

Abstract
Activation-induced cytidine deaminase (AID3) initiates a process generating DNA mutations and breaks in germinal center (GC) B cells that are necessary for somatic hypermutation and class switch recombination. GC B cells can “tolerate” DNA damage while rapidly proliferating due to partial suppression of the DNA damage response by BCL6. Here, we develop a model to study the response of mouse GC B cells to endogenous DNA damage. We show that the base excision repair protein apurinic/apyrimidinic endonuclease 2 (APE2) protects activated B cells from oxidative damage in vitro. APE2-deficient mice have smaller germinal centers and reduced antibody responses compared to wild-type mice. DNA double-strand breaks are increased in the rapidly dividing GC centroblasts of APE2-deficient mice, which activate a p53-independent cell-cycle checkpoint and a p53-dependent apoptotic response. Proliferative and/or oxidative damage and AID-dependent damage are additive stresses that correlate inversely with GC size in WT, AID- and APE2-deficient mice. Excessive DSBs lead to decreased expression of BCL6, which would enable DNA repair pathways but limit GC cell numbers. These results describe a non-redundant role for APE2 in the protection of GC cells from AID-independent damage and, although GC cells uniquely tolerate DNA damage, we find that the DNA damage response can still regulate GC size through pathways that involve p53 and BCL6.
Keywords: B Cells, Germinal Center, DNA damage, DNA repair
Introduction
B-lymphocytes face the unique challenge of undergoing rapid cellular proliferation coupled with developmentally programmed DNA rearrangements. During B cell development in the bone marrow, the recombinase-activating genes (RAGs) cause DNA double-strand breaks (DSB) in pro- and pre-B cells that are necessary for assembly of the antigen receptor (1). During an immune response, mature B cells proliferating in germinal centers (GCs) are subject to mutations and DSBs induced by activation-induced cytidine deaminase (AID) that are necessary for the processes of somatic hypermutation (SHM) and antibody class switch recombination (CSR), respectively, which improve the quality of the humoral immune response (2, 3). In addition to these programmed DNA breaks, increased metabolic activity results in a dramatic increase in the level of intracellular reactive oxygen species (ROS) that can cause oxidative damage to DNA (4, 5). Understanding the pathways that regulate the response to DNA damage while simultaneously allowing these cells to tolerate programmed DNA breaks is key to elucidating mechanisms of lymphomagenesis.
Expression of the transcriptional repressor BCL6, which is essential for GC formation, suppresses the DNA damage response in GC cells by inhibiting p53, p21, CHK1, and ATR (6–9), allowing these cells to tolerate DNA damage from AID. BCL6 also inhibits differentiation, promoting the rapid proliferative phenotype of GC centroblasts (10–12). It has been proposed that activation of the DNA damage sensor ATM might terminate the proliferative phase and trigger differentiation of B cells both during development and in GCs (13–15), suggesting that a feedback loop dependent on DNA damage might limit the expansion of these cells. Given that some components of the DNA damage response are suppressed in GC cells, we sought to determine how GC cells respond to endogenous DNA damage in vivo by developing a model where the effect of damage from AID as well as that due to AID-independent endogenous DNA damage associated with proliferation could be studied.
DNA bases damaged by oxidation are primarily repaired by the base excision repair (BER) pathway (5, 16). Glycosylases remove the damaged base, leaving an abasic site that can be cleaved by AP endonucleases (APEs), filled in by DNA polymerase β and sealed by DNA ligase. Previously, it was shown that two glycosylases, OGG1 and NEIL1, which remove oxidized DNA bases, are highly expressed in GC cells and that deficiency of NEIL1 results in a decreased frequency of GC B cells (17–19), suggesting that protection from oxidative damage is important for GC B cells. Surprisingly, and in contrast to cultured cells, we recently found that expression of the major mammalian AP-endonuclease, APE1, is dramatically decreased in GC B cells, where expression of a much less efficient homologue, APE2, is markedly increased (Stavnezer E.K. Linehan, M.R. Thompson, G. Habboub, A. Ucher, T. Kadungure, D. Tsuchimoto, Y. Nakabeppu and C.E. Schrader, submitted). We showed that the unique expression pattern of APE1 and APE2 in the GC contributes to SHM. APE2 shares extensive functional overlap with APE1, which is ubiquitously expressed and considered essential for abasic site repair (20–22). Although the endonuclease activity of APE2 is much lower than that of APE1, APE2 has 3′- to 5′-exonuclease and 3′-phosphodiesterase activities that are more efficient than those of APE1 (22–24), the latter of which could be important for removing 3′-phosphoglycolate blocking groups such as those made by direct attack of ROS on the DNA backbone (25). This activity could be important in rapidly dividing cells with high metabolic rates that generate intracellular ROS.
APE2 is important during B- and T-cell development (26, 27). APE2-deficient mice show a partial block at the pro- to pre-B cell transition, and in addition, defective expansion of earlier progenitor populations is observed during recovery of the bone marrow from chemotherapeutic treatment with 5-fluorouracil (27). Thymic cellularity is reduced five fold in APE2-deficient mice, and the loss of cells in both thymus and bone marrow appears to involve a p53-dependent pathway (26, 28). APE2 does not appear to be important for the process of V(D)J-recombination (27). These results indicate that APE2 is important for the repair of oxidative damage to DNA that occurs in rapidly dividing cells, such as during bursts of proliferation in developing lymphocytes. Loss of this repair function is consistent with the diminished production of B-cell progenitors observed in the bone marrow of APE2-deficient mice, the decreased ability of pro-B cells to expand in vitro, and the increase in γH2AX foci indicative of DNA breaks in APE2-deficient thymocytes (27, 28). Such a role would predict that APE2 is also important in the rapid proliferation that occurs during immune responses in B cells in the GC, especially given the new finding that APE1 expression is very low there, but the importance of APE2 in the function of mature peripheral lymphocytes in vivo has not previously been examined.
In addition to a more global role in DNA repair, APE2 has a direct role in mature B cells during CSR (29–31) and SHM (30)(Stavnezer et al, submitted), two processes that involve programmed DNA damage initiated by AID and that occur in germinal centers. Both APE1 and APE2 are expressed in splenic B cells activated in vitro (29), and both are important for efficient CSR, creating nicks that become DSBs in switch region DNA in response to abasic sites generated by AID deamination of dC and removal of the resulting dU by UNG (29, 32). Although APE2 contributes to CSR in spleen B cells, APE1 is sufficient for CSR, particularly in cell lines that undergo CSR (30, 33), and APE1 was recently shown to associate with AID, dependent on phosphorylation of AIDS38 (34). However, in contrast to cultured cells, it is not yet clear how low APE1 expression in the GC impacts CSR in vivo. APE2 expression in the GC contributes to the hypermutation phenotype of GC cells (J. Stavnezer et al, submitted), as the frequency of mutations (30), and the percent of mutations at A:T bp are reduced in its absence. The data suggest that APE2 is involved in making DNA single-strand breaks that contribute to Phase II mutations.
