

Abstract
Interleukin 17(IL-17) is a signature cytokine of Th17 cells. We previously reported that deletion of NF-κ B activator 1(Act1), the key transducer of IL-17R signaling, from the neuroectodermal lineage in mice (neurons, oligodendrocytes, astrocytes)results in attenuated severity of experimental autoimmune encephalomyelitis (EAE). Here we examined the cellular basis of this observation. EAE disease course was unaffected by deleting Act1 from neurons or mature oligodendrocytes and Act1 deletion from astrocytes only modestly affected disease course. Deletion of Act1 from NG2+ glia resulted in markedly reduced EAE severity. Furthermore, IL-17 induced characteristic inflammatory mediator expression in NG2+ glial cells. Additionally, IL-17 also exhibited strong inhibitory effects on the maturation of oligodendrocyte lineage cells in vitro and reduced their survival. These data identify NG2+ glia as the major CNS cellular target of IL-17 in EAE. The sensitivity of oligodendrocyte lineage cells to IL-17-mediated toxicity further suggests a direct link between inflammation and neurodegeneration in multiple sclerosis (MS).
INTRODUCTION
Multiple sclerosis (MS) is a T cell-mediated inflammatory demyelinating disease of the human central nervous system (CNS)1. Inflammatory demyelination is coupled with neurodegeneration during the course of MS, although precise mechanisms remain uncertain2–4. Many components of the myelin sheath have been injected into animals to induce experimental autoimmune encephalomyelitis (EAE). Studies of the EAE model have helped define the sequence of immunopathogenic events involved in the development of autoimmune CNS-directed inflammatory diseases5. During the initiation stage of EAE, in addition to T cell activation and expansion, the APCs produce cytokines to regulate the differentiation of effector CD4 T cells, including the classical Th1 (producing IFNγ and TNFα) and Th17 (producing IL-17, IL-6 and TNFα) T cell lineage. Importantly, recent data demonstrate that both Th1 and Th17 cells can independently induce EAE possibly through different mechanisms6, 7. Th17 cells are generated as a discrete lineage following priming in the presence of TGFβ and IL-6 and acquisition of encephalitogenicity following expansion in the presence of IL-238–10. EAE is markedly suppressed in mice lacking IL-17 or IL-17 receptor and IL-17-specific inhibition attenuates inflammation, indicating that IL-17-mediated signaling plays a critical role in the effector stage of EAE9, 11, 12. However, the precise mechanism by which IL-17 participates in EAE development and pathogenesis remains unclear.
A two-wave hypothesis has been proposed for Th17-mediated effector stage of EAE. After priming in peripheral lymph nodes, antigen-specific Th17 cells traffic through the choroid plexus into the subarachnoid space, where they encounter antigen, presented by macrophages (meningeal APCs), are restimulated and undergo clonal expansion. As a consequence of productive T cell/APC interactions, Th17 signature cytokines, including IL-17, are produced and impinge on the adjacent CNS tissue. Following activation of the parenchymal vasculature by this cytokine flux, perivascular leukocyte infiltrates accumulate leading to the explosive inflammatory cascade associated with the onset of EAE. These re-activated Th17 cells subsequently migrate across the glia limitans basement membrane, deep into the parenchymal CNS white matter, and initiate tissue destruction including demyelination and axonal injury.
Act1 is an essential intracellular adaptor for IL-17 signaling13–16. Considering the essential role of Act1 in IL-17 signaling, we used Act1-deficient mice as a model system to investigate the cellular mechanism of IL-17 signaling. We have previously found that Th17 cells efficiently infiltrate the Act1-deficient CNS but fail to recruit additional lymphocytes, neutrophils, and macrophages from the bloodstream into the CNS, indicating a critical role of IL-17-induced Act1-mediated signaling in the conversion of Wave 1 to Wave 2 during the effector stage of EAE. Surprisingly, targeted Act1 deficiency in neuroectoderm-derived CNS-resident cells (in NesCreAct1fl/− mice), but not in endothelial cells or macrophages and microglia, significantly delayed EAE onset and reduced EAE17. These data established that IL-17 signaling directly to neuroectodermal cells was required for the pathogenic inflammation which occurs during EAE. It is important to note that while IL-17 signaling activates transcription factor NFkB, inhibition of NFkB in the neuroectodermal cells also ameliorates EAE18, 19.
NG2+ glial cells, a distinct macroglial population in the adult CNS separate from astrocytes or oligodendrocytes20, are closely related to progenitors which give rise to myelinating oligodendrocytes during development. Following varied types of CNS injury, NG2+ glia proliferate and demonstrate a reactive phenotype including increased expression of NG2, a surface proteoglycan. Their fate in vivo thereafter has been a matter of some controversy, but it seems clear that they play a role in myelin repair and mainly give rise to oligodendrocytes21. NG2+ glia have not previously been implicated in generating pathogenic inflammatory responses. In the present study we sought to define the neuroectodermal cellular target(s) of IL-17 action during EAE. After a series of negative studies, we deleted Act1 from NG2+ glial cells (in NG2CreAct1fl/− mice) and virtually abrogated Th17 cell-induced EAE recapitulating the EAE phenotype of NestinCre Act1fl/− mice which lacked Act1 in all neuroectodermal cells17. Inflammatory gene expression in response to IL-17 was nearly absent from the CNS of NG2CreAct1fl/− mice after Th17-cell transfers, accounting for the lack of clinical disease.
RESULTS
Ablation of Act1 from astrocytes only modestly reduces autoimmune encephalomyelitis
Deletion of Act1 in neuroectoderm-derived cells in CNS ameliorated the severity of Th17-induced EAE17. Neuroectoderm-derived cells include astrocytes, neurons, NG2+ glia and oligodendrocytes. Astrocytes with processes in the glia limitans, and around cerebral blood vessels are positioned to transduce signals from meningeal Th17 cells to activate the BBB endothelium and drive perivascular leukocyte infiltration (Wave 2) and consequent explosive inflammatory cascade associated with the onset of EAE. We generated astrocyte-specific Act1-deficient mice, by breeding Act1−/− mice with GFAPCre transgenics to generate GFAPCreAct1+/− mice, and these mice were further bred onto Act1fl/fl mice to generate control mice (GFAPCreAct1fl/+) and astrocyte-specific Act1-deficient mice (GFAPCreAct1fl/−). Western analysis showed that Act1 expression was completely ablated in astrocytes derived from GFAPCreAct1fl/− mice (Fig. 1a).
Figure 1.
