

Abstract
Objective:
We aimed to investigate the relationship between plasma and CSF progranulin (PGRN) levels.
Methods:
Plasma and CSF PGRN were measured in a cohort of 345 subjects from the Mayo Clinic Study of Aging by ELISA. Single nucleotide polymorphism genotyping was performed using TaqMan assays. Associations between PGRN and sex, age at sample collection, diagnosis, single nucleotide polymorphism genotypes (GRN, SORT1, and APOE), and Pittsburgh compound B score were explored separately in CSF and plasma using single variable linear regression models. Pearson partial correlation coefficient was used to estimate the correlation of PGRN in CSF and plasma.
Results:
Plasma (p = 0.0031) and CSF (p = 0.0044) PGRN significantly increased with age, whereas plasma PGRN levels were 7% lower (p = 0.0025) and CSF PGRN levels 5% higher (p = 0.0024) in male compared with female participants. Correcting for age and sex, higher plasma PGRN was associated with higher CSF PGRN (partial r = 0.17, p = 0.004). In plasma, both rs5848 (GRN; p = 0.002) and rs646776 (SORT1; p = 3.56E-7) were associated with PGRN, while only rs5848 showed highly significant association in CSF (p = 5.59E-14). Age, sex, rs5848 genotype, and plasma PGRN together accounted for only 18% of the variability observed in CSF PGRN.
Conclusions:
While some correlation exists between plasma and CSF PGRN, age, sex, and genetic factors differently affect PGRN levels. Therefore, caution should be taken when using plasma PGRN to predict PGRN changes in the brain. These findings further highlight that plasma PGRN levels may not accurately predict clinical features or response to future frontotemporal lobar degeneration therapies.
Progranulin (PGRN) is a cysteine-rich protein encoded by the GRN gene on chromosome 17q21. Initially, PGRN research uncovered its role in cellular processes such as proliferation, growth, and tumorigenicity.1 In 2006, GRN mutations were discovered as a major cause of frontotemporal lobar degeneration (FTLD). To date, approximately 70 different GRN mutations have been found in at least 231 dementia families. A range of mutation types have been described, most causing PGRN haploinsufficiency due to the introduction of a premature stop codon and subsequent GRN messenger RNA (mRNA) nonsense-mediated decay.
PGRN ELISAs were successful in predicting GRN mutation status in serum, plasma, and CSF of patients with FTLD.2–6 However, PGRN levels vary greatly, even in non-GRN mutation carriers, suggesting other genetic and/or environmental PGRN regulators. Also, while GRN mutations ubiquitously alter PGRN expression in all cell types and allow for diagnosis using blood PGRN levels, other modifying factors might regulate PGRN in specific compartments. This is important because of increasing interest in identifying regulators of PGRN levels and/or function for FTLD therapies and in using plasma PGRN levels to monitor disease treatment. To better understand the relationship between plasma and CSF PGRN levels, we performed the first comparative study of PGRN levels in plasma and CSF using a large cohort of individuals ascertained in the Mayo Clinic Study of Aging. Furthermore, we provide data to suggest that patient characteristics as well as single nucleotide polymorphisms (SNPs) rs5848 (GRN) and rs646776 (sortilin 1 [SORT1]) differently regulate plasma vs CSF PGRN.
METHODS
Study population.
This study included all subjects (n = 345) recruited into the Mayo Clinic Study of Aging (MCSA) with plasma and/or CSF, and blood samples for DNA available for study.7 All subjects were between 70 and 89 years of age upon study entry and were residents of Olmsted County, MN. Study design details and participant recruitment are provided elsewhere.7,8 All MCSA subjects undergo a clinical and cognitive assessment every 15 months that includes a nurse interview, neurologic evaluation, and neuropsychological battery. Cognitive domain impairment was assessed by comparing the person's domain score with that of normal subjects from the same population. A score of ≤−1.0 SD below the age-specific mean in the general population was considered possible cognitive impairment. However, a decision about impairment in a cognitive domain was not based on an algorithm but on a consensus agreement among the examining physician, nurse, and neuropsychologist, taking into account years of education, prior occupation, and visual or hearing deficits.7,8 Mild cognitive impairment (MCI) was diagnosed according to the following published criteria: cognitive concern by subject, informant, nurse, or physician; impairment in ≥1 of the 4 cognitive domains; essentially normal functional activities; and absence of dementia.9 Blood was collected in a fasting state using standard venipuncture procedures. The blood was immediately centrifuged and the plasma was stored at −80°C until analyses were conducted. CSF was also collected in a fasting state using a 22-gauge needle and stored at −80°C in polypropylene tubes in 0.5-mL aliquots until analysis. Pittsburgh compound B (PiB) uptake scores obtained at the time of CSF collection were available for 107 subjects.
