

Abstract
GLUT proteins are encoded by the SLC2 genes and are members of the major facilitator superfamily of membrane transporters. Fourteen GLUT proteins are expressed in the human and they are categorized into 3 classes based on sequence similarity. All GLUTs appear to transport hexoses or polyols when expressed ectopically, but the primary physiological substrates for several of the GLUTs remain uncertain. GLUTs 1–5 are the most thoroughly studied and all have well established roles as glucose and/or fructose transporters in various tissues and cell types. The GLUT proteins are comprised of ~500 amino acid residues, possess a single N-linked oligosaccharide, and have 12 membrane-spanning domains. In this review we briefly describe the major characteristics of the 14 GLUT family members.
Keywords: SLC2, GLUT, Membrane Transport, Glucose Transporters, Glucose Homeostasis
1. Introduction
The transport of monosaccharides, polyols, and other small carbon compounds across the membranes of eukaryotic cells is mediated by members of the GLUT family of integral membrane proteins that are encoded by the SLC2 genes and are members of the major facilitator superfamily (MFS) (see Table 1) [reviewed in (Augustin, 2010; Joost et al., 2002; Thorens and Mueckler 2010; Uldry and Thorens, 2004)]. The 14 human GLUT proteins possess various substrate specificities and are involved in the transport of several hexoses in addition to myo-inositol (Uldry et al., 2001), urate (Bibert et al., 2009; Matsuo et al., 2008; So and Thorens, 2010), glucosamine (Maher and Harrison, 1990), and ascorbate (Lee et al., 2010). All of the members of the GLUT family are facilitative transporters with the exception of HMIT, which is a H+/myo-inositol symporter (Uldry et al., 2001). It is highly likely that the major substrates for several GLUT proteins have not yet been identified.
Table 1.
SLC2 – Facilitative GLUT transporter family
| Human gene name |
Protein name | Aliases | Predominant substrates |
Transport type/ coupling ions*) |
Tissue distribution and cellular/ subcellular expression |
Link to disease |
Human gene locus |
Sequence accession ID |
Splice variants and their features |
|---|---|---|---|---|---|---|---|---|---|
| SLC2A1 | GLUT1 | glucose, galactose, mannose, glucosamine | F | erythrocytes, brain, blood-brain barrier, blood-tissue barrier, many fetal | paroxysmal exertion-induced dyskinesia, dystonia-18, Glut1 deficiency syndrome | 1p35-31.3 | NM_006516 | ||
| SLC2A2 | GLUT2 | glucose, galactose, fructose, mannose, glucosamine | F | liver, islet of Langerhans, intestine, kidney, brain | Fanconi-Bickel syndrome, (type 2 diabetes) | 3q26.1-q26.2 | NM_000340 | ||
| SLC2A3 | GLUT3 | glucose, galactose, mannose, xylose | F− | brain (neurons), testis | 12p13.3 | NM_006931 | |||
| SLC2A3P1 | GLUT3 pseudogene 1 | 5q35.1 | NG_008263 | ||||||
| SLC2A3P2 | GLUT3 pseudogene 2 | 1p31.3 | NG_005841 | ||||||
| SLC2A3P4 | GLUT3 pseudogene 4 | 8q21.3 | NG_009577 | ||||||
| SLC2A4 | GLUT4 | glucose, glucosamine | F | adipose tissue (white and brown), skeletal and cardiac muscle | (type 2 diabetes) | 17p13 | NM_001042 | ||
| SLC2A5 | GLUT5 | fructose | F | small intestine, kidney | 1p36.2 | NM_003039 | 2 splice variants | ||
| SLC2A6 | GLUT6 | GLUT9 | glucose | F | brain, spleen, leucocytes | 9q34 | NM_017585 | 2 splice variants | |
| SLC2A7 | GLUT7 | glucose, fructose | F | small intestine, colon, testis, prostate | 1p36.2 | NM_207420 | |||
| SLC2A8 | GLUT8 | GLUTX1 | glucose, fructose, galactose | F | testis, brain, adrenal gland, liver, spleen, brown adipose tissue, lung (intracellular) | 9q33.3 | NM_014580 | ||
| SLC2A9 | GLUT9 | GLUTX, URATv1 | urate (glucose, fructose) | kidney, liver, small intestine, placenta, lung and leucocytes | renal hypouricemia | 4p16-15.3 |
NM_020041 NM_001001290 |
2 splice variants | |
| SLC2A10 | GLUT10 | glucose, galactose | F | heart, lung, brain, liver, skeletal muscle, pancreas, placenta and kidney | arterial tortuosity syndrome | 20q13.1 | NM_030777 | ||
| SLC2A11 | GLUT11 | GLUT10 | glucose, fructose | F | heart, muscle | 22q11.2 |
NM_030807 NM_001024938 NM_001024939 |
3 splice variants | |
| SLC2A12 | GLUT12 | GLUT8 | glucose | F | heart, prostate, skeletal muscle, placenta | 6q23.2 | NM_145176 | ||
| SLC2A13 | HMIT | GLUT13 | myo-inositol | C/H+ | brain, adipose tissue | 12q12 | NM_052885 | ||
| SLC2A14 | GLUT14 | SLC2A3P3 | O | testis | 12p13.31 | NM_153449 | |||
| SLC2AXP1 | (pseudogene) | 2q11.2 | NG_011411 |
C: Cotransporter; E: Exchanger; F: Facilitated transporter; O: Orphan transporter
References:
Original version of the SLC table:
Uldry M, Thorens B. The SLC2 family of facilitated hexose and polyol transporters. Pflugers Arch. 2004 Feb;447(5):480–9
The 14 GLUT proteins are comprised of ~500 amino acid residues and can be categorized into 3 classes based on sequence similarity: Class 1 (GLUTs 1–4, 14); Class 2 (GLUTs 5, 7, 9, and 11); and Class 3 (GLUTs 6, 8, 10, 12, and HMIT). All GLUT proteins appear to possess 12 transmembrane segments, a single site of N-linked glycosylation, a relatively large, central, cytoplasmic linker domain, and exhibit topologies with both their N and C termini positioned in the cytoplasm (Mueckler et al., 1985). The Class 1 and 2 GLUT proteins are structurally distinguishable from the Class 3 proteins by virtue of the location of their sites of N-linked glycosylation, which reside in the first exofacial linker domains of the Class 1 and 2 GLUTs and in the fifth exofacial linker domains of the Class 3 proteins.
One or more GLUT proteins are expressed in virtually every cell type of the human body. Eleven of the 14 members of the GLUT family are capable of transporting glucose under experimental conditions, although in several cases the principal physiologic substrates for the proteins have not been definitively identified (Thorens and Mueckler, 2010). The physiological explanation for the redundancy of proteins that transport glucose is most likely the critical nature of this sugar as a circulating fuel in humans and the consequent need for multiple glucose transporters with different kinetic and regulatory properties that are expressed in a cell-type specific fashion.
2. GLUT1
2.1 Transport Kinetics
GLUT1, encoded by the SLC2A1 gene, was one of the first membrane transporters to be purified (Baldwin and Lienhard, 1989; Kasahara and Hinkle, 1977) and cloned (Birnbaum et al., 1986; Mueckler et al., 1985) and is likely one of the most extensively studied of all membrane transport systems. The kinetics of glucose transport via GLUT1 have been explored since radioisotopic substrates became readily available in the early 1950s [reviewed in (Carruthers et al., 2009; Lowe and Walmsley, 1986)]. Two prominent characteristics of glucose transport have been observed in human erythrocytes. Firstly, the apparent affinity (the Km or half-saturation concentration) for transport is, under certain experimental conditions, higher at the outward substrate-binding site than at the cytoplasmic binding site, and secondly, transport occurs at a faster rate when substrate is present on the trans side of the membrane (the side of the membrane to which transport is being measured) as compared to zero trans conditions where substrate is present only in the aqueous compartment from which transport is being measured. The latter phenomenon is called trans or exchange acceleration and suggests that a conformational change involving the unloaded transporter is rate limiting for net transport to occur. Many investigators believe that most of the available kinetic and biophysical data are consistent with an alternating conformation mechanism for glucose transport whereby mutually exclusive substrate binding sites are sequentially exposed to either the exoplasm or the cytoplasm (Gorga and Lienhard, 1981; Lowe and Walmsley, 1989; Wheeler and Whelan, 1988) (see Figure 1). However, the kinetics of glucose transport as measured in the erythrocyte are complex and some experimental observations are not consistent with a simple asymmetric carrier type model (Cloherty et al., 1996). This has led to the proposal of alternative mechanisms, including fixed-site models in which multiple binding sites are simultaneously accessible from both sides of the membrane (Carruthers et al., 2009). It has been argued that asymmetric transporter distribution under equilibrium conditions with the standard carrier model violates energy conservation laws (Naftalin, 2008).
Figure 1. Simple carrier model for the mechanism of glucose transport.
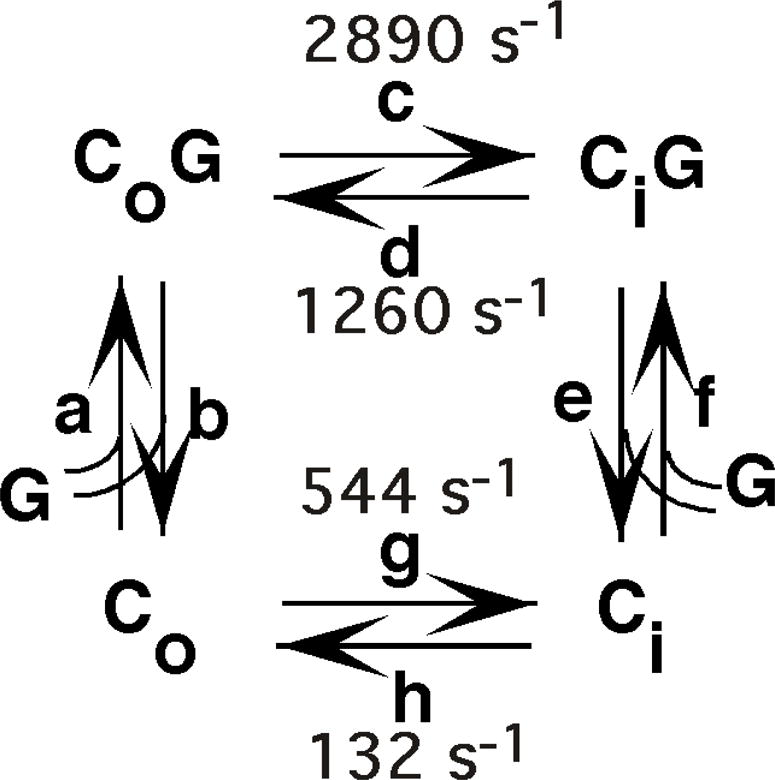
Co and Ci represent the transporter in the outward and inward-facing conformations, respectively, G is glucose, and a–h are the fundamental rate constants governing the conformational changes or the glucose binding and release steps. Estimates for rate constants c,d,g, and h at 20° C were calculated from the data of Lowe and Walmsley (Lowe and Walmsley, 1989).
However, abandonment of the carrier model for glucose transport is premature, in large part because of experimental difficulties associated with accurately measuring the kinetics of transport in erythrocytes. Questions concerning the validity of the alternately conformation model are based almost entirely on steady-state kinetic experiments. Glucose transporters are present at a very high concentration in the erythrocyte membrane (up to 10% of total integral membrane protein) (Gorga and Lienhard, 1982), and thus the rate of transport is extremely high. True initial rates of transport are at best difficult to measure using traditional methods, and the vast bulk of experimental findings that appear to contradict the alternating conformation mechanism may be the result of procedural difficulties and/or the fact that measurements were conducted at well below physiological temperatures. More sophisticated approaches are consistent with a simple carrier model that does not violate the laws of thermodynamics. For example, using a quenched-flow apparatus coupled with an automated syringe system, Lowe and Walmsley (Lowe and Walmsley, 1986) found little if any asymmetrical distribution of GLUT1 under equilibrium conditions at physiological temperatures. Using the Millipore-Swinnex filtering technique and the rapid continuous flow tube technique, which allow transport measurements within a fraction of a second, Brahm (Brahm, 1983) found that the transport kinetics of D-glucose at physiological temperatures in erythrocytes were consistent with that of a simple, symmetric carrier. The kinetics of transport via GLUT4 are also completely consistent with that of a symmetric carrier. Perhaps most importantly, the crystal structures of known members of the MFS are consistent with the presentation of a single substrate-binding site within a cavity exposed to only one aqueous compartment at a time (see below). An alternating conformation model thus remains the most viable description for the mechanism of glucose transport.
2.2 GLUT1 Structure
GLUT1 is comprised of 492 amino acid residues and possesses a single site of N-linked glycosylation at N45 (Mueckler et al., 1985). There are no other known post-translational modifications of the protein. A topology with 12 transmembrane segments and with cytoplasmic N and C termini is supported by hydrophobicity analysis and biochemical studies (Blodgett et al., 2008; Hresko et al., 1994; Mueckler et al., 1985) (see Figure 2). A low resolution, 2-dimensional model for the exofacial configuration of GLUT1 has been proposed based on a series of mutagenesis and solvent accessibility studies (Mueckler and Makepeace, 2009) (see Figure 3). GLUT proteins, like all eukaryotic members of the MFS, have thus far proven recalcitrant to crystallization, but the crystal structures of 4 bacterial members of the MFS have been determined to at least moderate resolution by x-ray diffraction analysis (Abramson et al., 2003; Dang et al., 2010; Huang et al., 2003; Yin et al., 2006). All 4 proteins share a common folding pattern with the first 6 transmembrane helices in a pseudo symmetrical configuration relative to the last 6 helices. Helices 1, 2, 4, 5, 7, 8, 10, and 11 form an inner bundle that is stabilized by the outer helices 3, 6, 9, and 12. The E. coli lactose permease (Abramson et al., 2003) and glycerol-3-P antiporter (Huang et al., 2003) were crystallized in their endofacial orientations with aqueous cavities containing their respective substrate binding sites exposed to the cytoplasm and closed off at their exofacial surfaces. No exoplasmic substrate binding sites have been identified in these structures. The E. coli fucose/proton symporter (Dang et al., 2010) was crystallized in its exofacial conformation with an aqueous cavity containing a single putative fucosedocking site exposed to the exoplasm and closed off at the cytoplasmic face. No endofacial fucose docking sites have been identified in the structure. Taken as a whole, these results strongly support an alternating conformation transport mechanism for MFS proteins and are inconsistent with any model in which multiple substrate binding sites that are exposed to both aqueous compartments exist simultaneously. A homology-based model of GLUT1 in its putative endofacial conformation is shown in Figure 4 (Salas-Burgos et al., 2004). However, the accuracy of this model is uncertain because the protein used as the template structure, the E. coli glycerol-3-P antiporter, shares little if any significant sequence identity with GLUT1, making it impossible to evaluate the accuracy of the sequence alignment (Forrest et al., 2006).
