, Baupere D. Carbohydr Res. 1997;303:185–191. [Google Scholar]
- 27.Prakash C, Saleh S, Blair IA. Tetrahedron Lett. 1989;30:19–22. [Google Scholar]
- 28.Sharma R, Lee J, Wang S, Milne GW, Lewin NE, Blumberg PM, Marquez VE. J Med Chem. 1996;39:29–35. doi: 10.1021/jm950277n. [DOI] [PubMed] [Google Scholar]
- 29.Nacro K, Sigano DM, Yan S, Nicklaus MC, Pearce LL, Lewin NE, Garfield SH, Blumberg PM, Marquez VE. J Med Chem. 2001;44:1892–1904. doi: 10.1021/jm010052e. [DOI] [PubMed] [Google Scholar]
- 30.Durgan J, Michael N, Totty N, Parker PJ. FEBS Lett. 2007;581:3377–3381. doi: 10.1016/j.febslet.2007.06.035. [DOI] [PubMed] [Google Scholar]
- 31.Aiba Y, Oh-Hora M, Kiyonaka S, Kimura Y, Hijikata A, Mori Y, Kurosaki T. Proc Natl Acad Sci U S A. 2004;101:16612–16617. doi: 10.1073/pnas.0407468101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng Y, Liu H, Coughlin J, Zheng J, Li L, Stone JC. Blood. 2005;105:3648–3654. doi: 10.1182/blood-2004-10-3916. [DOI] [PubMed] [Google Scholar]
- 33.Lorenzo PS, Kung JW, Bottorff DA, Garfield SH, Stone JC, Blumberg PM. Cancer Res. 2001;61:943–949. [PubMed] [Google Scholar]
- 34.Brodie C, Steinhart R, Kazimirsky G, Rubinfeld H, Hyman T, Ayres JN, Hur GM, Toth A, Yang D, Garfield SH, Stone JC, Blumberg PM. Mol Pharmacol. 2004;66:76–84. doi: 10.1124/mol.66.1.76. [DOI] [PubMed] [Google Scholar]
- 35.Teixeira C, Stang SL, Zheng Y, Beswick NS, Stone JC. Blood. 2003;102:1414–1420. doi: 10.1182/blood-2002-11-3621. [DOI] [PubMed] [Google Scholar]
- 36.Wang QJ, Bhattacharyya D, Garfield S, Nacro K, Marquez VE, Blumberg PM. J Biol Chem. 1999;274:37233–37239. doi: 10.1074/jbc.274.52.37233. [DOI] [PubMed] [Google Scholar]
- 37.Wang QJ, Fang TW, Fenick D, Garfield S, Bienfait B, Marquez VE, Blumberg PM. J Biol Chem. 2000;275:12136–12146. doi: 10.1074/jbc.275.16.12136. [DOI] [PubMed] [Google Scholar]
- 38.Kedei N, Lewin NE, Géczy T, Selezneva J, Braun DC, Chen J, Herrmann MA, Heldman MR, Lim L, Mannan P, Garfield SH, Poudel YB, Cummins TJ, Rudra A, Blumberg PM, Keck GE. ACS Chem Biol. 2013;8:767–777. doi: 10.1021/cb300671s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kedei N, Lubart E, Lewin NE, Telek A, Lim L, Mannan P, Garfield SH, Kraft MB, Keck GE, Kolusheva S, Jelinek R, Blumberg PM. Chembiochem A Eur J Chem Biol. 2011;12:1242–1251. doi: 10.1002/cbic.201100064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kedei N, Telek A, Czap A, Lubart ES, Czifra G, Yang D, Chen J, Morrison T, Goldsmith PK, Lim L, Mannan P, Garfield SH, Kraft MB, Li W, Keck GE, Blumberg PM. Biochem Pharmacol. 2011;81:1296–1308. doi: 10.1016/j.bcp.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Illinger D, Duportail G, Mely Y, Poirel-Morales N, Gerard D, Kuhry JG. Biochim Biophys Acta - Biomembr. 1995;1239:58–66. doi: 10.1016/0005-2736(95)00135-p. [DOI] [PubMed] [Google Scholar]
- 42.Lentz BR. Chem Phys Lipids. 1993;64:99–116. doi: 10.1016/0009-3084(93)90060-g. [DOI] [PubMed] [Google Scholar]
- 43.Burnett D, Audus LJ. Phytochemistry. 1964;3:395–415. [Google Scholar]
- 44.Lakowicz JR. Principles of Fluorescence Spectroscopy. 2. Kluwer Academic/Plenum; New York: 1999. p. 954. [Google Scholar]
- 45.Tanizaki S, Feig M. J Chem Phys. 2005;122:124706. doi: 10.1063/1.1865992. [DOI] [PubMed] [Google Scholar]
- 46.Persson S, Antoinette Killian J, Lindblom G. Biophys J. 1998;75:1365–1371. doi: 10.1016/s0006-3495(98)74054-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yau WM, Wimley WC, Gawrisch K, White SH. Biochemistry. 1998;37:14713–14718. doi: 10.1021/bi980809c. [DOI] [PubMed] [Google Scholar]