Standard protocol approvals, registrations, and patient consents.
The study protocols were approved by the Mayo Clinic and Olmsted Medical Center Institutional Review Boards. All subjects provided signed informed consent to participate in the study and in the CSF protocols.
Genetic analyses.
Whole blood DNA was extracted using standard procedures. APOE SNPs rs429358 and rs7412, as well as SNPs rs5848 and rs646776 genotyping was performed with a TaqMan allelic discrimination assay with Assay by Design probes (Applied Biosystems, Foster City, CA) and an ABI 7900 PCR system. Genotyping calls were made using SDS version 2.2 software (Applied Biosystems).
PGRN ELISA.
To determine plasma and CSF PGRN expression levels, we used the Human Progranulin ELISA Kit (Adipogen Inc., Seoul, Korea) per the manufacturer's instructions using CSF diluted 1:2 and plasma diluted 1:100 in the provided dilution buffer. All samples were analyzed in duplicate. The provided recombinant human PGRN was used as a standard.
Statistical analyses.
Statistical analyses were performed using SAS version 9.3 (SAS Institute, Cary, NC) and R statistical software version 2.14 (R Foundation for Statistical Computing, Vienna, Austria). PGRN levels in both CSF and plasma were multiplied by the dilution factor of 2 and 100, respectively, and transformed on the natural logarithm (LOG) scale because of their skewed distributions. Separately for CSF and plasma, a linear mixed-effects regression model with a fixed intercept and separate random effects for ELISA plate pair and subject was fit for LOG-transformed PGRN; so-called normalized PGRN levels were obtained by adding the intercept and random subject effect. Initially, we explored the association between PGRN levels (dependent) and potentially confounding variables (sex, age at sample collection, diagnosis at sample collection, APOE genotype, and PiB score) separately in CSF and plasma using a single variable linear regression model. Variables that were significantly associated with PGRN levels (p < 0.05) in the single variable analyses (sex and age at sample collection) were controlled for in all subsequent analyses examining the associations with normalized PGRN.
In the subset of subjects for which both plasma and CSF were obtained, the association between normalized plasma PGRN and normalized CSF PGRN was summarized by a Pearson partial correlation coefficient (partial r) controlling for sex and age at CSF collection. Exploratory analyses were performed to assess the effect of diagnosis and time interval between plasma and CSF collection on the association between normalized plasma PGRN and normalized CSF PGRN using linear regression with the inclusion of an interaction term with normalized plasma PGRN. In addition, a sensitivity analysis was performed excluding potential outliers (subjects with normalized PGRN levels >3 SD from the mean).
The association of 2 genetic variants, rs5848 and rs646776, with normalized PGRN levels (dependent) was first done separately for CSF and plasma using a multivariable linear regression model controlling for sex and age at sample collection. In the subset of subjects with both plasma and CSF collections, we evaluated the associated effects of rs5848 and rs646776 individually with normalized CSF PGRN (dependent) by linear regression models in which we adjusted for sex, age at CSF collection, and normalized plasma PGRN. These models were constructed for the purpose of association analysis. Separately for each variant, we report the p values that address whether adding the genetic variant to the model with sex, age at CSF collection, and normalized plasma PGRN is significant. Because PGRN levels were considered on the natural logarithm scale, regression coefficients were exponentiated and are presented as multiplicative effects, which are interpreted as the multiplicative increase on the median PGRN.
RESULTS
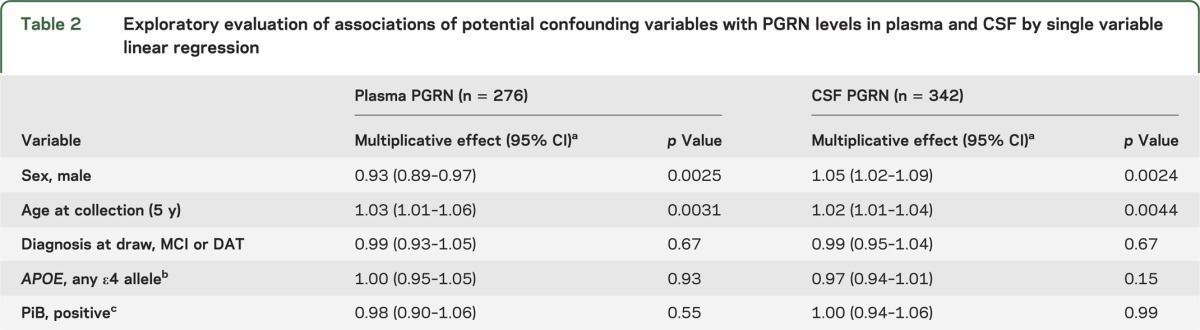
Using an ELISA, we obtained plasma PGRN levels for 276 individuals and CSF PGRN levels for 342 individuals from our cohort of 345 participants enrolled in the MCSA (subject characteristics are included in table 1). We first studied the effect of age and sex on PGRN levels separately in both plasma and CSF. There was an estimated 3% increase (p = 0.0031) in plasma PGRN and 2% increase (p = 0.0044) in CSF PGRN per 5-year increase in age. Moreover, PGRN levels in male participants were 7% lower in plasma (p = 0.0025) and 5% higher in CSF (p = 0.0024) than those in female participants. While all 345 subjects were normal at study entry, 58 individuals converted to MCI or dementia of the Alzheimer type (DAT) at the time of plasma and CSF collection. Therefore, while not a primary study objective, we also explored whether diagnosis, PiB uptake scores, or APOE genotypes were significantly associated with PGRN levels in either plasma or CSF. None of these variables showed an association with PGRN levels in either fluid (table 2).