Figure 2. Amino acid sequence and membrane topology of human GLUT1.
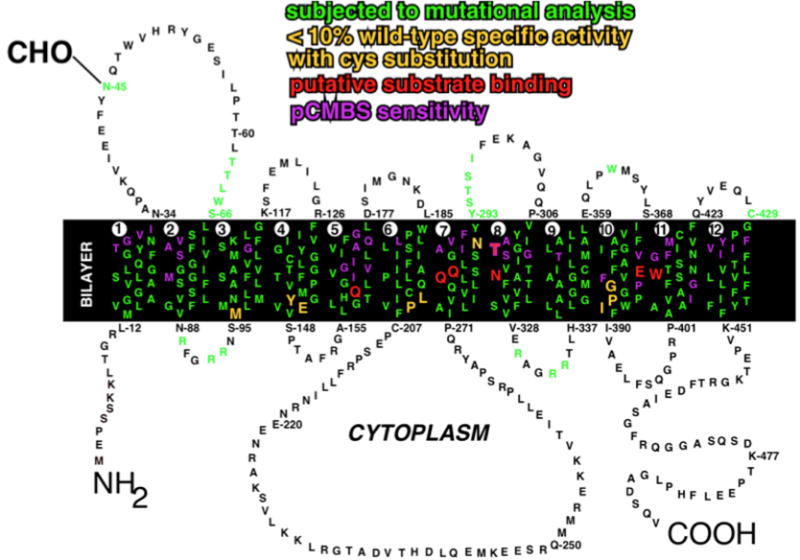
Amino acid residues are designated by the single letter code. The transmembrane segments are numbered 1–12. The linkage of the N-linked oligosaccharide at N45 is shown. Positions that have been analyzed by site-directed mutagenesis are in green. Residues that are believed to play a direct role in substrate binding are shown in red. Residues that are accessible to pCMBS from the external solvent are shown in purple. Residues that appear to be critical for transport activity are shown in yellow. Adapted from (Mueckler and Makepeace, 2009).
Figure 3. Model of the exoplasmic substrate-binding site of GLUT1 (Mueckler and Makepeace, 2009).
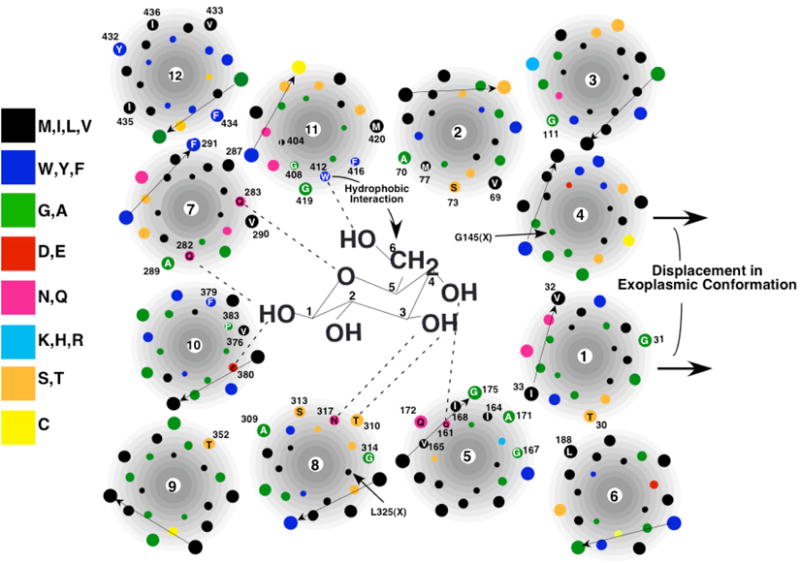
Glucose is not drawn to scale. The arrangement of helices is shown in a simplistic fashion for clarity. Amino acid residues that are in contact with solvent in the aqueous cavity are numbered and identified by the single-letter code. Dotted lines represent putative hydrogen bonds.
Figure 4. Localization of pCMBS-reactive and putative substrate-binding residues in a homology based model of GLUT1.
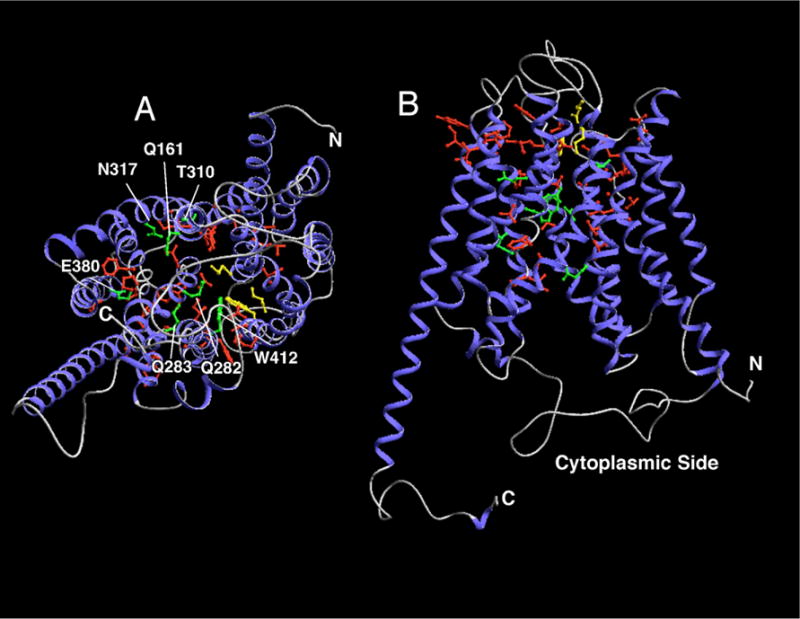
The molecular diagrams were created using DeepView (Swiss Institute of Bioinformatics) based on the coordinates published in (Salas-Burgos et al., 2004). The homology model is based on the crystal structure of the glycerol-3-P antiporter of E. coli in its cytoplasmic-facing conformation. The 12 transmembrane helices are drawn as blue ribbons and non-helical regions as gray lines. pCMBS-reactive side chains are shown in green. The 3 side chains near the exoplasmic face of the membrane that were identified as a putative glucose docking site are shown in yellow. Possible substrate-binding side chains based on mutagenesis experiments are presented in green and identified by the single letter amino acid code. A) View perpendicular to the membrane from inside the cell. B) Transverse view.
2.3. GLUT1 Tissue Distribution and Physiology
The principal physiological substrate of GLUT1 is clearly glucose but it is also capable of transporting mannose, galactose, glucosamine, and reduced ascorbate, and its activity is inhibited by cytochalasin B and phloretin (Carruthers et al., 2009). GLUT1 is expressed in many cell types, often in conjunction with one or more additional GLUT isoforms. It is expressed at its highest level in the human erythrocyte membrane, through which glucose freely equilibrates between the serum and the red cell cytoplasm. The function of the high level of GLUT1 expression in erythrocytes may be to effectively increase the glucose carrying capacity of the blood. GLUT1 plays a critical role in cerebral glucose uptake as the major GLUT isoform expressed in brain endothelial cells (Koranyi et al., 1991; Simpson et al., 2001; Yeh et al., 2008). Under normal physiological conditions the brain is absolutely dependent on glucose as a fuel source, and transport across the blood-brain barrier is rate limiting for brain glucose metabolism (Simpson et al., 1994). GLUT1 is also expressed in brain astrocytes, which have been proposed to supply glycolytically-derived lactate to neurons as a major fuel source (Pellerin et al., 2002). GLUT1 is responsible for mediating materno-placental transfer of glucose in the human and levels of placental GLUT1 are altered under various pathological conditions that affect the fetus [reviewed in (Illsley, 2000)]. GLUT1 plays an important role in the survival of the pre-implantation embryo (Moley, 2001; Moley et al., 1998) and throughout much of fetal development, and consequently homozygous GLUT1 null mice do not survive beyond embryonic day 14 (Wang et al., 2006).
2.4 GLUT1 Pathophysiology
Haplodeficiency of SLC2A1 is the cause of an autosomal dominant genetic disease called GLUT1 deficiency syndrome (GDS) [reviewed in (De Vivo et al., 2002)]. Several dozen distinct de novo mutations in the SLC2A1 structural gene have been identified in patients with this disorder. Patients with GDS experience seizures beginning in early infancy and exhibit developmental delay, ataxia, and neurobehavioral symptoms during development. The only current treatment for the disease is a rigid ketogenic diet, which supplies the nervous system with an alternative fuel source. However, ketogenic diet therapy does not prevent or reverse all of the symptoms associated with GDS and there is a need to develop alternative, more effective, therapies.
Upregulation of GLUT1 expression is observed in a wide variety of tumors and is likely to be an essential process in tumor progression (Yamamoto et al., 1990; Younes et al., 1996). Tumors almost universally become highly dependent on the substrate level generation of ATP via aerobic glycolysis (the Warburg Effect), thus explaining the necessity for the increased expression of glucose transporters (Bannasch et al., 2008). The isoform-specific inhibition of glucose transport may be an excellent novel pharmacological approach to cancer therapy.
3. GLUT2
GLUT2 was first characterized by cDNA cloning the Slc2a2/SLC2A2 gene from rat and human liver cDNA libraries (James et al., 1988; Kayano et al., 1988). GLUT2 has the unique characteristic among glucose transporters to have a low apparent affinity for glucose (Km ~17mM). It can also transport galactose (Km ~ 92 mM), mannose (Km ~125 mM) and fructose (Km ~76 mM) with low affinity. Interestingly, it has a very high affinity for glucosamine (Km 0.8 mM) (Uldry et al., 2002). The high Km for glucose indicates that the rate of glucose transport by GLUT2 varies as a function of glucose concentrations under physiological and even diabetic conditions. Cellular GLUT2 expression is usually very high and the rate of glucose uptake is not limiting for the process of glucose utilization.
GLUT2 is the major glucose transporter of hepatocytes. It is also expressed by intestinal absorptive cells, by cells forming the kidney proximal convoluted tubule, by pancreatic beta-cells (Thorens et al., 1990), and in a small number of neurons dispersed in many brain structures and in astrocytes (Arluison et al., 2004; Mounien et al., 2010); it is also expressed by tanycytes, special cells lining the bottom part and the floor of the third ventricle (Garcia et al., 2003).
In hepatocytes GLUT2 is involved in glucose uptake and release in the fed and fasted states, respectively. However, although it is indispensable for glucose uptake, glucose release from hepatocytes does not require the presence of GLUT2 as an alternate, membrane-traffic based system appears to release glucose in the case of stimulated hepatic glucose production (Guillam et al., 1998).
In the intestine, GLUT2 is the major basolateral glucose transporter isoform. Glucose uptake depends on the presence of the Na+/glucose co-transporter SGLT1 present in the apical membrane (Hediger et al., 1987). This can transport both glucose and galactose; the uptake of fructose is mediated by GLUT5 also present in the apical membrane. It has been shown that the expression of GLUT2 is dispensable for normal intestinal glucose absorption. Evidence has been presented that a similar membrane-based traffic system as described for liver also operates in enterocytes that allows release of glucose on the serosal side of the epithelial cells (Stumpel et al., 2001).
More recently, it has been shown that GLUT2 can be induced to translocate to the apical membrane of enterocytes to participate in glucose absorption (Kellett et al., 2008). This mechanism depends on the presence of glucose in the intestinal lumen, its absorption by SGLT1, which induces membrane depolarization and Ca++ entry, followed by phospholipase ßII activation-dependent translocation of GLUT2. This process may be important for maximizing glucose uptake in the presence of high luminal glucose concentrations. Insulin has been reported to induce the internalization of apically expressed GLUT2, a process that is impaired in insulin resistance, contributing to increased glucose absorption (Tobin et al., 2008).
In the kidney, GLUT2 is present on the basolateral membrane of the epithelial cells involved in glucose reabsorption. Its expression is required for glucose absorption. In contrast to the situation in intestinal cells, suppression of GLUT2 expression by Slc2a2 gene knockout induces massive glucosuria (Guillam et al., 1997) indicating this transporter is absolutely required for the renal glucose reabsorption process.
In rodent pancreatic beta cells GLUT2 is the major glucose transporter, whereas in human islets GLUT1 and GLUT3 are also expressed (De Vos et al., 1995; Thorens et al., 1988). Glucose uptake is the first step in the metabolic-dependent signaling pathway that controls glucose-stimulated insulin secretion (GSIS). The glucose transport rate is ~ 20–50-fold greater than the rate of glucose phosphorylation by glucokinase, the rate-limiting step in GSIS. Thus, under most conditions, glucose uptake is a permissive step in GSIS. The expression of GLUT2 is, however, regulated by glucose and lipids in beta cells, and glucolipotoxicity (high glucose and free fatty acid concentrations) reduce its surface expression (Gremlich et al., 1997; Reimer and Ahren, 2002). In both mouse and human beta cells, the cell surface expression of GLUT2 depends on its interaction with galectin 9, a cell surface lectin that binds to a specific N-linked glycan structure (Ohtsubo et al., 2011; Ohtsubo et al., 2005). Under glucolipotoxic conditions the glycosyltransferase involved in generating this structure is down-regulated and prevents the normal formation of the GLUT2 N-glycan, leading to its internalization; the reduction in cell surface expression of GLUT2 may then be sufficient to impair GSIS.
Slc2a2 gene knockout prevents beta-cell glucose uptake in mice and suppresses GSIS, leading to death of the newborn mice around the time of weaning. As transgenic expression of Slc2a1 in Slc2a2 knockout mice normalizes GSIS and allows these mice to live and reproduce normally, this indicates that if there is isoform specificity in GLUT2 function, it may not be linked to its role in allowing glucose uptake for normal GSIS, but may be related to a regulatory function that has not yet been elucidated.
Because GLUT2 as well as glucokinase are present in brain nuclei involved in the control of energy homeostasis, such as different hypothalamic and brainstem nuclei, it has been postulated that GLUT2 may be associated with glucose sensing mechanisms that control feeding, energy expenditure, and counterregulation (Marty et al., 2007).