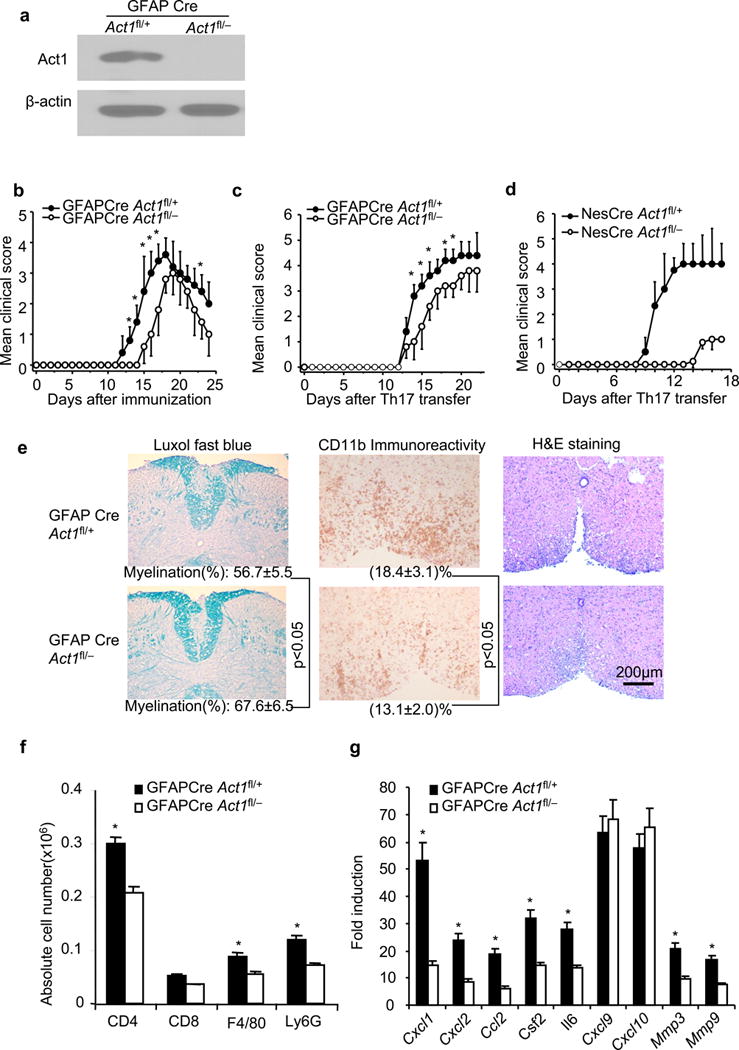
Ablation of Act1 in astrocytes ameliorates autoimmune encephalomyelitis. (a) Immunoblot analysis for the Act1 expression in cultured astrocytes from neonatal GFAPCreAct1fl/− and control GFAPCreAct1fl/+ mice. Full-length blots are presented in Supplementary Figue 5. (b) Mean clinical score of EAE in GFAPCreAct1fl/− and GFAPCreAct1fl/+ mice induced by active immunization with MOG35–55. (c) Mean clinical score of EAE in GFAPCreAct1fl/− and GFAPCreAct1fl/+ mice induced by Th17 adoptive transfer. (d) Mean clinical score of EAE in NesCreAct1fl/− and NesCreAct1fl/+ mice induced by Th17 adoptive transfer (p<0.05, two-way ANOVA). (e) Luxol fast blue, anti-CD11b and H&E staining of lumber spinal cords of GFAPCreAct1fl/− and GFAPCreAct1fl/+ mice 20 days after Th17 adoptive transfer. Data are representative of 5 mice per group. (f) Absolute cell numbers of infiltrated immune cells in the brains of GFAPCreAct1fl/− and GFAPCreAct1fl/+ mice (20 days after Th17 cell transfer. (g) Real-time PCR analysis of inflammatory gene expression in spinal cords of GFAPCreAct1fl/− and GFAPCreAct1fl/+ mice 20 days after Th17 adoptive transfer. Data are representative of three independent experiments. n=5/group in each experiment. Error bars, SEM; *, p<0.05.
EAE was induced in GFAPCreAct1fl/− mice and control mice (GFAPCreAct1fl/+) by active immunization with MOG33–55. The astrocyte-specific Act1-deficient mice (GFAPCreAct1fl/−) showed a delay in the onset and reduced disease severity compared to the control mice (GFAPCreAct1fl/+). However, the EAE phenotype in the astrocyte-specific Act1-deficient mice was not nearly as dramatic as in the CNS-restricted Act1-deficient mice (NesCreAct1fl/−) (Fig. 1b)17. When EAE was induced by adoptive transfer of MOG35–55-specific wild-type Th17 cells, the GFAPCreAct1fl/− mice also showed modest but reproducible reduction in disease severity compared to controls (Fig. 1c), whereas the CNS-restricted Act1-deficient mice had delayed onset and greatly reduced EAE (Fig. 1d). Consistent with reduced clinical disease, mononuclear cell infiltrates were decreased in white matter of spinal cords from GFAPCreAct1fl/− (Fig. 1e–f). During EAE, signature IL-17-responsive inflammatory genes (cytokines, chemokines and matrix metalloproteinases) are significantly induced in CNS2, 22, 23. Induction of the IL-17-regulated inflammatory genes (including Cxcl1, Cxcl2, Csf2, Il6, Mmp3 and Mmp9) in the spinal cord was substantially reduced in GFAPCreAct1fl/− mice somewhat out of proportion to the clinical effect (Fig. 1g). Overall, results were similar to those reported using lentiviral shRNA to reduce Act1 expresion in astrocytes24. As the EAE phenotype of astrocyte-specific Act1-deficient mice failed to reproduce that of the CNS-restricted Act1-deficient mice, other neuroectoderm-derived cells must also participate in Th17-mediated EAE pathogenesis.
Neuronal Act1 is dispensable for EAE development
A recent study suggested that Th17 cells directly interact with neurons, mediating neuronal dysfunction and contributing to the development of EAE25. To determine whether neuronal Act1 is critical for EAE pathogenesis, we generated neuronal Act1-deficient mice by breeding the Act1fl/− mice onto Enolase2Cre transgenic mice. Western analysis showed that Act1 expression was abolished in neurons from the Enolase2CreAct1fl/− mice (Fig. 2a). The onset and disease severity of EAE were similar in Enolase2CreAct1fl/− and Enolase2CreAct1fl/+ mice after either active immunization with MOG35–55 or adoptive transfer of MOG35–55-specific Th17 cells(Fig. 2b–c). Furthermore, inflammatory cell infiltration in CNS of the two experimental groups were also comparable after Th17 adoptive transfer (Fig. 2d). Neuronal Act1 deficiency had no substantial impact on inflammatory gene expression in the spinal cords of Th17-induced EAE mice (Fig. 2e). These data indicate that neuronal Act1 is dispensable for Th17-mediated EAE pathogenesis.
Figure 2.
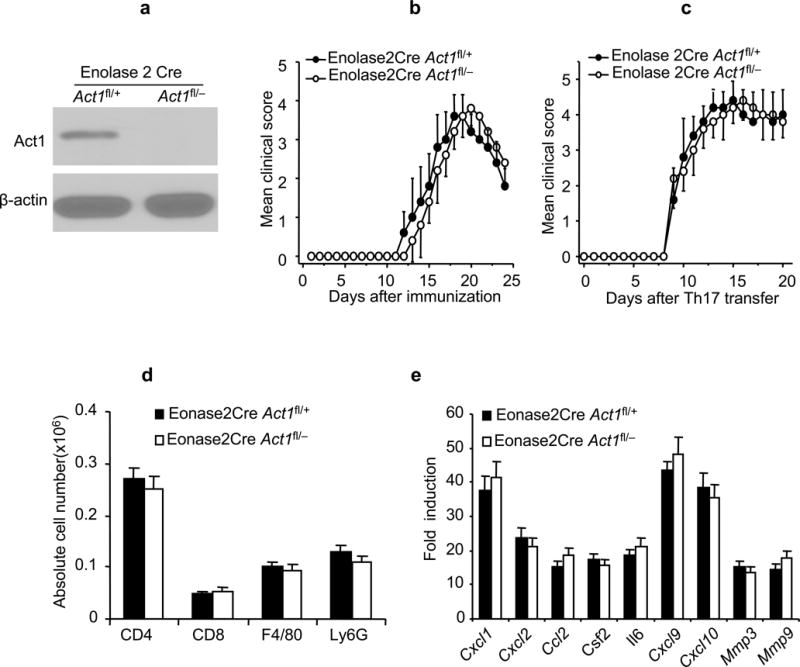
Neuronal Act1 is dispensable for EAE development. (a) Immunoblot analysis for the Act1 expression in cultured neuronal cells from embryonic brains (E14) of neuron-specific Act1-deficient (Enolase2CreAct1fl/−) and control mice (Enolase2CreAct1fl/+). Full-length blots are presented in Supplementary Figure 5. (b) Mean clinical score of EAE in Enolase2CreAct1fl/− and Enolase2CreAct1fl/+ mice induced by active immunization with MOG35–55. (c) Mean clinical score of EAE in the Enolase2CreAct1fl/− and Enolase2CreAct1fl/+ mice induced by MOG35–55-specific Th17 cells. (d) Flow cytometric analysis of immune cell infiltration in the brains of Th17-induced Enolase2CreAct1fl/− and Enolase2CreAct1fl/+ EAE mice (7 days after disease onset). (e) Real-time PCR analysis of inflammatory gene expression in spinal cords of Enolase2CreAct1fl/− and Enolase2CreAct1fl/+ mice transferred with MOG35–55-specific Th17 cells. Data are representative of three independent experiments, n=5/group in each experiment. Error bars, SEM. P>0.05 at b–e.