Table 1.
Subject characteristics
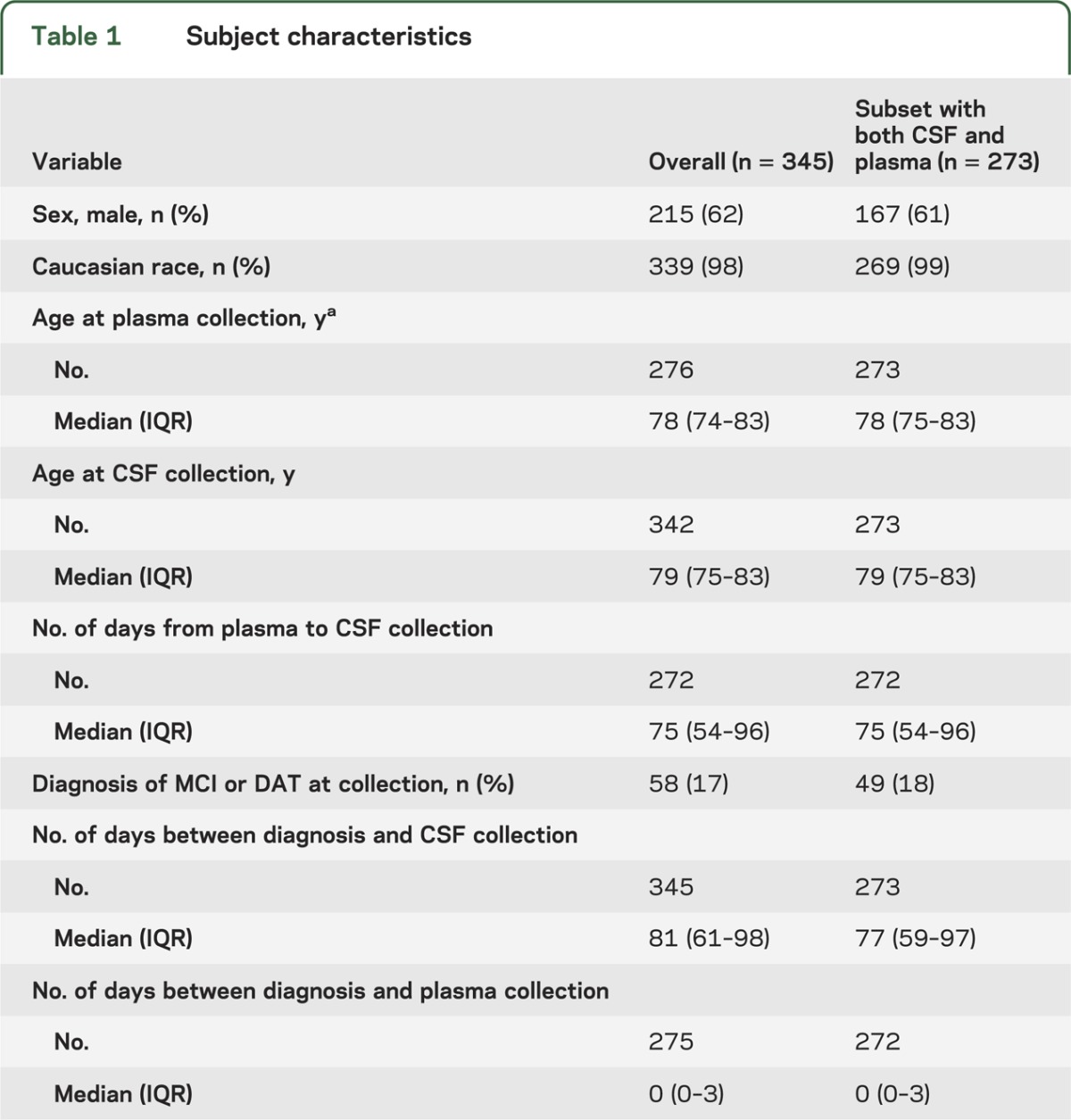
Table 2.