Studies with Slc2a2 knockout mice (with rescued beta-cell function) indeed showed that the absence of GLUT2 resulted in abnormal feeding behavior during the fasting-to-fed transition, or in response to intracerebroventricular injections of glucose or of the glucose anti-metabolite 2-deoxy-D-glucose (Bady et al., 2006). Further experiments with these mice also indicated that central GLUT2-dependent glucose sensing is involved in glucagon secretion in response to hypoglycemia (Marty et al., 2005) and in the control of thermogenesis by brown adipose tissue, probably secondary to a regulation of the activity of NPY and POMC neurons of the hypothalamic arcuate nucleus controlling the melanocortin pathway and the sympathetic innervation of brown fat (Mounien et al., 2010). In two human cohorts it has been shown that a mutation in the SLC2A2 gene is associated with a preference for sugar feeding (Eny et al., 2008), indicating that GLUT2 may also control processes related to food preference. Further studies are required to define in more detail the exact GLUT2-expressing cells involved in these regulatory mechanisms.
In the human, SLC2A2 gene defects are the cause of Fanconi-Bickel syndrome (Fanconi and Bickel, 1949; Foretz and Thorens, 2003; Santer et al., 1997). Patients suffering from this disease show the same phenotype as Slc2a2 knockout mice. Intestinal glucose absorption is not impaired, hepatic glucose production is normal but they display hepatomegaly and a renal syndrome with glucosuria and aminoaciduria. In contrast to the knockout mice, Fanconi-Bickel patients show only slightly impaired or normal insulin secretion, perhaps due to the expression of GLUT1 and GLUT3 in human islets.
4. GLUT3
The SLC2A3 gene encoding GLUT3 was first cloned from a human fetal skeletal muscle cell line (Kayano et al., 1988). It shares ~64% sequence identity with SLC2A1. GLUT3 has a higher apparent affinity (lower Km) and a higher maximum turnover number for glucose than the other Class 1 GLUT proteins, and its principal physiological substrate is clearly D-glucose (Manolescu et al., 2007). The kinetic characteristics of GLUT3 may explain its role as the primary mediator of glucose uptake into neurons (Leino et al., 1997), as the concentration of cerebral glucose is considerably lower than that in the blood. GLUT1 and GLUT3 are thus critical delivery systems for the major fuel substrate utilized by parenchymal cells of the brain. Although it has been proposed that lactate produced by glycolysis in astrocytes is the primary fuel substrate for neurons, this hypothesis has been seriously challenged (Simpson et al., 2007).
GLUT3 is also expressed in several other cell types, although in a species-specific manner. It is abundantly expressed in the murine sperm flagellum (Urner and Sakkas, 1999), but is absent in bovine sperm where GLUT5 is the most abundant GLUT transporter (Angulo et al., 1998). This difference presumably reflects the concentrations of glucose versus fructose in the seminal fluid of different species. GLUT3 is also critical for development of the preimplantation mouse embryo (Pantaleon et al., 1997; Pantaleon and Kaye, 1998; Pantaleon et al., 2008) as well as later in embryonic development (Ganguly et al., 2007), and is a critical component, along with GLUT1, of the transplacental supply of glucose to the fetus (Shin et al., 1997). Streptozotocin-induced maternal diabetes in the mouse results in a down-regulation of GLUT3 in the preimplantation embryo resulting in abnormal apoptosis and increased fetal resorptions (Wyman et al., 2008). Interestingly, GLUT3 is abundantly expressed in human white blood cells, where in resting cells it is largely confined to intracellular storage vesicles and translocates to the plasma membrane in response to various proliferative stimuli [reviewed in (Simpson et al., 2008)].
5. GLUT4
5.1 Tissue Distribution and Physiology
GLUT4 was first identified by a monoclonal antibody screen for proteins in rat adipocytes that translocate from an intracellular membrane fraction to a plasma membrane fraction in response to insulin (James et al., 1988). The Slc2a4 gene was cloned soon thereafter from rat adipose tissue (Birnbaum, 1989; Charron et al., 1989; James et al., 1989) and shares ~65% sequence identity with Slc2a1. GLUT4 is expressed most prominently in adipocytes, skeletal muscle, and cardiomyocytes, where it functions as the so-called insulin-responsive glucose transporter [reviewed in (Huang and Czech, 2007)]. Under basal (low insulin) conditions, GLUT4 resides primarily in intracellular membrane compartments. When circulating insulin levels rise after the ingestion of a carbohydrate meal, intracellular glucose transporters redistribute to the plasma membrane, thus increasing glucose uptake and metabolism in these tissues and preventing chronic elevations in blood glucose levels. A defect in this insulin-mediated translocation of GLUT4 to the plasma membrane is known as peripheral insulin resistance, which, in conjunction with a defect in insulin secretion from pancreatic beta cells and insulin resistance in the liver, results in type 2 diabetes mellitus (Kahn, 1996). Because the bulk of blood glucose is taken up by skeletal muscle in the presence of elevated insulin and GLUT4 transport activity is rate-limiting for this process, GLUT4 plays a critical role in the regulation of whole body glucose homeostasis (Mueckler, 1995). The results of transgenic overexpression studies (Hansen et al., 2000; Marshall and Mueckler, 1994; Ren et al., 1995) and adipose and/or muscle specific knock-out of Slc2a4 genes in mice (Abel et al., 2001; Kotani et al., 2004; Zisman et al., 2000) confirm an important role for GLUT4 expression in both of these tissues. The results also suggest that GLUT4 in adipose and muscle tissues plays an as yet poorly defined role in metabolic cross-talk among fat, muscle, and liver tissues in the regulation of whole body glucose homeostasis. Consistent with these animal models, enhanced insulin sensitivity in humans resulting from exercise is associated with increased expression of SLC2A4 in skeletal muscle at the transcriptional level (Ren et al., 1994), and insulin-resistant obese patients exhibit reduced GLUT4 expression levels in adipocytes (Garvey et al., 1991). GLUT4 is a specific target of HIV protease inhibitors that are essential components of combination drug therapy used to treat HIV infection (Murata et al., 2000). HIV protease inhibitors appear to bind directly to and inhibit GLUT4 activity in muscle and fat cells (Hresko and Hruz, 2011), thus causing acute peripheral insulin resistance (Hruz et al., 2002; Vyas et al.). This effect likely contributes to the metabolic syndrome and increased incidence of type 2 diabetes of patients treated with HIV protease inhibitors.
GLUT4 is also expressed in a subset of neurons, especially in cholinergic neurons of the rat forebrain, and is often co-expressed in conjunction with GLUT3 (Apelt et al., 1999). It has been speculated that GLUT4 may function to rapidly increase glucose uptake into specific neurons in response to an increased energy demand.
5.2 GLUT4 Subcellular Trafficking
In 3T3L1 adipocytes under basal conditions newly synthesized GLUT4 traverses the endoplasmic reticulum, the Golgi, and the trans-Golgi network in a normal manner before it enters what appears to be a tortuously complex subcellular trafficking pattern that likely involves the general endosomal pathway, specialized GLUT4 storage vesicles (GSVs), a subdomain of the treans-Golgi network, and probably the plasma membrane [reviewed in (Huang and Czech, 2007; Larance et al., 2008). The steady-state distribution of the protein is determined by rate constants controlled by accessory proteins governing these steps that act to retain GLUT4 in intracellular compartments. Insulin binding to its receptor, in an unknown manner, changes the rate-constants of various steps presumably by altering the activities of various proteins that in turn brings about a redistribution of about one-half of the total intracellular GLUT4 to the plasma membrane (Muretta et al., 2008; Yeh et al., 1995). This redistribution likely occurs through both the general endosomal pathway and via direct fusion of GSVs with the plasma membrane, combined with a decrease in the rate of endocytosis. A number of sorting and adaptor molecules have been implicated in GLUT4 trafficking and this subject has been recently reviewed in detail (Larance et al., 2008). Structurally, several linear targeting motifs have been implicated in GLUT4 trafficking, including a cytosolic N-terminal FQQI motif (Piper et al., 1993), and 3 distinct motifs within the cytoplasmic C-terminal domain including a dileucine motif (Verhey and Birnbaum, 1994), an acidic motif (TELEY) (Shewan et al., 2000), and the so-called IRM (insulin-responsive motif) motif (Song et al., 2008). A recent study (Blot and McGraw, 2008) in 3T3L1 adipocytes suggests that the FQQI and the TELEY motifs are involved in the intracellular retention of GLUT4 in the basal state via recycling between endosomal compartments and the trans-Golgi network and GSVs, respectively. Mutation of either motif causes a partial redistribution of GLUT4 to the cell surface and thus blunts the relative magnitude of insulin-induced redistribution to the cell surface. The di-leucine motif appears to be involved in the rapid internalization of GLUT4 from the cell surface after insulin withdrawal. The IRM appears to be involved in an early post-Golgi step because mutations in the motif completely ablate insulin responsiveness and prevent GLUT4 from reaching the cell surface under basal or insulin-stimulated conditions (see Figure 5). Furthermore, expression of the IRM mutant in 3T3L1 adipocytes appears to cause the formation of a novel membrane compartment consisting of large dispersed vesicles that are highly enriched in Ras GTPase-activating protein binding proteins 1 and 2 (G3bp1 and 2) and Caprin-1 (Song and Mueckler, Unpublished). G3bp-1 and Caprin-1 are both mRNA-binding proteins that are known to interact in cytoplasmic stress granules that represent large clusters of messenger ribonucleoprotein particles that are induced by various types of cellular stress and are not bounded by membranes (Kolobova et al., 2009; Solomon et al., 2007). The precise role of stress granules and of G3pb and Caprin-1 in their formation is unclear. G3bps have also been associated with the regulation of signaling by the Ras family of GTPases and have been proposed to be involved in a variety of other cellular functions. One possible explanation for the association of the IRM mutant with stress-granule associated proteins is that the mutant is funneled into a pathway by which miss-folded or miss-targeted proteins are sequestered in a specific membrane compartment, associated with stress granules, for subsequent degradation.
Figure 5. Colocalization of GFP-tagged wild type GLUT4 and HA-tagged IRM mutant GLUT4 by immunofluorescence laser confocal microscopy.
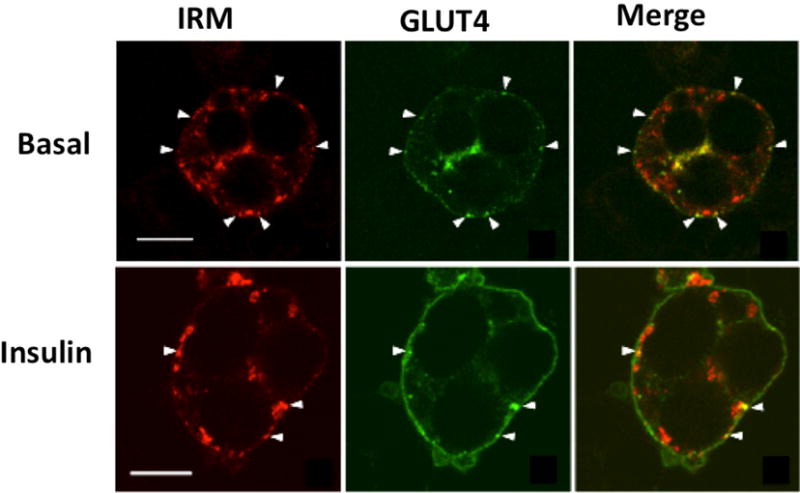
3T3L1 adipocytes were co-infected with recombinant adenoviruses encoding the GFP-tagged wild-type GLUT4 and a HA-tagged IRM mutant (see text). 48 h later the cells were serum-starved for two hours, and then either exposed to insulin for 30 min or maintained in the basal state. Tagged wild-type GLUT4 is shown in the middle panels in green, the tagged mutants are shown in the left panels in red, and the merged images are shown in the right panels with colocalization between the two coexpressed proteins presented in yellow. The scale bars represent 10 μM. Note the redistribution of wild-type GLUT4 to the plasma membrane after insulin treatment and the lack of redistribution in the IRM mutant. Additionally, the mutant and wild type transporter are largely present in distinct intracellular membranes in the absence or presence of insulin. Adapted from (Song et al., 2008).
5.3. Signaling Pathways Mediating GLUT4 Translocation
Upstream signaling to GLUT4 from either the insulin receptor or as the result of muscle contraction appears to be mediated by at least 2 distinct pathways in adipocytes and skeletal muscle. These pathways have been extensively reviewed but are still far from being completely understood (Herman and Kahn, 2006; Rose and Richter, 2005; Thong et al., 2005; Watson et al., 2004). In adipocytes and skeletal muscle, insulin binding to its receptor results in the dimerization and trans-phosphorylation of the receptor beta subunits, causing the activation of intrinsic tyrosine kinase activity leading to the recruitment and tyrosine phosphorylation of insulin receptor substrate-2 (IRS-2). The tyrosine phosphorylated SH2 domain of IRS-2 then binds to and activates PI 3-kinase, leading to the production of phosphatidylinositol 3,4,5 triphosphate (PIP3) in the membrane. Increased PIP3 levels recruit two distinct serine/threonine kinases, PDK1 and mTORC2 to the membrane, which then activate the pivotal protein kinase Akt via phosphorylations within its catalytic and hydrophobic domains. Akt exists at a central hub in the regulation of cellular growth, survival, and fuel metabolism and has several known direct substrates. There are 3 Akt isoforms and Akt2 appears to mediate, at least in part, the effect of insulin on GLUT4 translocation in adipocytes via the phosphorylation of AS160 (TBC1D4) (Eguez et al., 2005; Kane et al., 2002). AS160 is a Rab GTPase activating protein that acts on several putative downstream Rabs to convert them to their GDP-bound inactive forms. Thus, activated Akt appears to inactivate AS160, which, in turn, presumably results in the activation of one or more Rabs that are, at least in part (Gonzalez and McGraw, 2006), responsible for the recruitment of GLUT4 to the plasma membrane. Rab10 has been specifically implicated in this process, but its activation is unlikely to account fully for the redistribution of intracellular GLUT4 to the plasma membrane (Sano et al., 2007). Atypical PKCλ/ζ have also been implicated in insulin stimulated Glut4 translocation in adipocytes (Farese et al., 2007), but their precise role remains elusive. Likewise, a PI 3-kinase independent pathway involving APS, c-Cbl, CAP, CrkII, and TC10 has been proposed to be participate in GLUT4 redistribution to the plasma membrane in adipocytes (Saltiel and Pessin, 2003), but some evidence appears to contradict this hypothesis (Mitra et al., 2004).
