

Abstract
Protein lysine deacetylases (KDACs), including the classic Zn2+-dependent histone deacetylases (HDACs) and the nicotinamide adenine dinucleotide (NAD+)-requiring sirtuins, are enzymes that play critical roles in numerous biological processes, particularly the epigenetic regulation of global gene expression programs in response to internal and external cues. Dysregulation of KDACs is characteristic of several human diseases, including chronic metabolic, neurodegenerative, and cardiovascular diseases and many cancers. This has led to the development of KDAC modulators, two of which (HDAC inhibitors vorinostat and romidepsin) have been approved for the treatment of cutaneous Tcell lymphoma. By their nature, existing KDAC modulators are relatively nonspecific, leading to pan-KDAC changes and undesired side effects. Given that KDACs are regulated at many levels, including transcriptional, posttranslational, subcellular localization, and through their complexation with other proteins, it should be possible to affect specific KDAC activity through manipulation of endogenous signaling pathways. In this Minireview, we discuss our present knowledge of the cellular controls of KDAC activity and examples of their pharmacologic regulation.
Keywords: epigenetics, histone deacetylases (HDACs), homeostasis, lysine deacetylases (KDACs), multiprotein complexes, pharmacologic regulation, sirtuins
Introduction
Protein lysine acetylation, involving enzymatic transfer of an acetyl group from the cofactor acetyl coenzyme A (acetyl-CoA) to the terminal amine present on lysine side chains, is among the most important post-translational modifications of proteins.[1, 2] Catalyzed by lysine acetyltransferases, acetylation not only eliminates the normal positive charge present on the primary amine under physiological conditions, but also prevents alternative lysine modifications, including methylation, biotinylation, ubiquitination, SUMOylation, NEDDylation, glycation, among others.[3, 4] Given this panoply of lysine modifications, lysine acetylation can exert a host of effects on proteins, affecting protein structure and activity, protein–protein and protein– nucleic acid interactions, protein subcellular localization and trafficking, and subsequent protein modifications, and stability. Therefore, protein acetylation, one of the most common posttranslational modifications, is a major regulator of protein function in organisms ranging from bacteria to humans.[5, 6]
As might be expected for a regulatory protein modification, lysine acetylation is readily reversible. Deacetylations are carried out by a second group of enzymes, lysine deacetylases (KDAC), which are present in all organisms.[7, 8] Numerous lysine acetylases and deacetylases are typically present, even in the simplest of organisms, and the full spectrum of proteins affected by these enzymes is quite large, comprising thousands of different proteins in higher organisms.[9, 10] Some of the main targets of acetylation are the lysine residues present in the protruding N-terminal tails of nucleosomal histone proteins, whereby lysine deacetylation generally favors chromatin compaction and decreased levels of gene transcription, and lysine acetylation the converse.[11] Thus, KDACs are among the primary epigenetic repressors of gene expression in all organisms.
Elevated levels of histone deacetylation are evident in several chronic human diseases, particularly cancer and certain neurodegenerative diseases.[12–14] The molecular basis for their involvement spans from the transcriptional repression of critical tumor suppressor genes to inhibition of cellular responses to misfolded and aggregated protein accumulation.[15] This has led to the development of KDAC inhibitors as a therapeutic approach for these diseases.[16, 17] In contrast, activation of certain KDACs has been found to suppress aging and increase longevity in model organisms.[18–20] Thus, the search for KDAC activators is also being actively pursued.[21]
While some success has been achieved in the development and clinical application of both KDAC inhibitors and activators, much remains to be done. A significant limitation of existing therapeutics is their relative lack of isoform selectivity. This is to be expected given the ubiquitous nature of KDACs and their importance in multiple biological processes. While KDAC modulators with increased selectivity remain a worthwhile goal, there is an increasing appreciation that alternatives to direct catalytic effectors need be pursued. In this Minireview, we provide an overview of different human KDAC—their structural features, post-translational modifications, associations into functional complexes, and biological regulation. Emphasis is placed on those KDACs primarily involved in epigenetic regulation. Following that we explore studies demonstrating effects on specific KDACs through pharmacologic modulation of their regulatory pathways and provide an overview of future directions for selective KDAC modulation research.
KDAC Classifications
Globally, KDACs may be thought of as belonging to two superfamilies: those incorporating a bound Zn2+ ion and commonly referred to as “classic” histone deacetylases (HDACs) and those requiring a nicotinamide-adenine dinucleotide (NAD+) cofactor and are usually called sirtuins.[22, 23] We will continue to use this naming convention throughout the remainder of this Minireview when referring to specific deacetylases or subsets thereof, with KDACs being reserved for the set of all lysine deacetylases. In humans, 18 KDACs are known and these can be grouped into classes I–IV based on their structural homology (Figure 1). Class I members include human HDAC1, 2, 3, and 8. Class II members are organized into two subclasses: IIa (HDAC4, 5, 7, and 9), which possess highly homologous N-terminal extensions involved in specific protein–HDAC interactions, and IIb (HDAC6 and 10), which lack these extensions. Class III members include the human sirtuins SIRT1 through 7 and have been further subclassified based on their phylogeny. Class IV has a single member, HDAC11, which possesses features characteristic of both class I and class II HDAC. In each class, the primary region of homology is the conserved deacetylase domain.[10] This is aptly shown in Figure 1, where the percentage amino acid identity and similarity in the deacetylase domain is indicated between each KDAC class member and its archetype, either HDAC1 (class I), HDAC5 (class II), or SIRT1 (class III). As the class IV HDAC11 exhibits homology with both class I and class II HDACs, identity and similarity values are provided against both archetypes.
Figure 1.

Schematic representations of human histone deacetylases (HDACs) with the locations of their conserved deacetylase domains; their respective superfamilies are indicated through the color of their deacetylase domains. Both classical HDACs and sirtuins are organized by homology class. Shown are Zn2+ (blue) and NAD+-dependent (red) deacetylase domains, their percentage identity and similarity to a class archetype (e.g., HDAC1, HDAC5, SIRT1), and total protein length in amino acids. HDAC11 has homology to both HDAC1 and HDAC5; the latter values are shown in parentheses. Also indicated are the locations of validated KDAC post-translation modifications including acetylation (orange A, top), SUMOylation (green S), ubiquitination (black U), and phosphorylation (red inverted P, bottom).
KDAC Enzymatic Mechanisms
Classic HDAC enzymes catalyze the deacetylation of acetyl-l-lysine side chains in proteins through a general base-promoted nucleophilic attack by a water molecule bound to an activesite-coordinated Zn2+ (Figure 2a).[24, 25] The access to the active site is a tubular channel 11 Å in depth, which exhibits a high degree of sequence conservation among all HDACs. While this channel is sufficiently large to accept a variety of small molecules, proteins are the preferred substrates of KDACs given stabilizing interactions between their peptide backbone and amino acids present at the pocket exterior. Finally, while all human HDACs can function as lysine deacetylases, they do not catalyze deacetylation with equal efficiency. In vitro experiments with recombinant HDACs and a luminogenic peptide substrate provide a ranking of HDAC2 > 1 > 6 > 3 > 4, 5, 7, 8, 10 > 11 > 9 with regards to intrinsic deacetylase activity.[26] However, how representative these values are of protein activities in vivo remains questionable. Generally, class I HDACs are thought to exhibit greater deacetylase activity than class IIa HDACs, although this may be highly dependent on the context they reside within (e.g., whether they are part of a multiprotein complex).[27]
Figure 2.

Protein deacetylation by a) classic histone deacetylases (HDACs) or b) sirtuins.
Sirtuins catalyze the formation of an ADP-ribose-imidate intermediate between a chemically reactive enzyme-bound NAD+ and the N-ε-amino group of acetyl-l-lysine in proteins and generate nicotinamide (Figure 2b).[28] Subsequent attack by a base-activated water yields the novel product 2’-O-acetyl-adenosine-diphosphate-ribose (2’-AADPR), which can nonenzymatically interconvert to 3’-AADPR in solution.[29] As enzyme-bound NAD+ is a quite reactive species, it can potentially be attacked by nucleophiles other than peptidyl-acetyl-lysine.[30] In fact, sirtuins were originally reported to ADP-ribosylate proteins.[31, 32] However, other than the example of mitochondrial SIRT4 ADP-ribosylating glutamate dehydrogenase, most other examples occur at far lower frequency than observed deacety-lation events and might actually reflect nonenzymatic ADP-ribosyltransfer by intrinsically reactive AADPR.[33–35] Finally, sirtuins have different levels of reported intrinsic deacetylase activity, with SIRT1–3 being more catalytically active than SIRT6 and 7.[36] Notably, SIRT5 preferentially acts on acyl lysines other than acetyl-lysine, including malonyl-lysine and succinyl-lysine, which are among the known spectrum of acyl lysine modifications known to exist in vivo.[4, 37]
KDAC Tissue Specificity and Subcellular Localization
In general, KDACs are fairly ubiquitous with regards to their distribution in different tissues, with comparable levels of mRNA present in all cell types investigated.[38, 39] Information regarding their expression levels is provided in Table 1. However, this is not to say that different KDACs are expressed at equal levels. For example, levels of HDAC7, HDAC8, and SIRT2 mRNA are very high in most all cells, while those of HDAC4, 5, and 10 and SIRT7 are typically found at 100-fold lower levels. Likewise, there is some tissue specificity in expression observed for certain KDACs. Examples include increased levels of HDAC1 mRNA in more proliferative tissues (blood, small intestine, colon), HDAC4 in blood and brain, SIRT1 and 7 in blood, SIRT3 in brain, and SIRT2 in heart, striated muscle, and brain. These elevated levels most likely reflect an increased requirement for their encoded proteins in the proper function of these tissues. While this is a reasonable hypothesis, corresponding data regarding their absolute protein levels is at present incomplete, and a complete understanding of their full complement of substrates and their cellular acetylation status has yet to be achieved.[40, 41] mRNA levels, while presently tractable, is but a first step towards understanding KDAC levels and their biological roles in different tissues.
