

This article describes recently evolved testis-specific miRNAs in Drosophila. Evidence is provided that these miRNAs are expressed at much higher levels than non-testis-specific miRNAs of similar age. They also appear to have greater regulatory capacity than do other young miRNAs.
Keywords: positive selection, tissue specific, testis specific, microRNA clusters, Drosophila
Abstract
The propensity of animal miRNAs to regulate targets bearing modest complementarity, most notably via pairing with miRNA positions ∼2–8 (the “seed”), is believed to drive major aspects of miRNA evolution. First, minimal targeting requirements have allowed most conserved miRNAs to acquire large target cohorts, thus imposing strong selection on miRNAs to maintain their seed sequences. Second, the modest pairing needed for repression suggests that evolutionarily nascent miRNAs may generally induce net detrimental, rather than beneficial, regulatory effects. Hence, levels and activities of newly emerged miRNAs are expected to be limited to preserve the status quo of gene expression. In this study, we unexpectedly show that Drosophila testes specifically express a substantial miRNA population that contravenes these tenets. We find that multiple genomic clusters of testis-restricted miRNAs harbor recently evolved miRNAs, whose experimentally verified orthologs exhibit divergent sequences, even within seed regions. Moreover, this class of miRNAs exhibits higher expression and greater phenotypic capacities in transgenic misexpression assays than do non-testis-restricted miRNAs of similar evolutionary age. These observations suggest that these testis-restricted miRNAs may be evolving adaptively, and several methods of evolutionary analysis provide strong support for this notion. Consistent with this, proof-of-principle tests show that orthologous miRNAs with divergent seeds can distinguish target sensors in a species-cognate manner. Finally, we observe that testis-restricted miRNA clusters exhibit extraordinary dynamics of miRNA gene flux in other Drosophila species. Altogether, our findings reveal a surprising tissue-directed influence of miRNA evolution, involving a distinct mode of miRNA function connected to adaptive gene regulation in the testis.
INTRODUCTION
MicroRNAs (miRNAs) are an abundant class of endogenous ∼22-nucleotide (nt) RNAs that derive from hairpin precursor transcripts. In animals, the majority of miRNAs are generated by a canonical pathway involving sequential cleavage of primary transcript containing one or more hairpin foldbacks, first by nuclear Drosha (to release pre-miRNA hairpins) and then by cytoplasmic Dicer (which cuts the hairpins into duplexes). Although several noncanonical miRNA pathways that utilize other ribonucleases have been characterized, the strong majority of miRNA species in animal cells are generated by the Drosha–Dicer pathway (Yang and Lai 2011). As both of these are RNase III enzymes, the resultant small RNA duplexes exhibit characteristic ∼2-nt overhangs at both 3′ ends. These duplexes are loaded into an Argonaute protein and matured to a single-stranded ribonucleoprotein complex that is guided to complementary targets (Meister 2013). The sequence and structural features of a small RNA duplex usually dictate asymmetry in the maturation of the effector complex (Czech and Hannon 2010). The strand that accumulates to a higher level is operationally termed the mature miRNA; its complementary strand, the “miRNA*” (or star) strand (Ambros et al. 2003).
Animal miRNAs are capable of mediating substantial regulation via 6–7 nt complements to their 5′ ends, preferably positions 2–8 of the mature miRNA (Lai 2002; Doench and Sharp 2004; Brennecke et al. 2005; Lewis et al. 2005). Such miRNA “seed” complements are experimentally sufficient to mediate substantial regulation, and whole-genome comparisons show that ∼7-nt seed complementary sites are frequently and specifically subject to purifying selection (Bartel 2009). Bioinformatic studies now strongly support that a substantial fraction of animal transcripts is subject to evolutionarily constrained miRNA seed targeting (Krek et al. 2005; Lewis et al. 2005; Ruby et al. 2007; Friedman et al. 2009), with additional functional target sites that exhibit noncanonical pairing to miRNAs (Ha et al. 1996; Vella et al. 2004; Shin et al. 2010; Chi et al. 2012; Loeb et al. 2012). Although most functional studies focus on “mature” miRNAs, star strands do not behave simply as bystander passenger strands. While some star strands accumulate nucleotide divergence, consistent with a structural role only to maintain a duplex, most well-conserved Drosophilid and vertebrate miRNA loci exhibit strong constraint of both hairpin arms. This is attributable to the frequent incorporation of both duplex strands into regulatory networks that are subject to evolutionary constraint (Okamura et al. 2008; Yang et al. 2011). A consequence of this is that alignments of miRNA orthologs almost always exhibit a “saddle shape” conservation profile, in which the terminal loop evolves much more quickly than either hairpin arm (Lai et al. 2003; Berezikov et al. 2005).
The deep conservation of target sites of well-conserved miRNAs reflects regulatory interactions that are of sufficient benefit to be preserved during evolution. This is not to say that miRNA target sites necessarily need be well conserved to have a functional impact. Transcriptomic and proteomic studies support the notion that there is a detectable impact, even if generally modest, of a broad pool of recently evolved target sites (Lim et al. 2005; Giraldez et al. 2006; Baek et al. 2008; Selbach et al. 2008; Guo et al. 2010). Conversely, among miRNAs that are conserved across distant animal clades, it has been empirically observed that most targets that are under selection within a clade (e.g., among Drosophilids, nematodes, or vertebrates) have turned over across larger evolutionary distances (Chen and Rajewsky 2006). These findings indicate fluidity in the aggregate pool of animal miRNA target sites, and the regulation of some recently evolved targets of well-conserved miRNAs can have phenotypic impact (Clop et al. 2006; Mor and Shomron 2013).
Another source of regulatory novelty derives from recently evolved miRNAs, which by definition must mediate species-specific events. The impact of evolutionarily nascent miRNAs is difficult to judge, since computational inference of purifying selection on predicted sites requires that they be reasonably well conserved. Only a minority of total miRNA annotations (http://www.mirbase.org/) are sufficiently conserved to permit reasonably specific target predictions. Given the permissive pairing requirements of animal miRNAs, it is easy to speculate about the potential targets of the large number of mostly unstudied, poorly conserved miRNAs. Tempering this scenario, though, is the fact that well-conserved miRNAs are generally expressed at much higher levels than recently evolved miRNAs (Landgraf et al. 2007; Ruby et al. 2007; Chiang et al. 2010). Since miRNA function is intimately tied to cellular concentration (Wee et al. 2012), it is relevant to ask whether newly evolved miRNAs ever achieve sufficient levels to mediate meaningful regulation.
Indeed, it has been posited that miRNAs necessarily have low expression levels at “birth” since (1) they may not have acquired all the features that could endow efficient biogenesis, and (2) most of their incidental targeting interactions are expected to be detrimental (Bartel and Chen 2004). In this scenario, most evolutionarily nascent miRNAs are predicted to be removed by negative selection but may occasionally be selected on the basis of rare beneficial target interactions. Over time, an evolutionarily successful miRNA may acquire additional beneficial targets, while the genome is concomitantly purged of its detrimental targets (Chen and Rajewsky 2007). One can imagine that these events fuel a feed-forward cycle that permits the miRNA to be selected for higher expression and thus capacity for more targets, as well as increasingly stereotyped processing to yield a specific mature small RNA. Such a cycle could explain why “young” miRNAs usually have low expression and/or imprecise processing, while “old” miRNAs usually have high expression and/or precise processing. In addition, it helps explain the strong selection pressure to maintain precise mature miRNA sequences that are increasing locked into a growing target network that is reliant upon a specific seed.
In this study, we examine tissue-specific expression in Drosophila melanogaster and unexpectedly find that the testis harbors a disproportionate number of recently evolved miRNAs that are arranged in genomic clusters. These miRNAs have significantly higher expression and greater capacity to induce mutant phenotypes when misexpressed in transgenic animals, relative to other miRNAs of comparable evolutionary age. We show that remarkably many of these testis-restricted, recently evolved loci have characteristic evidence for typical miRNA/star duplex processing in species closely related to D. melanogaster, yet they defy typical miRNA divergence patterns and instead frequently exhibit seed divergence. Comparison of single-nucleotide polymorphism data and species data provide clear evidence for positive selection on testis-restricted, recently evolved miRNAs. These findings are complemented by our finding that testis-restricted miRNA clusters are extraordinary breeding grounds for miRNA emergence in other Drosophilid species. Altogether, our data define surprising trajectories for miRNA evolution in testis-expressed clusters and define a new principle for the adaptive evolution of miRNA function in this tissue.