Contraction of skeletal muscle also induces translocation of GLUT4 from intracellular membrane compartments to the plasma membrane and to transverse tubules (T-tubules), possibly via both PI 3-kinase dependent and independent pathways (Jessen and Goodyear, 2005). Increased cytosolic Ca+2/calmodulin levels and an increase in the [AMP]/[ATP] ratio have been implicated as upstream mediators of contraction-induced translocation. The latter presumably occurs via the activation of AMPK kinase, which is believed to act as a sensor for available intracellular energy stores. The former may be mediated by the tyrosine kinase, ErB4.
6. GLUT5
GLUT5 was the first of the class 2 GLUT proteins to be identified and the SLC2A5 gene was initially cloned from human small intestine (Kayano et al., 1990). It has a high specificity for fructose (Burant et al., 1992) and one of its primary functions is to mediate the uptake of dietary fructose across the apical membrane of the small intestine [reviewed in (Douard and Ferraris, 2008)]. Fructose is then released into the bloodstream via GLUT2 in the intestinal basolateral membrane. SLC2A5 expression in the intestine is regulated at the transcriptional level by the presence of fructose in the gut (Jiang et al., 2001) and by diurnal rhythm independent of fructose availability (Corpe and Burant, 1996). Even though fructose is consumed at high levels in many countries in the form of sucrose and high fructose corn syrup, levels of circulating fructose are generally ~10–100 times lower than that of glucose. This is because most dietary fructose is rapidly metabolized after absorption by the intestine, liver (via GLUT2 uptake) and kidney (also via GLUT2 uptake) (Douard and Ferraris, 2008). GLUT5 is also expressed in the kidney, fat, skeletal muscle, brain, and the sperm of certain species. Because of the very low levels of fructose in peripheral blood coupled with the relatively low apparent affinity of GLUT5 for fructose, the physiological significance of GLUT5 expression in these other tissues is uncertain, and it is even questionable whether fructose crosses the blood/brain barrier. The possibility remains that GLUT5 transports one or more additional physiological substrates other than fructose that might explain its tissue distribution and response to diabetes.
There has been much interest generated about fructose metabolism since correlations between increased consumption of dietary fructose and the rising incidence of type 2 diabetes mellitus, metabolic syndrome, and obesity in many countries became apparent (Havel, 2005). However, a cause and effect relationship between fructose consumption and these pathological states has not been established, and the correlation between the incidence of obesity and type 2 diabetes is just as strong when one compares it to total caloric intake and a sedentary lifestyle. Interestingly, GLUT5 levels are significantly upregulated in skeletal muscle and intestine of type 2 diabetic patients (Dyer et al., 2002; Stuart et al., 2007), but the physiological significance of these observations is not known.
7. GLUT8
Sequences for human, rat and mouse GLUT8 (also initially called GLUTX1), encoded by the SLC2A8/Slc2a8 gene, were first identified by database mining, cloning and functional characterization (Carayannopoulos et al., 2000; Doege et al., 2000; Ibberson et al., 2000). This transporter is only expressed in an intracellular compartment and to study its transport characteristics, surface expression was induced by mutating a dileucine internalization or intracellular retention motif (Ibberson et al., 2000). This mutated form of GLUT8 is entirely expressed at the cell surface of Xenopus oocytes or of mammalian cells. GLUT8 has a high affinity for glucose (Km ~2mM); fructose and galactose compete with glucose transport activity, which is also inhibited by cytochalasin B.
SLC2A8 mRNA is expressed at high level in the testis and at lower levels in the cerebellum, adrenal gland, liver, spleen, brown adipose tissue, and lung. In the testis, GLUT8 is expressed in differentiating spermatocytes at stage 1 (Ibberson et al., 2002) and has also been reported to be present in the acrosome of mouse and human mature spermatozoa (Joost et al., 2002). In the brain, GLUT8 is found in hippocampus, in dentate gyrus, amygdala and primary olfactory cortex, hypothalamic nuclei and the nucleus of the tractus solitarius (Ibberson et al., 2002; Reagan et al., 2002). High GLUT8 levels are present in the supraoptico-hypohyseal tract where it has been localized to synaptic vesicles and to vasopressin-containing secretory granules in the posterior pituitary. GLUT8 is also present in blastocysts, where it may play a crucial role in glucose metabolism since its suppression by antisense oligolucleotides leads to increased rates of apoptosis (Pinto et al., 2002).
Slc2a8 knockout mice display a very mild phenotype. In one study, an increase in the proliferation of hippocampal neurons and a small increase in heart P-wave duration was observed (Membrez et al., 2006). A reduction in sperm motility and in mouse locomotor activity were also reported in Slc2a8 knockout mice (Schmidt et al., 2009). As GLUT8 is only found in intracellular compartments and since there is no evidence for any significant intracellular free glucose concentration in most cells, glucose may not be the primary physiological substrate for this transporter.
8. GLUT9
GLUT9 is a type II transporter isoform that is expressed from two alternatively spliced variants of the SLC2A9 gene, which encode different amino-terminal cytoplasmic tails (Phay et al., 2000) (Augustin et al., 2004; Keembiyehetty et al., 2006). Human GLUT9a is 540 amino acids in length and is encoded by 12 exons whereas GLUT9b is comprised of 512 amino acids and is encoded by 13 exons. In both humans and mice GLUT9b is expressed only in the liver and kidney whereas Glut9a is present in many more tissues, including liver, kidney, intestine, leukocytes, and chondrocytes (Mobasheri et al., 2005). The different amino-terminal tails are important for the differential targeting of GLUT9 to opposite poles of epithelial cells. Whereas GLUT9a is expressed in the basolateral membrane, GLUT9b is targeted to the apical pole (Augustin et al., 2004). In kidney, GLUT9 is expressed in the proximal tubule (Augustin et al., 2004), whereas in mice it is present in the basolateral and apical membranes of distal convoluted tubules (Mounien et al., 2010).
Although initially considered a glucose or fructose transporter (Carayannopoulos et al., 2004) (Manolescu et al., 2007), it is now established that GLUT9 is a urate transporter (Anzai et al., 2008; Bibert et al., 2009). GLUT9a and GLUT9b transport urate with the same kinetics (Km for urate ~0.6 mM), and transport is not competed for by the presence of excess glucose or fructose. Transport is electrogenic and depends on membrane potential (Bibert et al., 2009). Urate transport can be inhibited by the uricosuric agents benzbromarone and losartan, and marginally by pyrazinoate. Transport can be partially inhibited by phloretin but not by cytochalasin B.
8.1 GLUT9 and the control of uricemia and gout
GLUT9 substrate specificity was revealed from genome wide association studies aimed at finding gene loci associated with uric acid levels. These studies found that SLC2A9 was the major locus, explaining up to 3.5% of serum uric acid level variations (Li et al., 2007) (Caulfield et al., 2008; Dehghan et al., 2008; Doring et al., 2008; Vitart et al., 2008; Wallace et al., 2008). This locus was also found to be significantly associated with gout (Doring et al., 2008). As uric acid levels have been associated with the metabolic syndrome, genetic studies have searched for association between SLC2A9 and the different components of this syndrome, i.e., insulin resistance, body mass index, hypertension, and coronary heart diseases. No strong associations have yet been established for coronary artery disease (Stark et al., 2009) or hypertension (Caulfield et al., 2008) although using Mendelian randomization analysis, a possible link between SLC2A9 variants and blood pressure has been established (Parsa et al., 2012).
Inactivating mutations in SLC2A9 have been identified in rare patients with hypouricemia (Anzai et al., 2008; Dinour et al., 2009). Such mutations also cause the large excretion of urate, often associated with renal uric acid crystals and nephropathy that characterizes the Dalmatian dog (Bannasch et al., 2008; Simkin, 2005).
In mice, genetic inactivation of Slc2a9 induces moderate hyperuricemia and massive renal excretion of urate (Preitner et al., 2009). This is associated with early-onset nephropathy, characterized by obstructive lithiasis, tubulointerstitial inflammation, and progressive inflammatory fibrosis of the cortex. Adult mice with liver-specific inactivation of GLUT9 (Mounien et al., 2010) display severe hyperuricemia associated with a urine concentration defect but with only a small increase in urine volume. Together these data indicate that liver GLUT9 is required for urate access to hepatic uricase and conversion to allantoin and for urate reabsorption in the kidney.
9. HMIT
HMIT is a H+/myo-inositol co-transporter encoded by the SLC2A13 gene (Uldry et al., 2001). Ectopic expression in Xenopus oocytes and in mammalian cells has been used for functional studies. However, for maximal plasma membrane expression two internalization motifs and one ER retention signal had to be mutated. Transport activity is specific for myo-inositol and strongly activated by acidifying the extracellular medium; this increased Vmax without changing the Km for myo-inositol (~100μM). Transport is inhibited by phloretin, phlorizin and cytochalasin B. No glucose transport activity could be measured.
SLC2A13 transcripts are expressed predominantly in the brain, with high expression in the hippocampus, hypothalamus, cerebellum and brainstem and at a low level in white and brown adipose tissues and in kidney. In brain, HMIT is found both in neurons and glial cells. In neurons HMIT is present in intracellular vesicles that can be induced to translocate and fuse with the plasma membrane to increase myo-inositol uptake. HMIT translocation occurs at growth cones and synapses and is triggered by neuronal activation and increased Ca++ influx or by activation of protein kinase C (Uldry et al., 2004). These data suggest a possible role of HMIT in regulating processes that require high levels of myo-inositol or its various phosphorylated derivatives, such as membrane recycling and growth cone dynamics and synaptic vesicle exocytosis.
10. GLUT Family Members Whose Physiological Roles Remain Unclear
GLUTs 6, 7, 10, 11, 12, and 14 were all ultimately identified as a result of the sequencing of the human genome and are encoded by the SLC2A6, SLC2A7, SLC2A10, SLC2A11, SLC2A12 and SLC2A14 genes respectively. Although they are all capable of transporting hexoses with varying efficiencies when expressed ectopically in Xenopus oocytes (Manolescu et al., 2007), the primary physiological substrates for most of these proteins have not been definitively identified.
GLUT14 is a class 1 protein whose gene (SLC2A14) shares 95% sequence identity with the SLC2A3 gene and therefore appears to be encoded by a gene duplication (Wu and Freeze, 2002). Its expression is largely confined to the testis and it has no rodent orthologue.
GLUTs 7 and 11 are class 2 proteins that share ~40–50% sequence identity with the fructose transporter, GLUT5. GLUT7 is most prominently expressed in the apical membranes of the small intestine and colon (Cheeseman, 2008). When expressed in Xenopus oocytes it displays a fairly high apparent affinity (Km ~ 0.3 mM) for both fructose and glucose, but turnover rates are low, thus calling into question whether these hexoses represent the true physiological substrates of this protein. Like GLUT7, GLUT11 (Doege et al., 2001) also transports both glucose and fructose with a fairly low Km and low turnover activity when expressed in Xenopus oocytes (Manolescu et al., 2007) and has no rodent orthologue. It is expressed as 3 sequence variants that differ at their N-termini and that probably result from differential promoter usage. The 3 GLUT11 variants are differentially expressed in heart, skeletal muscle, kidney, adipose tissue and pancreas (Sasaki et al., 2001).
GLUTs 6, 10, and 12 are class 3 proteins that share ~35% sequence identity. GLUT6 [originally named Glut9 (Phay et al., 2000)] is expressed primarily in spleen, brain, and leukocytes and displays an intracellular localization that may be due to the presence of a dileucine motif (Lisinski et al., 2001). It displays low glucose transport activity when reconstituted into proteoliposomes. The function of this protein may be to transport hexose molecules or related compounds across intracellular organellar membranes.
GLUT10 exhibits a very wide tissue distribution and is expressed in pancreas, placenta, heart, lung, liver, brain, fat, muscle, and kidney (McVie-Wylie et al., 2001). When ectopically expressed in Xenopus oocytes it exhibits a relatively low Km for 2-deoxyglucose transport (0.3 mM) but also exhibits very low intrinsic transport activity (Dawson et al., 2001). The GLUT10 gene, SLC2A10, lies within a possible susceptibility locus for type 2 diabetes mellitus (Ji et al., 1997), but no polymorphisms in the gene have yet been reported that are linked to the disease. Homozygous mutations in the SLC2A10 gene appear to be the cause of a rare genetic disorder called arterial tortuosity syndrome (ATS) (Coucke et al., 2006). Symptoms of the disease include extensive morphological abnormalities in arteries including elongation, aneurysms, and tortuosity.
GLUT12 is also widely distributed and is expressed in heart, skeletal muscle, prostate, and small intestine (Rogers et al., 2002), and is highly upregulated in breast ductal cell carcinoma (Rogers et al., 2003). It exhibits low glucose transport activity when expressed in Xenopus oocytes that is inhibited by cytochalasin B, fructose, and galactose. GLUT12 appears to localize to the Golgi apparatus and to the plasma membrane when ectopically expressed in Chinese Hamster Ovary cells (Flessner and Moley, 2009), and its subcellular targeting is influenced by a dileucine motif within its N-terminal cytoplasmic domain (Aerni-Flessner et al., 2011). Insulin has been reported to acutely stimulate the translocation of GLUT12 from intracellular membrane compartments to the plasma membrane in human skeletal muscle (Stuart et al., 2009), and transgenic overexpression of the protein enhances insulin sensitivity in mice (Purcell et al., 2011). However, what role GLUT12 plays in glucose homeostasis under normal or pathophysiological conditions remains unknown at present.
11. Concluding Remarks
A summary of the role of various Glut proteins in the regulation of glucose delivery and metabolism is provided schematically in Figure 6. The early studies on the glucose transporters GLUT1-4 revealed many facets of their involvement in the control of glucose homeostasis and provided molecular tools to understand some of the defects associated with insulin sensitivity and insulin secretion, two cardinal features of type 2 diabetes. Subsequent studies have revealed that several rare congenital disorders are due to mutations in various SLC2 genes. However, there is still much to be learned about the physiological functions of GLUT family members. Additionally, the precise mechanism of transport via these proteins has yet to be determined and awaits the resolution of three-dimensional structures for at least one of these transporters in multiple conformational states.
Figure 6. Role of GLUT Proteins in the Maintenance of glucose homeostasis.