Table 1.
KDAC characteristics.
| KDAC | Tissue specificity[a] | Subcellular localization | Post-translational modifications, biological effects[b] |
|---|---|---|---|
| HDAC1 | u + +; blood, intestines, colon, thymus, thyroid + + + |
Nucleus | [P CK2A1 S421,423] enz↑, →Sin3A [SU (−SUSENP1) K444,476] enz↑ [UbCHFR,KCTD11] pro↓ |
| HDAC2 | u++ | Nucleus | [P CK2A1 S394] enz↑ [P S422,424] enz↓ |
| HDAC3 | u++ | Nucleus > cytoplasm | [P CK2A1 S424] enz↑ |
| HDAC8 | u +++ | Nucleus > cytoplasm | [P PKACA S39] enz↓ |
| HDAC4 | u ∼; blood, brain + | Nucleus/cytoplasm | [P CaMK4,SIK1 S246,467,632] →14-3-3, enz↓ |
| HDAC5 | u∼ | Nucleus/cytoplasm | [P AMPK,CaMK4 S259,498] →14-3-3, enz↓ [P AurB S278,279] 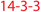 , enz↑ , enz↑ |
| HDAC7 | u +++ | Nucleus/cytoplasm | [P PKD1 S155,358,486] →cyto |
| HDAC9 | 4 | 4 | [P CaMK1 S220,451] →14-3-3, enz↓ [P PRK1 S253] 
|
| HDAC6 | u++ | Cytoplasm > nucleus | [P EGFR Y570] enz↓ |
| HDAC10 | u+ | Cytoplasm > nucleus | |
| HDAC11 | u∼ | Nucleus/cytoplasm | |
| SIRT1 | u ∼; blood + + | Nucleus/cytoplasm | [P JNK1 S27,47, T530] →nucl, enz↑ [P CDK1 T530, S540] →nucl, enz↑ [-SUSENP1] enz↓ |
| SIRT2 | u + + +; heart, muscle, brain + + + + |
Cytoplasm | [P CDK1,CDK2 S368] enz↓ [P S372] enz↑ |
| SIRT3 | u +; brain + + | Mitochondria | |
| SIRT4 | u+ | Mitochondria | |
| SIRT5* | u+ | Mitochondria | |
| SIRT6 | u+ | Nucleus | |
| SIRT7 | u ∼; blood + | Nucleolar |
u, ubiquitous. +, low level mRNA.
P, phosphorylation; SU, SUMOylation; Ub, ubiquitination. Enzyme involved indicated in superscript. Negative sign indicates removal of modification. Residue involved indicated if known. Enz, enzyme activity; pro, protein level; ↑, increased; ↓, decreased; →, associate with/transport to; 
 , does not associate with/transport to.
, does not associate with/transport to.
HDAC5 may not be a deacetylase but rather a related lysine demalonylase and desuccinylase.
Given their original definition as HDACs and the primary localization of histone proteins in chromatin, one might be led to believe that, at least with regards to HDACs, these are mostly nuclear proteins. At a first assumption level, this is generally correct.[42] Class I HDACs are primarily nuclear proteins, as are SIRT6 and 7. Information regarding the subcellular localization of KDACs is provided in Table 1.[43] However, it should also be noted that certain KDACs, including class IIb HDAC6 and 10 and SIRT2 are primarily cytoplasmic, while SIRT3–5 are located exclusively in mitochondria. Thus, it is highly unlikely that histone acetylation and epigenetic regulation is significantly affected by these KDACs. Most interesting are those KDACs, including HDAC3 and 8, most class IIa/IV HDAC, and SIRT1, that can shuttle between nuclear and cytoplasmic compartments. For these proteins, their subcellular localization is often controlled through post-translational modifications resulting from different signal transduction pathways and upstream cues. One example is the phosphorylation of conserved Ser/Thr residues upstream of the deacetylase domain in class IIa HDACs by Ca2+/calmodulin-dependent kinases, protein kinase D, or checkpoint kinase-1 and IIa HDAC sequestration in the cytoplasm by 14-3-3 proteins.[44–48] In addition, subcellular localization can be a primary means of affecting their biological function, either through limited their accessibility to partner proteins or target substrates.
KDAC Modifications and Effects
As important regulatory proteins, it is not surprising that KDACs can be subjected to many post-translational modifications. Primary among these are phosphorylations and various modifications of lysine residues, including acetylation, methylation, SUMOylation and ubiquitination.[42] Sites of reported post-translational modifications for the different KDACs are shown in Figure 1.[43, 49, 50] Emphasis has been made to include only those modification sites in human proteins that have been independently verified by multiple methods. While our current understanding of KDAC modifications is hardly exhaustive, it is nonetheless possible to ascertain certain trends from the available data. For example, phosphorylation sites in both classic HDACs and sirtuins are primarily located outside of their conserved deacetylase domains. Such is less consistent for other post-translational modifications, for example, acetylations and ubiquitinations, which can be found both within and outside of the conserved deacetylase domains. As might be expected from the high degree of homology observed between different members of a particular KDAC class, there is significant complementarity between the modification sites present in different KDACs. Examples include the cluster of phosphorylation sites located towards the C terminus of class I HDAC1 and 2 and the phosphorylation sites immediately downstream of the deacetylase domain in subclass I SIRT1 and 2. However, it is also quite evident from the available data that significant differences do exist between the post-translational modifications observed in different KDACs. These differences provide a possible means by which cells specifically regulate the activity of particular KDACs. In addition, these differences could provide a possible approach for the pharmacological control of specific KDAC levels and/or activity.
At present, our understanding as to the biological importance of specific post-translational modifications on KDACs is far from complete. For that subset of post-translational modifications that have been well characterized in human proteins by multiple methods, available information including modification, responsible enzyme, sites and their biological effects is provided in Table 1.[43, 49, 50] Given their considerable interest in the pharmaceutical field, most information regarding KDAC post-translational modifications involves phosphorylations.[42] These have been shown to increase the enzymatic activity of KDACs (e.g., HDAC1, HDAC6 and SIRT1), facilitating their association into multiprotein complexes (HDAC1 into mSin3), partitioning them into appropriate subcellular compartments (SIRT1 to the nucleus), and increasing protein stability (SIRT1).[51–56] Conversely, certain phosphorylation events can partition class IIa HDACs (HDAC4, 5, 7, 9) into the cytoplasm, where they are unable to interact with histones or be directly involved with epigenetic regulation, or inhibit KDAC association with substrates, thereby inhibiting their deacetylase activity.[44, 45, 57, 58]
Phosphorylation is not the only post-translational modification reported for KDACs. Others include lysine modifications such as SUMOylation, which typically facilitates nuclear transport and/or enzyme activation (HDAC1, SIRT1), and ubiquitination, which typically but not always results in increased protein degradation.[59–64] Curiously, while acetylation has also been reported for certain KDACs, its persistence and function beyond competing with other lysine modifications is as yet not fully understood.[65, 66]
KDAC Complexes and Substrates
In order to exert biological effects, KDACs must interact with their specific target acetylated proteins. This entails more than just being at the right place and time. With regards to epigenetic regulation, the desired outcome typically involves changing chromatin structure for a subset of genes, thereby affecting a change in a program of gene expression. Achieving such specificity requires site recognition beyond that which is possible with a single polypeptide. While several KDACs form simple complexes with sequence-specific DNA-binding transcription factors, several KDACs primarily function as parts of larger, multiprotein complexes.[67] This is well recognized for the essentially nuclear class I HDACs, including HDAC1–3. HDAC1 and 2 are members of the multiprotein complexes Sin3, Mi-2/NuRD, CoREST, CtBP, and MiDAC, whereas HDAC3 is a member of N-CoR/SMRT complexes.[7, 67–71] Constituents of these complexes include histone binding proteins (RBBP4/7), modified histone recognition proteins (ING1/2), methylated DNA recognition proteins (MBD2/3), histone demethylases (KDM1A), DNA methyltransferases (DNMT1), chromatin remodeling ATPases (CHD3/4), and transcriptional repressors (SIN3A/ B; p66A/B, NCoR), among others. Notably, these proteins have obvious functions in chromatin recognition and modification, as expected by the central role of these complexes in epigenetic regulation. Curiously, several of these complexes incorporate multiple KDAC, for example HDAC1 and 2 in Sin3, Mi-2/ NuRD and MiDAC complexes, HDAC1-3 in CoREST and CtBP. The need for redundancy in these cases is not fully known, although complexes containing HDAC3 and either HDAC7 or 9 may exist to provide more effective overall deacetylase activity, given the weak intrinsic catalytic activity observed for the latter class IIa HDACs.[27, 72]
Not all KDACs are members of multiprotein complexes or are multiprotein complex members at all times. This is quite evident with those KDACs that shuttle between different subcellular compartments. For example, the class IIa HDAC4, 5, and 7 can interact with the N-CoR complex when present in the nucleus but interact with 14-3-3 proteins once phosphorylated and present in the cytoplasm. Similarly, KDACs can form simple complexes with individual transcription factors, such as HDAC1 with Sp1 or E2F1/Rb and SIRT1 with p53, thereby affecting their stability, nuclear localization, or transcriptional activity with resulting downstream effects on specific programs of gene expression.[73–75] In fact, the full spectrum of interacting partners for individual KDACs is just beginning to be fully understood. Mass-spectrometry-based proteomic analysis of proteins associating with epitope-tagged KDACs and shRNA-based synthetic lethality genetic screens have only recently been applied towards identifying proteins that physically or functionally associate with individual KDACs.[67, 76] This has led to a wealth of data regarding the biological processes affected by individual KDACs. While chromatin modification and control of gene expression are major functions of many KDACs, even the known nuclear KDACs have significant functions in other processes, including cell cycle, RNA processing, ubiquitination control, signal transduction, protein and ion transport, and protein folding. Most important among all are KDAC roles in various metabolic processes. This would be highly expected, given the shared role of the metabolic cofactor NAD+ in sirtuin catalytic function. However, even ostensibly nuclear class I HDACs were also found to have significant roles in metabolic regulation, for example, regulation of adenosine monophosphate-activated protein kinase by HDAC1.[76]
KDAC Pharmacological Modulation
As both specific classic HDACs and sirtuins have been ascribed to various human diseases, this has led to the identification of pharmacological modulators of KDAC activity. In the case of HDACs involved in epigenetic regulation, diseases such as cancer and neurodegenerative diseases typically report increased HDAC activity. Thus, the search has been exclusively for HDAC inhibitors (HDACi).[24] In the case of SIRT1, augmenting its activity has been reported to increase lifespan in multiple organisms, while either SIRT activation or inhibition has been found to have antitumor activity in different cancer models. Presently both SIRT activators and inhibitors are being actively pursued.[21]
HDACi are all active site-binding competitive inhibitors, incorporating a Zn2+-chelating moiety, an 11 Å linker spanning the enzyme access channel, and a “cap” group interacting with the exterior of the protein. Chelating moieties explored include hydroxamates, benzamides or thiolate anions (Figure 3).[77–79]
Figure 3.