RESULTS
Tissue-specific expression of Drosophila miRNAs
We sought insights into the function of Drosophila miRNAs by querying their tissue specificity. Although we and others have generated hundreds of Drosophila small RNA libraries, the small size of this animal has precluded broad assessment of tissue-specific libraries, to the extent that has been done with mammals. Only a few fly tissues have been subjected to small RNA sequencing, particularly the adult head, ovary, and testis (Berezikov et al. 2011). To identify spatially restricted miRNAs, we compared small RNA libraries from these tissues with other whole-animal data (Supplemental Table 1), including various stages of embryonic development. We ranked miRNAs by the log fold-change in expression between a given tissue and its aggregate expression in all other libraries using the R Bioconductor package limma (see Materials and Methods). All miRNAs that were present in a given tissue at a cutoff of 10 RPM were classified as being “expressed.” However, we called tissue-restricted miRNAs using the conservative criterion that they be significantly higher-expressed in the tissue of choice versus all other libraries. For example, expression of dme-mir-982 was exclusively testis restricted, whereas dme-mir-986 was classified as testis expressed due to its substantial accumulation in multiple non-testis libraries (Fig. 1A,B; Supplemental Fig. 2, Supplemental Table 3). We were similarly able to distinguish head-restricted from head-expressed miRNAs, the latter exhibiting expression in the head and at least some other tissue.
FIGURE 1.

Distinctive properties of Drosophila testis-restricted miRNAs. (A) mir-982 and mir-210 are examples of testis- and head-restricted miRNAs, respectively, because they portray significantly higher, aggregate expression within the head or testes libraries. Contrarily, tissue-expressed miRNAs such as mir-986 and mir-124 showed non-negligible (>10 RPM) and nonexclusive expression within testes or head libraries. (B) We identified tissue-restricted miRNAs (i.e., in testes and heads) by comparing the log2(fold-change) in expression between the tissue of interest vs. other condition-specific libraries using statistical differential expression analysis. Tissue-restricted miRNAs are flagged if they show significantly higher expression in the head or testes libraries than in libraries of other conditions considered (left of arrow). Clustered miRNAs of tissues-restricted delegation are also labeled as tissue-restricted (right of arrow). (C) The majority of testis-restricted miRNAs are genomically clustered, unlike testes-expressed and head-restricted miRNAs. (D) Similarly, the majority of testes-restricted miRNAs are recently evolved.
We identified substantial numbers of miRNAs that exhibited strong specificity for the testis or the adult head (Fig. 1B). Four miRNAs that exhibited clear testis bias but did not fully meet the very strict criteria for tissue exclusivity were included as “testis-restricted” because they either clustered with other testes-restricted miRNAs (mir-972, -975, -2499) or were significantly higher in the testes compared with other animal tissues instead of embryo timepoints (mir-4976). We recognize that the categorization of “lowly expressed” miRNAs does not distinguish if such loci are simply not transcribed in a given tissue or if they might be expressed robustly but only in a spatially restricted cell subset. Nevertheless, the distinctively restricted expression of these miRNA cohorts in the head or testis suggests that they may play specialized roles in these tissues. For example, as many neural miRNAs play ongoing and continuous roles in the adult CNS (Sun and Lai 2013), one may hypothesize that this set of adult head–restricted miRNAs may be relevant for Drosophila neurophysiology or behavior.
Testis-restricted miRNAs are strongly enriched in clusters bearing recently evolved loci
We noticed several properties of the testis-restricted miRNAs that distinguished them from head-restricted miRNAs and from other miRNAs expressed in the testis. For example, many testis-restricted miRNAs are members of genomic clusters. In and of itself, this is not particularly notable since 54% of D. melanogaster miRNAs are clustered (Ruby et al. 2007; Berezikov et al. 2011; Mohammed et al. 2013), and we detected the specific expression of clustered miRNAs in other tissues. However, the representation of clustered miRNAs among testis-restricted loci (84%) was notably much higher compared with the number of clustered miRNAs among head-restricted loci (21%) or with clustered Drosophila miRNAs in general (Fig. 1C). More remarkable was the fact that the testis clusters had a strong representation of recently evolved miRNAs, as defined by loci whose orthologs were restricted to the closely related group of melanogaster-group species (i.e., D. melanogaster, Drosophila simulans, Drosophila sechellia, Drosophila yakuba, Drosophila erecta, and Drosophila ananassae), which radiated ∼8 million yr ago from other Drosophilids. By use of this species cutoff, 58% of the testis-restricted miRNAs were classified as recently evolved, whereas only 5% of head-restricted miRNAs were (Fig. 1D). The majority of these recently evolved, testis-restricted miRNAs were also members of genomic clusters, so that combining these features further highlighted that this class appears to be a specific attribute of the testis. We refer to these testis-restricted, recently evolved, clustered loci as TRC miRNAs.
TRC miRNAs exhibit conserved processing often defy typical miRNA divergence patterns
Nearly all conserved miRNAs with known biological activity produce similar mature species across their orthologs, and these inevitably exhibit invariant seed regions. The rationale for this is believed to be well established: miRNAs that are subject to conserved processing have been concomitantly selected for conserved seed-pairing to target cohorts. Moreover, a substantial fraction of animal miRNA loci have been selected for regulatory potential of small RNAs from both hairpin arms (Okamura et al. 2008; Yang et al. 2011). These features have had a strong impact on the manner in which conserved miRNA genes evolve. Alignments of miRNA orthologs show that terminal loops nearly always exhibit greater divergence than the hairpin duplex regions, and thus they exhibit a “saddle-shaped” pattern of divergence (Lai et al. 2003; Berezikov et al. 2005). Even some recently evolved miRNAs exhibit this loop-preferred divergence pattern (Fig. 2A), which may be taken as evidence that they may have successfully acquired beneficial regulatory functions as miRNAs. Only rarely do miRNAs exhibit duplex regions that diverge similarly or faster than their terminal loops, and such a pattern usually reflects an atypical feature of miRNA processing or function.
FIGURE 2.

Atypical divergence patterns of testis-restricted, recently emerged, clustered (TRC) miRNAs. (A) Example of a recently evolved miRNA that evolves like a well-conserved miRNA, that is, with loop-preferred divergence and preservation of the hairpin arms. (B) Examples of melanogaster-group, testis-restricted, clustered miRNAs with divergent seed regions. These 11 miRNAs belonged to three clusters (dme-mir-4966-2 not shown). Only the mir-984→982 cluster contained only newly evolved miRNAs, but other clusters contained both pan-Drosophilid and melanogaster subgroup miRNAs. All miRNA orthologs, even seed-divergent ones, attained hairpin secondary structures (species with green highlight) and small RNA evidence indicative of microprocessor cleavage. This evidence suggests adaptation to a newly emergent function. (C) mir-2499 is a melanogaster-group miRNA that provides a detailed illustration of this secondary structure and small RNA evidence.
With this in mind, we were surprised to observe that many TRC miRNAs do not abide by the typical saddle-shaped divergence pattern but instead exhibit diverged nucleotides that were scattered throughout the hairpin, frequently including both miRNA and star regions (Fig. 2B). This is not intrinsically due to the possibility that recently evolved miRNAs cannot be selected for regulatory capacity. Indeed, we earlier noted specific examples of recently evolved miRNAs that exhibit preferred loop divergence (Okamura et al. 2007), and subsequently we showed that recently evolved miRNAs exhibit overall similar patterns of divergence as more deeply conserved miRNAs in Drosophila (Mohammed et al. 2013). Instead, our current observations indicate that recently evolved, testis-restricted miRNAs comprise a distinct class of loci with unique evolutionary properties.
An alternate, and less compelling, interpretation is that the purported orthologs of these loci might not actually be functionally processed miRNAs. In this scenario, the “random” divergence patterns might simply reflect neutral evolution. To assess this possibility, we prepared small RNA libraries from the male bodies across the Drosophila-subgroup species, namely, D. simulans, D. sechellia, D. yakuba, and D. erecta. We sequenced these to a depth of 21 million to 32 million reads (Supplemental Table 2), affording a deep perspective of newly evolved miRNAs in these different species. These data confirm that all of the TRC miRNA loci annotated in D. melanogaster were specifically processed into mature miRNAs in multiple other species (Fig. 2C; Supplemental Fig. 1). Notably, the availability of small RNA data directly shows that orthologous miRNAs in this set frequently exhibit divergent seeds, and also included examples of shifts in dominant 5′ ends. Heterogeneities of miRNA 5′ termini have been previously observed (Fernandez-Valverde et al. 2010; Pantano et al. 2010; Berezikov et al. 2011), although these are rarely truly species specific. Rather, there turn out to be collective heterogeneities present in different libraries from individual species that are as a whole preserved in different species. In short, this set of testis-restricted miRNAs rates among the most rapidly evolving miRNAs ever shown explicitly to have processed orthologs.