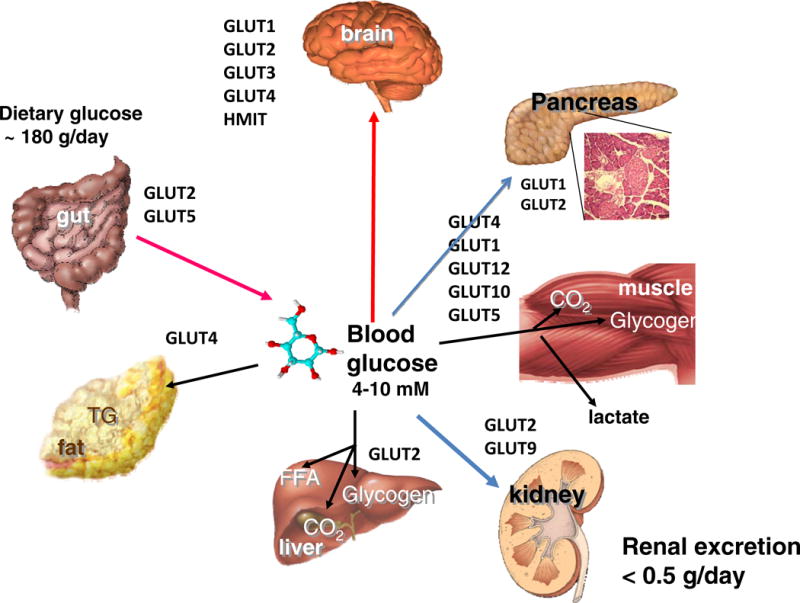
Glucose enters the hepatic portal system by transport across the gut via SGLT1 and GLUT2. In humans about one-third of blood glucose is carried within the red cell cytoplasm due to the very high level of expression of Glut1 and subsequent equilibration that occurs across the red cell membrane. Fructose crosses the gut via GLUT5. Most absorbed glucose escapes permanent catabolism by the hepatic system (which it enters via GLUT2) except when hepatic glycogen levels are low. Dietary fructose is mostly metabolized in the gut and liver and consequently circulating levels are very low. In the resting state most circulating glucose is oxidized by the central nervous after crossing the blood brain barrier via GLUT1 and enters parenchymal cells of the brain via GLUT3 (neurons) and GLUT1 (astrocytes). HMIT transports inositol against its concentration gradient in many brain cells. Other Glut proteins are expressed at lower levels and/or in smaller numbers of brain parenchymal cells and some, such as GLUT2 and GLUT4, may participate in fuel sensing by individual neurons. During exercise skeletal muscle consumes the bulk of circulating glucose via uptake by Glut1 in the endothelium and hence via Glut4 into muscle fibers. Glucose entering resting muscle is mostly converted to glycogen. Most glucose taken up into fat depots (via GLUT4) provides the glycerol moieties for the synthesis of triglycerides. Adipose tissue is also a critical endocrine organ with respect to glucose homeostasis and secretes numerous adipokines and cytokines that regulate this process. The most important organ of all with respect to the regulation of whole body glucose homeostasis is the endocrine pancreas, especially the insulin-secreting beta cells that sense blood glucose levels after initial uptake via GLUT1 (humans) or GLUT2 (rodents). Glucose is efficiently retained by the human body and very little is lost to urinary excretion, due to the combined actions of SGLT2 and GLUT2. (Figure is courtesy of Ernest Wright, UCLA Medical School).
This transporter family has now been expanded to include 14 members in the human. It is clear that many of these newly identified transporters have physiological substrates other than glucose, such as urate for GLUT9 and myo-inositol for HMIT (SLC2A13). The physiological substrates and functional roles for most of the other transporters still need to be discovered. What has been fascinating so far in the study of glucose transporters is that their careful study has generated new knowledge in various fields of research, from transporter function, cellular biology, integrated physiology, and molecular mechanisms of various diseases. Further study of these transporters will undoubtedly yield many more exciting discoveries in the years to come.
Acknowledgments
Work in the authors’ laboratories is supported by grants from the National Institutes of Health (R0143695, to M. Mueckler), the Diabetes Research and Training Center and Nutritional Sciences Center, and the Digestive Disease Center at Washington University (M. Mueckler) and the Swiss National Science Foundation (31003A-113525), Juvenile Diabetes Research Foudation Program Project 7-2005-1158, the Integrated Project Eurodia LSHM-CT-2006 518153, and Framework Programme 6 [FP6] of the European Community to B. Thorens.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abel ED, Peroni O, Kim JK, Kim YB, Boss O, Hadro E, Minnemann T, Shulman GI, Kahn BB. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. [see comments] Nature. 2001;409(6821):729–733. doi: 10.1038/35055575. [DOI] [PubMed] [Google Scholar]
- Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata S. Structure and mechanism of the lactose permease of Escherichia coli.[see comment] Science. 2003;301(5633):610–615. doi: 10.1126/science.1088196. [DOI] [PubMed] [Google Scholar]
- Aerni-Flessner LB, Otu MC, Moley KH. The amino acids upstream of NH(2)-terminal dileucine motif play a role in regulating the intracellular sorting of the Class III transporters GLUT8 and GLUT12. Mol Membr Biol. 2011;28(1):30–41. doi: 10.3109/09687688.2010.508196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angulo C, Rauch MC, Droppelmann A, Reyes AM, Slebe JC, Delgado-Lopez F, Guaiquil VH, Vera JC, Concha II. Hexose transporter expression and function in mammalian spermatozoa: cellular localization and transport of hexoses and vitamin C. Journal of Cellular Biochemistry. 1998;71(2):189–203. [PubMed] [Google Scholar]
- Anzai N, Ichida K, Jutabha P, Kimura T, Babu E, Jin CJ, Srivastava S, Kitamura K, Hisatome I, Endou H, Sakurai H. Plasma urate level is directly regulated by a voltage-driven urate efflux transporter URATv1 (SLC2A9) in humans. J Biol Chem. 2008;283(40):26834–26838. doi: 10.1074/jbc.C800156200. [DOI] [PubMed] [Google Scholar]
- Apelt J, Mehlhorn G, Schliebs R. Insulin-sensitive GLUT4 glucose transporters are colocalized with GLUT3-expressing cells and demonstrate a chemically distinct neuron-specific localization in rat brain. Journal of Neuroscience Research. 1999;57(5):693–705. [PubMed] [Google Scholar]
- Arluison M, Quignon M, Nguyen P, Thorens B, Leloup C, Penicaud L. Distribution and anatomical localization of the glucose transporter 2 (GLUT2) in the adult rat brain–an immunohistochemical study. J Chem Neuroanat. 2004;28(3):117–136. doi: 10.1016/j.jchemneu.2004.05.009. [DOI] [PubMed] [Google Scholar]
- Augustin R. The protein family of glucose transport facilitators: It’s not only about glucose after all. IUBMB life. 2010;62(5):315–333. doi: 10.1002/iub.315. [DOI] [PubMed] [Google Scholar]
- Augustin R, Carayannopoulos MO, Dowd LO, Phay JE, Moley JF, Moley KH. Identification and characterization of human glucose transporter-like protein-9 (GLUT9): alternative splicing alters trafficking. J Biol Chem. 2004;279(16):16229–16236. doi: 10.1074/jbc.M312226200. [DOI] [PubMed] [Google Scholar]
- Bady I, Marty N, Dallaporta M, Emery M, Gyger J, Tarussio D, Foretz M, Thorens B. Evidence from glut2-null mice that glucose is a critical physiological regulator of feeding. Diabetes. 2006;55(4):988–995. doi: 10.2337/diabetes.55.04.06.db05-1386. [DOI] [PubMed] [Google Scholar]
- Baldwin SA, Lienhard GE. Purification and reconstitution of glucose transporter from human erythrocytes. Methods In Enzymology. 1989;174:39–50. doi: 10.1016/0076-6879(89)74008-8. [DOI] [PubMed] [Google Scholar]
- Bannasch D, Safra N, Young A, Karmi N, Schaible RS, Ling GV. Mutations in the SLC2A9 gene cause hyperuricosuria and hyperuricemia in the dog. PLoS Genet. 2008;4(11):e1000246. doi: 10.1371/journal.pgen.1000246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibert S, Hess SK, Firsov D, Thorens B, Geering K, Horisberger JD, Bonny O. Mouse GLUT9: evidences for a urate uniporter. Am J Physiol Renal Physiol. 2009;297(3):F612–619. doi: 10.1152/ajprenal.00139.2009. [DOI] [PubMed] [Google Scholar]
- Birnbaum MJ. Identification of a novel gene encoding an insulin-responsive glucose transporter protein. Cell. 1989;57(2):305–315. doi: 10.1016/0092-8674(89)90968-9. [DOI] [PubMed] [Google Scholar]
- Birnbaum MJ, Haspel HC, Rosen OM. Cloning and characterization of a cDNA encoding the rat brain glucose-transporter protein. Proc Natl Acad Sci U S A. 1986;83(16):5784–5788. doi: 10.1073/pnas.83.16.5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blodgett DM, Graybill C, Carruthers A. Analysis of glucose transporter topology and structural dynamics. J Biol Chem. 2008;283(52):36416–36424. doi: 10.1074/jbc.M804802200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blot V, McGraw TE. Molecular Mechanisms Controlling GLUT4 Intracellular Retention. Molecular Biology of the Cell. 2008;19(8):3477–3487. doi: 10.1091/mbc.E08-03-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahm J. Kinetics of glucose transport in human erythrocytes. J Physiol. 1983;339:339–354. doi: 10.1113/jphysiol.1983.sp014720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burant CF, Takeda J, Brot LE, Bell GI, Davidson NO. Fructose transporter in human spermatozoa and small intestine is GLUT5. J Biol Chem. 1992;267(21):14523–14526. [PubMed] [Google Scholar]
- Carayannopoulos MO, Chi MMY, Cui Y, Pingsterhaus JM, McKnight RA, Mueckler M, Devaskar SU, Moley KH. GLUT8 is a glucose transporter responsible for insulin-stimulated glucose uptake in the blastocyst. Proceeding of the National Academy of Sciences USA. 2000;97:7313–7318. doi: 10.1073/pnas.97.13.7313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carayannopoulos MO, Schlein A, Wyman A, Chi M, Keembiyehetty C, Moley KH. GLUT9 is differentially expressed and targeted in the preimplantation embryo. Endocrinology. 2004;145(3):1435–1443. doi: 10.1210/en.2003-1264. [DOI] [PubMed] [Google Scholar]
- Carruthers A, DeZutter J, Ganguly A, Devaskar SU. Will the original glucose transporter isoform please stand up! Am J Physiol Endocrinol Metab. 2009;297(4):E836–848. doi: 10.1152/ajpendo.00496.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caulfield MJ, Munroe PB, O’Neill D, Witkowska K, Charchar FJ, Doblado M, Evans S, Eyheramendy S, Onipinla A, Howard P, Shaw-Hawkins S, Dobson RJ, Wallace C, Newhouse SJ, Brown M, Connell JM, Dominiczak A, Farrall M, Lathrop GM, Samani NJ, Kumari M, Marmot M, Brunner E, Chambers J, Elliott P, Kooner J, Laan M, Org E, Veldre G, Viigimaa M, Cappuccio FP, Ji C, Iacone R, Strazzullo P, Moley KH, Cheeseman C. SLC2A9 is a high-capacity urate transporter in humans. PLoS Med. 2008;5(10):e197. doi: 10.1371/journal.pmed.0050197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charron MJ, Brosius FD, Alper SL, Lodish HF. A glucose transport protein expressed predominately in insulin-responsive tissues. Proc Natl Acad Sci U S A. 1989;86(8):2535–2539. doi: 10.1073/pnas.86.8.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseman C. GLUT7: a new intestinal facilitated hexose transporter. American Journal of Physiology – Endocrinology & Metabolism. 2008;295(2):E238–241. doi: 10.1152/ajpendo.90394.2008. [DOI] [PubMed] [Google Scholar]
- Cloherty EK, Heard KS, Carruthers A. Human erythrocyte sugar transport is incompatible with available carrier models. Biochemistry. 1996;35(32):10411–10421. doi: 10.1021/bi953077m. [DOI] [PubMed] [Google Scholar]
- Corpe CP, Burant CF. Hexose transporter expression in rat small intestine: effect of diet on diurnal variations. American Journal of Physiology. 1996 doi: 10.1152/ajpgi.1996.271.1.G211. [DOI] [PubMed] [Google Scholar]
- Coucke PJ, Willaert A, Wessels MW, Callewaert B, Zoppi N, De Backer J, Fox JE, Mancini GM, Kambouris M, Gardella R, Facchetti F, Willems PJ, Forsyth R, Dietz HC, Barlati S, Colombi M, Loeys B, De Paepe A. Mutations in the facilitative glucose transporter GLUT10 alter angiogenesis and cause arterial tortuosity syndrome.[see comment] Nature Genetics. 2006;38(4):452–457. doi: 10.1038/ng1764. [DOI] [PubMed] [Google Scholar]
- Dang S, Sun L, Huang Y, Lu F, Liu Y, Gong H, Wang J, Yan N. Structure of a fucose transporter in an outward-open conformation. Nature. 2010;467(7316):734–738. doi: 10.1038/nature09406. [DOI] [PubMed] [Google Scholar]
- Dawson PA, Mychaleckyj JC, Fossey SC, Mihic SJ, Craddock AL, Bowden DW. Sequence and functional analysis of GLUT10: a glucose transporter in the Type 2 diabetes-linked region of chromosome 20q12-13.1. Molecular genetics and metabolism. 2001;74(1–2):186–199. doi: 10.1006/mgme.2001.3212. [DOI] [PubMed] [Google Scholar]
- De Vivo DC, Wang D, Pascual JM, Ho YY. Glucose transporter protein syndromes. International Review of Neurobiology. 2002;51:259–288. doi: 10.1016/s0074-7742(02)51008-4. [DOI] [PubMed] [Google Scholar]
- De Vos A, Heimberg H, Quartier E, Huypens P, Bouwens L, Pipeleers D, Schuit F. Human and rat beta cells differ in glucose transporter but not in glucokinase gene expression. Journal of clinical investigation. 1995;96:2489–2495. doi: 10.1172/JCI118308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehghan A, Kottgen A, Yang Q, Hwang SJ, Kao WL, Rivadeneira F, Boerwinkle E, Levy D, Hofman A, Astor BC, Benjamin EJ, van Duijn CM, Witteman JC, Coresh J, Fox CS. Association of three genetic loci with uric acid concentration and risk of gout: a genome-wide association study. Lancet. 2008;372(9654):1953–1961. doi: 10.1016/S0140-6736(08)61343-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinour D, Gray NK, Campbell S, Shu X, Sawyer L, Richardson W, Rechavi G, Amariglio N, Ganon L, Sela BA, Bahat H, Goldman M, Weissgarten J, Millar MR, Wright AF, Holtzman EJ. Homozygous SLC2A9 Mutations Cause Severe Renal Hypouricemia. J Am Soc Nephrol. 2009 doi: 10.1681/ASN.2009040406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doege H, Bocianski A, Scheepers A, Axer H, Eckel J, Joost HG, Schurmann A. Characterization of human glucose transporter (GLUT) 11 (encoded by SLC2A11), a novel sugar-transport facilitator specifically expressed in heart and skeletal muscle. The Biochemical journal. 2001;359(Pt 2):443–449. doi: 10.1042/0264-6021:3590443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doege H, Schürmann A, Bahrenberg G, Brauers A, Joost HG. GLUT8, a novel member of the sugar transport facilitator family with glucose transport activity. Journal of Biological Chemistry. 2000;275:16275–16280. doi: 10.1074/jbc.275.21.16275. [DOI] [PubMed] [Google Scholar]