Histone deacetylase inhibitors (HDACi). Coordination of enzyme active site Zn2+ ion is indicated.
As the active site and access channel are highly conserved among HDACs, achieving specificity has historically been directed towards changes in cap groups, although changes in the chelating moiety (e.g., hydroxamate to aminobenzamide) can drive specificity for certain HDACs.[80, 81] Presently, paninhibition of class I HDACs by the carboxylate valproate, selective inhibition of HDAC1 and HDAC9 by the benzamide entinostat (MS-275), and preferential inhibition of HDAC1 and HDAC2 by romidepsin have been reported.[82, 83] Selective inhibition of HDAC8 and HDAC6 has also been described, but these inhibitors are also found not to affect general histone acetylation levels, suggesting that these HDACs may not be greatly involved in epigenetic regulation.[84, 85] Rather, the clinically more useful HDACi have generally been pan-HDACi, including vorinostat and romidepsin, although these can be fraught with untoward side effects, believed to be a consequence of their limited selectivity.[86–88]
Based on their differing cofactor requirements and chemistry, sirtuins are not susceptible to the chemical inhibitors used to target classical HDACs. Rather, both the cofactor NAD+ and the reaction product nicotinamide (NAM) greatly affect the rate of lysine deacetylation by sirtuins, making sirtuins directly responsive to cellular levels of these important products of metabolism and energy homeostasis.[89, 90] Additional modulators of sirtuin activity have been identified and are currently under investigation. These include sirtuin inhibitors sirtinol, salermide, EX-527, and AGK-2, and sirtuin activators resveratrol and imidazo[1,2-b] thiazoles SRT1720 and SRT2104 (Figure 4a, b).[91–98] Specificity for individual sirtuins has yet to be fully investigated. However, SIRT1 and 2 are often found to both be affected by different inhibitors, with EX-527 exhibiting a high degree of specificity for SIRT1 and AGK-2 preferentially inhibiting SIRT2.[21, 93, 94] From a structural perspective, sirtuin inhibitors have only the slightest resemblance to nicotinamide, and their mechanisms of action are not fully understood, although it appears that β-napthols like sirtinol and salermide do not bind to the NAD+ site but rather affect substrate binding at a proximal site and indoles like EX-527 stabilize the closed enzyme conformation and prevent product release.[99, 100] However, circumstances are even more clouded with sirtuin activators, suggesting that they may not actually involve direct effects on sirtuin enzymatic activity but rather on ancillary effects, including post-translational modifications affecting protein trafficking/stability or altering cellular NAD+ levels.[101]
Figure 4.

Sirtuin a) inhibitors and b) activators.
Alternative Regulation of KDAC Activity
Example 1: Selective HDAC1 inhibition
HDAC1 and its close compatriot, HDAC2, are arguably the most important deacetylases involved in epigenetic regulation.[15, 102] Invariably nuclear in localization, their primary targets are acetylated histones, although many nonhistone nuclear proteins have also been identified.[16, 103] Homozygous germline deletion of HDAC1 is embryonic lethal at day 9.5, resulting in proliferation defects.[104] Notably, loss of HDAC1 function cannot be compensated for by overexpression of alternative HDACs, including HDAC2 or 3.[105] Similarly, cell type-specific knockouts of HDAC1 alone or with HDAC2 demonstrated increased proliferation and decreased differentiation, often leading to early lethality.[3, 102]
HDAC1 overexpression is observed in several cancers, including colorectal, gastric, and pancreatic, and increased expression typically correlates with decreased survival.[106–110] However, this was not the case with breast cancer, wherein increased HDAC1 expression correlated with elevated expression of estrogen and progesterone receptors, which rendered the cancer more amenable to treatment.[111] HDAC1 primary mechanism of action in cancer cells is thought to arise through its suppression of cyclin-dependent kinase (CDK) inhibitors p21WAF1/CIP1 and p27KIP1 expression, thereby resulting in a loss of cell cycle controls and promoting increased proliferation.[104, 112] HDAC1 overexpression also can suppress apoptosis by intrinsic but not extrinsic pathways and suppress autophagy, rendering cancer cells refractory to many anticancer treatments.[113–116] Alternatively, HDAC1 as part of multiprotein complexes can be recruited by chromosomal translocation-derived chimeric transcription factors to downregulate genes involved in differentiation. Examples include RARα/PLZF in acute promyelocytic leukemia and AML1-ETO and CBFb-SMMHC in acute myeloid leukemia.[117–120]
Suppressing HDAC1 activity has been an important goal in cancer chemotherapy research. To date, those HDACi that have been approved for clinical use (e.g., vorinostat and romidepsin, for the treatment of cutaneous T cell lymphoma) and those most promising agents in advanced clinical trials are all HDAC1 inhibitors.[121, 122] However, they are also either pan-HDACi or class I-selective HDACi and have exhibited multiple significant adverse effects, including gastrointestinal symptoms, bone marrow suppression, cardiac toxicity, thrombocytopenia, neutropenia, diarrhea, vomiting, and fatigue, albeit often at decreased severity as compared with other antineoplastic agents.[88] This is understandable given the importance of class I HDACs in multiple biological processes and in multiple cell types. Several laboratories have sought to design an HDAC1-specific inhibitor. While HDACi with some preference for HDAC1 compared with other class I HDACs have been described (e.g., entinostat), this goal has been difficult to achieve, given the structural similarity in the active sites of all HDACs and especially between HDAC1 and 2.[24, 25]
Notably, HDACs do not often exist as independent molecules but as part of multiprotein complexes. This is especially true for HDAC1, which is often found in chromatin remodeling complexes such as CoREST, mSin3, and NuRD.[7, 68–70] These typically consist of a stable core consisting of HDAC1, HDAC2, and the histone-binding proteins RBBP4 and RBBP7, as well as additional more labile members that facilitate complex association with specific sites in chromatin.[67] As it is through these complexes that HDAC1 acts epigenetically upon specific programs of gene expression, both in normal cells and in cancer, investigators have begun to use isolated chromatin remodeling complexes to identify molecules that selectively bind to them and interfere with their deacetylase activities.[123] While these studies are at an early stage, such an approach offers the potential of identifying more functionally selective HDACi. However, this will only be true if the search is expanded to include molecules that disrupt complex function and not just those that block acetyl-lysine access to the enzyme active site, as this may not be sufficiently altered in chromatin remodeling complexes to allow selective recognition by small molecules.
HDAC1 does not originate as a member of multiprotein complexes, and nor is its residence among them permanent. Formation of these complexes is a post-translational event and, while relatively stable entities, their maintenance can be affected by cellular cues. Unsurprisingly, various signal transduction pathways and post-translational modifications can regulate the homeostasis of HDAC1-containing complexes. Perhaps the best example of this is phosphorylation of HDAC1 at Ser 421 or Ser 423 by casein kinase II (CK2) or cyclic AMP-dependent protein kinase (PKA), leading to recruitment into repressor complexes and increased activity.[51, 124] Such phosphorylation events can be reversed; for example, genotoxic stress activation of ataxia telangiectasia mutated (ATM) leads to activation of protein phosphatase 1 (PP1), thereby removing the Ser 421/423 phosphorylations from HDAC1 and causing its dissociation from Rb complexes.[125]
Phosphorylation changes in and of themselves can affect HDAC activities and delivery but these effects can be shortlived and are readily reversible. More robust changes require additional modifications that affect overall cellular levels of HDAC proteins. An example of this is ubiquitination, the ligation of a 76-amino-acid polypeptide onto lysine residues of target proteins.[126] Polyubiquitination, in which sequential polymerization of ubiquitin occur through their Lys48 residues, is a marker for protein degradation by the 26S proteasome. Notably, ubiquitination of HDAC1 at any of several lysine residues promotes it degradation.[60] Several ubiquitin E3 ligases have been identified that ubiquitinate HDAC1, including CHFR, KCTD11, and MDM2.[62, 63, 127, 128] Recovery of HDAC activity necessitates minimally translation of existing HDAC1 mRNA and reconstitution of HDAC1-containing complexes—lengthy events compared with phosphorylation changes on the time scale of most cells.