Evidence for functional activity of TRC miRNAs
Since recently evolved miRNAs are generally believed not to be endowed with substantial regulatory capacity, we assessed the functional properties of “young” Drosophila miRNAs. We segregated these loci according to whether or not they were testis-restricted and first compared their expression levels. Recently evolved miRNAs generally exhibit far lower expression levels than more deeply conserved loci (Ruby et al. 2007). This could be due to a variety of reasons. For example, it may be that evolutionarily nascent miRNAs have not fully acquired all the structural or sequence characteristics that permit efficient biogenesis. An alternative, and not mutually exclusive, proposal is that evolutionary nascent miRNAs may be more likely to induce detrimental rather than beneficial regulatory consequences. Therefore, high expression of evolutionarily nascent miRNAs may actually be under negative selection (Bartel and Chen 2004; Chen and Rajewsky 2007).
We compared the levels of “young” Drosophila miRNAs, taking care to analyze cohorts of miRNAs of similar evolutionary age. For this analysis, we selected 14 TRC D. melanogaster miRNAs and 76 other miRNAs, whose sole alignable hairpin orthologs were restricted to the melanogaster group. This places their collective birth sometime within the last ∼6 million to 10 million yr. We quantified the counts per million miRNA reads for each of these loci across 39 D. melanogaster animal tissue libraries (Supplemental Table 1). We then plotted the maximal expression level of these loci in any single library. We reasoned that this approach provided a fairer comparison than, say, taking the average expression level of miRNAs across libraries, which would tend to dilute the representation of tissue- or cell-restricted miRNAs. Plotting these results provided a clear result that the TRC miRNAs collectively achieved significantly higher expression levels than did other miRNAs of comparable evolutionary age (Mann-Whitney test P-value <10−7) (Fig. 3A). Therefore, this higher accumulation suggests that TRC miRNAs could be endowed with regulatory activities on par with more conserved miRNAs.
FIGURE 3.
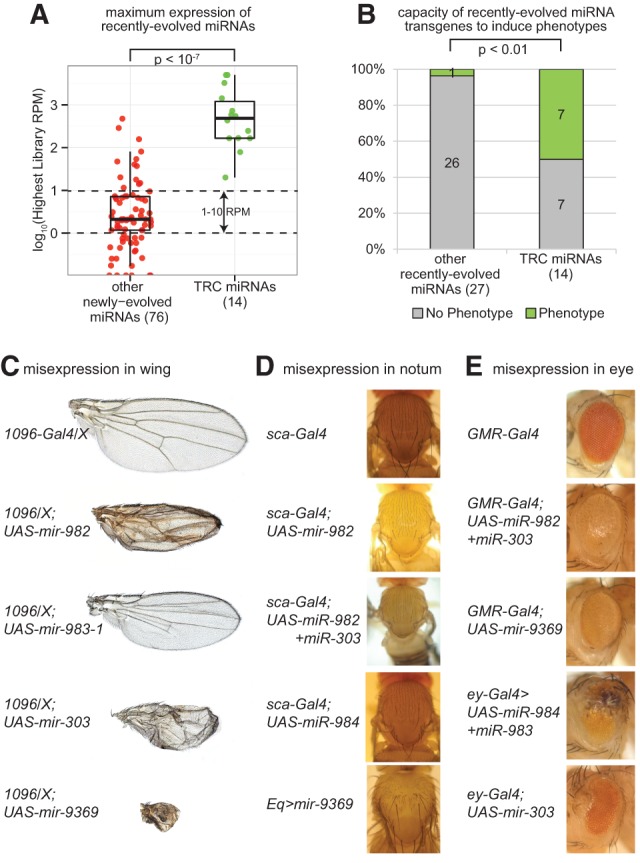
Evidence for in vivo function of TRC miRNAs. (A) TRC miRNAs are expressed at significantly higher levels compared with other recently evolved miRNAs. Plotted are data for the maximal expression of a given miRNA in any library analyzed. (B) Half of TRC miRNAs induced phenotypes when misexpressed using the Gal4-UAS system, whereas barely any recently evolved miRNAs that were not testis-restricted had such capacity. (C–E) Examples of TRC miRNAs whose misexpression generated mutant phenotypes in somatic tissues. (C) Phenotypes induced in the wing upon misexpression using 1096-Gal4. (D) Phenotypes induced in the notum mechanosensory bristle field upon misexpression using Eq-Gal4. (E) Phenotypes induced in the eye upon misexpression with GMR-Gal4 or ey-Gal4. Note also that several additional TRC miRNAs induced lethality; the full phenotypic descriptions are tabulated in Supplemental Table 4 and depicted in Supplemental Figure 3.
We extended these expression tests by performing functional analyses in transgenic animals. We expanded our recently published collection of conditional UAS-DsRed-miRNA expression transgenes (Bejarano et al. 2012) by generating 28 additional transgenic lines (Supplemental Tables 4, 5), mostly composed of newly evolved miRNAs that we had annotated more recently (Berezikov et al. 2011; Chung et al. 2011). We then selected all the transgenes for newly evolved miRNAs and performed a systematic screen of all the independent insertions for each construct against nine Gal4 drivers, including ubiquitous (da-Gal4), eye specific (GMR-Gal4, ey-Gal4), wing specific (Sd-Gal4), wing and notum (1096-Gal4), anterior posterior compartment boundary (dpp-Gal4 and ptc-Gal4), notum specific (Eq-Gal4), and neuron specific (elav-Gal4). Our intent was to survey broadly for the capacity of recently evolved miRNAs to induce any type of gain-of-function phenotype by examining a variety of tissues.
In general, while our previous survey of well-conserved Drosophila miRNAs showed that the vast majority (>80%) could induce mutant phenotypes when ectopically expressed (Bejarano et al. 2012), most recently evolved miRNAs did not share this capacity. However, by segregating newly evolved miRNAs according to whether they were testis restricted or not, we observed that the former set exhibit dramatically greater phenotypic capacity (Fig. 3B). That is, half of the TRC miRNAs could induce mutant phenotypes, whereas <4% of the remaining recently evolved miRNAs could do so. Examples of defective wing, notum, and eye phenotypes caused by misexpression of these miRNAs are shown in Figure 3, C through E, and all such data are summarized in Supplemental Fig. 3. Because these phenotypes were generated by misexpression of testis miRNAs in noncognate somatic tissues, we can infer that they are due to the suppression of genes that are present more broadly.
Overall, our expression and phenotypic profiling data demonstrate that recently evolved miRNAs differ substantially in their properties according to their endogenous setting of expression. In particular, those recently evolved miRNAs that are mostly restricted to the testis prove to be much higher expressed and have a much greater capacity to induce dominant in vivo phenotypic aberrations.
Adaptive evolution of TRC miRNAs
While essentially all other miRNAs studied with orthologs in multiple species exist to regulate target genes via conserved sites, our experimental tests support a model in which most testis-restricted miRNAs are functional regulatory RNAs whose orthologs may regulate genes via divergent sites. We attempted to identify such putatively evolving targets that might drive the unusual evolutionary properties of TRC miRNA genes. In silico tests to identify coevolving targets that mimic the seed substitution patterns of these special miRNAs were inconclusive because they performed similar to background alignments of random sequences (see Materials and Methods). Next, we turned our attention to three classical computational methods for detecting evidence of natural selection: (1) a divergence-only test, which measures the branch length scale ratios between a neutral phylogeny and one fitted to putatively functional DNA elements; (2) an extension to the McDonald-Kreitman test (generalized MKT) that facilitates noncoding DNA (McDonald and Kreitman 1991); and (3) INSIGHT, a recent, probabilistic graphical model that offers several enhancements from the MKT (Gronau et al. 2013). Both the MKT and INSIGHT improve upon the divergence-only test by incorporating population polymorphism data, which enables inference of recent selection with better precision. We utilized the recent Drosophila Genetic Reference Panel (DGRP) Freeze 2 population data set, which provides polymorphism data from 205 D. melanogaster lines (Mackay et al. 2012). Moreover, INSIGHT improves upon the MKT by accounting for mutational heterogeneity and incorporating allele frequency information.
The three tests for natural selection all agreed and indicated strong positive selection within the miR strand for TRC miRNAs (Fig. 4; Supplemental Fig. 4). In the divergence-only test, we observed a branch-length scale factor (ρ) greater than one for TRC miRNAs, a canonical signature of accelerated divergence and potentially adaptive evolution (Fig. 4A). Interestingly, the signature of accelerated divergence was strongest in TRC miRNAs and was progressively weaker within other testes-restricted supersets, such as all clustered, testes-restricted miRNAs, and was near absent within other testes-expressed miRNAs. This observation indicates that the signature of positive selection is specific to TRC miRNAs. The generalized MKT offered analogous evidence for positive selection within TRC miRNAs (Fig. 4B). In the generalized MKT, an excess in divergences to polymorphisms in functional elements (DE/PE) significantly beyond that of adjacent, neutrally evolving flanking regions (DNet/PNet) is an indication of adaptive evolution. For TRC miRNAs, we observed many divergences between D. simulans and D. melanogaster for TRC miRNAs, but a total lack of polymorphism. This suggests that all divergences observed in D. melanogaster sweep to fixation rapidly, and the complete lack of polymorphisms indicates a recent constraint by forces of weak negative selection. We observed, similar to the divergence-only test, reduced D/P ratios within other supersets of testis-restricted miRNAs, suggesting the signal of adaptive evolution is strongest within TRC miRNAs.