Deletion of Act1 in oligodendrocyte-lineage cells substantially attenuates autoimmune encephalomyelitis
A distinct population of glial cells which express OPC markers NG2 and PDGFR-alpha persists throughout the adult CNS20, 26. These glia (termed NG2 glia or polydendrocytes) can give rise to myelinating oligodendrocytes in vitro or in vivo. They show brisk morphological responses to injury of the CNS. Their physiological role(s) in the adult CNS have been extensively studied but remain incompletely understood21, 27–29. NG2Cre transgenic mice20, 30 were used to mediate specific deletion of Act1 in oligodendrocyte lineage including developmental OPCs, mature oligodendrocytes and NG2 glia of the adult CNS. The Act1−/− mice were first bred onto NG2Cre transgenic mice to generate NG2CreAct1+/− mice. These mice were further bred onto Act1fl/fl mice to generate NG2CreAct1fl/+ and NG2CreAct1fl/− mice. Immunoblot showed that Act1 expression was completely ablated in NG2+ cells derived from NG2CreAct1fl/− mice(Fig. 3a).
Figure 3.
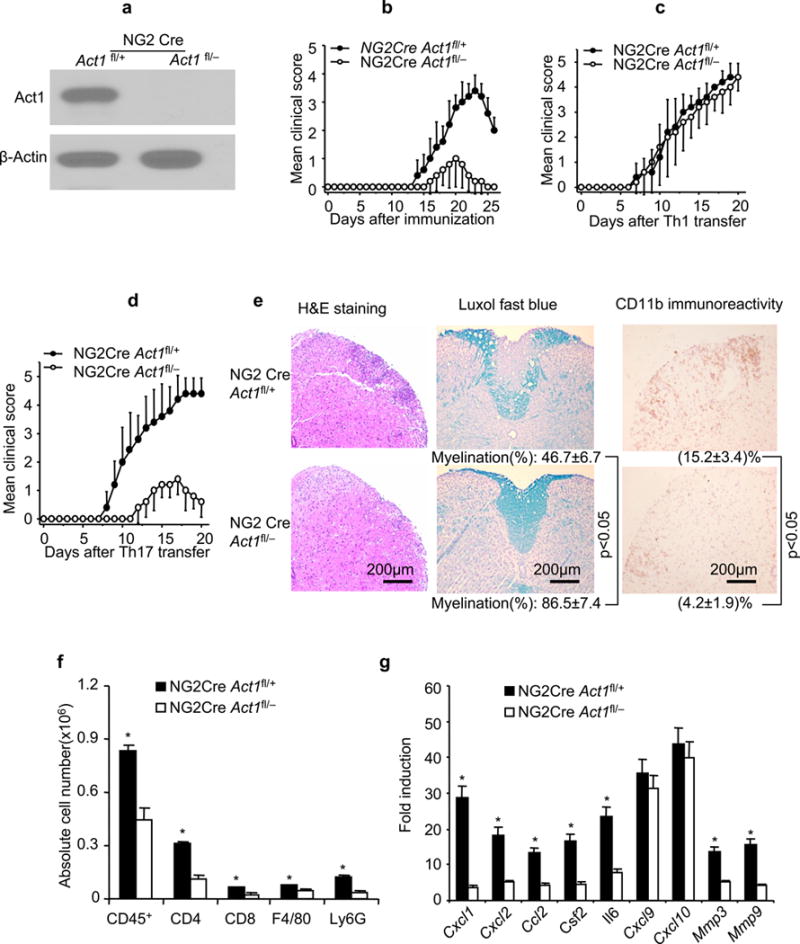
Ablation of Act1 in oligodendrocyte lineage ameliorates autoimmune encephalomyelitis. (a) Immunoblot analysis for the Act1 expression in cultured NG2+ OPCs from embryonic brains. Full-length blots are presented in Supplementary Figure 5. (b) Mean clinical score of EAE in NG2CreAct1fl/− and NG2CreAct1fl/+ mice induced by active immunization with MOG35–55(p<0.05, two-way ANOVA). (c) Mean clinical score of EAE in NG2CreAct1fl/− and NG2CreAct1fl/+ mice induced by Th1 adoptive transfer (p>0.05, two-way ANOVA). (d) Mean clinical score of EAE in NG2CreAct1fl/− and NG2CreAct1fl/+ mice induced by Th17 adoptive transfer (p<0.05, two-way ANOVA). (e) H&E, Luxol fast blue, anti-CD11b staining of lumber spinal cords of NG2CreAct1fl/− and NG2CreAct1fl/+ mice 20 days after Th17 adoptive transfer. Data are representative of 5 mice per group. (f) Absolute cell numbers of infiltrated immune cells in the brains of NG2CreAct1fl/− and NG2CreAct1fl/+ mice (20 days after Th17 cell transfer). (g) Real-time PCR analysis of inflammatory gene expression in spinal cords of NG2CreAct1fl/− and NG2CreAct1fl/+ mice transferred with MOG35–55-specific Th17 cells. Data are representative of three independent experiments. n=5/group in each experiment. Error bars, SEM; *, p<0.05.
EAE was induced in NG2CreAct1fl/− mice and control mice (NG2CreAct1fl/+) by active immunization with MOG33–55. Remarkably, Act1 deficiency in NG2+ glia delayed the onset of EAE and greatly reduced the disease severity (Fig. 3b), similar to NesCreAct1fl/− mice. It is important to note that the MOG-induced T cell priming was comparable in NG2Cre Act1fl/− and NG2CreAct1fl/+ mice (Supplementary. Fig. 1b). Furthermore, Th17 cells from MOG-immunized NG2Cre Act1fl/− and NG2CreAct1fl/+ mice induced similar EAE in wild-type recipient mice (Supplementary. Fig. 1c–e). These data suggest that the resistance of NG2Cre Act1fl/− mice to EAE induced by active immunization with MOG33–55(Fig. 3b) was probably at the effector stage of EAE. To confirm this interpretation, both NG2CreAct1fl/+ and NG2CreAct1fl/− mice were used as recipients of activated wild-type MOG35–55-specific Th1 or Th17 cells. Whereas the Th1 cells induced a similar EAE phenotype in both groups of mice, the onset and severity of Th17 cell-induced EAE were greatly reduced in NG2Cre Act1fl/− compared to that in control mice (Fig. 3c–d). The NG2Cre Act1fl/− mice had fewer inflammatory cells in CNS, including CD4+ and CD8+ T cells, macrophages, and neutrophils, than did control mice at the peak of disease after Th17 cell transfer (Fig. 3e–f and Supplementary. Fig. 1a). Th17-associated but not Th1-related inflammatory gene expression levels in the spinal cords of NG2CreAct1fl/− mice were dramatically reduced compared to that in the controls (Fig. 3g). Collectively, these data indicate among the neuroectoderm-derived cells, NG2+ glia are pivotal for the occurrence of Th17-mediated EAE.
What is the impact of Act1 deficiency on NG2+ cells and mature oligodendrocytes during EAE? The numbers of NG2+ cells and mature oligodendrocytes were comparable in the spinal cords of naïve NG2CreAct1fl/+ and NG2CreAct1fl/− mice (Supplementary. Fig. 2a–b). In EAE mice, mature GST-π+ oligodendrocytes were substantially reduced in the spinal cords of NG2Cre Act1fl/+ mice (Supplementary. Fig. 2a). By contrast, NG2+ cells were dramatically increased in NG2Cre Act1fl/+ mice while NG2+ glial cell numbers were unchanged in NG2Cre Act1fl/− mice (Supplementary. Fig. 2b). Consistently, BrdU incorporation in NG2+ cells was much more in the spinal cords of NG2CreAct1fl/+ mice than in NG2CreAct1fl/− mice (Supplementary. Fig. 2c). These results suggest that while NG2+ cells were activated during EAE (as evident by increased BrdU incorporation and NG2+ cell numbers), indicating the possibility that these cells might participate in the inflammatory cascade which underlies EAE pathogenesis.