Selective cellular depletion of HDACs was well demonstrated by the specific loss of cellular HDAC1 protein following exposure of cells to proinflammatory cytokines. Activation of the NF-κB signal transduction pathway by cytokines, such as TNFα, IL-1β or LPS, caused a substantial cellular depletion of HDAC1 in several cancer cell lines.[129] Cellular levels of other class I HDACs were unaffected. Depletion occurred through ubiquitination of HDAC1 and subsequent proteasomal degradation. Kinase activity from the IκB kinase (IKK) signalsome was required for this degradation to occur, although it was not shown to directly phosphorylate HDAC1. Interestingly, HDAC1-containing species normally associated with the p21WAF1/CIP1 promoter region, presumably Sp1-localized mSin3A complexes or the like, were absent following proinflammatory cytokine treatment. Such was evident by the local loss of both HDAC1 and HDAC2, whereas overall cellular HDAC2 levels were relatively unaffected. These data are consistent with a model in which IKK signaling labialized HDAC1-containing multiprotein complexes, leading to HDAC1 polyubiquitination and proteasomal degradation. Similar results were observed following cellular treatment with the proinflammatory cytokine IFNg and presumably involve JAK/STAT1 signaling.[130] At present, the exact proteins responsible for these HDAC1 post-translational modifications, presumably one or more kinases and/or phosphatases and a ubiquitin E3 ligase, remain unknown.
Selectively depleting HDAC1 levels through proinflammatory cytokine exposure might be feasible, but highly undesirable from a cancer therapy point of view. Inflammation, especially mediated through NF-κB signaling, is generally thought to be a critical component of cancer progression, fostering proliferation, antiapoptosis, angiogenesis, and metastasis.[131, 132] Countering inflammation is also a valuable facet of cancer prevention and treatment. As part of our characterization of NF-κB signaling involved in HDAC1 depletion, we investigated selective small-molecule inhibitors. Curiously, we discovered that the reported IKK2-specific kinase inhibitor parthenolide was capable of inducing HDAC1 depletion in the absence of proinflammatory cytokines.[133] This effect was specific for HDAC1; other class I and class II HDACs investigated were not affected. As before, HDAC1 depletion involved polyubiquitination and proteasomal degradation, and required sequences in the C terminus (432–482 aa) of HDAC1. Yet while dissociation of transcriptionally repressive complexes associated with the p21WAF1/CIP1 promoter was observed, these effects did not require the IKK signalsome or other members of the NF-κB signaling pathway. Rather, the genotoxic stress mediator ATM was required for parthenolide-induced HDAC1 depletion. Given what is now known regarding ATM and HDAC1, we now believe that parthenolide activates ATM, which in turn phosphorylates and activates PP1. Activated PP1 facilitates the removal of Ser 421/423 phosphorylations from HDAC1, thereby leading to its depletion from multiprotein complexes.[134] Parthenolide-activated ATM also directly phosphorylates the tumor suppressor p53 and the E3 ligase MDM2, thereby dissociating this complex.[135] With MDM2 no longer occupied with p53, it is interesting to speculate that it can then be responsible for de-phosphorylated HDAC1 ubiquitination and degradation, much as has been observed for MDM2-mediated HDAC1 ubiquitination when complexed with the androgen receptor transcription factor.[63] It should be noted that while sesquiterpene lactones such as parthenolide cause multiple cellular changes, including glutathione depletion, increasing reactive oxygen species, and apoptosis induction, the specific depletion of HDAC1 by parthenolide might not be a direct consequence of these effects.[136] Similarly, other sesquiterpene lactones tested do not promote specific HDAC1 depletion nor do other molecules that activate ATM through DNA-damage induction (E. Chanchorn, unpublished observations). HDAC1 depletion by parthenolide likely requires coordinate effects on multiple pathways and proteins. Identification of additional molecules that act similarly will require careful assay design to observe such cooperativity in effects.
Example 2: Selective SIRT1 activation
SIRT1 is probably one of the most important sirtuins involved in epigenetic regulation.[8, 21, 137] As a NAD+-dependent deacetylase, it communicates the cellular metabolic state to the regulation of targeted gene expression. Mainly resident nuclear, the primary targets of SIRT1 include acetylated histone residues, including H3K9, H3K56, and H4K16, as well as several nonhistone nuclear proteins, including transcription factors p53, FOXO1/3/ 4, and PGC-1α.[87] Mice that possess homozygous germline deletion of SIRT1 are smaller at birth and often die shortly there-after.[138] Whole-body SIRT1-overexpressing mice exhibited several positive phenotypes, including protection against development of glucose intolerance, obesity, and fatty liver disease induced by diet and aging, as well as reductions in several age-related and metabolic syndrome-associated cancers.[139–141] Such is very similar to results in lower eukaryotes demonstrating increased longevity with activation of their SIRT1 homologues.
SIRT1 has a mixed story with regards to its role in cancer. SIRT1 is well known to deacetylate p53, thereby leading to its inactivation.[142] Given the important role of p53 as a tumor suppressor, this would suggest that SIRT1 acts as an oncogene, promoting cell proliferation and suppressing apoptosis. Similarly, the effects of SIRT1 on chromatin promote the silencing of important tumor suppressors, including CDH1, GATA5, and SFRP1, again supporting the role of SIRT1 as an oncogene.[143] However, SIRT1 is also known to deacetylate the transcription factor proto-oncogene c-Myc, thereby inhibiting its function, as well as deacetylating and inhibiting HIF-1α, an important promoter of cell growth and angiogenesis under hypoxic conditions.[144, 145] Knockout and overexpression studies in mice tend to favor a tumor suppressor role for SIRT1 in some, but not all, cancers.[146] Examination of tumor samples from a variety of human cancers has also shown mixed results, with both elevated and decreased levels of SIRT1 being reported in different cancers.[147–149] Thus, the role of SIRT1 in individual cancers remains uncertain and both SIRT1 activation and inhibition are being explored for the treatment of cancer.
With regards to the well-recognized roles of SIRT1 in aging, metabolic diseases and age-related cancers, efforts have been made to identify compounds that activate endogenous SIRT1 activity. Foremost among these is resveratrol, a polyphenolic flavonoid antioxidant found in grapes, red wine and other plant products.[150] Resveratrol was originally identified in a high-throughput biochemical screen of compounds that promoted SIRT1-mediated deacetylation of a fluorescent peptide substrate.[151] Evidence was obtained indicating that resveratrol decreased the Km value of both the peptide substrate and NAD+ without affecting the Vmax value of the reaction, consistent with it being an allosteric activator of SIRT1. This effect was specific for SIRT1 and was not observed for SIRT2.[96] However, later studies found that resveratrol did not affect the binding of native peptide substrates, suggesting that the stabilization could be an artefact of the coumarin fluorophore used in the original assays.[95, 96, 152] Resveratrol was found to significantly extend the lifespan of yeast, the nematode Caenorhabditis elegans, flies, and certain mouse model systems, consistent with sirtuin overexpression studies.[151, 153–155] However, resveratrol interacts with many different molecules in vivo and affects a multitude of different biological processes, including those involving mitochondrial biogenesis, various metabolic pathways, and inflammation.[150] Teasing out a single cause-and-effect might not be possible for this molecule. Most interesting are recent studies demonstrating that 5’ AMP-activated protein kinase (AMPK), an enzyme critically involved in metabolism control, is activated by resveratrol, potentially through direct inhibition of phosphodiesterase 4 and/or mitochondrial ATP synthase.[156–158] AMPK activation results in increased levels of NAD+ and thus could activate SIRT1, albeit indirectly.[159]
Additional small-molecule activators of SIRT1 have now been synthesized by Sirtris Pharmaceuticals and other companies.[21] Perhaps the best characterized is the imidazo[1,2-b] thiazole SRT1720.[97] Preclinical studies with this molecule demonstrated significantly improved metabolic parameters in a mouse model of diet-induced obesity, decreased blood glucose in a leptin-deficient ob/ob mouse model, and improved insulin sensitivity in a Zucker fa/fa model of diabetes. Most important, treatment of animals with SRT1720 demonstrated increased acetylation of known SIRT1 targets, including FOXO1, p53, and PGC-1α.[101] Several cell-based siRNA/shRNA SIRT1 knockdown studies have been performed that support the contention that the biological effects of SRT1720 are mediated, at least in part, through SIRT1.[101, 160] However, in vitro studies have not always shown high-affinity direct binding between SRT1720 and SIRT1.[97, 161] Additionally, like resveratrol, SRT1720 is also well recognized to have potent anti-inflammatory properties, decreasing levels of inflammatory cytokines like TNFα and IL-6, and the chemokine MCP-1.[160] Thus, a clear causal connection between direct pharmacologic SIRT1 activation and various biological effects remains elusive.
Outlook
It is well recognized that KDACs are a worthwhile target for pharmacologic modulation in several chronic human diseases, including diabetes, aging, various neurodegenerative disorders, and cancer. To date, some success has been observed in clinical trials with different KDAC modulators and two HDACi, vorinostat and romidepsin, have been approved for the treatment of one form of cancer, cutaneous T cell lymphoma. Clinically successful KDAC modulators exhibit many undesirable side effects, some of which can be life threatening. This is understandable given that the typical KDAC modulator is not highly specific, affecting multiple KDAC isoforms in both normal and disease cells. Ideally what is desired is a KDAC modulator that is specific for a critically important KDAC present in a unique context in the targeted cell. One example might be a HDACi specific for the NCoR/SMRT multiprotein complexes repressing retinoic acid-inducible myeloid differentiation gene expression through their association with the RARα/PML chimeric transcription factor product of a t(15;17)(q22;q21) chromosomal translocation commonly present in acute promyleocytic leukemia neutrophilic promyleocytes.[162] Alternatively, even a celltype or isoform-specific KDAC modulator could be sufficiently selective such that its benefits could outweigh those potentially decreased side effects that may still arise. The search for more specific/selective KDAC modulators presently continues.