FIGURE 4.

Evidence for adaptive evolution of testis-restricted miRNAs. Tests for signatures of natural selection were conducted via three independent tests: (A) a divergence-only phylogenetic model (DIVMOD); (B) a generalized McDonald-Kreitman test (MKT); and (C,D) INSIGHT. All three tests showed signatures of positive selection for testes-restricted miRNAs alone in their respective framework, which include (A) a larger D. melanogaster branch length for TRC miRNAs compared with a neutral phylogeny in the DIVMOD, (B) a higher divergence/polymorphism ratio in miRs compared with flanking neutral sites in the MKT, and (C) a significant excess in the estimated number of divergences under strong positive selection using INSIGHT. Closer examination of testes-restricted miRNAs subdivided by clustering status and age showed that only clustered, recently evolved cases contained the signature of adaptive evolution. (D) Analysis of the major partitions of the miRNA hairpin (loop, lower-stem, mature, and star arms) revealed that the signature of adaptive evolution is specific to the mature region within the set of TRC miRNAs.
In the MKT, all polymorphisms contribute equally; however, an excess of high-frequency polymorphisms is typically an indication of weak negative selection. If not dealt with separately, these variants may bias the results of the MKT. Recently, we developed a new method, called INSIGHT, that effectively partitions the site-frequency spectrum in distinct frequency regimes as one of several enhances over the MKT, in order to obtain a more accurate measures selection. Similar to previous tests, INSIGHT reported the strongest signal of positive selection within the TRC miRNAs, via the highest expected number of divergences under strong positive selection (E[Dp/Kbp]) (Fig. 4C). Finally, we examined the relative strength of selection among varied partitions of the miRNA hairpin and the precursor miRNA using INSIGHT. Importantly, the mass of divergences driven by strong positive selection was concentrated within the mature strand and was virtually absent within the complementary star region or lower-stem region (Fig. 4D). Even though the terminal loop showed an indication of positively selected sites, the error within this estimation was large and the overall result was nonsignificant. Given the lack of adaptively evolving sites within these hairpin partitions, we conclude that the significant signature of positive selection is primarily concentrated within the mature sequence. By use of an assortment of comparative and population genetics tests for natural selection ranging in complexity, we conclude that only the mature regions of TRC miRNAs are adaptively evolving, which further supports their role as new regulators of gene expression.
Adaptive functional capacity of newly evolved, testis-restricted miRNAs
Our data thus far support the notion that TRC miRNAs evolve adaptively, in contrast to the prevailing view that conserved miRNAs are predominantly under negative selection to resist nucleotide changes, especially within the seed regions. It is not yet possible to specifically recognize those conserved miRNA targets that mediate phenotypically substantial biology, and the situation is even less promising for poorly conserved miRNA targets, especially those of poorly conserved miRNA loci (Bartel 2009). We attempted to examine whether recently emerged, testis-specific miRNAs were distinct from other newly emerged miRNAs with respect to bioinformatic predictions of seed matches. We did not observe any striking differences between these groups, nor did we identify a clear signature of putative targets bearing cognate changes that matched seed divergences of the rapidly evolving testis miRNAs (data not shown). However, it is difficult to make conclusions from these tests, since there is a strong likelihood of false-positive interactions when solely comparing seed matches in closely related species.
Nevertheless, as we have shown clear evidence that newly emerged testis miRNAs frequently have sufficient processing capacity to induce morphologically mutant phenotypes, we directly tested the model whether they might have detectable adaptive regulatory capacity. Specifically, we assayed whether synthetic targets bearing multimerized seed matches could be distinctly repressed by orthologous miRNAs in a species-specific manner (Fig. 5; Supplemental Table S6). We tested such sensors for the D. melanogaster (dme) and D. erecta (der) orthologs of mir-2498 and observed that dme-mir-2498 indeed suppressed its cognate sensor better than did der-mir-2498 and vice versa. We performed another set of assays to compare the activities of D. melanogaster and D. sechellia (dse) mir-303 and again observed species-specific regulation of these sensors by the cognate miRNA orthologs. These data are notable in that they constitute first proof of principle that orthologous miRNAs can have capacity to discriminate targets in a species-specific manner, consistent with the hypothesis of their adaptive evolution.
FIGURE 5.
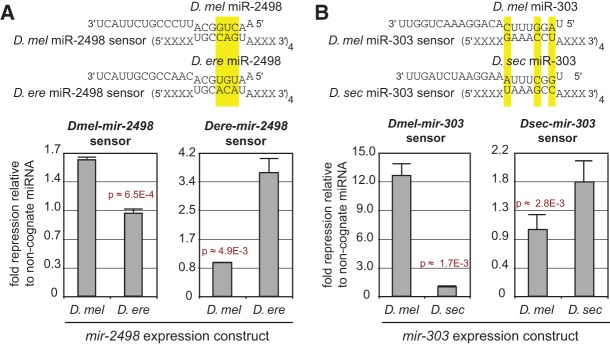
Proof of principle for adaptive regulatory activity of newly evolved, testis-restricted miRNAs. Luciferase sensor assay tests for binding and regulatory potential of two seed-divergent, testes-specific, recently evolved miRNAs: (A) mir-2498 and (B) mir-303. (Top) miR homologs with three seed mismatches were assayed for regulatory potential to congruent sensor targets. (Bottom) In all cases, miRs significantly repressed their cognate targets but not noncognate ones. This test validates that divergent miRs of the same family (1) have regulatory potential for gene regulation and (2) base-identity constraint to repress only a set of specific-specific target genes.
Highly dynamic gene flux of testis-restricted miRNA clusters in other Drosophilids
To date, the vast majority of miRNA annotations in fruitflies have been anchored using D. melanogaster, that is, loci that are well conserved between this species and other sequenced Drosophilids or that were recognized on the basis of short RNAs cloned from D. melanogaster. A limited amount of de novo annotation has been done in D. simulans and Drosophila pseudoobscura, with an earlier study purporting a very high rate of miRNA dynamics in these species on the basis of modest sequencing depth (Lu et al. 2008). This was tempered by a subsequent study using much more extensive small RNA data, which revealed more restricted dynamics in the flux of miRNA loci supported by cloned small RNA features of genuine RNase III products (Berezikov et al. 2010). Recently, we found that 11 clustered miRNAs of 18 total clustered and solo miRNA genes experienced atypical, lineage-specific, or clade-specific miRNA death events (Mohammed et al. 2013). In fact, 10 of these cases resided within testis-restricted clusters, a disproportion in miRNA loss within testis-restricted clusters that prompted an updated investigation of miRNA flux.
Using our new small RNA data sets (Supplemental Table 2), we performed detailed analysis of potential miRNA loci in the vicinity of the 20 D. melanogaster miRNA clusters and their syntenic regions in five fruitfly genomes (Supplemental Table 7). We identified de novo birth based on precursor sequence identity of <60% in all-by-all pairwise comparisons of all novel miRNA predictions. Of the 45 de novo miRNAs identified (27 confident novel and 18 candidate miRNAs), which excludes unannotated orthologs of D. melanogaster annotated miRNAs, only three were annotated within somatically expressed clusters (Fig. 6A; Supplemental Fig. 5). These three miRNAs include dse-mir-9685 and dvi-mir-9716 in the mir-317/277/34 cluster and dvi-mir-9717 in the mir-994/318 cluster. However, we observed a striking number of new miRNA annotations (42 in total) within the three testis-restricted clusters. The disproportion of new miRNA emergence within testes-restricted compared with somatic clusters was observed even after normalization to account for the total number of conserved D. melanogaster orthologs, which may confer an advantage for miRNA emergence (Fig. 6A). Of these de novo, testis-restricted clustered miRNAs, we observed varied modes of miRNA emergence, including duplication, new hairpin emergence, and duplication followed by rapid sequence evolution. The mir-972→979 cluster provides two examples of miRNA duplication. Within this cluster, we observed varying copy number of the newly evolved mir-4966 miRNA in D. melanogaster (two copies), D. simulans (three copies), and D. sechellia (two sense copies, two antisense copies) (Supplemental Fig. 6A). Duplication events were not limited to recently evolved miRNAs either, since we observed two copies of mir-974 in Drosophila virilis. Of the de novo miRNAs, many were species-specific and interlaced between duplicated miRNAs and long-standing miRNAs. Moreover, D. virilis, the most distantly related species to D. melanogaster in our comparison, harbored the most de novo miRNAs.
FIGURE 6.