However, it also remained plausible that the ability of NG2+ glia to participate at later time points in myelin repair might be perturbed by IL-17-induced Act1-mediated signaling as shown by decreased mature oligodendrocytes. Notably, upon Th1 induction of EAE, NG2-CreAct1fl/− mice had similar levels of demyelination as compared with their controls by MBP staining (Supplementary. Fig. 2d).
NG2+ glial cells, a distinct macroglial population in the adult CNS separate from astrocytes or oligodendrocytes20, are closely related to progenitors which give rise to myelinating oligodendrocytes during development or in response to CNS injury. Our results suggested that IL-17 signaling in NG2+ cells might incorporate these glia into the inflammatory pathogenesis of EAE. In particular, the dramatic reactive phenotype of increased proliferation and cell number supported this interpretation. It remained important to exclude the possibility that the EAE-resistant phenotype of NG2CreAct1fl/− mice could be attributed in whole or in part to deletion of Act1 from NG2+ vessel-associated pericytes, Olig2 Cre transgenic mice were used to mediate the deletion of Act1 in oligodendrocyte lineage31–33 (Supplementary. Fig. 3a). Act1 deficiency in Olig2+ cells delayed the onset of EAE and greatly reduced the disease severity upon active immunization with MOG35–55 (Supplementary. Fig. 3b). Furthermore, the onset and severity of Th17 cell-induced EAE were greatly reduced in Olig2CreAct1fl/− mice compared to that in control mice, whereas the Th1 cells induced similar EAE phenotype in both groups of mice (Supplementary. Fig. 3c–d). The Olig2CreAct1fl/− mice had fewer inflammatory cells in CNS than control mice at the peak of disease after Th17 cell transfer (Supplementary. Fig. 3e). Th17-associated but not Th1-related inflammatory gene expression levels in the spinal cords of Olig2CreAct1fl/− mice were dramatically reduced compared to that in the controls (Supplementary. Fig. 3f). The EAE phenotypes of Nestin CreAct1fl/− mice, NG2CreAct1fl/− mice and Olig2CreAct1fl/− mice were virtually identical, supporting the hypothesis that the neuroepithelial cell whose response to IL-17 was crucial for EAE pathogenesis was the NG2+ glial cell.
Mature oligodendrocyte-derived Act1 is dispensable for EAE
Since NG2Cre mediates deletion of floxed sequences in oligodendrocyte-lineage cells during development, as well as in NG2+ glia in the adult, we asked whether mature oligodendrocyte-derived Act1 is important for EAE development. CNP1/CNPase promoter activity is low in OPCs but gradually increases during OL differentiation and remains high in mature oligodendrocytes26, 32, 34, 35. Therefore, CNP1/CNPase Cre transgenic mice have been invaluable to delete target genes in mature oligodendrocytes35–37. Immunohistochemical staining indicated that Act1 expression was completely ablated in CNPase+ cells derived from CNPaseCre Act1fl/− mice (Fig. 4a). Both immunization with MOG35–55 and adoptive transfer of activated MOG35–55 specific Th17 cells resulted in similar onset and disease severity of EAE in the CNPaseCreAct1fl/− mice compared to that in the control mice (Fig. 4b–c). Cell infiltration was also analyzed by flow cytometry after Th17 adoptive transfer. As shown in Fig. 4d, there were comparable levels of cell infiltration in the CNS tissues derived from these two groups of mice. Furthermore, real-time PCR showed similar levels of induction of inflammatory genes in the spinal cords of CNPaseCreAct1fl/− mice and CNPaseCreAct1fl/+ controls (Fig. 4e). Together, these data demonstrate that mature oligodendrocyte-derived Act1 is dispensable for the pathogenesis of EAE.
Figure 4.
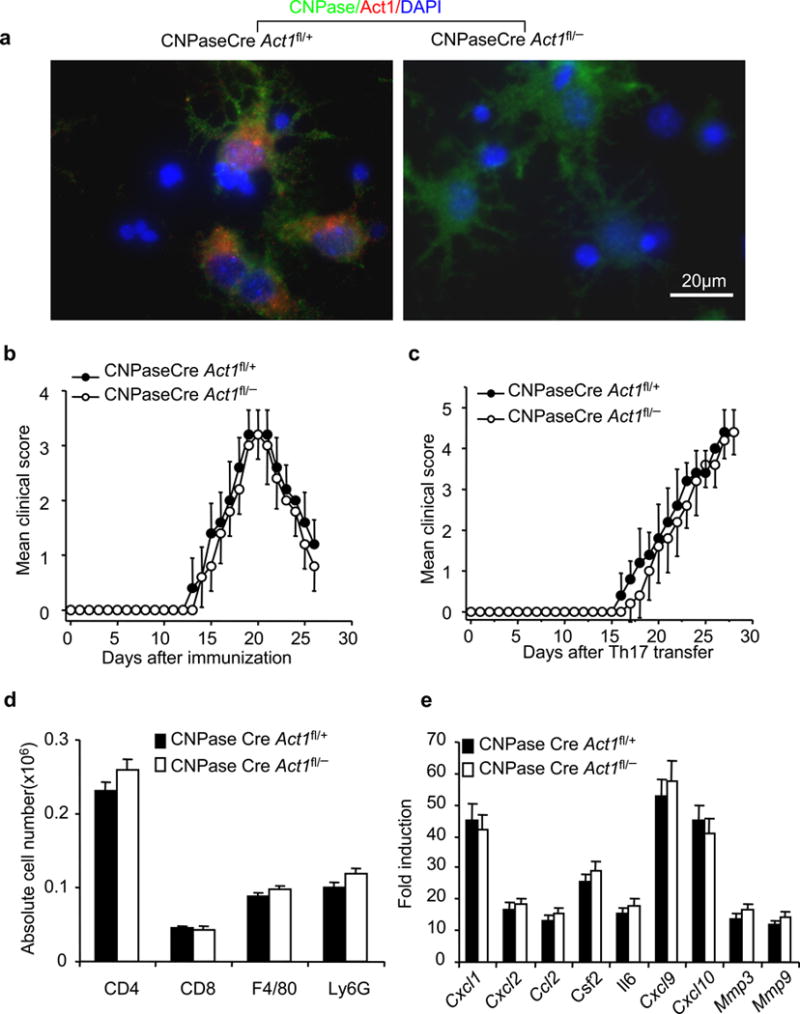
Act1 in mature oligodendrocytes is dispensible for the pathogenesis of EAE. (a) Co-staining of Act1 (Red) and CNPase (Green) in cultured oligodendrocytes from CNPaseCreAct1fl/− and control mice (CNPaseCreAct1fl/+). (b) Mean clinical score of EAE in CNPaseCreAct1fl/− and CNPaseCreAct1fl/+ mice induced by active immunization with MOG35–55. (c) Mean clinical score of EAE in the CNPaseCre Act1fl/− and CNPaseCre Act1fl/+ mice induced by MOG35–55-specific Th17 cells. (d) Absolute number of immune cell infiltration in the brains of Th17-induced CNPaseCreAct1fl/− and CNPaseCreAct1fl/+ EAE mice (20 days after Th17 transfer). (e) Real-time PCR analysis of inflammatory gene expression in spinal cords of CNPaseCreAct1fl/− and CNPaseCreAct1fl/+ mice transferred with MOG35–55-specific Th17 cells. Data are representative of three independent experiments. n=5/group in each experiment. Error bars, SEM. P>0.05 at b–e.
Inflammatory and cytotoxic effects of IL-17-induced Act1-mediated signaling on NG2+ glia
Deletion of Act1 from mature oligodendrocytes showed no effect on Th17-mediated EAE, while Act1 deficiency in NG2+ glia decisively altered EAE pathogenesis. What responses of NG2+ glia to IL-17 might mediate these effects on EAE severity? To answer this question, we cultured embryonic neurospheres to generate oligodendrocyte progenitor cells (OPCs) and mature oligodendrocytes. After 4 days of culture in cytokine cocktail of FGF+PDGF, the neural stem cells were differentiated into OPCs (>98% Olig2+CNPase−). The OPCs were then induced to mature into oligodendrocytes by withdrawing growth factors. At the end of 2 days of maturation, >95% cells were mature oligodendrocytes (CNPase+ GFAP− β-tubulin-III−)(Supplementary. Fig. 4a–b). IL-17 induced robust inflammatory responses in OPCs, whereas we detected little inflammatory response in mature oligodendrocytes(Fig. 5a,b). This inflammatory response in OPCs was Act1-dependent. These results were consistent with hypothesis that NG2+ glia constitute an essential component of the CNS inflammatory response in Th17-EAE.