Obviously, it is not a simple endeavor to design an isoform-specific KDAC modulator, let alone one that is also cell type or context specific. Humans have 18 different KDACs expressed ubiquitously throughout their bodies, most of which are performing important biological functions ranging from regulating specific gene expression to controlling specific protein stability and activity. These can be subdivided into two large families, 11 classic HDACs and seven sirtuins, based on the structure of their active sites and the chemistry used to catalyze acetyl-lysine hydrolysis. Present KDAC modulators typically bind at or near these enzyme active sites. Thus, given the high degree of structural homology between different HDAC or sirtuin active sites, it is quite difficult to achieve isoform selectivity with small molecules that interact with only a small, conserved, surface on these proteins. Complicating this is the observation that many KDACs do not predominantly exist in cells as isolated proteins but rather as members of large, multiprotein complexes. It has recently been recognized that these multiprotein complexes should be the targets of drug discovery and not naked KDAC alone.[123] However, the limitations of successful drug design suggest that small molecules will still find it difficult to discriminate between different KDACs, let alone different multiprotein complexes, so long as these drugs are also targeting KDAC active sites.
While certain KDACs spend most of their existence in tight complex with other proteins, for example the core deacetylase complex consisting of HDAC1, HDAC2, RBBP4, and RBBP7 present in the epigenetic regulatory multiprotein complexes CoREST, mSin3, and NuRD, this is definitely not always the case.[67] KDACs need to be initially loaded into these complexes and, like all things in biology, these complexes have finite lifespans. KDAC association in multiprotein complexes is dynamic and is affected by post-translational modifications, such as phosphorylations, which can readily change as a result of temporal or environmental cues. We have observed during the course of cell treatment with proinflammatory cytokines that HDAC1 levels were severely depleted, including those HDAC1 proteins present in multiprotein complexes.[129] This would suggest that pharmacologic modulation of the signaling pathways responsible for specific multiprotein complex homeostasis might be a more facile approach towards achieving specificity in targeting individual KDACs and their biological functions. A schematic model outlining this approach is shown in Figure 5. Note that such an approach also offers the potential of achieving cell-type specificity in regulating KDAC activity, given that their interacting partners and regulatory pathways might differ in different cell types.
Figure 5.

Schematic model of specific KDAC targeting through pharmaco-logic modulation of its regulatory pathway. Example shown is postulated from the depletion of HDAC1 following cell treatment with the proinflam-matory cytokine TNFa.
It is the associations between KDACs and other proteins that confer the unique specificity of their biological functions. Thus, these associations should be the target of drug interventions. While a direct approach targeting protein–protein interfaces might potentially be feasible, an alternative approach directed towards the pharmacologic regulation of existing signaling pathways is more akin to the biology that regulates KDAC activity in vivo and is a worthwhile avenue to pursue for the treatment of many chronic human diseases. Such would be analogous to high-dose retinoic acid treatment, which promotes the dissociation of the HDAC3-containing NCoR/SMRT multiprotein complex from its directing oncogenic transcription factor RARα/PML, which has been found to be an effective treatment for many patients with acute promyelocytic leukemia.[163]
Acknowledgements
M.V.D. is supported by the US National Institutes of Health (NIH) (GM 104833) and by the Kennesaw State University Foundation (Kennesaw, GA, USA). The author thanks ChemAxon (Budapest, Hungary) for a license to use MarvinSketch 6.1.2.
References
- 1.Roth SY, Denu JM, Allis CD. Annu. Rev. Biochem. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- 2.Aka JA, Kim GW, Yang XJ. Handb. Exp. Pharmacol. 2011;206:1–12. doi: 10.1007/978-3-642-21631-2_1. [DOI] [PubMed] [Google Scholar]
- 3.Yang XJ, Seto E. Mol. Cell. 2008;31:449–461. doi: 10.1016/j.molcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu Z, Wang Y, Gao T, Pan Z, Cheng H, Yang Q, Cheng Z, Guo A, Ren J, Xue Y. Nucleic Acids Res. 2014;42:D531–D536. doi: 10.1093/nar/gkt1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 6.Eichler J, Adams MW. Microbiol. Mol. Biol. Rev. 2005;69:393–425. doi: 10.1128/MMBR.69.3.393-425.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang XJ, Seto E. Nat. Rev. Mol. Cell Biol. 2008;9:206–218. doi: 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Imai S, Guarente L. Trends Pharmacol. Sci. 2010;31:212–220. doi: 10.1016/j.tips.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang L, Tang Y, Cole PA, Marmorstein R. Curr. Opin. Struct. Biol. 2008;18:741–747. doi: 10.1016/j.sbi.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gregoretti IV, Lee YM, Goodson HV. J. Mol. Biol. 2004;338:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 11.Haberland M, Montgomery RL, Olson EN. Nat. Rev. Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fraga M, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, Iyer N, Perez-Rosado A, Calvo E, Lopez J, Cano A, Calasanz M, Colomer D, Piris M, Ahn N, Imhof A, Caldas C, Jenuwein T, Esteller M. Nat. Genet. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- 13.Barneda-Zahonero B, Parra M. Mol. Oncol. 2012;6:579–589. doi: 10.1016/j.molonc.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Donmez G, Outeiro TF. EMBO Mol. Med. 2013;5:344–352. doi: 10.1002/emmm.201302451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Witt O, Deubzer HE, Milde T, Oehme I. Cancer Lett. 2009;277:8–21. doi: 10.1016/j.canlet.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 16.Minucci S, Pelicci P. Nat. Rev. Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 17.Bolden JE, Peart MJ, Johnstone RW. Nat. Rev. Drug Discovery. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 18.Sinclair DA, Guarente L. Cell. 1997;91:1033–1042. doi: 10.1016/s0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- 19.Tissenbaum HA, Guarente L. Nature. 2001;410:227–230. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- 20.Rogina B, Helfand S. Proc. Natl. Acad. Sci. USA. 2004;101:15998–16003. doi: 10.1073/pnas.0404184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blum CA, Ellis JL, Loh C, Ng PY, Perni RB, Stein RL. J. Med. Chem. 2011;54:417–432. doi: 10.1021/jm100861p. [DOI] [PubMed] [Google Scholar]
- 22.De Ruijter A, Van Gennip A, Caron H, Kemp S, Van Kuilenburg A. Biochem. J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sauve AA, Wolberger C, Schramm VL, Boeke JD. Annu. Rev. Biochem. 2006;75:435–465. doi: 10.1146/annurev.biochem.74.082803.133500. [DOI] [PubMed] [Google Scholar]
- 24.Bertrand P. Eur. J. Med. Chem. 2010;45:2095–2116. doi: 10.1016/j.ejmech.2010.02.030. [DOI] [PubMed] [Google Scholar]
- 25.Lombardi PM, Cole KE, Dowling DP, Christianson DW. Curr. Opin. Struct. Biol. 2011;21:735–743. doi: 10.1016/j.sbi.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Halley F, Reinshagen J, Ellinger B, Wolf M, Niles AL, Evans NJ, Kirkland TA, Wagner JM, Jung M, Gribbon P, Gul S. J. Biomol. Screening. 2011;16:1227–1235. doi: 10.1177/1087057111416004. [DOI] [PubMed] [Google Scholar]
- 27.Fischle W, Dequiedt F, Hendzel M, Guenther M, Lazar M, Voelter W, Verdin E. Mol. Cell. 2002;9:45–57. doi: 10.1016/s1097-2765(01)00429-4. [DOI] [PubMed] [Google Scholar]
- 28.Sauve AA. Biochim. Biophys. Acta Proteins Proteomics. 2010;1804:1591–1603. doi: 10.1016/j.bbapap.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jackson MD, Denu JM. J. Biol. Chem. 2002;277:18535–18544. doi: 10.1074/jbc.M200671200. [DOI] [PubMed] [Google Scholar]