High miRNA flux within testis-restricted cluster. (A) De novo miRNA birth is enriched within testes-restricted clusters compared with somatically expressed clusters. This enrichment is observed after normalizing to account for cluster size as measured by total number of conserved D. melanogaster miRNA orthologs. (B) Emergence of new miRNA within the mir-982-894 cluster via de novo hairpin emergence and gene duplication. A mir-982 subcluster containing three miRNAs (two mir-982 copies and mir-2582) duplicated in the D. simulans and D. sechellia lineage. Within D. sechellia, the duplicated copies remained similar; however, those of D. simulans experience gene loss and rapid sequence evolution. (C) De novo emergence of testis-restricted cluster in D. virilis. The 17 sense-strand miRNAs of this large cluster are testis-restricted (D) and interspersed among tRNA genes, while antisense members are predominantly expressed in other somatic tissues.
Most interesting to our analysis of miRNA flux within testes-restricted clusters was the identification of several new miRNAs that arose via duplication immediately succeeded by rapid evolution. This rapid evolution modulated distinct miRNA birth via seed region divergences and miRNA death. Within two testis-restricted clusters, we observed species- and clade-specific, subcluster duplications. First, within the mir-984→982 cluster we observed a three-member miRNA subcluster containing the mir-982 ortholog in the D. simulans and D. sechellia sister species (Fig. 6B). This subcluster is duplicated in both species; however, both subclusters rapidly evolved in D. simulans despite near identity of the D. sechellia copies. Duplicated members within D. simulans experienced seed region substitutions and one death event. Without confident D. simulans polymorphism data sets, it is unclear if this rapid evolution is an indication of adaptive evolution or gene death; however, the lower expression of duplicated members within this subcluster, compared with “progenitor” members, may indicate the latter. Second, we observed that the entire D. erecta mir-992→310 cluster is duplicated (Supplemental Fig. 6B). We questioned whether this duplication in the D. erecta genome might be a genome assembly artifact, but the presence of numerous diverged positions between these cluster copies supported their existence as distinct sequences. Many paralogous genes in both clusters preserved their seed sequence (mir-992, 991, 313→310); however, two miRNAs underwent rapid divergence (mir-2498, mir-9681), which gave rise to unique seed sequences and, consequently, distinct miRNAs.
The availability of extensive small RNA data from D. virilis gonads (Rozhkov et al. 2010) provided us an opportunity to study the dynamics of miRNA clusters outside of the melanogaster subgroup. Among novel miRNA annotations from this species (J Mohammed and EC Lai, in prep.) we discovered five miRNA clusters that are entirely specific to D. virilis (Fig. 6C; Supplemental Fig. S7). A notably sizable cluster contained 17 miRNAs that are transcribed from the sense genomic strand and five from the antisense strand. All the sense-strand members of this miRNA super-cluster are dominantly expressed in the testis of D. virilis, compared with small RNA libraries prepared from ovaries, heads, and embryos (Fig. 6D). The limited testis expression of antisense members of this cluster, and the four other smaller clusters identified, offers additional evidence of tissue-specific expression of sense and antisense miRNA transcripts similar to the sex dimorphisms between mir-978/979 and their antisense miRNAs (Berezikov et al. 2011).
In summary, analysis of tissue-specific small RNA data demonstrates the highly accelerated emergence of miRNAs among testis-restricted genomic clusters across the Drosophilid phylogeny and corroborates our functional and evolutionary data that support the concept for adaptive functions of miRNAs in the testis.
CONCLUSIONS
Distinct rates and classes of miRNA divergence patterns
The “saddle-shaped” pattern of nucleotide divergence for conserved miRNA hairpins is one of the most characteristic features of miRNA evolution (Lai et al. 2003; Berezikov et al. 2005) and differentiates them from structural hairpins whose duplex regions typically accumulate compensatory mutations (Stark et al. 2007; Parker et al. 2011; Will et al. 2013). The relatively few cases that defy this evolutionary principle have usually been indicative of novel aspects of miRNA biogenesis and/or regulation. For example, some cases of highly invariant terminal loops have led to an appreciation of post-transcriptional regulation of miRNA processing (Michlewski et al. 2008) or of functional terminal loops (Okamura et al. 2013). On the other hand, conserved vertebrate mir-451 exhibits a highly constrained terminal loop and a diverging 3′ hairpin arm (Yang et al. 2010), and this atypical pattern proved to reflect that its pre-miRNA hairpin is not a Dicer substrate (Cheloufi et al. 2010; Cifuentes et al. 2010; Yang et al. 2010).
We recently reported that miRNAs do not exhibit uniform rates of evolution and instead are influenced by biogenesis route (i.e., canonical pathway vs. splicing-derived pathway) and genomic arrangement (singleton vs. operon) (Berezikov et al. 2010; Mohammed et al. 2013). In the current study, we further refine the properties of miRNA evolutionary patterns and find that the intersection of tissue specificity and genomic arrangement yields a substantial class of atypically evolving miRNAs. Instead of the dominant saddle-shaped pattern observed in nearly all the alignments of miRNA orthologs previously studied, we observed a distributed divergence pattern across the available orthologs of TRC miRNAs.
As we show using computational and experimental strategies, the unusual properties of the TRC miRNAs are linked to their adaptive evolution, presumably to evolve in concert with specific target genes. We showed proof of principle that orthologous TRC miRNAs can indeed preferentially regulate species-specific target genes, unlike the case for all other cases of orthologous miRNAs that would have highly overlapping target specificity owing to their de facto conserved seed. We were not able to identify statistically significant evidence for such coevolving targets (data not shown), likely due to the limited signal afforded by miRNA seed matching. Indeed, it is not currently possible to identify biologically relevant, newly evolved targets of well-conserved miRNAs using computational methods. However, it is possible that signals for this may emerge in the future with increased Drosophila population sequencing.
De novo birth of testis protein-coding genes and short regulatory RNAs
The Drosophila testis has long been recognized as a tissue bearing a distinct gene regulatory landscape. For example, the testis expresses specialized paralogs of general regulatory machineries ranging from TBP-associated factors (TAFs) that regulate the core transcriptional initiation machinery (Hiller et al. 2001; Chen et al. 2005), certain ribosome subunits and translational initiation factors (Kearse et al. 2011; Hernandez et al. 2012), and even tissue-specific paralogs of proteasome subunits (Yuan et al. 1996; Zhong and Belote 2007). Concomitant with this is the observation that the testis exhibits a particularly distinct expression profile, including many genes that are not expressed in other tissues or cell types (Arbeitman et al. 2002; Brown et al. 2014). Indeed, the testis has long been recognized to have particular propensity to harbor newly emerged and/or rapidly evolving protein-coding genes, and these contribute to diverse processes such as male fertility fitness, intraspecies sperm competition and specialization, and speciation (Nurminsky et al. 1998; Ting et al. 1998; Begun et al. 2007; Haerty et al. 2007; Tao et al. 2007a,b; Chen et al. 2013; Reinhardt et al. 2013).
Our current study extends the notion of the accelerated gene birth and evolution in the testis to noncoding transcripts and, in particular, to newly emerged, testis-restricted, miRNA operons. Rapid evolution of an X-linked, testis-expressed miRNA cluster was previously reported (Zhang et al. 2007). However, this was studied as an individual case, and it was later suggested that the rapid evolution of mammalian testis miRNAs was associated with their residence on the X chromosome and not because of their testis expression per se (Guo et al. 2009). We note that two of the major clusters of rapidly evolving, testis-restricted miRNAs in Drosophila (mir-303 cluster and mir-972 cluster) are located on the X, suggesting that X-linkage is a preferred property of adaptively evolving TRC miRNAs. However, it is also certainly not required for their unique evolutionary behavior, since we observe similarly rapid and atypical divergence patterns (i.e., within mature species and within seed regions for the autosomal mir-991 TRC cluster). Moreover, we identified extensive, de novo, autosomal TRC clusters in D. virilis, which indicates that it is their deployment in the testis that best explains the unusual evolutionary flux of miRNAs. The phenomenon of species-specific and/or adaptive functions of miRNAs that we uncovered in the testis bears striking similarity to the distinct behavior of protein-coding genes in this tissue (Chen et al. 2013).
A notable feature of the TRC miRNAs that distinguishes them from bulk recently evolved miRNAs is that they are higher expressed than they “ought” to be, and they have greater capacity to induce in vivo mutant phenotypes when misexpressed in the animal. Therefore, in contrast to the supposition that recently evolved miRNAs should only “creep” into existence, the testis is a privileged location for the birth and functional activity of newly born miRNAs. We infer that these properties could only be associated if there is a feedback, namely, for selection for TRC miRNAs to become better processed and/or more suited to regulate gene expression than bulk neutrally evolving miRNAs or, in fact, negatively selected hairpins whose function is detrimental. Overall, our findings reveal a novel evolutionary intermediate that is largely restricted to the testis by which recently evolved miRNAs may be in position to actively coevolve with targets. The identification and biological utility of such targets may be facilitated in the future by the application of CLIP-sequencing (Mittal and Zavolan 2014) and CRISPR-Cas9 engineering (Gratz et al. 2013) across Drosophila species.