Given the involvement of APE2 in CSR and SHM, and the low APE1 expression in the GC, we sought to analyze the effect of APE2-deficiency on the ability of B cells to proliferate and survive in the germinal center. We find an important and non-redundant role for APE2 in the repair of endogenously acquired AID-independent DNA damage in GC B cells and that APE2 is necessary for B cell expansion during the GC reaction. We show here that APE2-deficient GC B cells have increased DSBs, and study the response of GC B cells in situ to endogenous DNA damage. We report that despite suppression of the DNA damage response by BCL6, DNA damage in GC cells can activate both p53-dependent and p53-independent damage response pathways, reduce levels of BCL6, and limit the expansion of these cells.
Materials and Methods
Mice
All mouse strains were backcrossed to C57BL/6 for more than 8 generations. Because apex2 is on the X chromosome, we used male apex2Y/− mice and apex2Y/+ littermates in all experiments. apex2Y/− mice were described previously (35). apex1+/− mice were obtained from E. Friedberg (36) (University of Texas Southwestern Medical Center, Dallas, TX). tp53+/− mice were obtained from Stephen Jones (U. Mass. Medical School) and were previously described (37). Ogg1−/− mice were obtained from Chris Hollander (NIH). OT-II ovalbumin-specific TcR-transgenic mice are available from Jackson Labs. aid−/− mice were from T. Honjo. Heterozygotes of all strains were bred to generate KO, double KO, or doubly-deficient (apex1+/− apex2Y/−) mice. B6.SJL mice originally purchased from Jackson Labs (Bar Harbor, ME) were bred in our facility. Mice were housed in the Institutional Animal Care and Use Committee-approved specific pathogen-free facility at the University of Massachusetts Medical School. The mice were bred and used according to the guidelines from the University of Massachusetts Animal Care and Use Committee.
ROS sensitivity
1×105 T-depleted spleen B cells were cultured in triplicate as in (29) in 1mL with LPS and IL-4 for two days with buthionine sulfoximide (BSO, Sigma). Cultures were harvested after two days, washed in PBS and fixed in 70% ethanol before staining with propidium iodide for cell cycle analysis, as described below. Late stage apoptotic cells that degrade their DNA were quantified as having less than the DNA content of normal cells in G1 phase (sub-G1) by propidium iodide staining. These cells are smaller, but are still identifiable as cells by forward and side scatter analysis. Results were confirmed using Live/Dead Blue® staining (Invitrogen)(not shown).
Flow cytometry
Spleen or Peyer’s patch cells were gently dispersed mechanically and filtered through 70 μm nylon mesh and re-suspended in staining media containing biotin, flavin-, and phenol red-deficient RPMI 1640 (Irvine Scientific), 10 mM HEPES, pH 7.2, 0.2% sodium azide, 1 mM EDTA, and 2% fetal bovine serum. Cells were incubated with anti-CD16/32 antibody (eBioscience) for 10 min. on ice to block Fc receptors, primary antibodies for 30 min., washed before the addition of streptavidin-Pacific Blue (Invitrogen) secondary reagent for 15 min., and then washed three more times. After the final wash, cells were resuspended in 7-AAD or 1 μg/ml propidium iodide to exclude dead cells. Primary antibodies included B220-APC (RA3-6B2), GL7-FITC, CD95-PE, CXCR4-biotin, CD23-PE, and CD21-FITC (all from eBioscience or BD Pharmingen). All cell populations were gated on viable cells by propidium iodide or 7-AAD exclusion and singlets by forward scatter area and height. For intracellular staining, cells were first stained with Live/Dead Blue® (Invitrogen), then surface stained with combinations of B220-AF647, B220-biotin, GL7-FITC, CXCR4-bio and SA-Pacific blue, fixed in 2% paraformaldehyde and then in Permeabilization buffer (eBioscience) before addition of anti-BCL6-PE or anti-γH2AX-AF647 or labeled istoype control antibodies (BD Pharmingen). Flow cytometry was performed on a 3 laser, 9 detector LSRII (Becton Dickinson). Data were analyzed with FlowJo software (Tree Star).
Analysis of DNA Synthesis and Cell Cycle
Cell-cycle status was determined on PP cells that were surface stained with B220-AF647 (Invitrogen), GL7-FITC, CXCR4-biotin and Pacific Blue-streptavidin and then fixed in 70% ethanol overnight prior to incubation with RNaseA and propidium iodide (50 μg/ml each, Sigma) for 20 min. at 37°C. Cells were then analyzed by flow cytometry and after gating on GC cells (B220+GL7+) or centroblasts (B220+GL7+CXCR4HI), the Watson Pragmatic model was applied to fit the curves to the stages of the cell-cycle. Turnover and production rates of cells were determined by continuous BrdU labeling of in vivo DNA synthesis. Mice were injected i.p. with 1 mg BrdU (Sigma) in 0.2 ml PBS every 12 hours for 1, 2 or 3 days and 6 hours prior to sacrifice. PP cells were harvested and surface stained as above. Cells were permeabilized in 70% ethanol overnight, washed, fixed in 1% paraformaldehyde in PBS with 0.2% Tween-20 for 1 h, washed, and treated with 5 U/ml DNase1 (Roche) in 0.15 M NaCl, 4.2 mM MgCl2 for 30 min. at 37°C. After one wash with deficient RPMI/5% FBS/0.1% Tween-20, cells were incubated with anti-BrdU-AF594 (Invitrogen) for 30 min. at room temperature and washed twice more before analysis by flow cytometry. Renewal rates (turnover) and production rates were calculated as described (38).
Cell transfers
1×107 T-depleted spleen cells from WT or APE2-deficient mice (C57BL/6, CD45.2+, n=2 each, pooled, immunized 6 months previously with 50 μg NP-CGG (Biosearch Technologies) in alum i.p.) were transferred along with 1.5×106 nylon wool-purified OT-II Tg cells i.v. into B6.SJL (CD45.1+) recipients that had been sub-lethally irradiated (900 rad) 24 h previously. Recipient mice were challenged with 50 μg NP-OVA (Biosearch Technologies) in alum i.p. the next day, and maintained on antibiotics for 14d, when spleens were analyzed by flow cytometry, as described above, with B220-A700, anti-CD45.2-APC (ebioscience), CD95-PE and GL7-FITC.
Spectratyping and RT-PCR
Bone marrow cells were prepared and immature B cells were surface stained as in (27), purified by FACS and collected into Trizol for RNA preparation. Primers used for RT-PCR for bax were Bax-F, 5′-GCCTCCTCTCCTACTTCGGG-3′ and Bax-R, 5′-TGAGGACTCCAGCCACAAAGA-3′ and primers used for spectratype analysis (VH1_F and Cμ-FAM_R) are published (39).
Immunizations and ELISAs
Mice were immunized intraperitoneally (i.p.) with 0.2 mL of washed 10% sRBC (Innovative Research, Inc.) in PBS, 50 μg NP-CGG in alum, or 50 μg NP-Ficoll (Biosearch Technologies) in PBS. Boosted mice received 50 μg NP-CGG in PBS i.p. 6 months after primary immunization. 3-fold serial dilutions of serum from immunized mice were assayed on plates coated with antigen: sRBC membranes prepared as described (40), NP4-BSA, or NP14-BSA (Biosearch Technologies). Bound Ab was detected with anti-IgM-biotin or anti-IgG-biotin and streptavidin-alkaline phosphatase (Southern Biotech.). A high-affinity purified IgG anti-NP standard (generous gift from Dr. T. Iminishi-Kari) was used to determine anti-NP Ab concentration using SoftMax Pro 5.3 software (Molecular Devices).