Exploratory evaluation of associations of potential confounding variables with PGRN levels in plasma and CSF by single variable linear regression
To evaluate to what extent a subject's CSF PGRN level can be predicted using their plasma PGRN measurement, we estimated the correlation between normalized PGRN levels in these 2 compartments using a linear regression model. We included data obtained from the 273 subjects with both plasma and CSF PGRN levels available and used sex and age at CSF collection as covariates (figure 1). This statistical model revealed that higher plasma PGRN was associated with higher CSF PGRN (partial r = 0.17; 95% confidence interval: 0.05–0.29; p = 0.004; n = 273). To ensure that other confounding variables were not significantly affecting the correlation, we performed exploratory analyses to determine whether this correlation differed between diagnosis groups (normal vs MCI/DAT) or between subjects with short (≤75 days; n = 139) vs long (>75 days; n = 133) time periods between plasma and CSF collection. The calculated interaction p values were not significantly different for either analysis (interaction p value = 0.68 and 0.56, respectively). In addition, the primary model's Pearson partial r did not change and significance was not lost when outliers were excluded (p = 0.007; n = 269).
Figure 1. Correlation between PGRN levels in plasma and CSF.
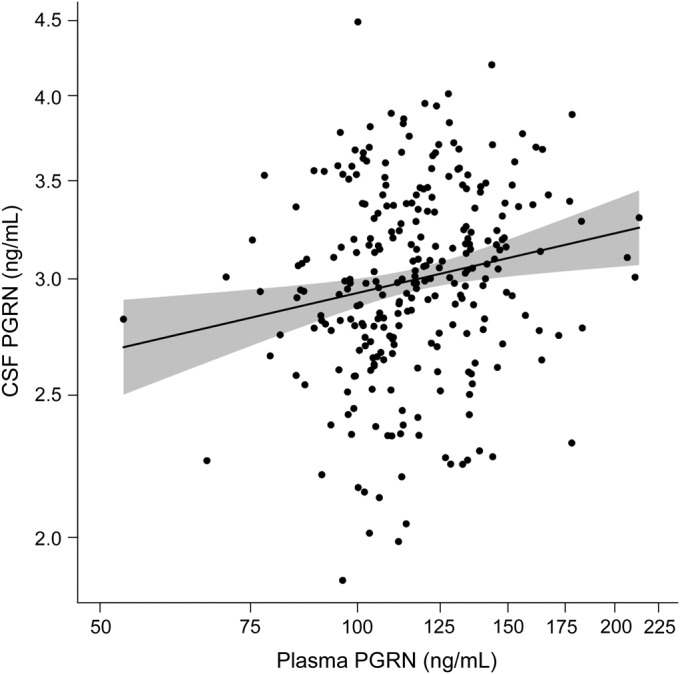
Scatter plot depicting the correlation between plasma and CSF protein levels in 273 individuals. The trend line indicates the estimated mean log-transformed CSF progranulin (PGRN) levels as a function of log-transformed plasma PGRN levels while controlling for sex and age at CSF collection. The shaded region represents the 95% confidence interval. Values on the axes are back-transformed to represent PGRN values as their original units.
Although CSF PGRN levels increased as plasma PGRN levels increased, our model accounting for age, sex, and plasma PGRN levels only explained 6.2% of the variability in CSF PGRN levels (R2 = 0.062); therefore, other components might contribute to the variability observed between plasma and CSF PGRN levels. Previous reports identified variants rs5848 and rs646776 as important genetic modifiers of PGRN levels in plasma and/or serum.10,11 To confirm these previous studies and to assess to what extent these variants associate with CSF PGRN levels, we stratified our cohort by rs5848 and rs646776 genotypes (summary of results in table 3). Association analyses revealed a significant effect on plasma PGRN levels for variant rs5848, with a 5% decrease in PGRN per minor (T) allele (figure 2A). For clarity, this reflects a 5% lower median level of plasma PGRN in the CT genotype as compared with the CC genotype, with the same proportional difference applying to those with the TT as compared with the CT genotype. The association of rs5848 with CSF PGRN levels was also observed with each minor allele contributing to an 8% decrease in PGRN levels (figure 2B). Association analyses also showed a significant 9% decrease in plasma PGRN levels for each rs646776 minor (C) allele (figure 2C). However, variant rs646776 showed no significant effect on PGRN levels in the CSF despite the strong association of rs646776 with PGRN in plasma (figure 2D). To determine whether accounting for rs5848 or rs646776 genotypes improved our CSF prediction model, we reevaluated the linear regression using rs5848 or rs646776 genotype as a covariate. SNP rs646776 did not account for further variability in CSF PGRN levels (R2 = 0.062); however, rs5848 improved the prediction model by 12.4% (R2 = 0.188).