MATERIALS AND METHODS
Drosophila small RNA data sets
D. melanogaster miRNA annotations were downloaded from miRBase, revision 19 (ftp://mirbase.org/pub/mirbase/19/) (Kozomara and Griffiths-Jones 2011). From these annotations, we defined primary-miRNA hairpins as the Drosha-cropped pre-miRNAs, based on dominant mature and star reads, supplemented by 15 nt of sequence on either side to capture the lower stem. To identify tissue-specific miRNAs, we used in-house data (Chung et al. 2008; Berezikov et al. 2011; Wen et al. 2014) and other public small RNA libraries from the Short Read Archive (http://www.ncbi.nlm.nih.gov/sra) (Supplemental Table 1). These library types comprise diverse developmental timepoints and tissues, including the testes, ovary, head, embryo 0–2 h (hr), embryo 2–6 h, embryo 6–10 h, and embryo 12–24 h.
To analyze miRNA evolution in other species, we obtained cultures of the sequenced D. simulans, D. sechellia, D. yakuba, and D. erecta strains from the Drosophila Species Center. We isolated ∼18–28 nt RNA from male bodies using polyacrylamide gel electrophoresis, and we prepared libraries as described. These were sequenced on Illumina GAxII or Hi-seq instruments. For identification of novel miRNAs and for miRNA expression analysis, reads were aligned to the reference genomes of each species using bowtie, allowing for no mismatch and multiple mapping to 20 genomic positions or less.
Identification of tissue-restricted miRNA cohorts
We utilized the Bioconductor R package limma adapted for RNA-seq data to identify tissue-restricted miRNAs (Smyth 2004, 2005). limma required the raw number of reads per library per D. melanogaster miRNA gene and the total number of mapped reads per library. Normalization was performed internally via the “voom” method. Only reads contained within miR and miR* sequences ±2 nt were counted per miRNA gene. For each miRNA and for each condition (tissue or developmental timepoint), we evaluated the differential expression between all libraries in that condition to an aggregate of all other libraries. We repeated this “one-vs-all other” experiment for all conditions and tabulated log2 fold-change and P-values. Tissue-restricted miRNAs were classified as those miRNAs with a significantly higher expression in the tissue under consideration versus all other tissues. Tissue-expressed miRNAs were classified as all other miRNAs with more than 10 reads per million in the tissue of consideration.
Computational tests for selection
We used three methods to test for evidence of adaptive evolution: (1) a divergence-only model based on phylogenetic branch- and tree-scaling factors between a neutral phylogenetic model and a phylogenetic model inferred from test elements, such as miRNAs; (2) the generalized MKT, which measures the statistical significances in the excess of divergence to polymorphism ratio among test and neutral element classes (McDonald and Kreitman 1991); and (3) INSIGHT (Gronau et al. 2013), a recent method based upon a probabilistic graphical model that offers advantages over the MKT, including the accommodation of ancestral uncertainty and explicit modeling of weak negative selection by utilizing allele frequency information. In addition, INSIGHT was recently shown to be well suited for short functional elements, such as transcription factor binding sites (Arbiza et al. 2013) and miRNAs (Gronau et al. 2013). While all three tests are designed to capture evidence of both negative and positive selection, estimates of positive selection from the MKT and INSIGHT are more confident because they utilize population-specific polymorphism data, which inform of selection happening along a shorter evolutionary period and hence can better detect recent adaptation. In the divergence-only test, we computed the scaling factors (ρ) for each partition of miRNA genes using phyloFit in the RPHAST R statistical package (Pollard et al. 2010; Hubisz et al. 2011). ρ was computed for the D. melanogaster branch alone.
For all tests, we estimated local neutral evolutionary rates based on a neutral set of sites flanking miRNA genes. To define this neutral set we filtered (1) highly conserved regions defined as sites with 100% phastCons conservation score and 25 nt of flanking sequence; (2) exons, UTR genic regions, and 50 nt of flanking sequence; (3) 10-nt flanking sequences from exon, UTR, or conserved regions; (4) repeat-masked and simple repeat region inferred from Tandem Repeat Finder and RepeatMasker; (5) segmental duplications; and (6) CpG sites. D. melanogaster gene annotations (r5.46) were downloaded from Flybase (www.flybase.org).
After filtering, ∼25% of the genome remained within our neutral set. In the MKT framework, we wrote custom scripts to tabulate polymorphic and divergence counts for the element and neutral sets, ensuring that sites both polymorphic and divergent were removed as prescribed by the MKT. P-values for deciding the significance in divergence/polymorphism ratios for both the element and neutral sites were computed by Fisher's Exact test. Polymorphism and divergence information were processed into the suitable format for use with INSIGHT and are made available as a web resource (http://compgen.bscb.cornell.edu/INSIGHT/).
Polymorphism and divergence
We obtained polymorphism data for 205 D. melanogaster lines from the DGRP (Freeze 2: ftp://ftp.hgsc.bcm.edu/DGRP/freeze2_Feb_2013/) of Raleigh, North Carolina, populations (Mackay et al. 2012) and post-processed these for use within the MKT (McDonald and Kreitman 1991) and INSIGHT (Gronau et al. 2013) frameworks. Briefly, we removed polymorphic sites which were triallelic, INDELs, or sites with more than five individuals with missing data. Missing data include sites without a reported base or with an ambiguous base assignment (N's). In order to ensure a sample size of 400 chromosomes (or 200 individuals) per polymorphic site, we uniformly subsampled chromosomes without replacement for sites with greater than 400 nonambiguous bases. Sites that were nonpolymorphic or unfiltered were assumed to be monomorphic within the sample population. In total, ∼14.6% of sites were filtered, which yielded 4,149,065 total, usable SNPs.
To infer divergence events and compute ancestral prior probabilities for selection analysis, we utilized genome assemblies of D. melanogaster (dm3), D. sechellia (droSec1), D. yakuba (droYak2), and D. erecta (droEre2) from the UCSC genome browser, as well as a recently improved genome assembly from the D. simulans w[501] strain (Hu et al. 2012) (http://genomics.princeton.edu/AndolfattoLab/w501_genome.html). We created a multiple-species alignment of the five melanogaster-subgroup species using LASTZ (Blanchette et al. 2004; Harris 2007) and MULTIZ (Blanchette et al. 2004). We used a five-species phylogeny derived from pruning non-melanogaster-subgroup species from the 12-Drosophila phylogeny (Drosophila 12 Genomes Consortium 2007; Stark et al. 2007). Ancestral posterior probabilities for the internal parent node of the D. melanogaster, D. simulans, and D. sechellia extant species were computed using phyloFit in the RPHAST package after soft-masking the D. melanogaster base (Pollard et al. 2010; Hubisz et al. 2011).
Generation and phenotypic analysis of inducible miRNA transgenes
We followed our previous strategy for cloning 400–500 nt of pri-miRNA fragments into the 3′ UTR of a UAS-DsRed P element transformation vector (Bejarano et al. 2012). The primer sets used to clone 28 such transgenes are listed in Supplemental Table 5. We crossed all (three to eight) independent insertions to the ubiquitous driver da-Gal4 to determine the typical activities of these miRNAs, since this usually resulted in a binary lethal/viable output. We subsequently crossed two typical insertions of each miRNA to additional tissue-specific Gal4 driver strains, including da-Gal4, ey-Gal4, GMR-Gal4, sd-Gal4, bx-Gal4, and Eq-Gal4. We compared these phenotypes with the effects of expressing other UAS-DsRed-miRNA transgenes from our published collection (Bejarano et al. 2012). All of the phenotypes were recorded using a controlled vocabulary and are presented in full in Supplemental Table 4.
Luciferase sensor tests
Renilla luciferase sensors for D. melanogaster miR-2498, D. melanogaster miR-303, D. erecta miR-2498, and D. sechellia miR-303 were created using a modified version of psiCheck2 (Promega). DNA fragments bearing four elements complementary to nucleotides 2–8 of each miRNA along with an “A” residue corresponding to the first base of the miRNA were synthesized from long overlapping primers using PCR (Supplemental Table 6). Both vector and the “71A” sensor fragments were digested with Not1 and Xho1 and ligated together to create the luciferase-based sensors. Expression constructs for each of the miRNAs listed above were generated from pUASt-dsRed. Purified genomic DNA samples from the three species were used as templates to amplify either ∼500-bp regions containing only the miRNA of interest for the solo constructs or extended genomic regions encoding additional miRNAs for the cluster constructs. The PCR products were inserted into pUASt-dsRed, downstream from the dsRed ORF, using the cold fusion cloning kit. Luciferase assays were performed as previously described. Plasmids listed above were transfected into S2-R+ cells (Dasgupta Lab) in a 96-well plate format, followed by luciferase expression detection via the DualGlo kit (Promega) and a luminometer (Turner Biosciences). In each condition, three vectors were transfected: one expressing GAL4 driven by the Ubiquitin promoter, a psiCheck2 sensor for one of the four miRNAs above, and a pUASt-dsRed-miRNA expression construct.