Statistics
Statistical tests were performed with GraphPad Prism software. All experiments that involved the comparison of 3 or more genotypes were analyzed using one-way ANOVA with Tukey’s Multiple Comparison Test. Experiments that compared only 2 groups were analyzed by unpaired Student t test, unless noted otherwise.
Results
We hypothesized that APE2 protects against oxidative damage caused by ROS that are generated as byproducts of cellular respiration. To test this, we assessed whether APE2-deficiency confers hypersensitivity to intracellular ROS in activated, proliferating mature B cells. We activated splenic B cells with LPS and IL-4 to induce proliferation (conditions where both APE1 and APE2 are expressed (29)), in the presence of increasing doses of L-buthionine-sulfoximine (BSO), a compound that inhibits the anti-oxidant glutathione synthetase, thereby increasing intracellular H2O2. Wildtype (WT) B cells were unaffected by the doses of BSO added, while APE2-deficient B cells showed a dose-dependent increase in cell death, as measured by DNA-content analysis with propidium iodide staining (Fig. 1, left). APE2-deficient B cells that also lacked AID were equally sensitive to BSO, confirming that APE2 is important for repairing AID-independent damage (Fig. 1, right, APE2/AID). APE1 haplo-insufficiency did not confer sensitivity to BSO, and only slightly increased the sensitivity of APE2-deficient B cells (Fig. 1, right, APE1/APE2). We conclude that APE1 is not sufficient to protect rapidly proliferating mature B cells from intracellular ROS, and that APE2 is required. These data prompted us to investigate the effect of APE2 deficiency on peripheral mature B-cell subsets.
Figure 1. APE2-deficient B cells are hypersensitive to oxidative damage.
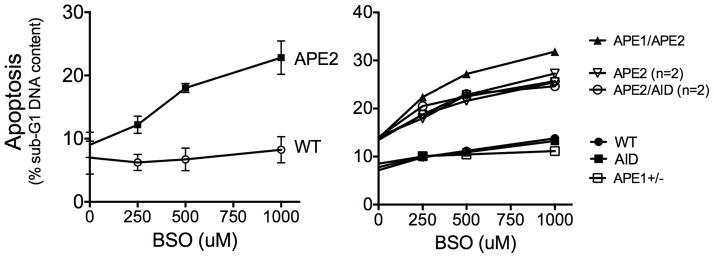
LPS-activated apex2Y/− spleen B cells undergo apoptosis when cultured with increasing amounts of BSO to increase ROS. Shown is the percent of cells with DNA content less than that of G1 phase cells, as determined by propidium iodide staining and flow cytometry after 48h (Left, mean ± SEM of 4 independent experiments; right, WT, aid−/−, apex1+/−, n = 1; apex2Y/− and apex2Y/− aid−/−, n = 2).
Reduced spleen B cell numbers and antibody responses in APE2-deficient mice
We first examined the peripheral B-cell compartment in the spleen of APE2-deficient mice (designated as APE2 in the figures, or as apex2Y/−, since the gene is on the X chromosome). Consistent with the decreased generation of pre-B cells we previously observed in the bone marrow (27), we find that the total number of splenocytes in apex2Y/− mice is approximately half of that in spleens from WT littermates (Fig. 2A). B220+ cells still constitute ~50% of the spleen, as in WT mice, but the total number of B cells is reduced twofold. The number of follicular and newly formed B cells is reduced twofold, while the total number of marginal zone (MZ) B cells is comparable to that in WT mice, consistent with the proportion of B-cell subsets we reported previously (29). By contrast, normal numbers of splenocytes and splenic B-cell subsets were found in apex2Y/−tp53−/− mice, indicating that the reduction in cellularity is likely due to DNA damage-induced p53-dependent apoptosis (Fig. 2A). Similarly, we previously found that the loss of pre-B cells in the bone marrow of APE2-deficient mice is p53-dependent (27).
Figure 2. Analysis of the B cell pool and antibody responses in APE2-deficient mice.

(A) Spleens from mice of the indicated genotypes were analyzed by flow cytometry for B cell subsets (n=3, *p<0.05 vs. WT). (B) Immature bone marrow B cells (B220+IgM+CD19+AA4.1+) were purified by FACS and the diversity of CDR3 region lengths was determined by spectratyping analysis (representative of 2 experiments). (C–E) WT (solid line) or apex2Y/− (grey dotted line) mice were immunized with sRBC (C,D; n=6 WT, 4 APE2) or NP-ficoll (E, n=4 each) and antibody titers were determined at the times indicated. n.d., no difference.
To determine whether the decrease in B-cell production in APE2 mice leads to a decrease in the diversity of clonotypes in the newly formed B cell population, we performed Ig heavy chain complementarity determining region 3 (CDR3) spectratype analysis. RNA was isolated from FACS-purified immature B cells (B220+IgM+CD19+AA4.1+) from bone marrow of apex2Y/− and WT mice. Spectratype analysis of the products from amplification of the Ig heavy chain CDR3 region by RT-PCR showed a Gaussian distribution, similar to that of WT cells, indicating that the reduced B-cell pool did not cause a skewed clonal distribution, and that a diverse population of B-cell clonotypes is available in the periphery to participate in an immune response (Fig. 2B). However, we found that the ability of APE2-deficient mice to mount an antibody response to thymus-dependent (TD) antigens is impaired. We observed reduced IgM and IgG titers in response to immunization with a potent TD antigen, sheep red blood cells (sRBC) (Fig. 2C,D). The decreased response to TD-antigens could be due to the reduced B-cell numbers, impaired T-cell help or a decreased GC response in APE2-deficient mice. In contrast, antibody production in response to the thymus-independent (TI) antigen nitrophenol (NP)-Ficoll was normal (Fig. 2E). The difference in requirement for APE2 in TD- and TI-responses may be owing to normal levels of MZ B cells in APE2-deficient mice, which is the major responding subset to TI-2 antigens, and/or to normal levels of APE1 expression in extra-follicular B cells, compared to the reduced levels of APE1 in the GC (J. Stavnezer et al, submitted).
GC cell numbers are reduced in APE2-deficient mice
To test whether APE2 is important for proliferating B cells in the GC, we first examined Peyer’s patches (PP) in the small intestine, where GC cells are constitutively present. By flow cytometric analysis, we found that the percent of B220+GL7+ GC cells in the PP is reduced by 2.5-fold (Fig. 3C). Similar results were obtained when CD95 was included as an additional GC cell marker (Fig. 3A). In addition, the PPs are visibly much smaller; with the total number of PP cells recovered in APE2-deficient mice reduced two-fold (Fig. 3B), resulting in an average 5-fold reduction in the total number of PP-GC B cells (Fig. 3D). APE1-deficiency cannot be studied in vivo since it causes early-embryonic lethality, but mice that are haploinsufficient in APE1 (apex1+/−) show impaired BER (36). GC size and cellularity is not reduced in apex1+/− mice, nor is it further decreased relative to apex2Y/− in mice that are apex2Y/−apex1+/− (Fig. 3E), consistent with the already low expression of APE1 in GC cells, and indicating that APE1 does not compensate for APE2-deficiency in the GC. In addition to PP GCs, the percent of GC B cells in the spleen is also reduced ~two-fold after immunization of apex2Y/− mice with NP-chicken gammaglobulin (CGG) (Fig. 3F). Upon staining of sections from immunized spleens for GCs with the lectin peanut agglutinin (PNA), very few GC of normal size were observed in spleens from apex2Y/− mice, and the percent of B-cell follicles that are PNA+ are also reduced ~two-fold relative to WT spleens (Fig. 3G,H).