- 30.Tanner K, Landry J, Sternglanz R, Denu J. Proc. Natl. Acad. Sci. USA. 2000;97:14178–14182. doi: 10.1073/pnas.250422697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frye R. Biochem. Biophys. Res. Commun. 1999;260:273–279. doi: 10.1006/bbrc.1999.0897. [DOI] [PubMed] [Google Scholar]
- 32.Tanny J, Dowd G, Huang J, Hilz H, Moazed D. Cell. 1999;99:735–745. doi: 10.1016/s0092-8674(00)81671-2. [DOI] [PubMed] [Google Scholar]
- 33.Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ, Valenzuela DM, Yancopoulos GD, Karow M, Blander G, Wolberger C, Prolla TA, Weindruch R, Alt FW, Guarente L. Cell. 2006;126:941–954. doi: 10.1016/j.cell.2006.06.057. [DOI] [PubMed] [Google Scholar]
- 34.Kowieski TM, Lee S, Denu JM. J. Biol. Chem. 2008;283:5317–5326. doi: 10.1074/jbc.M707613200. [DOI] [PubMed] [Google Scholar]
- 35.Cervantes-Laurean D, Jacobson EL, Jacobson MK. J. Biol. Chem. 1996;271:10461–10469. doi: 10.1074/jbc.271.18.10461. [DOI] [PubMed] [Google Scholar]
- 36.Michishita E, Park J, Burneskis J, Barrett J, Horikawa I. Mol. Biol. Cell. 2005;16:4623–4635. doi: 10.1091/mbc.E05-01-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, Kim J, Woo J, Kim JH, Choi BH, He B, Chen W, Zhang S, Cerione RA, Auwerx J, Hao Q, Lin H. Science. 2011;334:806–809. doi: 10.1126/science.1207861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takeda J, Yamasaki C, Murakami K, Nagai Y, Sera M, Hara Y, Obi N, Habara T, Gojobori T, Imanishi T. Nucleic Acids Res. 2013;41:D915–D919. doi: 10.1093/nar/gks1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu C, MacLeod I, Su AI. Nucleic Acids Res. 2013;41:D561–D565. doi: 10.1093/nar/gks1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kolker E, Higdon R, Haynes W, Welch D, Broomall W, Lancet D, Stanberry L, Kolker N. Nucleic Acids Res. 2012;40:D1093–D1099. doi: 10.1093/nar/gkr1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang M, Weiss M, Simonovic M, Haertinger G, Schrimpf SP, Hengartner MO, von Mering C. Mol. Cell. Proteomics. 2012;11:492–500. doi: 10.1074/mcp.O111.014704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sengupta N, Seto E. J. Cell. Biochem. 2004;93:57–67. doi: 10.1002/jcb.20179. [DOI] [PubMed] [Google Scholar]
- 43.Uni Prot Consortium. Nucleic Acids Res. 2011;39(Suppl. 1):D214–D219. [Google Scholar]
- 44.Yang XJ, Gregoire S. Mol. Cell. Biol. 2005;25:2873–2884. doi: 10.1128/MCB.25.8.2873-2884.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Verdin E, Dequiedt F, Kasler H. Trends Genet. 2003;19:286–293. doi: 10.1016/S0168-9525(03)00073-8. [DOI] [PubMed] [Google Scholar]
- 46.McKinsey TA, Zhang CL, Lu J, Olson EN. Nature. 2000;408:106–111. doi: 10.1038/35040593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vega R, Harrison B, Meadows E, Roberts C, Papst P, Olson E, McKinsey T. Mol. Cell. Biol. 2004;24:8374–8385. doi: 10.1128/MCB.24.19.8374-8385.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim M, Kim H, Brown AL, Lee M, Bae Y, Park J, Kwak J, Chung JH, Yun J. Exp. Mol. Med. 2007;39:205–212. doi: 10.1038/emm.2007.23. [DOI] [PubMed] [Google Scholar]
- 49.Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, Latham V, Sullivan M. Nucleic Acids Res. 2012;40:D261–D270. doi: 10.1093/nar/gkr1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lane L, Argoud-Puy G, Britan A, Cusin I, Duek PD, Evalet O, Gateau A, Gaudet P, Gleizes A, Masselot A, Zwahlen C, Bairoch A. Nucleic Acids Res. 2012;40:D76–D83. doi: 10.1093/nar/gkr1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pflum M, Tong J, Lane W, Schreiber S. J. Biol. Chem. 2001;276:47733–47741. doi: 10.1074/jbc.M105590200. [DOI] [PubMed] [Google Scholar]
- 52.Wen Y, Perissi V, Staszewski L, Yang W, Krones A, Glass C, Rose-nfeld M, Seto E. Proc. Natl. Acad. Sci. USA. 2000;97:7202–7207. doi: 10.1073/pnas.97.13.7202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA. Cell. 2007;129:1351–1363. doi: 10.1016/j.cell.2007.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ford J, Ahmed S, Allison S, Jiang M, Milner J. Cell Cycle. 2008;7:3091–3097. doi: 10.4161/cc.7.19.6799. [DOI] [PubMed] [Google Scholar]
- 55.Sasaki T, Maier B, Koclega KD, Chruszcz M, Gluba W, Stukenberg PT, Minor W, Scrable H. PLoS One. 2008;3:e4020. doi: 10.1371/journal.pone.0004020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nasrin N, Kaushik VK, Fortier E, Wall D, Pearson KJ, de Cabo R, Bordone L. PLoS One. 2009;4:e8414. doi: 10.1371/journal.pone.0008414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yuan F, Xie Q, Wu J, Bai Y, Mao B, Dong Y, Bi W, Ji G, Tao W, Wang Y, Yuan Z. J. Biol. Chem. 2011;286:6940–6945. doi: 10.1074/jbc.M110.182543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Back JH, Rezvani HR, Zhu Y, Guyonnet-Duperat V, Athar M, Ratner D, Kim AL. J. Biol. Chem. 2011;286:19100–19108. doi: 10.1074/jbc.M111.240598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kirsh O, Seeler J, Pichler A, Gast A, Müller S, Miska E, Mathieu M, Harel-Bellan A, Kouzarides T, Melchior F, Dejean A. EMBO J. 2002;21:2682–2691. doi: 10.1093/emboj/21.11.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.David G, Neptune M, DePinho R. J. Biol. Chem. 2002;277:23658–23663. doi: 10.1074/jbc.M203690200. [DOI] [PubMed] [Google Scholar]
- 61.Yang Y, Fu W, Chen J, Olashaw N, Zhang X, Nicosia SV, Bhalla K, Bai W. Nat. Cell Biol. 2007;9:1253–1262. doi: 10.1038/ncb1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gaughan L, Logan I, Neal D, Robson C. Nucleic Acids Res. 2005;33:13–26. doi: 10.1093/nar/gki141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oh YM, Kwon YE, Kim JM, Bae SJ, Lee BK, Yoo SJ, Chung CH, Deshaies RJ, Seol JH. Nat. Cell Biol. 2009;11:295–302. doi: 10.1038/ncb1837. [DOI] [PubMed] [Google Scholar]
- 64.Hook S, Orian A, Cowley S, Eisenman R. Proc. Natl. Acad. Sci. USA. 2002;99:13425–13430. doi: 10.1073/pnas.172511699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Luo Y, Jian W, Stavreva D, Fu X, Hager G, Bungert J, Huang S, Qiu Y. J. Biol. Chem. 2009;284:34901–34910. doi: 10.1074/jbc.M109.038356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu Y, Peng L, Seto E, Huang S, Qiu Y. J. Biol. Chem. 2012;287:29168–29174. doi: 10.1074/jbc.M112.371120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Joshi P, Greco TM, Guise AJ, Luo Y, Yu F, Nesvizhskii AI, Cristea IM. Mol. Syst. Biol. 2013;9:672. doi: 10.1038/msb.2013.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang Y, Iratni R, Bromage HE, Tempst P, Reinberg D. Cell. 1997;89:357–364. doi: 10.1016/s0092-8674(00)80216-0. [DOI] [PubMed] [Google Scholar]
- 69.Zhang Y, Ng H, Erdjument-Bromage H, Tempst P, Bird A, Reinberg D. Genes Dev. 1999;13:1924–1935. doi: 10.1101/gad.13.15.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.You SA, Tong J, Grozinger C, Schreiber S. Proc. Natl. Acad. Sci. USA. 2001;98:1454–1458. doi: 10.1073/pnas.98.4.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li J, Wang J, Wang J, Nawaz Z, Liu J, Qin J, Wong J. EMBO J. 2000;19:4342–4350. doi: 10.1093/emboj/19.16.4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fischle W, Dequiedt F, Fillion M, Hendzel MJ, Voelter W, Verdin E. J. Biol. Chem. 2001;276:35826–35835. doi: 10.1074/jbc.M104935200. [DOI] [PubMed] [Google Scholar]
- 73.Doetzlhofer A, Rotheneder H, Lagger G, Koranda M, Kurtev V, Brosch G, Wintersberger E, Seiser C. Mol. Cell. Biol. 1999;19:5504–5511. doi: 10.1128/mcb.19.8.5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Magnaghi-Jaulin L, Groisman R, Naguibneva I, Robin P, Lorain S, Le Villain J, Troalen F, Trouche D, Harel-Bellan A. Nature. 1998;391:601–605. doi: 10.1038/35410. [DOI] [PubMed] [Google Scholar]