Table 3.
Single SNP associations with PGRN levels in plasma and CSF from a linear regression model
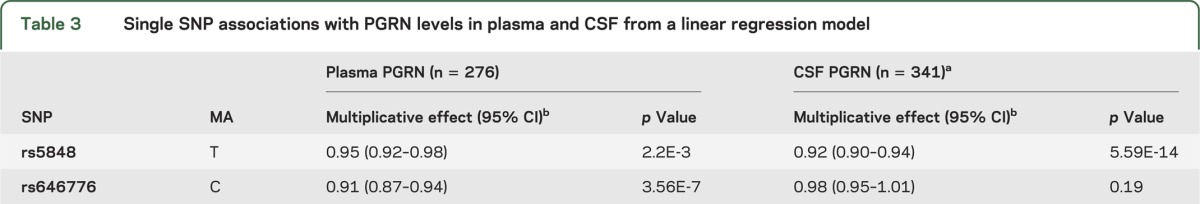
Figure 2. PGRN levels in plasma and CSF by rs5848 or rs646776 genotype.

(A, B) Scatter plots representing the log-transformed plasma (A) and CSF (B) progranulin (PGRN) levels in individuals expressing the different rs5848 genotypes. (C, D) Scatter plots representing log-transformed plasma (C) and CSF (D) PGRN levels in individuals expressing the different rs646776 genotypes. Values on the axes are back-transformed to represent PGRN values as their original units.
DISCUSSION
Since the discovery of GRN mutations as a significant FTLD cause, clinical interest has centered around plasma PGRN levels as a biomarker for FTLD-TDP risk assessment as well as a potential therapeutic readout for therapies focused on increasing PGRN levels once they become available. This, in part, results from consistent reports showing significantly reduced plasma PGRN levels as an accurate way to identify individuals with autosomal dominant GRN mutations that lead to the FTLD-TDP phenotype.3 However, GRN mutations result in PGRN haploinsufficiency in all cell types, whereas FTLD-TDP is a disease of the CNS. Therefore, determining whether relative plasma PGRN levels are representative of brain PGRN levels is important when considering the clinical applications of plasma PGRN measurements, especially in individuals without GRN mutations. A key question in this regard is whether baseline or pharmaceutical-induced changes in blood PGRN levels adequately represent PGRN levels in the CNS. To provide insight into this issue, this report provides the first correlation study between PGRN levels in plasma and CSF, a CNS-relevant fluid, in a large human cohort enrolled in a population-based study.
We first found that age and sex are important PGRN modifiers in both plasma and CSF. While plasma and CSF PGRN levels were similarly influenced by age, with an increase of approximately 2% to 3% per 5 years, opposing effects were observed when our cohort was stratified by sex, with male participants showing significant 7% lower levels of PGRN in plasma but 5% higher levels of PGRN in CSF as compared with female participants. Given that some patients had converted to MCI or DAT at the time of sample collection, we studied whether diagnosis, PiB uptake scores, or APOE genotypes were associated with PGRN levels. None of these factors significantly affected PGRN in either plasma or CSF. Determining PGRN levels in patients with DAT was not the primary objective of this study and the number of affected subjects that we analyzed was low; however, these findings are consistent with previous studies reporting no significant difference in plasma PGRN levels in patients with Alzheimer disease (AD) as compared with controls.3,12,13
Using plasma and CSF PGRN levels obtained from 273 individuals and accounting for age and sex, we found that high plasma PGRN levels were associated with high CSF PGRN levels. This correlation did not differ when we accounted for outliers, diagnosis, or time between CSF and plasma collection. This indicates that an individual's relative PGRN expression will tend to deviate from the average in the same direction in both sample types. However, we observed only a small correlation. In fact, age, sex, and plasma PGRN levels together accounted for only 6.2% of the variability observed in CSF, indicating that other variables are involved. Based on these findings, it is clear that using plasma as the predominant fluid for PGRN quantification for FTLD-TDP risk might not always represent that of the CNS. This would explain results from our earlier study in which we did not identify a correlation between plasma PGRN levels and the age of disease onset in GRN mutation carriers or non-GRN mutation carriers.3 These findings are comparable to publications that highlight the lack of correlation between plasma and CSF measurements for disease-specific biomarkers in other neurodegenerative disorders. For example, studies comparing tau and β-amyloid (Aβ) levels in AD cases compared with controls revealed discrepant results using plasma Aβ and tau measurements; however, reports have been more consistent to show increased total or phosphorylated tau, as well as decreased Aβ1–42 levels, in patients with AD compared with controls when quantifying these proteins in CSF, indicating the CSF as potentially a better fluid for biomarker studies in AD.14–20