Figure 3. Reduced germinal center formation in APE2-deficient Peyer’s patch and spleen.

(A) FACS plot of PP cells gated on B220+ cells (representative of data shown in B–E). (B) ex vivo PP cell numbers from mice of the indicated genotypes. (C) PP GL7+ GC cells as a percent of B220+ cells or (D,E) cell number recovered per mouse, as determined by flow cytometry. (B–D, n=4; E, n=3) (F) The percent of B220+ cells expressing GC markers (CD95+GL7+) in APE2-deficient spleens 10 d after immunization with NP-CGG is reduced (n=3, **p<0.01). (G,H) The percent of B cell follicles in NP-CGG immunized spleens that are PNA+ by immunohistochemistry is reduced (n = 424 follicles from 6 sections for WT and 217 follicles from 5 sections for apex2Y/−, **p<0.0001, Chi Square). NP-CGG immunized spleens (d 10) were fixed in paraformaldehyde, imbedded in paraffin and stained with hematoxylin (blue), and PNA-biotin (Vector Labs)/streptavadin-alkaline phosphatase (brown). Arrow indicates an example PNA+ GC (5x magnification, Leica DM IRE2 Phase Contrast Microscope, representative area shown for each genotype). (I) Mice were immunized with NP-CGG and boosted after 6 months. Anti-NP IgG titers are reduced in apex2Y/− mice in the primary response but indistinguishable from WT 10 d after the boost (secondary response). (J) Affinity maturation of the NP response as measured by the ratio of high affinity Ab that binds low-coupled NP4-BSA to that which binds high-coupled NP14-BSA was normal in apex2Y/− mice. (A,B, n=4 each genotype). n.d., no difference.
To evaluate memory B-cell responses and affinity maturation, we immunized mice with NP-CGG and then boosted the mice after six months. Similar to the anti-sRBC response (Fig. 2C,D), the primary response to the TD-antigen NP-CGG was also significantly reduced (Fig. 3I, left). Surprisingly, the memory or secondary response (Fig. 3I, right) and affinity maturation to NP, measured as the ratio of high affinity NP4-binding antibody to total NP14-binding antibody (Fig. 3J), are both normal. We conclude that while APE2-deficiency has a substantial effect on the size of GCs, it appears that selection and affinity maturation are minimally affected.
GC defect is not due to limiting T cell help or to AID-dependent DNA damage
Since GC formation is thymus-dependent and T-cell development is affected in APE2-deficient mice (26), the defective GC response we observe could be due to a reduction in T-cell help and/or reduced B cell numbers. To address this issue, we transferred ovalbumin-specific spleen T cells from OT-II transgenic mice together with CD45.2+ B cells from WT or APE2-deficient mice previously primed with NP-CGG into irradiated congenic CD45.1+ mice. The recipients were immunized with NP-ovalbumin and the spleens were analyzed for GC cells by flow cytometry. Despite equal numbers of input B cells and the provision of WT T-cell help, the GC response by CD45.2+ APE2-deficient B cells is still reduced (Fig. 4A,B), as compared to that of CD45.2+ WT B cells. The host response in each group of mice (CD45.1+ cells) is shown for comparison (Fig. 4B, white bars). This suggests that the decrease in GC cell numbers represents a B-cell intrinsic defect.
Figure 4. The reduction in germinal center size is not due to lack of T cell help or to AID-dependent damage.

(A,B) GC formation by WT or APE2-deficient B cells (CD45.2+) transferred into WT CD45.1+ congenic mice along with OT-II OVA-specific T helper cells. (A) Representative FACS plot of spleen B220+CD45.2+ cells, d 14. (B) GL7+CD95+ cells as a percent of spleen B220+ cells (n=4, *p=0.011). (C) GC size is increased in aid−/− mice, but is reduced in apex2Y/−aid−/− mice. FACS plots of PP B220+ cells are representative of three independent experiments and n=6 (total) of each genotype.
Since APE2-deficiency affects both CSR and SHM, it is possible that APE2 is needed in GC cells exclusively to repair AID-dependent DNA damage. We therefore generated apex2Y/−aid−/− mice and examined their PP GCs. As previously reported (41, 42), aid−/− mice have enlarged GCs, but in the absence of APE2, the percent GC cells is reduced by nearly three-fold relative to aid−/− mice (Fig. 4C). We conclude that APE2 is needed for GC B cell expansion even in the absence of AID-dependent damage. However, the observation that the numbers of apex2Y/−aid−/− GC B cells are intermediate between that of aid−/− and apex2Y/− GCs suggests that AID-dependent damage might be additive to that which occurs in the absence of APE2.
Analysis of proliferation rates and cell cycle profile in APE2-deficient GC cells
B cells undergo many rounds of proliferation, AID-dependent mutations and selection in GCs in order to achieve affinity maturation of the antibody response. To determine if APE2-deficiency impacts the proliferation of GC cells, we analyzed the turnover and production rates of GC cells by BrdU incorporation. We found no difference in the percent of the GC pool that incorporates BrdU into their DNA, and conclude that APE2-deficient GC cells proliferate at about the same rate as WT GC B cells (Fig. 5A). However, the production rate (the number of BrdU labeled GC cells accumulating over time) was dramatically reduced in apex2Y/− relative to WT mice (Fig. 5B). This could result from fewer B cells entering the GC reaction, or from increased cell death or exit from the GC compartment.
Figure 5. Proliferation and cell cycle profile of APE2-deficient GC cells.

(A,B) Incorporation of BrdU into GC B cells was determined by flow cytometry in WT (black circles) and APE2-deficient (gray squares) mice injected with BrdU every 12h for 3d (n=3 for each time point). (A) The percent of the GL7+ cells that are BrdU+ (turnover rate) and (B) the total number recovered per mouse (production rate) is shown. (C) Representative propidium iodide stain for DNA content of PP cells gated on B220+GL7+ cells shows slight G2/M block in APE2-deficient GC cells (mean ± SEM, n=3, p<0.05 for each cell cycle phase). (D) Representative cell cycle profile of PP B220+GL7+CXCR4HI cells from mice of the indicated genotypes (n=3).