- 75.Vaziri H, Dessain S, Eagon E, Imai S, Frye R, Pandita T, Guarente L, Weinberg R. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 76.Lin Y, Kiihl S, Suhail Y, Liu S, Chou Y, Kuang Z, Lu J, Khor CN, Lin C, Bader JS, Irizarry R, Boeke JD. Nature. 2012;482:251–255. doi: 10.1038/nature10804. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 77.Vannini A, Volpari C, Filocamo G, Casavola E, Brunetti M, Renzoni D, Chakravarty P, Paolini C, De Francesco R, Gallinari P, Steinkuhler C, Di Marco S. Proc. Natl. Acad. Sci. USA. 2004;101:15064–15069. doi: 10.1073/pnas.0404603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bressi JC, Jennings AJ, Skene R, Wu Y, Melkus R, De Jong R, O’Connell S, Grimshaw CE, Navre M, Gangloff AR. Bioorg. Med. Chem. Lett. 2010;20:3142–3145. doi: 10.1016/j.bmcl.2010.03.091. [DOI] [PubMed] [Google Scholar]
- 79.Cole KE, Dowling DP, Boone MA, Phillips AJ, Christianson DW. J. Am. Chem. Soc. 2011;133:12474–12477. doi: 10.1021/ja205972n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Estiu G, Greenberg E, Harrison CB, Kwiatkowski NP, Mazitschek R, Bradner JE, Wiest O. J. Med. Chem. 2008;51:2898–2906. doi: 10.1021/jm7015254. [DOI] [PubMed] [Google Scholar]
- 81.Moradei OM, Mallais TC, Frechette S, Paquin I, Tessier PE, Leit SM, Fournel M, Bonfils C, Trachy-Bourget M, Liu J, Yan TP, Lu A, Rahil J, Wang J, Lefebvre S, Li Z, Vaisburg AF, Besterinan JM. J. Med. Chem. 2007;50:5543–5546. doi: 10.1021/jm701079h. [DOI] [PubMed] [Google Scholar]
- 82.Khan N, Jeffers M, Kumar S, Hackett C, Boldog F, Khramtsov N, Qian X, Mills E, Berghs SC, Carey N, Finn PW, Collins LS, Tumber A, Ritchie JW, Jensen PB, Lichenstein HS, Sehested M. Biochem. J. 2008;409:581–589. doi: 10.1042/BJ20070779. [DOI] [PubMed] [Google Scholar]
- 83.Furumai R, Matsuyama A, Kobashi N, Lee K, Nishiyama N, Nakajima I, Tanaka A, Komatsu Y, Nishino N, Yoshida M, Horinouchi S. Cancer Res. 2002;62:4916–4921. [PubMed] [Google Scholar]
- 84.Haggarty S, Koeller K, Wong J, Grozinger C, Schreiber S. Proc. Natl. Acad. Sci. USA. 2003;100:4389–4394. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Balasubramanian S, Ramos J, Luo W, Sirisawad M, Verner E, Buggy JJ. Leukemia. 2008;22:1026–1034. doi: 10.1038/leu.2008.9. [DOI] [PubMed] [Google Scholar]
- 86.Duvic M, Vu J. Expert Opin. Invest. Drugs. 2007;16:1111–1120. doi: 10.1517/13543784.16.7.1111. [DOI] [PubMed] [Google Scholar]
- 87.Bertino EM, Otterson GA. Expert Opin. Invest. Drugs. 2011;20:1151–1158. doi: 10.1517/13543784.2011.594437. [DOI] [PubMed] [Google Scholar]
- 88.Bruserud O, Stapnes C, Ersvaer E, Gjertsen BT, Ryningen A. Curr. Pharm. Biotechnol. 2007;8:388–400. doi: 10.2174/138920107783018417. [DOI] [PubMed] [Google Scholar]
- 89.Landry J, Sutton A, Tafrov S, Heller R, Stebbins J, Pillus L, Sternglanz R. Proc. Natl. Acad. Sci. USA. 2000;97:5807–5811. doi: 10.1073/pnas.110148297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bitterman K, Anderson R, Cohen H, Latorre-Esteves M, Sinclair D. J. Biol. Chem. 2002;277:45099–45107. doi: 10.1074/jbc.M205670200. [DOI] [PubMed] [Google Scholar]
- 91.Grozinger C, Chao E, Blackwell H, Moazed D, Schreiber S. J. Biol. Chem. 2001;276:38837–38843. doi: 10.1074/jbc.M106779200. [DOI] [PubMed] [Google Scholar]
- 92.Lara E, Mai A, Calvanese V, Altucci L, Lopez-Nieva P, Martinez-Chantar ML, Varela-Rey M, Rotili D, Nebbioso A, Ropero S, Montoya G, Oyarzabal J, Velasco S, Serrano M, Witt M, Villar-Garea A, Inhof A, Mato JM, Esteller M, Fraga MF. Oncogene. 2009;28:781–791. doi: 10.1038/onc.2008.436. [DOI] [PubMed] [Google Scholar]
- 93.Napper A, Hixon J, McDonagh T, Keavey K, Pons J, Barker J, Yau W, Amouzegh P, Flegg A, Hamelin E, Thomas R, Kates M, Jones S, Navia M, Saunders J, DiStefano P, Curtis R. J. Med. Chem. 2005;48:8045–8054. doi: 10.1021/jm050522v. [DOI] [PubMed] [Google Scholar]
- 94.Outeiro TF, Kontopoulos E, Altmann SM, Kufareva I, Strathearn KE, Amore AM, Volk CB, Maxwell MM, Rochet J, McLean PJ, Young AB, Abagyan R, Feany MB, Hyman BT, Kazantsev AG. Science. 2007;317:516–519. doi: 10.1126/science.1143780. [DOI] [PubMed] [Google Scholar]
- 95.Kaeberlein M, McDonagh T, Heltweg B, Hixon J, Westman E, Cald-well S, Napper A, Curtis R, DiStefano P, Fields S, Bedalov A, Kennedy B. J. Biol. Chem. 2005;280:17038–17045. doi: 10.1074/jbc.M500655200. [DOI] [PubMed] [Google Scholar]
- 96.Borra M, Smith B, Denu J. J. Biol. Chem. 2005;280:17187–17195. doi: 10.1074/jbc.M501250200. [DOI] [PubMed] [Google Scholar]
- 97.Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, Bemis JE, Xie R, Disch JS, Ng PY, Nunes JJ, Lynch AV, Yang H, Galonek H, Israelian K, Choy W, Iffland A, Lavu S, Medvedik O, Sinclair DA, Olefsky JM, Jirousek MR, Elliott PJ, Westphal CH. Nature. 2007;450:712–716. doi: 10.1038/nature06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ng PY, Bemis JE, Disch JS, Vu CB, Oalmann CJ, Lynch AV, Carney DP, Riera TV, Song J, Smith JJ, Lavu S, Tornblom A, Duncan M, Yeager M, Kriksciukaite K, Gupta A, Suri V, Elliot PJ, Milne JC, Nunes JJ, Jirousek MR, Vlasuk GP, Ellis JL, Perni RB. Lett. Drug Des. Discovery. 2013;10:793–797. [Google Scholar]
- 99.Medda F, Russell RJM, Higgins M, McCarthy AR, Campbell J, Slawin AMZ, Lane DP, Lain S, Westwood NJ. J. Med. Chem. 2009;52:2673–2682. doi: 10.1021/jm8014298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gertz M, Fischer F, Nguyen GTT, Lakshminarasimhan M, Schutkowski M, Weyand M, Steegborn C. Proc. Natl. Acad. Sci. USA. 2013;110:E2772–E2781. doi: 10.1073/pnas.1303628110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Feige JN, Lagouge M, Canto C, Strehle A, Houten SM, Milne JC, Lambert PD, Mataki C, Elliott PJ, Auwerx J. Cell Metab. 2008;8:347–358. doi: 10.1016/j.cmet.2008.08.017. [DOI] [PubMed] [Google Scholar]
- 102.Reichert N, Choukrallah M, Matthias P. Cell. Mol. Life Sci. 2012;69:2173–2187. doi: 10.1007/s00018-012-0921-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Glozak M, Sengupta N, Zhang X, Seto E. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 104.Lagger G, O’Carroll D, Rembold M, Khier H, Tischler J, Weitzer G, Schuettengruber B, Hauser C, Brunmeir R, Jenuwein T, Seiser C. EMBO J. 2002;21:2672–2681. doi: 10.1093/emboj/21.11.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Montgomery RL, Davis CA, Potthoff MJ, Haberland M, Fielitz J, Qi X, Hill JA, Richardson JA, Olson EN. Genes Dev. 2007;21:1790–1802. doi: 10.1101/gad.1563807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Weichert W, Roeske A, Niesporek S, Noske A, Buckendahl A, Dietel M, Gekeler V, Boehm M, Beckers T, Denkert C. Clin. Cancer Res. 2008;14:1669–1677. doi: 10.1158/1078-0432.CCR-07-0990. [DOI] [PubMed] [Google Scholar]
- 107.Choi J, Kwon H, Yoon B, Kim J, Han S, Joo H, Kim D. Jpn. J. Cancer Res. 2001;92:1300–1304. doi: 10.1111/j.1349-7006.2001.tb02153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Weichert W, Roeske A, Gekeler V, Beckers T, Ebert MPA, Pross M, Dietel M, Denkert C, Roecken C. Lancet Oncol. 2008;9:139–148. doi: 10.1016/S1470-2045(08)70004-4. [DOI] [PubMed] [Google Scholar]
- 109.Miyake K, Yoshizumi T, Imura S, Sugimoto K, Batmunkh E, Kanemura H, Morine Y, Shimada M. Pancreas. 2008;36:e1–e9. doi: 10.1097/MPA.0b013e31815f2c2a. [DOI] [PubMed] [Google Scholar]
- 110.Wang W, Gao J, Man X, Li Z, Gong Y. Oncol. Rep. 2009;21:1439–1447. doi: 10.3892/or_00000372. [DOI] [PubMed] [Google Scholar]
- 111.Krusche C, Wulfing P, Kersting C, Vloet A, Bocker W, Kiesel L, Beier H, Alfer J. Breast Cancer Res. Treat. 2005;90:15–23. doi: 10.1007/s10549-004-1668-2. [DOI] [PubMed] [Google Scholar]
- 112.Gui C, Ngo L, Xu W, Richon V, Marks P. Proc. Natl. Acad. Sci. USA. 2004;101:1241–1246. doi: 10.1073/pnas.0307708100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Luo J, Su F, Chen D, Shiloh A, Gu W. Nature. 2000;408:377–381. doi: 10.1038/35042612. [DOI] [PubMed] [Google Scholar]
- 114.Senese S, Zaragoza K, Minardi S, Muradore I, Ronzoni S, Passafaro A, Bernard L, Draetta GF, Alcalay M, Seiser C, Chiocca S. Mol. Cell. Biol. 2007;27:4784–4795. doi: 10.1128/MCB.00494-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Inoue S, Mai A, Dyer MJS, Cohen GM. Cancer Res. 2006;66:6785–6792. doi: 10.1158/0008-5472.CAN-05-4563. [DOI] [PubMed] [Google Scholar]
- 116.Oh M, Choi IK, Kwon HJ. Biochem. Biophys. Res. Commun. 2008;369:1179–1183. doi: 10.1016/j.bbrc.2008.03.019. [DOI] [PubMed] [Google Scholar]