Other elements, including genetic modifiers, epigenetic aspects, epidemiologic influences, and/or environmental factors, are also likely to influence PGRN levels. For example, it is known that PGRN levels are elevated due to inflammation or other clinical conditions such as pregnancy or cancer21,22; however, whether these conditions similarly affect plasma and CSF PGRN levels is unknown. PGRN receptor expression levels and distribution will also have an important role and might be significantly different in the periphery as compared with the brain. Our new data clearly demonstrate that genetic influences on PGRN levels are different in plasma vs CSF. We first studied rs5848, a common variant located in the 3′-untranslated region of GRN. This SNP is predicted to alter a micro-RNA binding site, with the minor T allele resulting in lower PGRN levels through enhanced suppression of GRN translation.10,23 An association of rs5848 with plasma PGRN levels as well as with total brain PGRN levels from autopsy tissues was reported; however, its effect on CSF PGRN was not determined. Using the MCSA cohort, we now confirm the association of rs5848 with human plasma PGRN levels in a dose-dependent manner, and we show for the first time a significant association of rs5848 with CSF PGRN levels. More specifically, our data indicated that one's rs5848 genotype is predicted to account for approximately 12% of the variability in CSF PGRN levels in our cohort. The significant correlation of this SNP with PGRN levels in the CNS could explain why we previously observed that carriers homozygous for the T allele of rs5848 (associated with lower PGRN levels) have a 3.2-fold increased risk of developing FTLD-TDP compared with homozygous C-allele carriers.23
The second SNP studied, rs646776, was identified by a quantitative genome-wide association study of plasma PGRN levels and was shown to regulate PGRN levels by altering the expression of the PGRN receptor SORT1.11,24 While significantly associated with plasma PGRN levels, we did not observe an association between rs646776 and CSF PGRN levels in the MCSA cohort. This is important when considering whether plasma PGRN levels accurately depict CNS PGRN measurements. Furthermore, this would explain why adding the rs646776 genotype as a covariate in our correlation analyses had no effect in the linear regression used to predict CSF PGRN levels. As previously suggested, rs646776 (or its proxy rs12740374) most likely regulates SORT1 mRNA levels by affecting a liver-specific transcription factor binding site, likely explaining why its effect on PGRN was confined to the periphery.25 The lack of an association of rs646776 with CNS PGRN levels also clarifies why rs646776 was not associated with FTLD-TDP in a genome-wide association study26 and why no correlation was seen between rs646776 and brain SORT1 mRNA levels in our previous report.11
To date, no effective treatment or therapy has been developed to treat FTLD or slow its progression. However, given the importance of GRN mutations in FTLD, increasing PGRN levels to overcome disease risk and/or onset has been of much clinical interest. Recently, several promising compounds were identified in cellular models of PGRN loss and were shown to be effective in increasing PGRN levels in lymphoblastoid cell lines derived from GRN mutation carriers.27,28 Whether plasma PGRN levels could be useful to monitor the effect of these and other treatments is unknown and will depend on the drug's mechanism of action. Our current data suggest that it will be critical to validate any effects seen on plasma PGRN levels in the brain and to realize that the absence of an effect on plasma PGRN does not exclude an effect in the brain or a potential effect on PGRN function. Moreover, our present sample cohort did not allow us to study the variability associated with PGRN measurements within the same individual over time or the effect of other variables such as fasting, which are additional complicating factors that need to be studied before plasma PGRN levels could be reliably used in the clinic. However, we also cannot exclude the possibility that stronger correlations might be observed using subjects of different ethnicities, subjects of younger age, or subjects with plasma and CSF samples collected at the same time.
Our study identified differences in PGRN regulation in the plasma compared with CSF. These findings provide a clear rationale not to use plasma PGRN levels as a reliable diagnostic aid to predict clinical features such as age at disease onset or the risk of developing FTLD, with the one exception being the use of plasma PGRN levels to determine GRN mutation status. Furthermore, caution should be taken when using plasma PGRN levels to predict PGRN changes in the brain, especially when using compounds without a known mechanism of action.