We further analyzed the proliferation of GC B cells by propidium iodide staining for DNA content to determine cell-cycle profile. We found that the percent of APE2-deficient GC cells in the G2/M phase of the cell-cycle is increased 3-fold relative to WT GC cells (Fig 5C). This result suggests that APE2-deficient GC cells are activating cell-cycle checkpoints as a result of DNA damage. In the centroblast compartment, where the rapidly proliferating GC cells can be defined by high expression of CXCR4 (43), the percent of cells blocked in G2/M phase is even greater (Fig 5D). Compared to WT centroblasts (~40% in S/G2M phase), an additional 25% of the apex2Y/− centroblasts (total of ~67%) are in S/G2M phase, apparently having sufficient damage to activate this cell-cycle checkpoint. The G2/M cell-cycle block was also still apparent in the centroblasts from apex2Y/−aid−/− mice (Fig. 5D), showing that the cell-cycle block is not due solely to damage from AID-induced lesions. This indicates that damage from proliferation and/or oxidative stress in GC cells, where APE1 expression is very low, requires APE2 for its repair.
In vitro biochemical experiments have shown that APE2 can remove adenine inserted opposite the oxidatively damaged base 8-oxoG (24). 8-oxoG is normally removed by OGG1, a glycosylase that is highly expressed in human GC cells, but if 8-oxoG is encountered by polymerase it will be paired with dA. To determine if APE2 helps repair A:8-oxoG lesions in vivo, we created APE2- and OGG1-doubly deficient mice and asked if their GC defect became more severe. We found that the accumulation of cells in the S and G2/M phases of the cell cycle is slightly, but not significantly, increased in these mice relative to mice deficient in APE2 alone (Fig. S1). As such, repair of this particular lesion probably does not contribute significantly to the phenotype we observe, and while APE2 might serve as a backup mechanism for 8-oxoG repair in GC B cells, it probably repairs other types of oxidative damage as well.
Reduced BCL6 associated with DNA damage in GC cells
To detect DNA damage directly in GC cells by flow cytometry, we surface-stained PP cells for GC markers and then performed intracellular staining for γH2AX, an indicator of DSBs. GC cells positive for γH2AX were considerably increased in APE2-deficient PP (60% positive) compared to 32% of WT GC cells (Fig. 6A) and 28% of AID-deficient GC cells. The percent of GC cells with damage in APE2-deficient PP cells that lacked AID (apex2Y/−aid−/−, 52%) was also much higher than in WT (p<0.0001), but also significantly less than that in apex2Y/− cells (p<0.05). This result demonstrates that proliferative/oxidative damage and AID-dependent damage are additive stresses for GC cells.
Figure 6. DNA damage is associated with decreased BCL6 and GL7 expression.

(A) γ-H2AX staining for DSBs by flow cytometry in PP GC cells (black line) and WT non-GC control B cells (gray line). (B) Data from (A) plotted vs. GL7+ cells as percent of B220+ PP cells. (C) BCL6 and γ-H2AX intracellular staining in B220+CD95+GL7+ PP cells. Gates are set so that non-GC cells are ≤ 1% positive for H2AX or BCL6. Plots are representative of 3 or more independent experiments and gate frequencies shown are representative (C) or the mean ± SEM (A); n = 4 WT, 4 apex2Y/−, 5 aid−/− and 3 apex2Y/−aid−/− mice; ****p<0.0001, one-way ANOVA with Tukey’s multiple comparison test.
Expression of BCL6 is known to suppress the DNA damage response in GC cells, allowing them to proliferate and presumably tolerate AID-dependent damage. However, we find that increased amounts of DNA damage are associated with a decreased percentage of GC cells in aid−/−, WT, apex2Y/−aid−/− and apex2Y/− PP (Fig. 6B), suggesting that not all DNA damage is tolerated, and a response that limits GC size can be activated in the germinal center. Indeed, a mechanism wherein BCL6 is degraded upon DNA damage has been shown to occur in vitro (15), although it has not previously been demonstrated in ex vivo GC cells with endogenous DNA damage. To explore this further, we co-stained for intracellular BCL6 and DSB damage (γH2AX) and found that, while the DSBs appeared predominantly in CD95+GL7+ GC cells expressing high levels of BCL6, the population with the highest amount of damage (oval gate, Fig. 6C) includes cells with low or intermediate levels of BCL6. As previously proposed, down-regulation of BCL6 could restore the DNA damage response in these cells.
GC B-cell loss is p53-dependent
As mechanisms that suppress p53 in the GC have been reported (6, 44), we asked if the loss of GC B cells in APE2-deficient mice occurs by p53-dependent apoptosis. In apex2Y/−tp53−/− mice, we found that the percent of PP B cells that are GL7+ is restored to near WT levels (Fig. 7A), and the total number of GC cells recovered per mouse was partially restored (Fig. 7B). In addition, we find that expression of mRNA from the p53-dependent pro-apoptotic gene bax is increased almost 3-fold in FACS-purified apex2Y/− GC cells relative to WT GC cells (Fig. 7C), indicating that p53 is active. To determine whether activation of the cell-cycle checkpoint in APE2-deficient GC B cells is p53-dependent, we analyzed the cell-cycle profile of PP GC cells from apex2Y/−tp53−/− mice, and find that the G2/M block is still present in these cells (Fig. 7D), likely causing the slight decrease in GC cells in apex2Y/−tp53−/− mice (Fig. 7A,B). These combined results demonstrate that DNA damage, unrepaired in the absence of APE2, activates a p53-independent G2/M phase cell-cycle checkpoint. Furthermore, we propose that DNA damage in GCs is associated with decreased BCL6 expression, and if unrepaired, results in p53-dependent bax-mediated apoptosis.
Figure 7. The reduction in germinal center size is p53-dependent.

Germinal center formation is partially restored in APE2-deficient mice that are also deficient in p53. (A) FACS plot showing the percent of PP GC cells in mice of the indicated genotypes (representative of 3 independent experiments). (B) Partial recovery in the number of GC cells in apex2Y/− x tp53−/− mice (WT and APE2, n=3; p53 and APE2xp53, n=4; *p < 0.05 vs. WT; n.s., not significant). (C) qRT-PCR shows increased expression of the p53-dependent pro-apoptotic gene bax relative to HPRT expression in APE2-deficient FACS-purified PP GC B cells (mean ± SEM, n=3 independent sorts). (D) APE2-deficient B220+GL7+CXCR4HI centroblasts show a cell cycle block that is not dependent on p53 (representative FACS profile with mean ± SEM, n=3, except p53 n=2 and range is shown).
Discussion
Our results show that APE2 actively protects rapidly proliferating GC B cells from endogenous DNA damage, such as that caused by proliferation-associated oxidative stress. In contrast to the modest DNA repair activity observed for APE2 in in vitro biochemical assays, our analysis of GC B cells in vivo shows that APE2 is critical for their survival. In mice, the consequences of removing this protective enzyme include small GC size and poor primary T-dependent antibody responses in addition to the previously reported defects in B-cell development and in the thymus. Together with our recent finding that APE1 expression is dramatically reduced in GC B cells, where APE2 expression is strongly induced (J. Stavnezer et al, submitted), our data suggest that APE2 is the predominant AP endonuclease in GC B cells. APE2 is also protective in LPS-activated cultured B cells (where levels of APE1 are not limiting) when these cells are stressed by BSO (Fig. 1). Activated B cells undergo efficient CSR in culture, which requires APE1 (29, 30, 33), but these cells do not hyper-mutate their Ig variable region genes as do GC B cells. SHM may require down-regulation of APE1, since APE1 is very efficient at accurate repair. APE2 contributes to error-prone repair in the GC (30)(J. Stavnezer et al, submitted), and can interact with PCNA (21, 24), which is known to recruit translesion polymerases (45). It will be interesting to see how these findings will impact current models of CSR, a process long-associated with the GC. CSR may occur predominantly in extra-follicular regions and prior to GC formation, before APE1 expression is suppressed. Alternatively, or in addition, the small amount of APE1 we detect in GC cells might be recruited to interact with AID via AIDS38 phosphorylation (34). We think it is likely that APE2 works with the low levels of APE1 in the GC to effect CSR, since it contributes to CSR in activated spleen B cells (29). However, APE2 does not contribute to CSR in the CH12F3 cell line (30, 33). Our findings underscore the need for more in vivo studies.