- Doring A, Gieger C, Mehta D, Gohlke H, Prokisch H, Coassin S, Fischer G, Henke K, Klopp N, Kronenberg F, Paulweber B, Pfeufer A, Rosskopf D, Volzke H, Illig T, Meitinger T, Wichmann HE, Meisinger C. SLC2A9 influences uric acid concentrations with pronounced sex-specific effects. Nat Genet. 2008;40(4):430–436. doi: 10.1038/ng.107. [DOI] [PubMed] [Google Scholar]
- Douard V, Ferraris RP. Regulation of the fructose transporter GLUT5 in health and disease. American Journal of Physiology – Endocrinology & Metabolism. 2008;295(2):E227–237. doi: 10.1152/ajpendo.90245.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer J, Wood IS, Palejwala A, Ellis A, Shirazi-Beechey SP. Expression of monosaccharide transporters in intestine of diabetic humans. American journal of physiology. Gastrointestinal and liver physiology. 2002;282(2):G241–248. doi: 10.1152/ajpgi.00310.2001. [DOI] [PubMed] [Google Scholar]
- Eguez L, Lee A, Chavez JA, Miinea CP, Kane S, Lienhard GE, McGraw TE. Full intracellular retention of GLUT4 requires AS160 Rab GTPase activating protein. Cell Metabolism. 2005;2(4):263–272. doi: 10.1016/j.cmet.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Eny KM, Wolever TM, Fontaine-Bisson B, El-Sohemy A. Genetic variant in the glucose transporter type 2 is associated with higher intakes of sugars in two distinct populations. Physiol Genomics. 2008;33(3):355–360. doi: 10.1152/physiolgenomics.00148.2007. [DOI] [PubMed] [Google Scholar]
- Fanconi G, Bickel H. Die chronische Aminoacidurie bei der Glykogenose und der Cystinkrankheit. Helvetica Paediatrica Acta. 1949;4:359–396. [PubMed] [Google Scholar]
- Farese RV, Sajan MP, Yang H, Li P, Mastorides S, Gower WR, Jr, Nimal S, Choi CS, Kim S, Shulman GI, Kahn CR, Braun U, Leitges M. Muscle-specific knockout of PKC-lambda impairs glucose transport and induces metabolic and diabetic syndromes. J Clin Invest. 2007;117(8):2289–2301. doi: 10.1172/JCI31408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flessner LB, Moley KH. Similar [DE]XXXL[LI] motifs differentially target GLUT8 and GLUT12 in Chinese hamster ovary cells. Traffic. 2009;10(3):324–333. doi: 10.1111/j.1600-0854.2008.00866.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foretz M, Thorens B. The facilitative glucose transporter 2: pathophysiological role in mouse and human. In: Broër S, Wagner CA, editors. Membrane transporter diseases. Kluwer Academic/Plenum Publishers; New York, Boston, Dordrecht, London, Moscow: 2003. pp. 175–190. [Google Scholar]
- Forrest LR, Tang CL, Honig B. On the accuracy of homology modeling and sequence alignment methods applied to membrane proteins. Biophys J. 2006;91(2):508–517. doi: 10.1529/biophysj.106.082313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly A, McKnight RA, Raychaudhuri S, Shin BC, Ma Z, Moley K, Devaskar SU. Glucose transporter isoform-3 mutations cause early pregnancy loss and fetal growth restriction. American Journal of Physiology – Endocrinology & Metabolism. 2007;292(5):E1241–1255. doi: 10.1152/ajpendo.00344.2006. [DOI] [PubMed] [Google Scholar]
- Garcia MA, Millan C, Balmaceda-Aguilera C, Castro T, Pastor P, Montecinos H, Reinicke K, Zuniga F, Vera JC, Onate SA, Nualart F. Hypothalamic ependymal-glial cells express the glucose transporter GLUT2, a protein involved in glucose sensing. J Neurochem. 2003;86(3):709–724. doi: 10.1046/j.1471-4159.2003.01892.x. [DOI] [PubMed] [Google Scholar]
- Garvey WT, Maianu L, Huecksteadt TP, Birnbaum MJ, Molina JM, Ciaraldi TP. Pretranslational suppression of a glucose transporter protein causes insulin resistance in adipocytes from patients with non-insulin-dependent diabetes mellitus and obesity. J Clin Invest. 1991;87(3):1072–1081. doi: 10.1172/JCI115068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez E, McGraw TE. Insulin signaling diverges into Akt-dependent and -independent signals to regulate the recruitment/docking and the fusion of GLUT4 vesicles to the plasma membrane. Molecular Biology of the Cell. 2006;17(10):4484–4493. doi: 10.1091/mbc.E06-07-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorga FR, Lienhard GE. Equilibria and kinetics of ligand binding to the human erythrocyte glucose transporter. Evidence for an alternating conformation model for transport. Biochemistry. 1981;20(18):5108–5113. doi: 10.1021/bi00521a003. [DOI] [PubMed] [Google Scholar]
- Gorga FR, Lienhard GE. Changes in the intrinsic fluorescence of the human erythrocyte monosaccharide transporter upon ligand binding. Biochemistry. 1982;21(8):1905–1908. doi: 10.1021/bi00537a031. [DOI] [PubMed] [Google Scholar]
- Gremlich S, Bonny C, Waeber G, Thorens B. Fatty acids decrease IDX-1 expression in rat pancreatic islets and reduce GLUT2, glucokinase, insulin, and somatostatin levels. Journal of Biological Chemistry. 1997;272:30261–30269. doi: 10.1074/jbc.272.48.30261. [DOI] [PubMed] [Google Scholar]
- Guillam MT, Hümmler E, Schaerer E, Yeh JY, Birnbaum MJ, Beermann F, Schmidt A, Dériaz N, Thorens B. Early diabetes and abnormal postnatal pancreatic islet development in mice lacking GLUT2. Nature Genetics. 1997;17:327–330. doi: 10.1038/ng1197-327. [DOI] [PubMed] [Google Scholar]
- Guillam MT, Burcelin R, Thorens B. Normal hepatic glucose production in the absence of GLUT2 reveals an alternative pathway for glucose release from hepatocytes. Proc Natl Acad Sci U S A. 1998;95(21):12317–12321. doi: 10.1073/pnas.95.21.12317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen PA, Marshall BA, Chen M, Holloszy JO, Mueckler M. Transgenic overexpression of hexokinase II in skeletal muscle does not increase glucose disposal in wild-type or Glut1-overexpressing mice. J Biol Chem. 2000 doi: 10.1074/jbc.M001946200. [DOI] [PubMed] [Google Scholar]
- Havel PJ. Dietary fructose: implications for dysregulation of energy homeostasis and lipid/carbohydrate metabolism. Nutrition reviews. 2005;63(5):133–157. doi: 10.1301/nr.2005.may.133-157. [DOI] [PubMed] [Google Scholar]
- Hediger MA, Coady MJ, Ikeda TS, Wright EM. Expression cloning and cDNA sequencing of the Na+/glucose co-transporter. Natur. 1987;330:379–381. doi: 10.1038/330379a0. [DOI] [PubMed] [Google Scholar]
- Herman MA, Kahn BB. Glucose transport and sensing in the maintenance of glucose homeostasis and metabolic harmony. J Clin Invest. 2006;116(7):1767–1775. doi: 10.1172/JCI29027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hresko RC, Hruz PW. HIV protease inhibitors act as competitive inhibitors of the cytoplasmic glucose binding site of GLUTs with differing affinities for GLUT1 and GLUT4. PloS one. 2011;6(9):e25237. doi: 10.1371/journal.pone.0025237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hresko RC, Kruse M, Strube M, Mueckler M. Topology of the Glut 1 glucose transporter deduced from glycosylation scanning mutagenesis. Journal of Biological Chemistry. 1994;269(32):20482–20488. [PubMed] [Google Scholar]
- Hruz PW, Murata H, Qiu H, Mueckler M. Indinavir induces acute and reversible peripheral insulin resistance in rats. Diabetes. 2002;51(4):937–942. doi: 10.2337/diabetes.51.4.937. [DOI] [PubMed] [Google Scholar]
- Huang S, Czech MP. The GLUT4 glucose transporter. [Review] [238 refs] Cell Metabolism. 2007;5(4):237–252. doi: 10.1016/j.cmet.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Huang Y, Lemieux MJ, Song J, Auer M, Wang DN. Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli.[comment] Science. 2003;301(5633):616–620. doi: 10.1126/science.1087619. [DOI] [PubMed] [Google Scholar]
- Ibberson M, Riederer BM, Uldry M, Guhl B, Roth J, Thorens B. Immunolocalization of GLUTX1 in the testis and to specific brain areas and vasopressin-containing neurons. Endocrinology. 2002;143(1):276–284. doi: 10.1210/endo.143.1.8587. [DOI] [PubMed] [Google Scholar]
- Ibberson M, Uldry M, Thorens B. GLUTX1, a novel mammalian glucose transporter expressed in the central nervous system and insulin-sensitive tissues. Journal of Biological Chemistry. 2000;275:4607–4612. doi: 10.1074/jbc.275.7.4607. [DOI] [PubMed] [Google Scholar]
- Illsley NP. Glucose transporters in the human placenta. Placenta. 2000;21(1):14–22. doi: 10.1053/plac.1999.0448. [DOI] [PubMed] [Google Scholar]
- James DE, Brown R, Navarro J, Pilch PF. Insulin-regulatable tissues express a unique insulin sensitive glucose transport protein. Nature. 1988;333(6169):183–185. doi: 10.1038/333183a0. [DOI] [PubMed] [Google Scholar]
- James DE, Strube M, Mueckler M. Molecular cloning and characterization of an insulin regulatable glucose transporter. Nature. 1989;338(6210):83–87. doi: 10.1038/338083a0. [DOI] [PubMed] [Google Scholar]
- Jessen N, Goodyear LJ. Contraction signaling to glucose transport in skeletal muscle. [Review] [123 refs] Journal of Applied Physiology. 2005;99(1):330–337. doi: 10.1152/japplphysiol.00175.2005. [DOI] [PubMed] [Google Scholar]
- Ji L, Malecki M, Warram JH, Yang Y, Rich SS, Krolewski AS. New susceptibility locus for NIDDM is localized to human chromosome 20q. Diabetes. 1997;46(5):876–881. doi: 10.2337/diab.46.5.876. [DOI] [PubMed] [Google Scholar]
- Jiang L, David ES, Espina N, Ferraris RP. GLUT-5 expression in neonatal rats: crypt-villus location and age-dependent regulation. American Journal of Physiology – Gastrointestinal & Liver Physiology. 2001;281(3):G666–674. doi: 10.1152/ajpgi.2001.281.3.G666. [DOI] [PubMed] [Google Scholar]
- Joost HG, Bell GI, Best JD, Birnbaum MJ, Charron MJ, Chen YT, Doege H, James DE, Lodish HF, Moley KH, Moley JF, Mueckler M, Rogers S, Schurmann A, Seino S, Thorens B. Nomenclature of the GLUT/SLC2A family of sugar/polyol transport facilitators. Am J Physiol Endocrinol Metab. 2002;282(4):E974–976. doi: 10.1152/ajpendo.00407.2001. [DOI] [PubMed] [Google Scholar]
- Kahn BB. Lilly lecture 1995. Glucose transport: pivotal step in insulin action. [Review] [63 refs] Diabetes. 1996;45(11):1644–1654. doi: 10.2337/diab.45.11.1644. [DOI] [PubMed] [Google Scholar]
- Kane S, Sano H, Liu SC, Asara JM, Lane WS, Garner CC, Lienhard GE. A method to identify serine kinase substrates. Akt phosphorylates a novel adipocyte protein with a Rab GTPase-activating protein (GAP) domain. Journal of Biological Chemistry. 2002;277(25):22115–22118. doi: 10.1074/jbc.C200198200. [DOI] [PubMed] [Google Scholar]
- Kasahara M, Hinkle PC. Reconstitution and purification of the D-glucose transporter from human erythrocytes. J Biol Chem. 1977;252(20):7384–7390. [PubMed] [Google Scholar]
- Kayano T, Burant CF, Fukumoto H, Gould GW, Fan YS, Eddy RL, Byers MG, Shows TB, Seino S, Bell GI. Human facilitative glucose transporters. Isolation, functional characterization, and gene localization of cDNAs encoding an isoform (GLUT5) expressed in small intestine, kidney, muscle, and adipose tissue and an unusual glucose transporter pseudogene-like sequence (GLUT6) J Biol Chem. 1990;265(22):13276–13282. [PubMed] [Google Scholar]
- Kayano T, Fukumoto H, Eddy RL, Fan YS, Byers MG, Shows TB, Bell GI. Evidence for a family of human glucose transporter-like proteins. Sequence and gene localization of a protein expressed in fetal skeletal muscle and other tissues. J Biol Chem. 1988;263(30):15245–15248. [PubMed] [Google Scholar]
- Keembiyehetty C, Augustin R, Carayannopoulos MO, Steer S, Manolescu A, Cheeseman CI, Moley KH. Mouse glucose transporter 9 splice variants are expressed in adult liver and kidney and are up-regulated in diabetes. Mol Endocrinol. 2006;20(3):686–697. doi: 10.1210/me.2005-0010. [DOI] [PubMed] [Google Scholar]
- Kellett GL, Brot-Laroche E, Mace OJ, Leturque A. Sugar absorption in the intestine: the role of GLUT2. Annu Rev Nutr. 2008;28:35–54. doi: 10.1146/annurev.nutr.28.061807.155518. [DOI] [PubMed] [Google Scholar]