Figure 5.
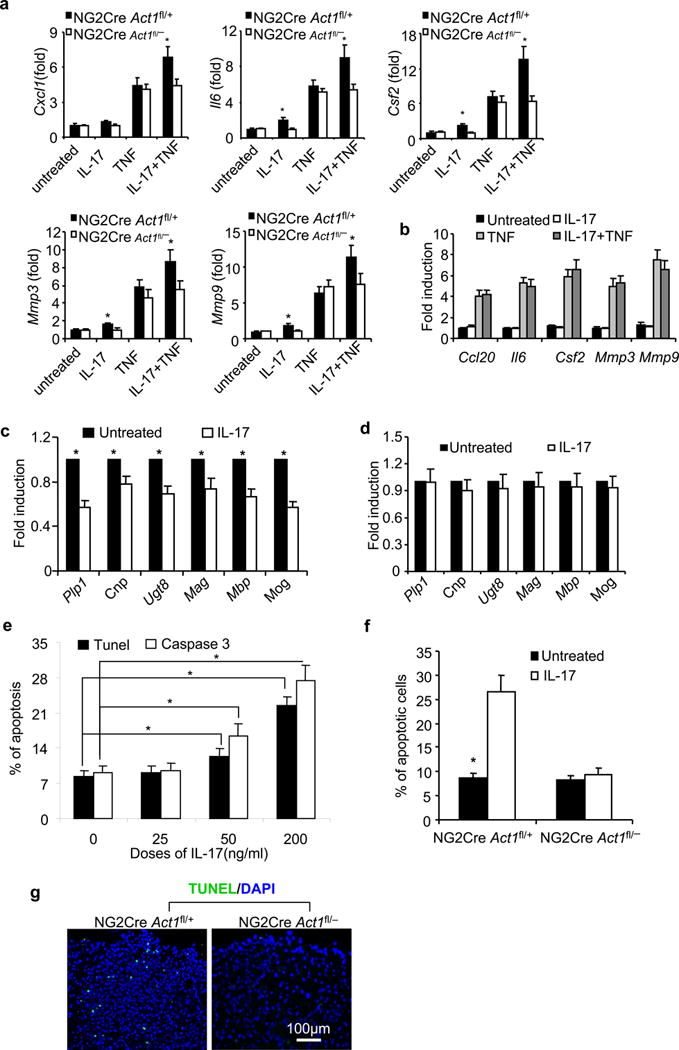
Cytotoxic and inflammatory effects of IL-17-induced Act1-mediated signaling on oligodendroglia. After 4-day differentiation culture of neural stem cells with cytokine cocktail of FGF+PDGF, >98% of cells became Olig2+CNPase− suggesting they are OPCs. Then cytokines were withdrawn to drive the maturation of OPC for 2 days, immunohistochemical staining suggested that >95% cells were CNPase+ GFAP− β-tubulin-III− suggesting they are mature oligodendrocytes. (a). OPCs from NG2CreAct1fl/+ and NG2CreAct1fl/− mice were treated with IL-17(25ng/ml), TNF(10ng/ml) or IL-17+TNF for 6 hours. Indicated gene expression was quantified by realtime PCR; (b). Mature oligodendrocytes from wild-type mice were treated with IL-17(25ng/ml), TNF(10ng/ml) or IL-17+TNF for 6 hours. Indicated gene expression was quantified by real-time PCR; (c–d). OPCs from NG2CreAct1fl/+ mice(c) and NG2Cre Act1fl/− mice(d) were cultured for another two days without FGF and PDGF, but with or without addition of IL-17(25ng/ml). Myelin-related gene expression was quantified by realtime PCR. (e). TUNEL and cleaved caspase 3 assays for apoptotic cells after 4 day cultures of wild-type OPCs with/without of IL-17 as indicated. 1500 cells were scored from 15 power field of 5 slices from 5 mice. (F). Data shown are the apoptotic percentages of OPCs from NG2Cre Act1fl/+ mice and NG2Cre Act1fl/− mice after 4 days culture with or without IL-17(200ng/mL). (g). TUNEL assay of apoptotic cells in the lumber spinal cord of NG2CreAct1fl/+ and NG2CreAct1fl/− mice 20 days after Th17 adoptive transfer. Apoptotic cells were counted from five sections of each mouse and then got the mean value per section. Data are representative of three independent experiments. n=5/group in each experiment. Error bars, SEM; *, p<0.05.
We also asked whether IL-17 action towards OPCs might affect myelin repair following demyelination. The addition of IL-17 to the OPCs from NG2CreAct1fl/+ mice (Fig. 5c), but not NG2CreAct1fl/− mice (Fig. 5d), substantially reduced the expression of myelin proteins and other markers associated with myelinating oligodendrocytes suggesting that commitment to an inflammatory program of gene expression might impair the ability of these cells to engage in the process of remyelination. Interestingly, we noticed that IL-17 also induced dose-dependent cell apoptosis during the differentiation of OPCs to oligodendrocytes (Fig. 5e and Supplementary. Fig. 4c), and this effect was abolished in NG2CreAct1fl/− OPCs (Fig. 5f). TUNEL assay showed increased cell death in the parenchyma of spinal cords from NG2CreAct1fl/+ mice as compared with NG2CreAct1fl/− mice(43.0±8.3 versus 5.0±3.3 apoptotic cells/section) after Th17 adoptive transfer (Fig. 5g). Taken together, these data demonstrate that IL-17 exerted neurotoxic effects on OPCs in vitro, and, by extension, could mediate such effects in vivo. As inflammation and neurodegeneration are the cardinal pathogenic processes of MS, these results may have important implications for the human disease. We also examined the impact of IL-17 treatment on cultured astrocytes and neurons. Consistent with our previous findings17, while IL-17 induced an inflammatory response in astrocytes from both NG2CreAct1fl/+ and NG2CreAct1fl/− mice, neurons from either group of mice were barely responsive to IL-17 (data not shown). It is intriguing to note that IL-17 also induced apoptosis in astrocytes from NG2CreAct1fl/+ and NG2CreAct1fl/− mice, indicating that IL-17-induced apoptosis is not restricted to OPCs (data not shown). On the other hand, IL-17 stimulation failed to induce apoptosis in neurons (data not shown). Thus, these ex vivo results are consistent with our in vivo EAE phenotypes: while ablation of Act1 from astrocytes modestly reduces autoimmune encephalomyelitis, neuronal Act1 is dispensable for EAE development.
Although much effort has been devoted to study IL-17-induced inflammatory response, it is equally important to understand how IL-17 mediates cell apoptosis. Interestingly, we found IL-17 induced caspase 3 cleavage in wild-type MEFs, but not in Act1-deficient MEFs (Fig. 6a), confirming that IL-17-induced apoptosis is not restricted to OPCs. Although a recent study suggested that IL-17 can synergize with TNF-α to induce apoptosis38, we found that TNF-α neutralization did not affect IL-17 induced apoptosis in MEFs (Fig. 6b). Furthermore, IL-17 induced apoptosis in TNFR1/p55- TNFR2/p75- single and double knockout kidney epithelial cells (Fig. 6c). These results indicate IL-17 induces apoptosis independent of TNF-α but left unanswered how Act1 signaling intersects with apoptotic pathways. We found that Act1 interacted with FADD, a death domain containing adaptor in the cell death pathway (Fig. 6d). A recent study showed that IL-25 (IL-17E) receptor interacts with the death domain of FADD though the SEFIR domain39. Since Act1 also contains a SEFIR domain and interacts with FADD, we hypothesized that IL-17R might recruit FADD through Act1’s SEFIR domain. Interestingly, we found the SEFIR domain of Act1 was indeed required for its interaction with FADD (Fig 6d), and IL-17 failed to induce apoptosis in OPCs from which FADD was depleted by shRNA-mediated knock-down (Fig. 6E). These data implicate the IL-17R-Act1-FADD axis as a new pathway for cell apoptosis.