In GC B cells, BCL6 is pivotal to the expression of genes that promote proliferation and suppress differentiation, and it also specifically suppresses the DNA damage response (46), necessary to accommodate programmed DNA damage by AID. To better understand the suppression of the DNA damage response, we used APE2-deficiency as a model to examine the effect of endogenous DNA damage in GCs. Our data show the first detailed analysis of the damage response in this unique cell type. We find that DNA damage in GC B cells can activate at least two pathways: a p53-independent G2/M phase cell-cycle checkpoint, and a p53-dependent cell death pathway. In addition, we see that DSB damage is associated with decreased expression of BCL6. A post-translational mechanism was previously reported to reduce the stability of BCL6 in Raji Burkitt lymphoma cells: DNA DSBs activate ATM, which phosphorylates Pin1, resulting in the ubiquitination and proteasomal degradation of BCL6 (15). A reduction in BCL6, whose expression is essential for GC centroblasts (10, 11), could end the proliferative phase and restore the DNA damage response, allowing cells with damage to repair their DNA or undergo apoptosis. Consistent with this important role as regulator of cellular expansion, differentiation, and the DNA damage response, BCL6 is a well-known tumor suppressor (reviewed in (47)).
We demonstrate that endogenous, proliferative and/or oxidative stress causes damage in GC cells that is additive to the programmed DNA damage caused by AID. That the amount of DNA damage in WT, aid−/− and apex2Y/− mice correlates inversely with the size of the GC suggests that these pathways comprise a DDR that is capable of limiting GC expansion. We found that endogenous DNA damage in the GC activates a G2/M phase cell-cycle checkpoint, and it is tempting to speculate that the G2/M phase block could represent a biologically significant link to differentiation. It was recently shown that signaling through the B-cell receptor is suppressed in GC cells by protein phosphatases, but signaling activity is regained in the G2 phase of the cell-cycle (48). Differentiation of GC B cells requires a positive selection signal based on the affinity of the B-cell receptor, and cell-cycle arrest, specifically via activation of ATM by DNA breaks, has been associated with differentiation in progenitor B cells and in human GC cells (13, 14). Incidentally, a recently identified pathway linking APE2 with ATR-Chk1 checkpoint signaling (34) is not likely to be active in GCs, since ATR and Chk1 are both directly repressed by BCL6.
The activity of p53 in germinal centers is not well understood. BCL6 can suppress the p53 promoter in vitro (6). In addition, IRF-8, which is expressed in GC cells, induces expression of MDM2, which inhibits p53 (Fig. 8)(44). We find that PP GCs from p53-deficient mice resemble those from WT mice in cell number, percentage of GC cells, cell-cycle profile (Fig. 7) and amount of DNA damage (C.E.S., unpublished observation). However, we find that the decrease in GC cell numbers in apex2Y/− mice is dependent on p53 and find increased expression of the p53-dependent pro-apoptotic factor BAX, which may be involved in a loss of GC B cells due to induction of apoptosis. We conclude that, while p53 activity may be repressed to some extent in GC cells, it does have a role in responding to DNA damage in GC cells that is likely dependent on the extent of the damage. We present a model of the DNA damage response in GCs, based on the literature (Fig. 8). Since the loss of APE2-deficient GC cells is mostly p53-dependent, defects in p53 would allow GC cells with DNA damage to survive and promote lymphomagenesis. It seems likely that ATM activates p53 in GC cells, but we are so far unable to confirm a role for ATM in these pathways since, like p53, GC size and cell-cycle profiles are not altered in ATM-deficient mice (C.E.S., unpublished observation). Perhaps this is due to redundancy with ATR or DNA-PKCS (49, 50), although ATR is directly repressed by BCL6 (7).
Figure 8. Model of the DNA damage response in GC cells.

Oxidative damage in GC cells activates p53 despite mechanisms reported to suppress it. p53 activation could occur directly through ATM, or may involve ATM-mediated down-regulation of the repressor BCL6. DNA damage has been reported to reduce BCL6 levels through protein degradation, which could relieve repression of the DNA damage response, and in addition cause cells to exit the proliferative phase allowing them to differentiate into memory B cells and plasma cells. According to this model, the amount of DNA damage that germinal center B cells sustain plays a part in determining the fate of these cells. Extensive DNA damage (such as in the absence of repair proteins such as APE2) would negatively impact on the germinal center output, whereas too little DNA damage (such as in the absence of AID) would have a similar outcome, since fewer B cells would exit the proliferative phase. CB, centroblast.
In summary, we show that endogenous proliferative and/or oxidative damage in rapidly proliferating B cells in vivo can affect GC size, and that this damage is suppressed by APE2. Although BCL6 expression allows GC cells to tolerate DNA damage in order to accommodate AID-induced mutations and DSBs, our data show that DNA damage is still detected and can regulate GC size through cell-cycle checkpoints, apoptosis and possibly differentiation by pathways that involve p53 and BCL6.
Supplementary Material
Acknowledgments
The authors wish to thank Anna Ucher, Monique Oud, the U. Mass. Flow Cytometry Core Facility and the U. Mass. DERC Pathology Core for excellent technical assistance, Dr. Madelyn Schmidt for helpful technical advice and Dr. Janet Stavnezer for helpful suggestions on the manuscript.
Footnotes
This work was supported by National Institute of Health Grants AI065639 and AI092528 (to C.E.S.), AI84800 (to R.T.W) and an Academic Medical Center Fellowship and the Innovational Research Incentives Scheme Vidi grant 016126355 from the Netherlands Organization for Scientific Research (Zon-MW) (to J.E.J.G.).
Abbreviations used: AID, Activation-induced cytidine deaminase; APE, apurinic/apyrimidinic endonuclease; BER, base excision repair; BSO, buthionine-sulfoximine; CGG, chicken gammaglobulin; CSR, class switch recombination; DSB, double-strand break; GC, germinal center; MZ, marginal zone; NP, nitrophenol; PNA, peanut-agglutinin; PP, Peyer’s patch; ROS, reactive oxygen species; TD, thymus-dependent and TI, thymus-independent.