DATA DEPOSITION
Raw data sets for sequencing data sets have been deposited in the NCBI Gene Expression Omnibus (GEO) repository (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE56244 (Supplemental Table 2).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
J.M. was supported in part by the Tri-Institutional Training Program in Computational Biology and Medicine (via NIH training grant 1T32-GM083937). Work in A.S.’s group was supported by a David and Lucile Packard Fellowship for Science and Engineering and NIH grant R01-GM102192. Work in E.C.L.’s group was supported by the Burroughs Wellcome Fund and NIH grant R01-GM083300.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.044644.114.
REFERENCES
- Ambros V, Bartel B, Bartel DP, Burge CB, Carrington JC, Chen X, Dreyfuss G, Eddy SR, Griffiths-Jones S, Marshall M, et al. 2003. A uniform system for microRNA annotation. RNA 9: 277–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbeitman MN, Furlong EE, Imam F, Johnson E, Null BH, Baker BS, Krasnow MA, Scott MP, Davis RW, White KP 2002. Gene expression during the life cycle of Drosophila melanogaster. Science 297: 2270–2275 [DOI] [PubMed] [Google Scholar]
- Arbiza L, Gronau I, Aksoy BA, Hubisz MJ, Gulko B, Keinan A, Siepel A 2013. Genome-wide inference of natural selection on human transcription factor binding sites. Nat Genet 45: 723–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP 2008. The impact of microRNAs on protein output. Nature 455: 64–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP 2009. MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP, Chen CZ 2004. Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat Genet 5: 396–400 [DOI] [PubMed] [Google Scholar]
- Begun DJ, Lindfors HA, Kern AD, Jones CD 2007. Evidence for de novo evolution of testis-expressed genes in the Drosophila yakuba/Drosophila erecta clade. Genetics 176: 1131–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejarano F, Bortolamiol-Becet D, Dai Q, Sun K, Saj A, Chou YT, Raleigh DR, Kim K, Ni JQ, Duan H, et al. 2012. A genome-wide transgenic resource for conditional expression of Drosophila microRNAs. Development 139: 2821–2831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezikov E, Guryev V, van de Belt J, Wienholds E, Plasterk RH, Cuppen E 2005. Phylogenetic shadowing and computational identification of human microRNA genes. Cell 120: 21–24 [DOI] [PubMed] [Google Scholar]
- Berezikov E, Liu N, Flynt AS, Hodges E, Rooks M, Hannon GJ, Lai EC 2010. Evolutionary flux of canonical microRNAs and mirtrons in Drosophila. Nat Genet 42: 6–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezikov E, Robine N, Samsonova A, Westholm JO, Naqvi A, Hung JH, Okamura K, Dai Q, Bortolamiol-Becet D, Martin R, et al. 2011. Deep annotation of Drosophila melanogaster microRNAs yields insights into their processing, modification, and emergence. Genome Res 21: 203–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette M, Kent WJ, Riemer C, Elnitski L, Smit AF, Roskin KM, Baertsch R, Rosenbloom K, Clawson H, Green ED, et al. 2004. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res 14: 708–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke J, Stark A, Russell RB, Cohen SM 2005. Principles of microRNA-target recognition. PLoS Biol 3: e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JB, Boley N, Eisman R, May G, Stoiber M, Duff M, Booth B, Park S, Suzuki A, Wan K, et al. 2014. Diversity and dynamics of the Drosophila transcriptome. Nature (in press) 10.1038/nature12962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheloufi S, Dos Santos CO, Chong MM, Hannon GJ 2010. A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature 465: 584–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Rajewsky N 2006. Deep conservation of microRNA-target relationships and 3′UTR motifs in vertebrates, flies, and nematodes. Cold Spring Harb Symp Quant Biol 71: 149–156 [DOI] [PubMed] [Google Scholar]
- Chen K, Rajewsky N 2007. The evolution of gene regulation by transcription factors and microRNAs. Nat Rev Genet 8: 93–103 [DOI] [PubMed] [Google Scholar]
- Chen X, Hiller M, Sancak Y, Fuller MT 2005. Tissue-specific TAFs counteract Polycomb to turn on terminal differentiation. Science 310: 869–872 [DOI] [PubMed] [Google Scholar]
- Chen S, Krinsky BH, Long M 2013. New genes as drivers of phenotypic evolution. Nat Rev Genet 14: 645–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi SW, Hannon GJ, Darnell RB 2012. An alternative mode of microRNA target recognition. Nat Struct Mol Biol 19: 321–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang HR, Schoenfeld LW, Ruby JG, Auyeung VC, Spies N, Baek D, Johnston WK, Russ C, Luo S, Babiarz JE, et al. 2010. Mammalian microRNAs: experimental evaluation of novel and previously annotated genes. Genes Dev 24: 992–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WJ, Okamura K, Martin R, Lai EC 2008. Endogenous RNA interference provides a somatic defense against Drosophila transposons. Curr Biol 18: 795–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WJ, Agius P, Westholm JO, Chen M, Okamura K, Robine N, Leslie CS, Lai EC 2011. Computational and experimental identification of mirtrons in Drosophila melanogaster and Caenorhabditis elegans. Genome Res 21: 286–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes D, Xue H, Taylor DW, Patnode H, Mishima Y, Cheloufi S, Ma E, Mane S, Hannon GJ, Lawson N, et al. 2010. A novel miRNA processing pathway independent of Dicer requires Argonaute2 catalytic activity. Science 328: 1694–1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clop A, Marcq F, Takeda H, Pirottin D, Tordoir X, Bibe B, Bouix J, Caiment F, Elsen JM, Eychenne F, et al. 2006. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat Genet 38: 813–818 [DOI] [PubMed] [Google Scholar]
- Czech B, Hannon GJ 2010. Small RNA sorting: matchmaking for Argonautes. Nat Rev Genet 12: 19–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Sharp PA 2004. Specificity of microRNA target selection in translational repression. Genes Dev 18: 504–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosophila 12 Genomes Consortium. 2007. Evolution of genes and genomes on the Drosophila phylogeny. Nature 450: 203–218 [DOI] [PubMed] [Google Scholar]
- Fernandez-Valverde SL, Taft RJ, Mattick JS 2010. Dynamic isomiR regulation in Drosophila development. RNA 16: 1881–1888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman RC, Farh KK, Burge CB, Bartel DP 2009. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19: 92–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraldez AJ, Mishima Y, Rihel J, Grocock RJ, Van Dongen S, Inoue K, Enright AJ, Schier AF 2006. Zebrafish MiR-430 promotes deadenylation and clearance of maternal mRNAs. Science 312: 75–79 [DOI] [PubMed] [Google Scholar]
- Gratz SJ, Wildonger J, Harrison MM, O'Connor-Giles KM 2013. CRISPR/Cas9-mediated genome engineering and the promise of designer flies on demand. Fly 7: 249–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronau I, Arbiza L, Mohammed J, Siepel A 2013. Inference of natural selection from interspersed genomic elements based on polymorphism and divergence. Mol Biol Evol 30: 1159–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Su B, Zhou Z, Sha J 2009. Rapid evolution of mammalian X-linked testis microRNAs. BMC Genomics 10: 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Ingolia NT, Weissman JS, Bartel DP 2010. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466: 835–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha I, Wightman B, Ruvkun G 1996. A bulged lin-4/lin-14 RNA duplex is sufficient for Caenorhabditis elegans lin-14 temporal gradient formation. Genes Dev 10: 3041–3050 [DOI] [PubMed] [Google Scholar]
- Haerty W, Jagadeeshan S, Kulathinal RJ, Wong A, Ravi Ram K, Sirot LK, Levesque L, Artieri CG, Wolfner MF, Civetta A, et al. 2007. Evolution in the fast lane: rapidly evolving sex-related genes in Drosophila. Genetics 177: 1321–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS 2007. “Improved pairwise alignment of genomic DNA.” PhD thesis, The Pennsylvania State University, University Park, PA [Google Scholar]
- Hernandez G, Han H, Gandin V, Fabian L, Ferreira T, Zuberek J, Sonenberg N, Brill JA, Lasko P 2012. Eukaryotic initiation factor 4E-3 is essential for meiotic chromosome segregation, cytokinesis and male fertility in Drosophila. Development 139: 3211–3220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller MA, Lin TY, Wood C, Fuller MT 2001. Developmental regulation of transcription by a tissue-specific TAF homolog. Genes Dev 15: 1021–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu TT, Eisen MB, Thornton KR, Andolfatto P 2012. A second generation assembly of the Drosophila simulans genome provides new insights into patterns of lineage-specific divergence. Genome Res 1: 89–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubisz MJ, Pollard KS, Siepel A 2011. PHAST and RPHAST: phylogenetic analysis with space/time models. Brief Bioinform 12: 41–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse MG, Chen AS, Ware VC 2011. Expression of ribosomal protein L22e family members in Drosophila melanogaster: rpL22-like is differentially expressed and alternatively spliced. Nucleic Acids Res 39: 2701–2716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S 2011. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 39(Database issue): D152–D157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, Macmenamin P, da Piedade I, Gunsalus KC, Stoffel M, et al. 2005. Combinatorial microRNA target predictions. Nat Genet 37: 495–500 [DOI] [PubMed] [Google Scholar]