- Kolobova E, Efimov A, Kaverina I, Rishi AK, Schrader JW, Ham AJ, Larocca MC, Goldenring JR. Microtubule-dependent association of AKAP350A and CCAR1 with RNA stress granules. Experimental cell research. 2009;315(3):542–555. doi: 10.1016/j.yexcr.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koranyi L, Bourey RE, James D, Mueckler M, Fiedorek FT, Permutt MA. Glucose transporter gene expression in rat brain: Pretranslational changes associated with chronic insulin-induced hypoglycemia, fasting, and diabetes. Mol Cell Neurosci. 1991;2:244–252. doi: 10.1016/1044-7431(91)90051-o. [DOI] [PubMed] [Google Scholar]
- Kotani K, Peroni OD, Minokoshi Y, Boss O, Kahn BB. GLUT4 glucose transporter deficiency increases hepatic lipid production and peripheral lipid utilization.[see comment] Journal of Clinical Investigation. 2004;114(11):1666–1675. doi: 10.1172/JCI21341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larance M, Ramm G, James DE. The GLUT4 code. Mol Endocrinol. 2008;22(2):226–233. doi: 10.1210/me.2007-0282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YC, Huang HY, Chang CJ, Cheng CH, Chen YT. Mitochondrial GLUT10 facilitates dehydroascorbic acid import and protects cells against oxidative stress: mechanistic insight into arterial tortuosity syndrome. Hum Mol Genet. 2010;19(19):3721–3733. doi: 10.1093/hmg/ddq286. [DOI] [PubMed] [Google Scholar]
- Leino RL, Gerhart DZ, van Bueren AM, McCall AL, Drewes LR. Ultrastructural localization of GLUT 1 and GLUT 3 glucose transporters in rat brain. Journal of Neuroscience Research. 1997;49(5):617–626. doi: 10.1002/(SICI)1097-4547(19970901)49:5<617::AID-JNR12>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Li S, Sanna S, Maschio A, Busonero F, Usala G, Mulas A, Lai S, Dei M, Orru M, Albai G, Bandinelli S, Schlessinger D, Lakatta E, Scuteri A, Najjar SS, Guralnik J, Naitza S, Crisponi L, Cao A, Abecasis G, Ferrucci L, Uda M, Chen WM, Nagaraja R. The GLUT9 gene is associated with serum uric acid levels in Sardinia and Chianti cohorts. PLoS Genet. 2007;3(11):e194. doi: 10.1371/journal.pgen.0030194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisinski I, Schurmann A, Joost HG, Cushman SW, Al-Hasani H. Targeting of GLUT6 (formerly GLUT9) and GLUT8 in rat adipose cells. Biochemical Journal. 2001;358(Pt 2):517–522. doi: 10.1042/0264-6021:3580517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe AG, Walmsley AR. The kinetics of glucose transport in human red blood cells. Biochimica et Biophysica Acta. 1986;857(2):146–154. doi: 10.1016/0005-2736(86)90342-1. [DOI] [PubMed] [Google Scholar]
- Lowe AG, Walmsley AR. The kinetics and thermodynamics of glucose transport in human erythrocytes. In: Agre P, Parker JC, editors. Red Blood Cell Membranes. Marcel Dekker, Inc.; New York: 1989. pp. 597–634. [Google Scholar]
- Maher F, Harrison LC. Hexose specificity for downregulation of HepG2/brain-type glucose transporter gene expression in L6 myocytes. Diabetologia. 1990;33(11):641–648. doi: 10.1007/BF00400564. [DOI] [PubMed] [Google Scholar]
- Manolescu AR, Augustin R, Moley K, Cheeseman C. A highly conserved hydrophobic motif in the exofacial vestibule of fructose transporting SLC2A proteins acts as a critical determinant of their substrate selectivity. Mol Membr Biol. 2007;24(5–6):455–463. doi: 10.1080/09687680701298143. [DOI] [PubMed] [Google Scholar]
- Marshall BA, Mueckler MM. Differential effects of GLUT-1 or GLUT-4 overexpression on insulin responsiveness in transgenic mice. American Journal of Physiology. 1994:E738–E744. doi: 10.1152/ajpendo.1994.267.5.E738. [DOI] [PubMed] [Google Scholar]
- Marty N, Dallaporta M, Foretz M, Emery M, Tarussio D, Bady I, Binnert C, Beermann F, Thorens B. Regulation of glucagon secretion by glucose transporter type 2 (glut2) and astrocyte-dependent glucose sensors. J Clin Invest. 2005;115(12):3545–3553. doi: 10.1172/JCI26309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty N, Dallaporta M, Thorens B. Brain glucose sensing, counterregulation and feeding behavior. Physiology (Bethesda) 2007;22:241–251. doi: 10.1152/physiol.00010.2007. [DOI] [PubMed] [Google Scholar]
- Matsuo H, Chiba T, Nagamori S, Nakayama A, Domoto H, Phetdee K, Wiriyasermkul P, Kikuchi Y, Oda T, Nishiyama J, Nakamura T, Morimoto Y, Kamakura K, Sakurai Y, Nonoyama S, Kanai Y, Shinomiya N. Mutations in glucose transporter 9 gene SLC2A9 cause renal hypouricemia.[erratum appears in Am J Hum Genet. 2008 Dec;83(6):795] Am J Hum Genet. 2008;83(6):744–751. doi: 10.1016/j.ajhg.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVie-Wylie AJ, Lamson DR, Chen YT. Molecular cloning of a novel member of the GLUT family of transporters, SLC2a10 (GLUT10), localized on chromosome 20q13.1: a candidate gene for NIDDM susceptibility. Genomics. 2001;72(1):113–117. doi: 10.1006/geno.2000.6457. [DOI] [PubMed] [Google Scholar]
- Membrez M, Hummler E, Beermann F, Haefliger JA, Savioz R, Pedrazzini T, Thorens B. GLUT8 is dispensable for embryonic development but influences hippocampal neurogenesis and heart function. Mol Cell Biol. 2006;26(11):4268–4276. doi: 10.1128/MCB.00081-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra P, Zheng X, Czech MP. RNAi-based analysis of CAP, Cbl, and CrkII function in the regulation of GLUT4 by insulin. Journal of Biological Chemistry. 2004;279(36):37431–37435. doi: 10.1074/jbc.C400180200. [DOI] [PubMed] [Google Scholar]
- Mobasheri A, Dobson H, Mason SL, Cullingham F, Shakibaei M, Moley JF, Moley KH. Expression of the GLUT1 and GLUT9 facilitative glucose transporters in embryonic chondroblasts and mature chondrocytes in ovine articular cartilage. Cell Biol Int. 2005;29(4):249–260. doi: 10.1016/j.cellbi.2004.11.024. [DOI] [PubMed] [Google Scholar]
- Moley KH. Hyperglycemia and apoptosis: mechanisms for congenital malformations and pregnancy loss in diabetic women. Trends in Endocrinology & Metabolism. 2001;12(2):78–82. doi: 10.1016/s1043-2760(00)00341-6. [DOI] [PubMed] [Google Scholar]
- Moley KH, Chi MM, Knudson CM, Korsmeyer SJ, Mueckler MM. Hyperglycemia induces apoptosis in pre-implantation embryos through cell death effector pathways.[comment][erratum appears in Nat Med 2002 Mar;8(3):303] Nature Medicine. 1998;4(12):1421–1424. doi: 10.1038/4013. [DOI] [PubMed] [Google Scholar]
- Mounien L, Marty N, Tarussio D, Metref S, Genoux D, Preitner F, Foretz M, Thorens B. Glut2-dependent glucose-sensing controls thermoregulation by enhancing the leptin sensitivity of NPY and POMC neurons. Faseb J. 2010 doi: 10.1096/fj.09-144923. [DOI] [PubMed] [Google Scholar]
- Mueckler M. Glucose transport and glucose homeostasis: New insights from transgenic mice. News in Physiological Sciences. 1995;10:22–29. [Google Scholar]
- Mueckler M, Caruso C, Baldwin SA, Panico M, Blench I, Morris HR, Allard WJ, Lienhard GE, Lodish HF. Sequence and structure of a human glucose transporter. Science. 1985;229(4717):941–945. doi: 10.1126/science.3839598. [DOI] [PubMed] [Google Scholar]
- Mueckler M, Makepeace C. Model of the exofacial substrate-binding site and helical folding of the human Glut1 glucose transporter based on scanning mutagenesis. Biochemistry. 2009;48(25):5934–5942. doi: 10.1021/bi900521n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata H, Hruz PW, Mueckler M. The mechanism of insulin resistance caused by HIV protease inhibitor therapy. Journal of Biological Chemistry. 2000;275(27):20251–20254. doi: 10.1074/jbc.C000228200. [DOI] [PubMed] [Google Scholar]
- Muretta JM, Romenskaia I, Mastick CC. Insulin releases Glut4 from static storage compartments into cycling endosomes and increases the rate constant for Glut4 exocytosis. Journal of Biological Chemistry. 2008;283(1):311–323. doi: 10.1074/jbc.M705756200. [DOI] [PubMed] [Google Scholar]
- Naftalin RJ. Alternating carrier models of asymmetric glucose transport violate the energy conservation laws. Biophys J. 2008;95(9):4300–4314. doi: 10.1529/biophysj.108.136366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsubo K, Chen MZ, Olefsky JM, Marth JD. Pathway to diabetes through attenuation of pancreatic beta cell glycosylation and glucose transport. Nat Med. 2011;17(9):1067–1075. doi: 10.1038/nm.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsubo K, Takamatsu S, Minowa MT, Yoshida A, Takeuchi M, Marth JD. Dietary and genetic control of glucose transporter 2 glycosylation promotes insulin secretion in suppressing diabetes. Cell. 2005;123:1307–1321. doi: 10.1016/j.cell.2005.09.041. [DOI] [PubMed] [Google Scholar]
- Pantaleon M, Harvey MB, Pascoe WS, James DE, Kaye PL. Glucose transporter GLUT3: ontogeny, targeting, and role in the mouse blastocyst. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(8):3795–3800. doi: 10.1073/pnas.94.8.3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantaleon M, Kaye PL. Glucose transporters in preimplantation development. Reviews of Reproduction. 1998;3(2):77–81. doi: 10.1530/ror.0.0030077. [DOI] [PubMed] [Google Scholar]
- Pantaleon M, Scott J, Kaye PL. Nutrient sensing by the early mouse embryo: hexosamine biosynthesis and glucose signaling during preimplantation development. Biology of Reproduction. 2008;78(4):595–600. doi: 10.1095/biolreprod.107.062877. [DOI] [PubMed] [Google Scholar]
- Parsa A, Brown E, Weir MR, Fink JC, Shuldiner AR, Mitchell BD, McArdle PF. Genotype-based changes in serum uric acid affect blood pressure. Kidney Int. 2012;81(5):502–507. doi: 10.1038/ki.2011.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, Bonvento G, Chatton JY, Pierre K, Magistretti PJ. Role of neuron-glia interaction in the regulation of brain glucose utilization. Diabetes, Nutrition & Metabolism – Clinical & Experimental. 2002;15(5):268–273. discussion 273. [PubMed] [Google Scholar]
- Phay JE, Hussain HB, Moley JF. Cloning and expression analysis of a novel member of the facilitative glucose transporter family, SLC2A9 (GLUT9) Genomics. 2000;66(2):217–220. doi: 10.1006/geno.2000.6195. [DOI] [PubMed] [Google Scholar]
- Pinto AB, Carayannopoulos MO, Hoehn A, Dowd L, Moley KH. Glucose transporter 8 expression and translocation are critical for murine blastocyst survival. Biology of Reproduction. 2002;66:1729–1733. doi: 10.1095/biolreprod66.6.1729. [DOI] [PubMed] [Google Scholar]
- Piper RC, Tai C, Kulesza P, Pang S, Warnock D, Baenziger J, Slot JW, Geuze HJ, Puri C, James DE. GLUT-4 NH2 terminus contains a phenylalanine-based targeting motif that regulates intracellular sequestration. Journal Of Cell Biology. 1993;121(6):1221–1232. doi: 10.1083/jcb.121.6.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preitner F, Bonny O, Laverriere A, Rotman S, Firsov D, Da Costa A, Metref S, Thorens B. Glut9 is a major regulator of urate homeostasis and its genetic inactivation induces hyperuricosuria and urate nephropathy. Proc Natl Acad Sci U S A. 2009;106(36):15501–15506. doi: 10.1073/pnas.0904411106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell SH, Aerni-Flessner LB, Willcockson AR, Diggs-Andrews KA, Fisher SJ, Moley KH. Improved insulin sensitivity by GLUT12 overexpression in mice. Diabetes. 2011;60(5):1478–1482. doi: 10.2337/db11-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reagan LP, Rosell DR, Alves SE, Hoskin EK, McCall AL, Charron MJ, McEwen BS. Glut8 glucose transporter is localized to excitatory and inhibitory neurons in the rat hippocampus. Brain Research. 2002;932:129–134. doi: 10.1016/s0006-8993(02)02308-9. [DOI] [PubMed] [Google Scholar]
- Reimer MK, Ahren B. Altered beta-cell distribution of pdx-1 and GLUT-2 after a short-term challenge with a high-fat diet in C57BL/6J mice. Diabetes. 2002;51(Suppl 1):S138–143. doi: 10.2337/diabetes.51.2007.s138. [DOI] [PubMed] [Google Scholar]
- Ren JM, Marshall BA, Mueckler MM, McCaleb M, Amatruda JM, Shulman GI. Overexpression of Glut4 protein in muscle increases basal and insulin-stimulated whole body glucose disposal in conscious mice. Journal of clinical investigation. 1995;95(1):429–432. doi: 10.1172/JCI117673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren JM, Semenkovich CF, Gulve EA, Gao J, Holloszy JO. Exercise induces rapid increases in GLUT4 expression, glucose transport capacity, and insulin-stimulated glycogen storage in muscle. Journal of Biological Chemistry. 1994;269(20):14396–14401. [PubMed] [Google Scholar]
- Rogers S, Docherty SE, Slavin JL, Henderson MA, Best JD. Differential expression of GLUT12 in breast cancer and normal breast tissue. Cancer Letters. 2003;193(2):225–233. doi: 10.1016/s0304-3835(03)00010-7. [DOI] [PubMed] [Google Scholar]
- Rogers S, Macheda ML, Docherty SE, Carty MD, Henderson MA, Soeller WC, Gibbs EM, James DE, Best JD. Identification of a novel glucose transporter-like protein-GLUT-12. American Journal of Physiology – Endocrinology & Metabolism. 2002;282(3):E733–738. doi: 10.1152/ajpendo.2002.282.3.E733. [DOI] [PubMed] [Google Scholar]