References
- 1.Schatz DG, Oettinger MA, Baltimore D. The V(D)J recombination activating gene, RAG-1. Cell. 1989;59:1035–1048. doi: 10.1016/0092-8674(89)90760-5. [DOI] [PubMed] [Google Scholar]
- 2.Stavnezer J, Guikema JE, Schrader CE. Mechanism and regulation of class switch recombination. Annu Rev Immunol. 2008;26:261–292. doi: 10.1146/annurev.immunol.26.021607.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 4.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 5.Barnes DE, Lindahl T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet. 2004;38:445–476. doi: 10.1146/annurev.genet.38.072902.092448. [DOI] [PubMed] [Google Scholar]
- 6.Phan RT, Dalla-Favera R. The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature. 2004;432:635–639. doi: 10.1038/nature03147. [DOI] [PubMed] [Google Scholar]
- 7.Ranuncolo SM, Polo JM, Dierov J, Singer M, Kuo T, Greally J, Green R, Carroll M, Melnick A. Bcl-6 mediates the germinal center B cell phenotype and lymphomagenesis through transcriptional repression of the DNA-damage sensor ATR. Nature immunology. 2007;8:705–714. doi: 10.1038/ni1478. [DOI] [PubMed] [Google Scholar]
- 8.Ranuncolo SM, Polo JM, Melnick A. BCL6 represses CHEK1 and suppresses DNA damage pathways in normal and malignant B-cells. Blood Cells Mol Dis. 2008;41:95–99. doi: 10.1016/j.bcmd.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Phan RT, Saito M, Basso K, Niu H, Dalla-Favera R. BCL6 interacts with the transcription factor Miz-1 to suppress the cyclin-dependent kinase inhibitor p21 and cell cycle arrest in germinal center B cells. Nature immunology. 2005;6:1054–1060. doi: 10.1038/ni1245. [DOI] [PubMed] [Google Scholar]
- 10.Ye BH, Cattoretti G, Shen Q, Zhang J, Hawe N, de Waard R, Leung C, Nouri-Shirazi M, Orazi A, Chaganti RS, Rothman P, Stall AM, Pandolfi PP, Dalla-Favera R. The BCL-6 proto-oncogene controls germinal-centre formation and Th2-type inflammation. Nat Genet. 1997;16:161–170. doi: 10.1038/ng0697-161. [DOI] [PubMed] [Google Scholar]
- 11.Dent AL, Shaffer AL, Yu X, Allman D, Staudt LM. Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science. 1997;276:589–592. doi: 10.1126/science.276.5312.589. [DOI] [PubMed] [Google Scholar]
- 12.Reljic R, Wagner SD, Peakman LJ, Fearon DT. Suppression of signal transducer and activator of transcription 3-dependent B lymphocyte terminal differentiation by BCL-6. J Exp Med. 2000;192:1841–1848. doi: 10.1084/jem.192.12.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bredemeyer AL, Helmink BA, Innes CL, Calderon B, McGinnis LM, Mahowald GK, Gapud EJ, Walker LM, Collins JB, Weaver BK, Mandik-Nayak L, Schreiber RD, Allen PM, May MJ, Paules RS, Bassing CH, Sleckman BP. DNA double-strand breaks activate a multi-functional genetic program in developing lymphocytes. Nature. 2008;456:819–823. doi: 10.1038/nature07392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sherman MH, Kuraishy AI, Deshpande C, Hong JS, Cacalano NA, Gatti RA, Manis JP, Damore MA, Pellegrini M, Teitell MA. AID-induced genotoxic stress promotes B cell differentiation in the germinal center via ATM and LKB1 signaling. Mol Cell. 2010;39:873–885. doi: 10.1016/j.molcel.2010.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Phan RT, Saito M, Kitagawa Y, Means AR, Dalla-Favera R. Genotoxic stress regulates expression of the proto-oncogene Bcl6 in germinal center B cells. Nature immunology. 2007;8:1132–1139. doi: 10.1038/ni1508. [DOI] [PubMed] [Google Scholar]
- 16.David SS, V, O’Shea L, Kundu S. Base-excision repair of oxidative DNA damage. Nature. 2007;447:941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuo FC, Sklar J. Augmented expression of a human gene for 8-oxoguanine DNA glycosylase (MutM) in B lymphocytes of the dark zone in lymph node germinal centers. J Exp Med. 1997;186:1547–1556. doi: 10.1084/jem.186.9.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klein U, Tu Y, Stolovitzky GA, Keller JL, Haddad J, Jr, Miljkovic V, Cattoretti G, Califano A, Dalla-Favera R. Transcriptional analysis of the B cell germinal center reaction. Proc Natl Acad Sci U S A. 2003;100:2639–2644. doi: 10.1073/pnas.0437996100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mori H, Ouchida R, Hijikata A, Kitamura H, Ohara O, Li Y, Gao X, Yasui A, Lloyd RS, Wang JY. Deficiency of the oxidative damage-specific DNA glycosylase NEIL1 leads to reduced germinal center B cell expansion. DNA Repair (Amst) 2009;8:1328–1332. doi: 10.1016/j.dnarep.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hadi MZ, Wilson DM., III Second human protein with homology to the Escherichia coli abasic endonuclease exonuclease III. Environ Mol Mutagen. 2000;36:312–324. [PubMed] [Google Scholar]
- 21.Tsuchimoto D, Sakai Y, Sakumi K, Nishioka K, Sasaki M, Fujiwara T, Nakabeppu Y. Human APE2 protein is mostly localized in the nuclei and to some extent in the mitochondria, while nuclear APE2 is partly associated with proliferating cell nuclear antigen. Nucleic Acids Res. 2001;29:2349–2360. doi: 10.1093/nar/29.11.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burkovics P, Szukacsov V, Unk I, Haracska L. Human Ape2 protein has a 3′-5′ exonuclease activity that acts preferentially on mismatched base pairs. Nucleic Acids Res. 2006;34:2508–2515. doi: 10.1093/nar/gkl259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hadi MZ, Ginalski K, Nguyen LH, Wilson DM., III Determinants in nuclease specificity of Ape1 and Ape2, human homologues of Escherichia coli exonuclease III. J Mol Biol. 2002;316:853–866. doi: 10.1006/jmbi.2001.5382. [DOI] [PubMed] [Google Scholar]
- 24.Burkovics P, Hajdu I, Szukacsov V, Unk I, Haracska L. Role of PCNA-dependent stimulation of 3′-phosphodiesterase and 3′-5′ exonuclease activities of human Ape2 in repair of oxidative DNA damage. Nucleic Acids Res. 2009;37:4247–4255. doi: 10.1093/nar/gkp357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Henle ES, Linn S. Formation, prevention, and repair of DNA damage by iron/hydrogen peroxide. J Biol Chem. 1997;272:19095–19098. doi: 10.1074/jbc.272.31.19095. [DOI] [PubMed] [Google Scholar]
- 26.Ide Y, Tsuchimoto D, Tominaga Y, Nakashima M, Watanabe T, Sakumi K, Ohno M, Nakabeppu Y. Growth retardation and dyslymphopoiesis accompanied by G2/M arrest in APEX2-null mice. Blood. 2004;104:4097–4103. doi: 10.1182/blood-2004-04-1476. [DOI] [PubMed] [Google Scholar]