GLOSSARY
- Aβ
β-amyloid
- AD
Alzheimer disease
- DAT
dementia of the Alzheimer type
- FTLD
frontotemporal lobar degeneration
- MCI
mild cognitive impairment
- MCSA
Mayo Clinic Study of Aging
- mRNA
messenger RNA
- PGRN
progranulin
- PiB
Pittsburgh compound B
- SNP
single nucleotide polymorphism
- SORT1
sortilin 1
- TDP
TAR DNA-binding protein
AUTHOR CONTRIBUTIONS
Alexandra Nicholson: acquisition of data, analysis or interpretation of data, drafting the manuscript for content, including writing for content. NiCole Finch, Aleksandra Wojtas, and Nicola Rutherford: acquisition of data, analysis or interpretation of data, revising the manuscript for content, including writing for content. Colleen Thomas: statistical analysis, revising the manuscript for content, including writing for content. Michelle Mielke: revising the manuscript for content, including writing for content. Rosebud Roberts: acquisition of data, revising the manuscript for content, obtaining funding. Bradley Boeve and David Knopman: revising the manuscript for content, including writing for content. Ronald Petersen: contribution of vital reagents/tools/subjects, revising the manuscript for content, including writing for content, obtaining funding. Rosa Rademakers: study concept or design, acquisition of data, analysis or interpretation of data, revising the manuscript for content, including writing for content, study supervision or coordination, obtaining funding.
STUDY FUNDING
This work was funded by NIH grants R01 NS065782, R01 NS076471, P50 AG016574, and UO1 AG006786, the Robert H. and Clarice Smith and Abigail Van Buren Alzheimer's Disease Research Program of the Mayo Foundation, and the Consortium for Frontotemporal Degeneration Research.
DISCLOSURE
A. Nicholson, N. Finch, C. Thomas, A. Wojtas, and N. Rutherford report no disclosures relevant to the manuscript. M. Mielke receives research support from NIH and the Alzheimer Drug Discovery Foundation. Dr. Mielke has served as a consultant for Eli Lilly. R. Roberts receives research funding from the Driskill Foundation and AbbVie Health Economics and Outcome Research. B. Boeve has served as an investigator for clinical trials sponsored by Cephalon, Inc., Allon Pharmaceuticals, and GE Healthcare. He receives royalties from the publication of a book titled Behavioral Neurology of Dementia (Cambridge Medicine, 2009). He serves on the scientific advisory board of the Tau Consortium. He has received honoraria from the American Academy of Neurology. He receives research support from the National Institute on Aging and the Mangurian Foundation. D. Knopman serves as deputy editor for Neurology®; served on a data safety monitoring board for Lilly Pharmaceuticals; served as a consultant to TauRx, was an investigator in clinical trials sponsored by Baxter, Elan Pharmaceuticals, and Forest Pharmaceuticals in the past 2 years; and receives research support from the NIH. R. Petersen chairs a data monitoring committee for Pfizer, Inc. and Janssen Alzheimer Immunotherapy, and is a consultant for GE Healthcare and Elan Pharmaceuticals. He receives royalties from Oxford University Press for Mild Cognitive Impairment. R. Rademakers receives research support from the NIH (R01 NS080882, R01 NS065782, R01 AG026251, R01 NS076471, and P50 AG16574), the ALS Therapy Alliance, and the Consortium for Frontotemporal Degeneration Research. Dr. Rademakers further received honoraria for lectures or educational activities not funded by industry; she serves on the medical advisory board of the Association for Frontotemporal Degeneration, on the board of directors of the International Society for Frontotemporal Dementia, and holds a patent on methods to screen for the hexanucleotide repeat expansion in the C9ORF72 gene. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Daniel R, He Z, Carmichael KP, Halper J, Bateman A. Cellular localization of gene expression for progranulin. J Histochem Cytochem 2000;48:999–1009 [DOI] [PubMed] [Google Scholar]
- 2.Van Damme P, Van Hoecke A, Lambrechts D, et al. Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J Cell Biol 2008;181:37–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Finch N, Baker M, Crook R, et al. Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain 2009;132:583–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghidoni R, Benussi L, Glionna M, Franzoni M, Binetti G. Low plasma progranulin levels predict progranulin mutations in frontotemporal lobar degeneration. Neurology 2008;71:1235–1239 [DOI] [PubMed] [Google Scholar]
- 5.Schofield EC, Halliday GM, Kwok J, Loy C, Double KL, Hodges JR. Low serum progranulin predicts the presence of mutations: a prospective study. J Alzheimers Dis 2010;22:981–984 [DOI] [PubMed] [Google Scholar]