Figure 6.
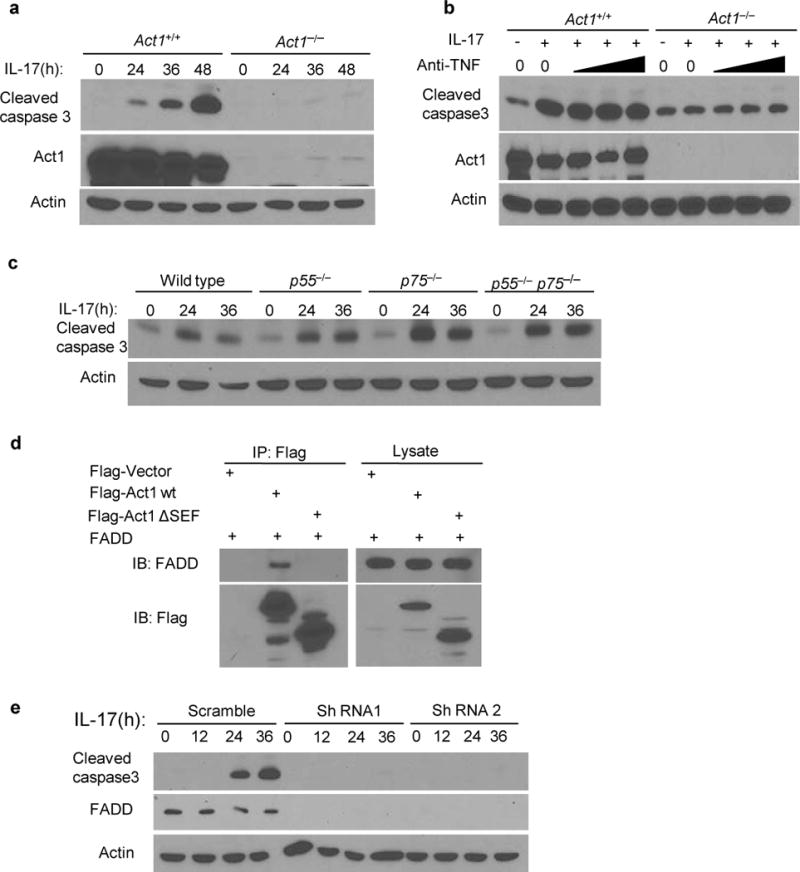
IL-17 induces cell apoptosis in an Act1 and FADD-dependent pathway. (a) Immunoassay of Act1+/+ or Act1−/− MEFs treated with IL-17 (50ng/ml) for the indicated times. Cells lysates were ran on SDS-PAGE gel and then probed for cleavaged caspase 3, Act1 and β-Actin. (b) MEFs treated with IL-17(50ng/ml) plus increasing amounts of anti-TNF-α neutralizing antibodies (1, 5, 10μg/ml). (c) Immunoblot analysis of wild type, p55-deficient, p75-deficient, p55 and p75 double-deficient kidney epithelial cells treated with IL-17 (200ng/ml) for the indicated times. (d) HEK293 cells were transfected with Flag-tagged vector as control, Flag -Act1 and – FADD. Lysates were immunoprecipitated with anti-Flag. (e) Immunoassay of OPCs transduced with lentivirus-expressing scrambled shRNA, or FADD-targeting shRNA1 or shRNA2. Cells were untreated or treated with IL-17 (200ng/ml). Cell lysates were blotted as indicated. Data are representative of at least three independent experiments. Full-length blots are presented in Supplementary Figure 6.
Discussion
The mechanisms by which IL-17 contributes to EAE pathogenesis have remained obscure. Our previous study showed that targeted Act1 deficiency in neuroectoderm-derived CNS resident cells resulted in markedly reduced severity in EAE17, suggesting that IL-17 operated within the CNS to cause disease manifestation. In this study, we used genetic models to address the cellular basis of this observation. We abrogated IL-17 signaling in astrocytes, oligodendroglial-lineage cells or neurons, to address directly how Th17-mediated demyelination and neurodegeneration proceed during EAE. Several notable observations emerged during these experiments. First this study provided definitive genetic evidence that IL-17 signaling to Act1 in astrocytes plays only a modest role in the severity of EAE. Since GFAP is also expressed by small fraction of neural stem cells, it raised the possibility that GFAP+ stem cells-derived NG2+ glia might partially account for the observed phenotype observed in GFAPCreAct1fl/− mice. However, western analysis showed that the sorted NG2+ glial cells from GFAP-Cre Act1fl/− mice had similar levels of Act1 compared to that in GFAP-Cre Act1fl/+ mice(Data not shown). Therefore, the reduced EAE phenotype observed in GFAP-CreAct1fl/− mice was probably due to the reduced inflammatory response of the astrocytes in these mice. Importantly, the deletion of Act1 from NG2+ glial cells dramatically reduced the severity of Th17 cell-induced EAE and recapitulated the EAE phenotype of mice which lacked Act1 in all neuroectodermal cells. Act1 deficiency in mature oligodendrocytes or neurons had no effect on EAE severity. We established that the EAE-resistant phenotype of NG2Cre Act1fl/− mice was attributable to IL-17 signaling to NG2+ glia rather than pericytes by showing an identical phenotype in Olig2CreAct1fl/− mice. NG2+ glial cell deficiency for Act1 coordinately reduced clinical severity and inflammatory infiltrates. Further, inflammatory gene expression analysis of tissues from NG2CreAct1fl/− mice with EAE showed a virtual absence of IL-17-induced genes such as Cxcl1 and Cxcl2, but no effect on IFN-γ-induced chemokines CXCL9 and CXCL10. The same gene expression pattern was observed in studies in vitro using cells derived from these mice. These results identify a novel and unexpected CNS-resident cellular target for IL-17-mediated EAE pathogenesis and initiate a discovery pathway for dissecting mechanisms of inflammatory neurodegeneration. It has been remarked on several occasions that NG2+ glia show morphological transformation in response to inflammation40, 41. To date, the microglia and astrocytes have been considered the major reactive glial elements of the CNS. The present study represents the first insight into specific pathogenic effects of NG2+ glial cell neuroinflammation.
Consistent with the in vivo EAE phenotype, IL-17 induced the induction of inflammatory gene expression in oligodendrocyte progenitors (OPCs) derived in vitro from neurosphere cells. While NG2 cells are also referred to as OPCs, about 85% of the cultured OPCs are NG2+ cells in our culture system. Taken with our in-vivo findings, IL-17 induced inflammatory response in OPCs strongly supports the interpretation that NG2+ cells in vivo are involved in IL-17-dependent inflammatory response during EAE. In support of this, we indeed found that NG2Cre-mediated Act1 deletion substantially reduced Th17-induced inflammatory gene expression in the spinal cords during EAE. Eliminating Act1 signaling from GFAP+ astrocytes also moderately reduced the severity of EAE and lessened expression of Th17- but not Th1-associated chemokines. Comparable effects were recently obtained by intrathecal lentiviral delivery of Act1 shRNA using an expression cassette that targeted astrocytes24.