- 27.Guikema JE, Gerstein RM, Linehan EK, Cloherty EK, Evan-Browning E, Tsuchimoto D, Nakabeppu Y, Schrader CE. Apurinic/apyrimidinic endonuclease 2 is necessary for normal B cell development and recovery of lymphoid progenitors after chemotherapeutic challenge. J Immunol. 2011;186:1943–1950. doi: 10.4049/jimmunol.1002422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dan Y, Ohta Y, Tsuchimoto D, Ohno M, Ide Y, Sami M, Kanda T, Sakumi K, Nakabeppu Y. Altered gene expression profiles and higher frequency of spontaneous DNA strand breaks in APEX2-null thymus. DNA Repair (Amst) 2008;7:1437–1454. doi: 10.1016/j.dnarep.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 29.Guikema JE, Linehan EK, Tsuchimoto D, Nakabeppu Y, Strauss PR, Stavnezer J, Schrader CE. APE1- and APE2-dependent DNA breaks in immunoglobulin class switch recombination. J Exp Med. 2007;204:3017–3026. doi: 10.1084/jem.20071289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sabouri Z, I, Okazaki M, Shinkura R, Begum N, Nagaoka H, Tsuchimoto D, Nakabeppu Y, Honjo T. Apex2 is required for efficient somatic hypermutation but not for class switch recombination of immunoglobulin genes. Int Immunol. 2009;21:947–955. doi: 10.1093/intimm/dxp061. [DOI] [PubMed] [Google Scholar]
- 31.Stavnezer J, Linehan EK, Thompson MR, Habboub G, Tsuchimoto D, Nakabeppu Y, Schrader CE. Somatic hypermutation of IgH variable region genes is reduced and the pattern is altered in APE1- and APE2-deficient mice 2012 [Google Scholar]
- 32.Schrader CE, Linehan EK, Mochegova SN, Woodland RT, Stavnezer J. Inducible DNA breaks in Ig S regions are dependent on AID and UNG. J Exp Med. 2005;202:561–568. doi: 10.1084/jem.20050872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Masani S, Han L, Yu K. Apurinic/apyrimidinic endonuclease 1 is the essential nuclease during immunoglobulin class switch recombination. Mol Cell Biol. 2013;33:1468–1473. doi: 10.1128/MCB.00026-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vuong BQ, Herrick-Reynolds K, Vaidyanathan B, Pucella JN, Ucher AJ, Donghia NM, Gu X, Nicolas L, Nowak U, Rahman N, Strout MP, Mills KD, Stavnezer J, Chaudhuri J. A DNA break- and phosphorylation-dependent positive feedback loop promotes immunoglobulin class-switch recombination. Nature immunology. 2013;14:1183–1189. doi: 10.1038/ni.2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ide Y, Tsuchimoto D, Tominaga Y, Iwamoto Y, Nakabeppu Y. Characterization of the genomic structure and expression of the mouse Apex2 gene. Genomics. 2003;81:47–57. doi: 10.1016/s0888-7543(02)00009-5. [DOI] [PubMed] [Google Scholar]
- 36.Meira LB, Devaraj S, Kisby GE, Burns DK, Daniel RL, Hammer RE, Grundy S, Jialal I, Friedberg EC. Heterozygosity for the mouse Apex gene results in phenotypes associated with oxidative stress. Cancer Res. 2001;61:5552–5557. [PubMed] [Google Scholar]
- 37.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 38.Cancro MP, Sah AP, Levy SL, Allman DM, Schmidt MR, Woodland RT. xid mice reveal the interplay of homeostasis and Bruton’s tyrosine kinase-mediated selection at multiple stages of B cell development. Int Immunol. 2001;13:1501–1514. doi: 10.1093/intimm/13.12.1501. [DOI] [PubMed] [Google Scholar]
- 39.Duy C, Yu JJ, Nahar R, Swaminathan S, Kweon SM, Polo JM, Valls E, Klemm L, Shojaee S, Cerchietti L, Schuh W, Jack HM, Hurtz C, Ramezani-Rad P, Herzog S, Jumaa H, Koeffler HP, de Alboran IM, Melnick AM, Ye BH, Muschen M. BCL6 is critical for the development of a diverse primary B cell repertoire. J Exp Med. 2010;207:1209–1221. doi: 10.1084/jem.20091299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kelly BS, Levy JG, Sikora L. The use of the enzyme-linked immunosorbent assay (ELISA) for the detection and quantification of specific antibody from cell cultures. Immunology. 1979;37:45–52. [PMC free article] [PubMed] [Google Scholar]
- 41.Fagarasan S, Muramatsu M, Suzuki K, Nagaoka H, Hiai H, Honjo T. Critical roles of activation-induced cytidine deaminase in the homeostasis of gut flora. Science. 2002;298:1424–1427. doi: 10.1126/science.1077336. [DOI] [PubMed] [Google Scholar]
- 42.Zaheen A, Boulianne B, Parsa JY, Ramachandran S, Gommerman JL, Martin A. AID constrains germinal center size by rendering B cells susceptible to apoptosis. Blood. 2009;114:547–554. doi: 10.1182/blood-2009-03-211763. [DOI] [PubMed] [Google Scholar]
- 43.Allen CD, Ansel KM, Low C, Lesley R, Tamamura H, Fujii N, Cyster JG. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nature immunology. 2004;5:943–952. doi: 10.1038/ni1100. [DOI] [PubMed] [Google Scholar]
- 44.Zhou JX, Lee CH, Qi CF, Wang H, Naghashfar Z, Abbasi S, Morse HC., 3rd IFN regulatory factor 8 regulates MDM2 in germinal center B cells. J Immunol. 2009;183:3188–3194. doi: 10.4049/jimmunol.0803693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garg P, Burgers PM. Ubiquitinated proliferating cell nuclear antigen activates translesion DNA polymerases eta and REV1. Proc Natl Acad Sci U S A. 2005;102:18361–18366. doi: 10.1073/pnas.0505949102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shaffer AL, Yu X, He Y, Boldrick J, Chan EP, Staudt LM. BCL-6 represses genes that function in lymphocyte differentiation, inflammation, and cell cycle control. Immunity. 2000;13:199–212. doi: 10.1016/s1074-7613(00)00020-0. [DOI] [PubMed] [Google Scholar]
- 47.Basso K, Dalla-Favera R. Roles of BCL6 in normal and transformed germinal center B cells. Immunological Reviews. 2012;247:172–183. doi: 10.1111/j.1600-065X.2012.01112.x. [DOI] [PubMed] [Google Scholar]
- 48.Khalil AM, Cambier JC, Shlomchik MJ. B cell receptor signal transduction in the GC is short-circuited by high phosphatase activity. Science. 2012;336:1178–1181. doi: 10.1126/science.1213368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gapud EJ, Dorsett Y, Yin B, Callen E, Bredemeyer A, Mahowald GK, Omi KQ, Walker LM, Bednarski JJ, McKinnon PJ, Bassing CH, Nussenzweig A, Sleckman BP. Ataxia telangiectasia mutated (Atm) and DNA-PKcs kinases have overlapping activities during chromosomal signal joint formation. Proc Natl Acad Sci U S A. 2011;108:2022–2027. doi: 10.1073/pnas.1013295108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stiff T, O’Driscoll M, Rief N, Iwabuchi K, Lobrich M, Jeggo PA. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004;64:2390–2396. doi: 10.1158/0008-5472.can-03-3207. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.