- 6.Sleegers K, Brouwers N, Van Damme P, et al. Serum biomarker for progranulin-associated frontotemporal lobar degeneration. Ann Neurol 2009;65:603–609 [DOI] [PubMed] [Google Scholar]
- 7.Roberts RO, Geda YE, Knopman DS, et al. The Mayo Clinic Study of Aging: design and sampling, participation, baseline measures and sample characteristics. Neuroepidemiology 2008;30:58–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petersen RC, Roberts RO, Knopman DS, et al. Prevalence of mild cognitive impairment is higher in men. The Mayo Clinic Study of Aging. Neurology 2010;75:889–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med 2004;256:183–194 [DOI] [PubMed] [Google Scholar]
- 10.Hsiung GY, Fok A, Feldman HH, Rademakers R, Mackenzie IR. rs5848 polymorphism and serum progranulin level. J Neurol Sci 2011;300:28–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carrasquillo MM, Nicholson AM, Finch N, et al. Genome-wide screen identifies rs646776 near sortilin as a regulator of progranulin levels in human plasma. Am J Hum Genet 2010;87:890–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Antonell A, Gil S, Sanchez-Valle R, et al. Serum progranulin levels in patients with frontotemporal lobar degeneration and Alzheimer's disease: detection of GRN mutations in a Spanish cohort. J Alzheimers Dis 2012;31:581–591 [DOI] [PubMed] [Google Scholar]
- 13.Piscopo P, Rivabene R, Galimberti D, et al. Gender effects on plasma PGRN levels in patients with Alzheimer's disease: a preliminary study. J Alzheimers Dis 2013;35:313–318 [DOI] [PubMed] [Google Scholar]
- 14.Andreasen N, Minthon L, Davidsson P, et al. Evaluation of CSF-tau and CSF-Abeta42 as diagnostic markers for Alzheimer disease in clinical practice. Arch Neurol 2001;58:373–379 [DOI] [PubMed] [Google Scholar]
- 15.Ida N, Hartmann T, Pantel J, et al. Analysis of heterogeneous A4 peptides in human cerebrospinal fluid and blood by a newly developed sensitive Western blot assay. J Biol Chem 1996;271:22908–22914 [DOI] [PubMed] [Google Scholar]
- 16.Kanai M, Matsubara E, Isoe K, et al. Longitudinal study of cerebrospinal fluid levels of tau, A beta1-40, and A beta1-42(43) in Alzheimer's disease: a study in Japan. Ann Neurol 1998;44:17–26 [DOI] [PubMed] [Google Scholar]
- 17.Motter R, Vigo-Pelfrey C, Kholodenko D, et al. Reduction of beta-amyloid peptide42 in the cerebrospinal fluid of patients with Alzheimer's disease. Ann Neurol 1995;38:643–648 [DOI] [PubMed] [Google Scholar]
- 18.Tamaoka A, Fukushima T, Sawamura N, et al. Amyloid beta protein in plasma from patients with sporadic Alzheimer's disease. J Neurol Sci 1996;141:65–68 [DOI] [PubMed] [Google Scholar]
- 19.Matsubara E, Ghiso J, Frangione B, et al. Lipoprotein-free amyloidogenic peptides in plasma are elevated in patients with sporadic Alzheimer's disease and Down's syndrome. Ann Neurol 1999;45:537–541 [PubMed] [Google Scholar]
- 20.Kuo YM, Emmerling MR, Lampert HC, et al. High levels of circulating Abeta42 are sequestered by plasma proteins in Alzheimer's disease. Biochem Biophys Res Commun 1999;257:787–791 [DOI] [PubMed] [Google Scholar]
- 21.Todoric J, Handisurya A, Perkmann T, et al. Circulating progranulin levels in women with gestational diabetes mellitus and healthy controls during and after pregnancy. Eur J Endocrinol 2012;167:561–567 [DOI] [PubMed] [Google Scholar]
- 22.Han JJ, Yu M, Houston N, Steinberg SM, Kohn EC. Progranulin is a potential prognostic biomarker in advanced epithelial ovarian cancers. Gynecol Oncol 2011;120:5–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rademakers R, Eriksen JL, Baker M, et al. Common variation in the miR-659 binding-site of GRN is a major risk factor for TDP43-positive frontotemporal dementia. Hum Mol Genet 2008;17:3631–3642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu F, Padukkavidana T, Vaegter CB, et al. Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron 2010;68:654–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Musunuru K, Strong A, Frank-Kamenetsky M, et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature 2010;466:714–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Deerlin VM, Sleiman PM, Martinez-Lage M, et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet 2010;42:234–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cenik B, Sephton CF, Dewey CM, et al. Suberoylanilide hydroxamic acid (vorinostat) up-regulates progranulin transcription: rational therapeutic approach to frontotemporal dementia. J Biol Chem 2011;286:16101–16108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Capell A, Liebscher S, Fellerer K, et al. Rescue of progranulin deficiency associated with frontotemporal lobar degeneration by alkalizing reagents and inhibition of vacuolar ATPase. J Neurosci 2011;31:1885–1894 [DOI] [PMC free article] [PubMed] [Google Scholar]