Multiple sclerosis(and the EAE model) results in both inflammatory and neurodegenerative damage to the CNS tissue. The relationship between inflammation and neurodegeneration has been controversial and difficult to resolve. Recent important reports indicated that removal of myelin renders axons metabolically non-viable, placing intense emphasis on understanding mechanisms by which myelin repair either occurs or fails42, 43. We also found that IL-17 affected survival and maturation of OPCs derived in vitro from neurosphere cells. Following varied types of CNS injury, NG2+ glia proliferate and play a role in myelin repair and mainly give rise to oligodendrocytes21. Intriguingly, we found that while NG2+ cells were indeed activated during EAE (as evident by increased BrdU incorporation and NG2+ cell numbers), mature oligodendrocytes were substantially reduced in the spinal cords of NG2CreAct1fl/+ mice compared to that in NG2CreAct1fl/− mice. These results suggest that the NG2+ glia may indeed function as precursors/progenitors (OPCs) in vivo and their ability to participate in the repair of demyelination might be perturbed by IL-17-induced Act1-mediated signaling. Taken together, these ex vivo and in vivo observations suggested that IL-17 might mediate neurotoxic effects towards NG2 glia in EAE, in addition to eliciting pronounced pathogenic inflammatory responses from these cells. Impaired remyelination has profound consequence for the progressive loss of demyelinated axons and functional deficits. The remyelination process involves the proliferation and maturation of NG2 cells, which are widely distributed through adult CNS and remain committed to the oligodendrocyte lineage44, 45.
The importance of cell apoptosis in the pathogenesis of EAE has been extensively studied. Both extrinsic and intrinsic pathways of apoptosis affect EAE development46–49. The death of oligodendrocytes not only causes direct demyelination but also releases “danger signals” which further induce sterile inflammation. Moreover, the debris from the dead cells provides abundant self-antigens which continuously reactivate infiltrated T cells. Thus inflammation and cell death constitute a positive feedback loop in CNS autoimmune inflammation50. In conclusion, our present in vitro data implicate neurotoxic effects of IL-17 in the pathogenesis of EAE and finding increased numbers of apoptotic cells in EAE tissues of mice with intact Act1 signaling in NG2 glia supports this hypothesis. Taken together our data suggest that the immune and neurotoxic effects of IL-17 towards glial cells (predominantly NG2 glia) provide a crucial link joining the inflammatory and neurodegenerative aspects of mutiple sclerosis.
Online Methods
Mice
Act1-deficient mice on the C57BL/6J background were generated as described51. C57BL/6-Tg(GFAP-Cre)8Gtm mice were purchase from National Cancer Institute52. CNPaseCre transgenic mice were provided by Dr. Klaus-Armin Nave (Max-Planck-Institute of Experimental Medicine). B6-Tg(Cspg4-cre)1Akik/J and B6.Cg-Tg(Eno2-cre)39Jme/J53 mice were purchased from Jackson laboratory. All the mice used in this study were female mice unless specified otherwise. For all experiments, mice were 6–12 week-old, age matched littermates between experimental groups. Mice were housed under specific pathogen-free conditions. Experimental protocols were approved by the Institutional Animal Care and Use Committee of Cleveland Clinic.
Induction and assessment of EAE
Active EAE was induced and assessed as previously described. Passive EAE was also induced as previously described. In brief, recipient mice were injected with 3.0 × 107 polarized MOG35–55-specific Th1 or Th17 cells/mouse 4 hours after 500-Rad sub-lethal irradiation. To prepare MOG35–55 specific polarized T cells, donor mice were immunized with MOG35–55 subcutaneously, draining lymph node cells were prepared from donor mice 10 days after immunization. Cells were cultured for 5 days with MOG35–55 at a concentration of 25 μg/ml under Th1 (20ng/ml rmIL-12[R&D], 2μg/ml αIL-23p19[eBioscience]) polarizing conditions or Th17(20ng/ml rmIL-23[R&D]) polarizing condition. The clinical score was performed in double-blinded manner. For the sample size, we performed power analysis for the clinical score of EAE, which has an average of 25% coefficient of variance. We determined that with n=15 we have 90% power to detect 30% difference between the groups.
Isolation and analysis of CNS inflammatory cells
Brains were homogenized in ice-cold tissue grinders, filtered through a 70 μm cell strainer and the cells collected by centrifugation at 400g for 5min at 4°C. Cells were resuspended in 10ml of 30% Percoll (Amersham Bioscience) and centrifuged onto a 70% Percoll cushion in 15-ml tubes at 800g for 30min. Cells at the 30–70% interface were collected and were subjected to flow cytometry. Fluorescence-conjugated CD4(Clone GK1.5), CD8(Clone 53–6.7), CD45(Clone 30-F11), Ly6G(Clone 1A8) monoclonal antibodies and isotype controls were purchased from BD Biosciences. F4/80(Clone Cl:A3-1) was obtained from Serotech. The antibodies were diluted at 1:100 when used.
Histological analysis
All the sections used here were 5 μm of thickness. For paraffin-embedded tissue, spinal cords collected from PBS-perfused mice were fixed in 10% formalin first, then dehydrated with 70% alcohol. Sections were stained with either hematoxylin and eosin (H&E) or luxol fast blue (LFB) to evaluate inflammation and demyelination, respectively. For frozen sections, fresh spinal cords were embedded in OCT (Tissue-Tek) and snap frozen in liquid nitrogen. Sections were incubated with anti-CD11b (BD Biosciences, M1/70, 1:200). Antigens were visualized following incubation with HRP conjugated secondary antibodies (Molecular probe). For cytohistochemical staining, cells were fixed by 2% paraformaldehyde for 10 minutes. Cells were further permeabilized by 0.1% Triton for 10 minutes, then incubated with primary antibody followed by fluorescence-conjugated secondary antibodies for microscopic analysis. NG2 antibodies were provided by William B. Stallcup (Burnham Institute for Medical Research, La Jolla, CA). Olig2 antibodies(AB9610, 1:500) were bought from Millipore. CNPase(SMI 91, 1:200) and MBP antibodies(SMI 99, 1:200) were bought from Convance Inc. Cleaved caspase 3 antibodies(5A1E, 1:200) were brought from Cell Signaling Technology and GST-π antibodies(ADI-MSA-102-E, 1:1000) from Enzo life science. The quantification of histology was performed in a double-blinded manner.
Primary culture of CNS resident cells
Astrocyte culture was prepared from 1 day-old neonatal mice. Briefly, brains freed of meninges were dissociated with 1ml pipettes. Debris was removed by filtration with a 70μm cell strainer (Falcon). Cells were cultured in DMEM plus 10% FBS supplemented with 50μg/ml penicillin and 50μg/ml streptomycin. After 10 days, Astrocytes were stained with anti- glial fibrillary acidic protein (GFAP) (Sigma, G3893, 1:500) and purity was >95%. Neurons were prepared from the pups at E15. Brains were dissociated with 1ml pipettes and the debris removed using a 70μm cell strainer (Falcon). Cells were cultured in Neuronbasal media (Invitrogen) plus B-27 (invitrogen) and 50μg/ml penicillin and 50μg/ml streptomycin. >90% of these cultured cells were positive for β-Tubulin III(a marker for neurons, anti-β-Tubulin III from Sigma, 2G10, 1:500). The culture of oligodendrocyte progenitor cells (OPCs) was done as described previously54, 55. In brief, neurospheres (Nphs) were prepared from embryos at 14.5 days of gestation obtained from timed pregnant females. Nph medium was DMEM/F12, B27 neuronal supplement (Invitrogen), and 10 ng/ml recombinant mouse EGF (R&D). Floating NPhs were passaged at 1:3 ratio in the same medium every 3–4 days. To produce pure OPCs, neurospheres were dissociated after 2–3 passages, dissociated cells were plated on poly-D-lysine-coated plates under the same medium of neurosphere but with 10ng/ml FGF+10ng/ml PDGF(Peprotech), instead of EGF. >98% cells were Olig2+ CNPase− cells after 4 days culture suggesting they were OPCs. Then cytokines were withdrawn to drive the maturation of OPC for 2 days, immunohistochemical staining suggested that >95% cells are CNPase+ GFAP−β-tubulin-III− suggesting they were mature oligodendrocytes. For genotyping of NG2CreAct1fl/− and NG2CreAct1fl/+ mice, OPCs from NG2CreAct1fl/− and NG2CreAct1fl/+ mice were cultured as stated above for 4 days, and were collected and stained with anti-NG2 antibodies, FITC-conjugated second antibodies were used for primary anti-NG2 antibodies. Following this, stained OPCs were subjected for sorting by flow cytometry. The purity of NG2+ cells after sorting was >98%. Sorted cells were lysed and western blot was used to assay Act1 expression NG2+ cells from NG2Cre Act1fl/− and NG2Cre Act1fl/+ mice.