- Lai EC 2002. microRNAs are complementary to 3′ UTR sequence motifs that mediate negative post-transcriptional regulation. Nat Genet 30: 363–364 [DOI] [PubMed] [Google Scholar]
- Lai EC, Tomancak P, Williams RW, Rubin GM 2003. Computational identification of Drosophila microRNA genes. Genome Biol 4: R42.41–R42.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, et al. 2007. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 129: 1401–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP 2005. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120: 15–20 [DOI] [PubMed] [Google Scholar]
- Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM 2005. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 433: 769–773 [DOI] [PubMed] [Google Scholar]
- Loeb GB, Khan AA, Canner D, Hiatt JB, Shendure J, Darnell RB, Leslie CS, Rudensky AY 2012. Transcriptome-wide miR-155 binding map reveals widespread noncanonical microRNA targeting. Mol Cell 48: 760–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Shen Y, Wu Q, Kumar S, He B, Shi S, Carthew RW, Wang SM, Wu CI 2008. The birth and death of microRNA genes in Drosophila. Nat Genet 40: 351–355 [DOI] [PubMed] [Google Scholar]
- Mackay TF, Richards S, Stone EA, Barbadilla A, Ayroles JF, Zhu D, Casillas S, Han Y, Magwire MM, Cridland JM, et al. 2012. The Drosophila melanogaster genetic reference panel. Nature 482: 173–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M 1991. Adaptive protein evolution at the Adh locus in Drosophila. Nature 351: 652–654 [DOI] [PubMed] [Google Scholar]
- Meister G 2013. Argonaute proteins: functional insights and emerging roles. Nat Rev Genet 14: 447–459 [DOI] [PubMed] [Google Scholar]
- Michlewski G, Guil S, Semple CA, Caceres JF 2008. Posttranscriptional regulation of miRNAs harboring conserved terminal loops. Mol Cell 32: 383–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal N, Zavolan M 2014. Seq and CLIP through the miRNA world. Genome Biol 15: 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed J, Flynt AS, Siepel A, Lai EC 2013. The impact of age, biogenesis, and genomic clustering on Drosophila microRNA evolution. RNA 19: 1295–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mor E, Shomron N 2013. Species-specific microRNA regulation influences phenotypic variability: perspectives on species-specific microRNA regulation. Bioessays 35: 881–888 [DOI] [PubMed] [Google Scholar]
- Nurminsky DI, Nurminskaya MV, De Aguiar D, Hartl DL 1998. Selective sweep of a newly evolved sperm-specific gene in Drosophila. Nature 396: 572–575 [DOI] [PubMed] [Google Scholar]
- Okamura K, Hagen JW, Duan H, Tyler DM, Lai EC 2007. The mirtron pathway generates microRNA-class regulatory RNAs in Drosophila. Cell 130: 89–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura K, Phillips MD, Tyler DM, Duan H, Chou YT, Lai EC 2008. The regulatory activity of microRNA* species has substantial influence on microRNA and 3′ UTR evolution. Nat Struct Mol Biol 15: 354–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura K, Ladewig E, Zhou L, Lai EC 2013. Functional small RNAs are generated from select miRNA hairpin loops in flies and mammals. Genes Dev 27: 778–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantano L, Estivill X, Marti E 2010. SeqBuster, a bioinformatic tool for the processing and analysis of small RNAs datasets, reveals ubiquitous miRNA modifications in human embryonic cells. Nucleic Acids Res 38: e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker BJ, Moltke I, Roth A, Washietl S, Wen J, Kellis M, Breaker R, Pedersen JS 2011. New families of human regulatory RNA structures identified by comparative analysis of vertebrate genomes. Genome Res 21: 1929–1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A 2010. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 20: 110–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt JA, Wanjiru BM, Brant AT, Saelao P, Begun DJ, Jones CD 2013. De novo ORFs in Drosophila are important to organismal fitness and evolved rapidly from previously non-coding sequences. PLoS Genet 9: e1003860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozhkov NV, Aravin AA, Zelentsova ES, Schostak NG, Sachidanandam R, McCombie WR, Hannon GJ, Evgen'ev MB 2010. Small RNA-based silencing strategies for transposons in the process of invading Drosophila species. RNA 16: 1634–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruby JG, Stark A, Johnston WK, Kellis M, Bartel DP, Lai EC 2007. Evolution, biogenesis, expression, and target predictions of a substantially expanded set of Drosophila microRNAs. Genome Res 17: 1850–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N 2008. Widespread changes in protein synthesis induced by microRNAs. Nature 455: 58–63 [DOI] [PubMed] [Google Scholar]
- Shin C, Nam JW, Farh KK, Chiang HR, Shkumatava A, Bartel DP 2010. Expanding the MicroRNA targeting code: functional sites with centered pairing. Mol Cell 38: 789–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK 2004. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3: Article3 [DOI] [PubMed] [Google Scholar]
- Smyth GK 2005. Limma: linear models for microarray data. In Bioinformatics and computational biology solutions using R and Bioconductor (ed. Gentleman R, et al. ), pp. 397–420 Springer, New York [Google Scholar]
- Stark A, Lin MF, Kheradpour P, Pedersen JS, Parts L, Carlson JW, Crosby MA, Rasmussen MD, Roy S, Deoras AN, et al. 2007. Discovery of functional elements in 12 Drosophila genomes using evolutionary signatures. Nature 450: 219–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Lai EC 2013. Adult-specific functions of animal microRNAs. Nat Rev Genet 14: 535–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y, Araripe L, Kingan SB, Ke Y, Xiao H, Hartl DL 2007a. A sex-ratio meiotic drive system in Drosophila simulans. II: an X-linked distorter. PLoS Biol 5: e293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y, Masly JP, Araripe L, Ke Y, Hartl DL 2007b. A sex-ratio meiotic drive system in Drosophila simulans. I: an autosomal suppressor. PLoS Biol 5: e292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting CT, Tsaur SC, Wu ML, Wu CI 1998. A rapidly evolving homeobox at the site of a hybrid sterility gene. Science 282: 1501–1504 [DOI] [PubMed] [Google Scholar]
- Vella MC, Choi EY, Lin SY, Reinert K, Slack FJ 2004. The C. elegans microRNA let-7 binds to imperfect let-7 complementary sites from the lin-41 3′UTR. Genes Dev 18: 132–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wee LM, Flores-Jasso CF, Salomon WE, Zamore PD 2012. Argonaute divides its RNA guide into domains with distinct functions and RNA-binding properties. Cell 151: 1055–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen J, Mohammed J, Bortolamiol-Becet D, Tsai H, Robine N, Westholm JO, Ladewig E, Dai Q, Okamura K, Flynt AS, et al. 2014. Diversity of miRNAs, siRNAs, and piRNAs across 25 Drosophila cell lines. Genome Res 24: 1236–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will S, Yu M, Berger B 2013. Structure-based whole-genome realignment reveals many novel noncoding RNAs. Genome Res 23: 1018–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JS, Lai EC 2011. Alternative miRNA biogenesis pathways and the interpretation of core miRNA pathway mutants. Mol Cell 43: 892–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JS, Maurin T, Robine N, Rasmussen KD, Jeffrey KL, Chandwani R, Papapetrou EP, Sadelain M, O'Carroll D, Lai EC 2010. Conserved vertebrate mir-451 provides a platform for Dicer-independent, Ago2-mediated microRNA biogenesis. Proc Natl Acad Sci 107: 15163–15168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JS, Phillips MD, Betel D, Mu P, Ventura A, Siepel AC, Chen KC, Lai EC 2011. Widespread regulatory activity of vertebrate microRNA* species. RNA 17: 312–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, Miller M, Belote JM 1996. Duplicated proteasome subunit genes in Drosophila melanogaster encoding testes-specific isoforms. Genetics 144: 147–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Peng Y, Wang W, Su B 2007. Rapid evolution of an X-linked microRNA cluster in primates. Genome Res 17: 612–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong L, Belote JM 2007. The testis-specific proteasome subunit Prosalpha6T of D. melanogaster is required for individualization and nuclear maturation during spermatogenesis. Development 134: 3517–3525 [DOI] [PubMed] [Google Scholar]