- 48.Gaede HC, Yau WM, Gawrisch K. J Phys Chem B. 2005;109:13014–13023. doi: 10.1021/jp0511000. [DOI] [PubMed] [Google Scholar]
- 49.Norman KE, Nymeyer H. Biophys J. 2006;91:2046–2054. doi: 10.1529/biophysj.105.080275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grossfield A, Woolf TB. Langmuir. 2002;18:198–210. [Google Scholar]
- 51.Kaiser RD, London E. Biochemistry. 1999;38:2610. doi: 10.1021/bi985065h. [DOI] [PubMed] [Google Scholar]
- 52.Chattopadhyay a, London E. Biochemistry. 1987;26:39–45. doi: 10.1021/bi00375a006. [DOI] [PubMed] [Google Scholar]
- 53.Goñi FM, Alonso A. Prog Lipid Res. 1999;38:1–48. doi: 10.1016/s0163-7827(98)00021-6. [DOI] [PubMed] [Google Scholar]
- 54.Eftink MR, Selvidge LA, Callis PR, Rehms AA. J Phys Chem. 1990;94:3469–3479. [Google Scholar]
- 55.Albinsson B, Nordén B. J Phys Chem. 1992;96:6204–6212. [Google Scholar]
- 56.Kang J, Benzaria S, Sigano DM, Lewin NE, Pu Y, Peach ML, Blumberg PM, Marquez VE. J Med Chem. 2006;49:3185–3203. doi: 10.1021/jm060011o. [DOI] [PubMed] [Google Scholar]
- 57.Geczy T, Peach ML, El Kazzouli S, Sigano DM, Kang J-H, Valle CJ, Selezneva J, Woo W, Kedei N, Lewin NE, Garfield SH, Lim L, Mannan P, Marquez VE, Blumberg PM. J Biol Chem. 2012;287:13137–13158. doi: 10.1074/jbc.M111.320010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kang J, Peach ML, Pu Y, Lewin NE, Nicklaus MC, Blumberg PM, Marquez VE. J Med Chem. 2005:5738–5748. doi: 10.1021/jm050352m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tamamura H, Sigano DM, Lewin NE, Peach ML, Nicklaus MC, Blumberg PM, Marquez VE. J Med Chem. 2004;47:4858–4864. doi: 10.1021/jm049723+. [DOI] [PubMed] [Google Scholar]
- 60.Sigano DM, Peach ML, Nacro K, Choi Y, Lewin NE, Nicklaus MC, Blumberg PM, Marquez VE. J Med Chem. 2003;46:1571–1579. doi: 10.1021/jm020476o. [DOI] [PubMed] [Google Scholar]
- 61.Pascher I. Curr Opin Struct Biol. 1996;6:439–448. doi: 10.1016/s0959-440x(96)80107-2. [DOI] [PubMed] [Google Scholar]
- 62.Lee J. Arch Pharm Res. 1998;21:452–457. doi: 10.1007/BF02974642. [DOI] [PubMed] [Google Scholar]
- 63.Goldberg EM, Zidovetzki R. Biophys J. 1997;73:2603–2614. doi: 10.1016/S0006-3495(97)78290-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cho W, Stahelin RV. Annu Rev Biophys Biomol Struct. 2005;34:119–151. doi: 10.1146/annurev.biophys.33.110502.133337. [DOI] [PubMed] [Google Scholar]
- 65.Pu Y, Perry NA, Yang D, Lewin NE, Kedei N, Braun DC, Choi SH, Blumberg PM, Garfield SH, Stone JC, Duan D, Marquez VE. J Biol Chem. 2005;280:27329–27338. doi: 10.1074/jbc.M414132200. [DOI] [PubMed] [Google Scholar]
- 66.Sanchez KM, Kang G, Wu B, Kim JE. Biophys J. 2011;100:2121–2130. doi: 10.1016/j.bpj.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Choi Y, Kang JH, Lewin NE, Blumberg PM, Lee J, Marquez VE. J Med Chem. 2003;46:2790–2793. doi: 10.1021/jm030082c. [DOI] [PubMed] [Google Scholar]
- 68.Moscho A, Orwar O, Chiu DT, Modi BP, Zare RN. Proc Natl Acad Sci U S A. 1996;93:11443–11447. doi: 10.1073/pnas.93.21.11443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brooks BR, Brooks CL, III, Mackerell AD, Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, Jr, Pastor RW. J Phys Chem B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, Mackerell AD., Jr J Comput Chem. 2010;31:671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.