- 117.David G, Alland L, Hong S, Wong C, DePinho R, Dejean A. Oncogene. 1998;16:2549–2556. doi: 10.1038/sj.onc.1202043. [DOI] [PubMed] [Google Scholar]
- 118.Hoemme C, Peerzada A, Behre G, Wang Y, McClelland M, Nieselt K, Zschunke M, Disselhoff C, Agrawal S, Isken F, Tidow N, Berdel WE, Serve H, Mueller-Tidow C. Blood. 2008;111:2887–2895. doi: 10.1182/blood-2007-03-079921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wang J, Hoshino T, Redner R, Kajigaya S, Liu J. Proc. Natl. Acad. Sci. USA. 1998;95:10860–10865. doi: 10.1073/pnas.95.18.10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Durst K, Lutterbach B, Kummalue T, Friedman A, Hiebert S. Mol. Cell. Biol. 2003;23:607–619. doi: 10.1128/MCB.23.2.607-619.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Venugopal B, Evans TRJ. Curr. Med. Chem. 2011;18:1658–1671. doi: 10.2174/092986711795471284. [DOI] [PubMed] [Google Scholar]
- 122.Giannini G, Cabri W, Fattorusso C, Rodriquez M. Future Med. Chem. 2012;4:1439–1460. doi: 10.4155/fmc.12.80. [DOI] [PubMed] [Google Scholar]
- 123.Bantscheff M, Hopf C, Savitski MM, Dittmann A, Grandi P, Michon A, Schlegl J, Abraham Y, Becher I, Bergamini G, Boesche M, Delling M, Duempelfeld B, Eberhard D, Huthmacher C, Mathieson T, Poeckel D, Reader V, Strunk K, Sweetman G, Kruse U, Neubauer G, Ramsden NG, Drewes G. Nat. Biotechnol. 2011;29:255–265. doi: 10.1038/nbt.1759. [DOI] [PubMed] [Google Scholar]
- 124.Cai R, Kwon P, Yan-Neale Y, Sambuccetti L, Fischer D, Cohen D. Bio-chem. Biophys. Res. Commun. 2001;283:445–453. doi: 10.1006/bbrc.2001.4786. [DOI] [PubMed] [Google Scholar]
- 125.Guo C, Mi J, Brautigan DL, Larner JM. Cell. Signalling. 2007;19:504–510. doi: 10.1016/j.cellsig.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 126.Kar G, Keskin O, Fraternali F, Gursoy A. Curr. Pharm. Des. 2013;19:3175–3189. doi: 10.2174/1381612811319180002. [DOI] [PubMed] [Google Scholar]
- 127.Lin —HK, Wang L, Yu YC, Altuwaijri S, Chang C. EMBO J. 2002;21:4037–4048. doi: 10.1093/emboj/cdf406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Canettieri G, Di Marcotullio L, Greco A, Coni S, Antonucci L, Infante P, Pietrosanti L, De Smaele E, Ferretti E, Miele E, Pelloni M, De Simone G, Pedone EM, Gallinari P, Giorgi A, Steinkuehler C, Vitaglia-no L, Pedone C, Schinina ME, Screpanti I, Gulino A. Nat. Cell Biol. 2010;12:132–142. doi: 10.1038/ncb2013. [DOI] [PubMed] [Google Scholar]
- 129.Vashisht Gopal YN, Arora TS, Van Dyke MW. EMBO Rep. 2006;7:291–296. doi: 10.1038/sj.embor.7400613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gopal YN, Van Dyke MW. Cell. Cycle. 2006;5:2738–2743. doi: 10.4161/cc.5.23.3522. [DOI] [PubMed] [Google Scholar]
- 131.Dolcet X, Llobet D, Pallares J, Matias-Guiu X. Virchows Arch. 2005;446:475–482. doi: 10.1007/s00428-005-1264-9. [DOI] [PubMed] [Google Scholar]
- 132.Sethi G, Sung B, Aggarwal BB. Exp. Biol. Med. 2008;233:21–31. doi: 10.3181/0707-MR-196. [DOI] [PubMed] [Google Scholar]
- 133.Gopal YN, Arora TS, Van Dyke MW. Chem. Biol. 2007;14:813–823. doi: 10.1016/j.chembiol.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 134.Canettieri G, Morantte I, Guzman E, Asahara H, Herzig S, Anderson S, Yates J, Montminy M. Nat. Struct. Biol. 2003;10:175–181. doi: 10.1038/nsb895. [DOI] [PubMed] [Google Scholar]
- 135.Gopal YN, Chanchorn E, Van Dyke MW. Mol. Cancer Ther. 2009;8:552–562. doi: 10.1158/1535-7163.MCT-08-0661. [DOI] [PubMed] [Google Scholar]
- 136.Zhang S, Won Y, Ong C, Shen H. Curr. Med. Chem. Anti-Cancer Agents. 2005;5:239–249. doi: 10.2174/1568011053765976. [DOI] [PubMed] [Google Scholar]
- 137.Haigis MC, Sinclair DA. Annu. Rev. Pathol. Mech. Dis. 2010;5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.McBurney M, Yang X, Jardine K, Hixon M, Boekelheide K, Webb J, Lansdorp P, Lemieux M. Mol. Cell. Biol. 2003;23:38–54. doi: 10.1128/MCB.23.1.38-54.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Banks AS, Kon N, Knight C, Matsumoto M, Gutierrez-Juarez R, Rossetti L, Gu W, Accili D. Cell Metab. 2008;8:333–341. doi: 10.1016/j.cmet.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Pfluger PT, Herranz D, Velasco-Miguel S, Serrano M, Tschop MH. Proc. Natl. Acad. Sci. USA. 2008;105:9793–9798. doi: 10.1073/pnas.0802917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Herranz D, MuÇoz-Martin M, CaÇamero M, Mulero F, Martinez-Pastor B, Fernandez-Capetillo O, Serrano M. Nat. Commun. 2010;1:3. doi: 10.1038/ncomms1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Luo J, Nikolaev A, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 143.Pruitt K, Zinn RL, Ohm JE, McGarvey KM, Kang SL, Watkins DN, Herman JG, Baylin SB. PLoS Genet. 2006;2:e40. doi: 10.1371/journal.pgen.0020040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Yuan J, Minter-Dykhouse K, Lou Z. J. Cell Biol. 2009;185:203–211. doi: 10.1083/jcb.200809167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lim J, Lee Y, Chun Y, Chen J, Kim J, Park J. Mol. Cell. 2010;38:864–878. doi: 10.1016/j.molcel.2010.05.023. [DOI] [PubMed] [Google Scholar]
- 146.Herranz D, Serrano M. Nat. Rev. Cancer. 2010;10:819–823. doi: 10.1038/nrc2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Jung-Hynes B, Nihal M, Zhong W, Ahmad N. J. Biol. Chem. 2009;284:3823–3832. doi: 10.1074/jbc.M807869200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Huffman DM, Grizzle WE, Bamman MM, Kim J, Eltoum IA, El-gavish A, Nagy TR. Cancer Res. 2007;67:6612–6618. doi: 10.1158/0008-5472.CAN-07-0085. [DOI] [PubMed] [Google Scholar]
- 149.Wang R, Sengupta K, Li C, Kim H, Cao L, Xiao C, Kim S, Xu X, Zheng Y, Chilton B, Jia R, Zheng Z, Appella E, Wang XW, Ried T, Deng C. Cancer Cell. 2008;14:312–323. doi: 10.1016/j.ccr.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Pervaiz S, Holme AL. Antioxid. Redox Signaling. 2009;11:2851–2897. doi: 10.1089/ars.2008.2412. [DOI] [PubMed] [Google Scholar]
- 151.Howitz K, Bitterman K, Cohen H, Lamming D, Lavu S, Wood J, Zipkin R, Chung P, Kisielewski A, Zhang L, Scherer B, Sinclair D. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 152.Beher D, Wu J, Cumine S, Kim KW, Lu S, Atangan L, Wang M. Chem. Biol. Drug Des. 2009;74:619–624. doi: 10.1111/j.1747-0285.2009.00901.x. [DOI] [PubMed] [Google Scholar]
- 153.Wood J, Rogina B, Lavu S, Howitz K, Helfand S, Tatar M, Sinclair D. Nature. 2004;430:686–689. doi: 10.1038/nature02789. [DOI] [PubMed] [Google Scholar]
- 154.Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 155.Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, Pistell PJ, Poosala S, Becker KG, Boss O, Gwinn D, Wang M, Ramaswamy S, Fish-bein KW, Spencer RG, Lakatta EG, Le Couteur D, Shaw RJ, Navas P, Puigserver P, Ingram DK, de Cabo R, Sinclair DA. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zheng J, Ramirez V. Br. J. Pharmacol. 2000;130:1115–1123. doi: 10.1038/sj.bjp.0703397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Gledhill JR, Montgomery MG, Leslie AGW, Walker JE. Proc. Natl. Acad. Sci. USA. 2007;104:13632–13637. doi: 10.1073/pnas.0706290104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hawley SA, Ross FA, Chevtzoff C, Green KA, Evans A, Fogarty S, Towler MC, Brown LJ, Ogunbayo OA, Evans AM, Hardie DG. CellMetab. 2010;11:554–565. doi: 10.1016/j.cmet.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Yoshizaki T, Schenk S, Imamura T, Babendure JL, Sonoda N, Bae EJ, Oh DY, Lu M, Milne JC, Westphal C, Bandyopadhyay G, Olefsky JM. Am. J. Physiol. Endocrinol. Metab. 2010;298:E419–E428. doi: 10.1152/ajpendo.00417.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Pacholec M, Bleasdale JE, Chrunyk B, Cunningham D, Flynn D, Garofalo RS, Griffith D, Griffor M, Loulakis P, Pabst B, Qiu X, Stockman B, Thanabal V, Varghese A, Ward J, Withka J, Ahn K. J. Biol. Chem. 2010;285:8340–8351. doi: 10.1074/jbc.M109.088682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Minucci S, Nervi C, Lo Coco F, Pelicci P. Oncogene. 2001;20:3110–115. doi: 10.1038/sj.onc.1204336. [DOI] [PubMed] [Google Scholar]
- 163.Castaigne S, Chomienne C, Daniel M, Ballerini P, Berger R, Fenaux P, Degos L. Blood. 1990;76:1704–1709. [PubMed] [Google Scholar]