- Rose AJ, Richter EA. Skeletal muscle glucose uptake during exercise: how is it regulated? Physiology (Bethesda) 2005;20:260–270. doi: 10.1152/physiol.00012.2005. [DOI] [PubMed] [Google Scholar]
- Salas-Burgos A, Iserovich P, Zuniga F, Vera JC, Fischbarg J. Predicting the three-dimensional structure of the human facilitative glucose transporter glut1 by a novel evolutionary homology strategy: insights on the molecular mechanism of substrate migration, and binding sites for glucose and inhibitory molecules. Biophys J. 2004;87(5):2990–2999. doi: 10.1529/biophysj.104.047886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltiel AR, Pessin JE. Insulin signaling in microdomains of the plasma membrane. Traffic. 2003;4(11):711–716. doi: 10.1034/j.1600-0854.2003.00119.x. [DOI] [PubMed] [Google Scholar]
- Sano H, Eguez L, Teruel MN, Fukuda M, Chuang TD, Chavez JA, Lienhard GE, McGraw TE. Rab10, a target of the AS160 Rab GAP, is required for insulin-stimulated translocation of GLUT4 to the adipocyte plasma membrane. Cell Metabolism. 2007;5(4):293–303. doi: 10.1016/j.cmet.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Santer R, Schneppenheim R, Dombrowski A, Götze H, Steinmann B, Schaub J. Mutations in GLUT2, the gene for the liver-type glucose transporter, in patients with Fanconi-Bickel syndrome. Nature Genetics. 1997;17:324–326. doi: 10.1038/ng1197-324. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Minoshima S, Shiohama A, Shintani A, Shimizu A, Asakawa S, Kawasaki K, Shimizu N. Molecular cloning of a member of the facilitative glucose transporter gene family GLUT11 (SLC2A11) and identification of transcription variants. Biochemical And Biophysical Research Communications. 2001;289(5):1218–1224. doi: 10.1006/bbrc.2001.6101. [DOI] [PubMed] [Google Scholar]
- Schmidt S, Joost HG, Schurmann A. GLUT8, the enigmatic intracellular hexose transporter. Am J Physiol Endocrinol Metab. 2009;296(4):E614–618. doi: 10.1152/ajpendo.91019.2008. [DOI] [PubMed] [Google Scholar]
- Shewan AM, Marsh BJ, Melvin DR, Martin S, Gould GW, James DE. The cytosolic C-terminus of the glucose transporter GLUT4 contains an acidic cluster endosomal targeting motif distal to the dileucine signal. Biochemical Journal. 2000;350(Pt 1):99–107. [PMC free article] [PubMed] [Google Scholar]
- Shin BC, Fujikura K, Suzuki T, Tanaka S, Takata K. Glucose transporter GLUT3 in the rat placental barrier: a possible machinery for the transplacental transfer of glucose. Endocrinology. 1997;138(9):3997–4004. doi: 10.1210/endo.138.9.5369. [DOI] [PubMed] [Google Scholar]
- Simkin PA. The Dalmatian defect: a hepatic endocrinopathy of urate transport. Arthritis Rheum. 2005;52(8):2257–2262. doi: 10.1002/art.21241. [DOI] [PubMed] [Google Scholar]
- Simpson IA, Carruthers A, Vannucci SJ. Supply and demand in cerebral energy metabolism: the role of nutrient transporters. J Cereb Blood Flow Metab. 2007;27(11):1766–1791. doi: 10.1038/sj.jcbfm.9600521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson IA, Dwyer D, Malide D, Moley KH, Travis A, Vannucci SJ. The facilitative glucose transporter GLUT3: 20 years of distinction. American Journal of Physiology – Endocrinology & Metabolism. 2008;295(2):E242–253. doi: 10.1152/ajpendo.90388.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson IA, Vannucci SJ, DeJoseph MR, Hawkins RA. Glucose transporter asymmetries in the bovine blood-brain barrier. Journal of Biological Chemistry. 2001;276(16):12725–12729. doi: 10.1074/jbc.M010897200. [DOI] [PubMed] [Google Scholar]
- Simpson IA, Vannucci SJ, Maher F. Glucose transporters in mammalian brain. [Review] Biochemical Society Transactions. 1994;22(3):671–675. doi: 10.1042/bst0220671. [DOI] [PubMed] [Google Scholar]
- So A, Thorens B. Uric acid transport and disease. J Clin Invest. 2010;120(6):1791–1799. doi: 10.1172/JCI42344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon S, Xu Y, Wang B, David MD, Schubert P, Kennedy D, Schrader JW. Distinct structural features of caprin-1 mediate its interaction with G3BP-1 and its induction of phosphorylation of eukaryotic translation initiation factor 2alpha, entry to cytoplasmic stress granules, and selective interaction with a subset of mRNAs. Molecular and cellular biology. 2007;27(6):2324–2342. doi: 10.1128/MCB.02300-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song XM, Hresko RC, Mueckler M. Identification of amino acid residues within the C terminus of the Glut4 glucose transporter that are essential for insulin-stimulated redistribution to the plasma membrane. Journal of Biological Chemistry. 2008;283(18):12571–12585. doi: 10.1074/jbc.M800838200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark K, Reinhard W, Grassl M, Erdmann J, Schunkert H, Illig T, Hengstenberg C. Common polymorphisms influencing serum uric acid levels contribute to susceptibility to gout, but not to coronary artery disease. PloS one. 2009;4(11):e7729. doi: 10.1371/journal.pone.0007729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart CA, Howell ME, Yin D. Overexpression of GLUT5 in diabetic muscle is reversed by pioglitazone. Diabetes Care. 2007;30(4):925–931. doi: 10.2337/dc06-1788. [DOI] [PubMed] [Google Scholar]
- Stuart CA, Howell ME, Zhang Y, Yin D. Insulin-stimulated translocation of glucose transporter (GLUT) 12 parallels that of GLUT4 in normal muscle. The Journal of clinical endocrinology and metabolism. 2009;94(9):3535–3542. doi: 10.1210/jc.2009-0162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpel F, Burcelin R, Jungermann K, Thorens B. Normal kinetics of intestinal glucose absorption in the absence of GLUT2: evidence for a transport pathway requiring glucose phosphorylation and transfer into the endoplasmic reticulum. Proc Natl Acad Sci U S A. 2001;98(20):11330–11335. doi: 10.1073/pnas.211357698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thong FS, Dugani CB, Klip A. Turning signals on and off: GLUT4 traffic in the insulin-signaling highway. [Review] [158 refs] Physiology. 2005;20:271–284. doi: 10.1152/physiol.00017.2005. [DOI] [PubMed] [Google Scholar]
- Thorens B, Cheng ZQ, Brown D, Lodish HF. Liver glucose transporter: a basolateral protein in hepatocytes and intestine and kidney cells. Am J Physiol. 1990;259(6 Pt 1):C279–285. doi: 10.1152/ajpcell.1990.259.2.C279. [DOI] [PubMed] [Google Scholar]
- Thorens B, Mueckler M. Glucose transporters in the 21st Century. Am J Physiol Endocrinol Metab. 2010;298(2):E141–145. doi: 10.1152/ajpendo.00712.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorens B, Sarkar HK, Kaback HR, Lodish HF. Cloning and functional expression in bacteria of a novel glucose transporter present in liver, intestine, kidney, and b-pancreatic islet cells. Cell. 1988;55:281–290. doi: 10.1016/0092-8674(88)90051-7. [DOI] [PubMed] [Google Scholar]
- Tobin V, Le Gall M, Fioramonti X, Stolarczyk E, Blazquez AG, Klein C, Prigent M, Serradas P, Cuif MH, Magnan C, Leturque A, Brot-Laroche E. Insulin internalizes GLUT2 in the enterocytes of healthy but not insulin-resistant mice. Diabetes. 2008;57(3):555–562. doi: 10.2337/db07-0928. [DOI] [PubMed] [Google Scholar]
- Uldry M, Ibberson M, Horisberger JD, Chatton JY, Riederer BM, Thorens B. Identification of a mammalian H+-myo-inositol symporter expressed predominantly in the brain. EMBO J. 2001;20:4467–4477. doi: 10.1093/emboj/20.16.4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uldry M, Ibberson M, Hosokawa M, Thorens B. GLUT2 is a high affinity glucosamine transporter. FEBS Lett. 2002;524(1–3):199–203. doi: 10.1016/s0014-5793(02)03058-2. [DOI] [PubMed] [Google Scholar]
- Uldry M, Steiner P, Zurich MG, Beguin P, Hirling H, Dolci W, Thorens B. Regulated exocytosis of an H+/myo-inositol symporter at synapses and growth cones. Embo J. 2004;23(3):531–540. doi: 10.1038/sj.emboj.7600072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uldry M, Thorens B. The SLC2 family of facilitated hexose and polyol transporters. Pflugers Arch. 2004;447(5):480–489. doi: 10.1007/s00424-003-1085-0. [DOI] [PubMed] [Google Scholar]
- Urner F, Sakkas D. A possible role for the pentose phosphate pathway of spermatozoa in gamete fusion in the mouse. Biology of Reproduction. 1999;60(3):733–739. doi: 10.1095/biolreprod60.3.733. [DOI] [PubMed] [Google Scholar]
- Verhey KJ, Birnbaum MJ. A Leu-Leu sequence is essential for COOH-terminal targeting signal of GLUT4 glucose transporter in fibroblasts. Journal of Biological Chemistry. 1994;269(4):2353–2356. [PubMed] [Google Scholar]
- Vitart V, Rudan I, Hayward C, Gray NK, Floyd J, Palmer CN, Knott SA, Kolcic I, Polasek O, Graessler J, Wilson JF, Marinaki A, Riches PL, Shu X, Janicijevic B, Smolej-Narancic N, Gorgoni B, Morgan J, Campbell S, Biloglav Z, Barac-Lauc L, Pericic M, Klaric IM, Zgaga L, Skaric-Juric T, Wild SH, Richardson WA, Hohenstein P, Kimber CH, Tenesa A, Donnelly LA, Fairbanks LD, Aringer M, McKeigue PM, Ralston SH, Morris AD, Rudan P, Hastie ND, Campbell H, Wright AF. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat Genet. 2008;40(4):437–442. doi: 10.1038/ng.106. [DOI] [PubMed] [Google Scholar]
- Vyas AK, Koster JC, Tzekov A, Hruz PW. Effects of the HIV protease inhibitor ritonavir on GLUT4 knock-out mice. J Biol Chem. 285(47):36395–36400. doi: 10.1074/jbc.M110.176321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace C, Newhouse SJ, Braund P, Zhang F, Tobin M, Falchi M, Ahmadi K, Dobson RJ, Marcano AC, Hajat C, Burton P, Deloukas P, Brown M, Connell JM, Dominiczak A, Lathrop GM, Webster J, Farrall M, Spector T, Samani NJ, Caulfield MJ, Munroe PB. Genome-wide association study identifies genes for biomarkers of cardiovascular disease: serum urate and dyslipidemia. Am J Hum Genet. 2008;82(1):139–149. doi: 10.1016/j.ajhg.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Pascual JM, Yang H, Engelstad K, Mao X, Cheng J, Yoo J, Noebels JL, De Vivo DC. A mouse model for Glut-1 haploinsufficiency. Hum Mol Genet. 2006;15(7):1169–1179. doi: 10.1093/hmg/ddl032. [DOI] [PubMed] [Google Scholar]
- Watson RT, Kanzaki M, Pessin JE. Regulated membrane trafficking of the insulin-responsive glucose transporter 4 in adipocytes. [Review] [362 refs] Endocrine Reviews. 2004;25(2):177–204. doi: 10.1210/er.2003-0011. [DOI] [PubMed] [Google Scholar]
- Wheeler TJ, Whelan JD. Infinite-cis kinetics support the carrier model for erythrocyte glucose transport. Biochemistry. 1988;27(5):1441–1450. doi: 10.1021/bi00405a008. [DOI] [PubMed] [Google Scholar]
- Wu X, Freeze HH. GLUT14, a duplicon of GLUT3, is specifically expressed in testis as alternative splice forms. Genomics. 2002;80(6):553–557. doi: 10.1006/geno.2002.7010. [DOI] [PubMed] [Google Scholar]
- Wyman A, Pinto AB, Sheridan R, Moley KH. One-cell zygote transfer from diabetic to nondiabetic mouse results in congenital malformations and growth retardation in offspring. Endocrinology. 2008;149(2):466–469. doi: 10.1210/en.2007-1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Seino Y, Fukumoto H, Koh G, Yano H, Inagaki N, Yamada Y, Inoue K, Manabe T, Imura H. Over-expression of facilitative glucose transporter genes in human cancer. Biochemical And Biophysical Research Communications. 1990;170(1):223–230. doi: 10.1016/0006-291x(90)91263-r. [DOI] [PubMed] [Google Scholar]
- Yeh JI, Verhey KJ, Birnbaum MJ. Kinetic analysis of glucose transporter trafficking in fibroblasts and adipocytes. Biochemistry. 1995;34(47):15523–15531. doi: 10.1021/bi00047a018. [DOI] [PubMed] [Google Scholar]
- Yeh WL, Lin CJ, Fu WM. Enhancement of glucose transporter expression of brain endothelial cells by vascular endothelial growth factor derived from glioma exposed to hypoxia. Mol Pharmacol. 2008;73(1):170–177. doi: 10.1124/mol.107.038851. [DOI] [PubMed] [Google Scholar]
- Yin Y, He X, Szewczyk P, Nguyen T, Chang G. Structure of the multidrug transporter EmrD from Escherichia coli. Science. 2006;312(5774):741–744. doi: 10.1126/science.1125629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younes M, Lechago LV, Somoano JR, Mosharaf M, Lechago J. Wide expression of the human erythrocyte glucose transporter Glut1 in human cancers. Cancer Research. 1996;56(5):1164–1167. [PubMed] [Google Scholar]
- Zisman A, Peroni OD, Abel ED, Michael MD, Mauvais-Jarvis F, Lowell BB, Wojtaszewski JF, Hirshman MF, Virkamaki A, Goodyear LJ, Kahn CR, Kahn BB. Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nature Medicine. 2000;6(8):924–928. doi: 10.1038/78693. [DOI] [PubMed] [Google Scholar]