Real-time PCR
Total RNA was extracted from spinal cord with TRIzol (Invitrogen) according to the manufacture’s instructions. All gene expression results are expressed as arbitrary units relative to expression of the gene encoding β-actin. Fold induction of gene expression in spinal cord after EAE induction was determined by dividing the relative abundance of experimental samples by the mean relative abundance of control samples from naïve mice. Primer sequences were described previously17.
TUNEL assay
Sections from paraffin-embeded spinal cords were processed for the terminal deoxynucleotidyl transferase biotin-dUTP nick-end labeling (TUNEL) staining using a kit from the manufacturer (Roche). For apoptosis analysis of cultured cells, cells were fixed by 2% paraformaldehyde. The other steps for TUNEL staining followed the manufacturer’s instruction. The cells were counter-stained with DAPI (4′, 6′-diamidino-2-phenylindole). Quantification was done in a double-blinded manner.
Immunoprecipitation
For co-immunoprecipitations, cell extracts were incubated with antibodies and protein A beads. After overnight incubation, beads were washed four times with lysis buffer, resolved by SDS-polyacrylamide gel electrophoresis (SDS-PAGE), and analyzed by Western blotting according to a standard procedure.
Statistics
Non-parametric statistics was applied to all data set. The p values of clinical scores were determined by two-way multiple-range analysis of variance (ANOVA) for multiple comparisons except for astrocyte-specific conditional knockout mice where mann-whitney test were used for each time point(GFAPCreAct1fl/+ versus GFAPCreAct1fl/−). Other p values were determined by Mann-Whitney test. P value of <0.05 was considered significant. Unless otherwise specified, all results are shown as mean and the s.e.m.
Supplementary Material
Acknowledgments
We gratefully acknowledge Dr. William B. Stallcup (Burnham Institute for Medical Research, La Jolla, CA) for providing antibodies to NG2. We thank Dr. K.-A. Nave for providing CNPase Cre transgenic mice(Max Planck Institute of Experimental Medicine, Germany). We also thank the Dana-Farber Cancer Institute for providing the Olig2-Cre transgenic mice. This investigation was supported by grants from US National Institutes of Health (R01NS071996 and P01HL103453).
Footnotes
Author contributions:
Z.K. designed and performed most of the experiments, interpreted the data, and wrote part of the manuscript. C.W. carried out most of the Western blotting. J. Zepp. did some western blotting and manuscript editing. L.W. performed some immunohistochemical staining. K.S. aided in western blotting, immunohistochemical staining and real-time PCR. J. Zhao. performed statistical analysis. U.C. executed mouse breeding and cell culture. P.E.D. helped with experimental design. B.D.T. also interpreted data. R.M.R. contributed with experimental design and manuscript writing. X.X.L. was integral in experimental design, manuscript writing, data interpretation, and project coordination.
References
- 1.Sawcer S, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–219. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Becher B, Bechmann I, Greter M. Antigen presentation in autoimmunity and CNS inflammation: how T lymphocytes recognize the brain. J Mol Med (Berl) 2006;84:532–543. doi: 10.1007/s00109-006-0065-1. [DOI] [PubMed] [Google Scholar]
- 3.Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- 4.Steinman L. Multiple sclerosis: a two-stage disease. Nat Immunol. 2001;2:762–764. doi: 10.1038/ni0901-762. [DOI] [PubMed] [Google Scholar]
- 5.Ransohoff RM. Animal models of multiple sclerosis: the good, the bad and the bottom line. Nat Neurosci. 2012;15:1074–1077. doi: 10.1038/nn.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stromnes IM, Cerretti LM, Liggitt D, Harris RA, Goverman JM. Differential regulation of central nervous system autoimmunity by T(H)1 and T(H)17 cells. Nat Med. 2008;14:337–342. doi: 10.1038/nm1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM. IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med. 2008;205:1535–1541. doi: 10.1084/jem.20080159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cua DJ, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 9.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 10.Mangan PR, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 11.Komiyama Y, et al. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- 12.Hu Y, et al. IL-17RC is required for IL-17A- and IL-17F-dependent signaling and the pathogenesis of experimental autoimmune encephalomyelitis. J Immunol. 2010;184:4307–4316. doi: 10.4049/jimmunol.0903614. [DOI] [PubMed] [Google Scholar]
- 13.Li X, et al. Act1, an NF-kappa B-activating protein. Proc Natl Acad Sci U S A. 2000;97:10489–10493. doi: 10.1073/pnas.160265197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leonardi A, Chariot A, Claudio E, Cunningham K, Siebenlist U. CIKS, a connection to Ikappa B kinase and stress-activated protein kinase. Proc Natl Acad Sci U S A. 2000;97:10494–10499. doi: 10.1073/pnas.190245697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qian Y, Zhao Z, Jiang Z, Li X. Role of NF kappa B activator Act1 in CD40-mediated signaling in epithelial cells. Proc Natl Acad Sci U S A. 2002;99:9386–9391. doi: 10.1073/pnas.142294499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qian Y, et al. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol. 2007;8:247–256. doi: 10.1038/ni1439. [DOI] [PubMed] [Google Scholar]
- 17.Kang Z, et al. Astrocyte-restricted ablation of interleukin-17-induced Act1-mediated signaling ameliorates autoimmune encephalomyelitis. Immunity. 2010;32:414–425. doi: 10.1016/j.immuni.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raasch J, et al. IkappaB kinase 2 determines oligodendrocyte loss by non-cell-autonomous activation of NF-kappaB in the central nervous system. Brain. 2011;134:1184–1198. doi: 10.1093/brain/awq359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Loo G, et al. Inhibition of transcription factor NF-kappaB in the central nervous system ameliorates autoimmune encephalomyelitis in mice. Nat Immunol. 2006;7:954–961. doi: 10.1038/ni1372. [DOI] [PubMed] [Google Scholar]
- 20.Nishiyama A, Komitova M, Suzuki R, Zhu X. Polydendrocytes (NG2 cells): multifunctional cells with lineage plasticity. Nat Rev Neurosci. 2009;10:9–22. doi: 10.1038/nrn2495. [DOI] [PubMed] [Google Scholar]
- 21.Richardson WD, Young KM, Tripathi RB, McKenzie I. NG2-glia as multipotent neural stem cells: fact or fantasy? Neuron. 2011;70:661–673. doi: 10.1016/j.neuron.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gold R, Linington C, Lassmann H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain. 2006;129:1953–1971. doi: 10.1093/brain/awl075. [DOI] [PubMed] [Google Scholar]
- 23.Weaver A, et al. An elevated matrix metalloproteinase (MMP) in an animal model of multiple sclerosis is protective by affecting Th1/Th2 polarization. FASEB J. 2005;19:1668–1670. doi: 10.1096/fj.04-2030fje. [DOI] [PubMed] [Google Scholar]
- 24.Yan Y, et al. CNS-specific therapy for ongoing EAE by silencing IL-17 pathway in astrocytes. Mol Ther. 2012;20:1338–1348. doi: 10.1038/mt.2012.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siffrin V, et al. In vivo imaging of partially reversible th17 cell-induced neuronal dysfunction in the course of encephalomyelitis. Immunity. 2010;33:424–436. doi: 10.1016/j.immuni.2010.08.018. [DOI] [PubMed] [Google Scholar]
- 26.Baumann N, Pham-Dinh D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev. 2001;81:871–927. doi: 10.1152/physrev.2001.81.2.871. [DOI] [PubMed] [Google Scholar]
- 27.Fancy SP, Chan JR, Baranzini SE, Franklin RJ, Rowitch DH. Myelin regeneration: a recapitulation of development? Annu Rev Neurosci. 2011;34:21–43. doi: 10.1146/annurev-neuro-061010-113629. [DOI] [PubMed] [Google Scholar]
- 28.Trotter J, Karram K, Nishiyama A. NG2 cells: Properties, progeny and origin. Brain Res Rev. 2010;63:72–82. doi: 10.1016/j.brainresrev.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Menn B, et al. Origin of oligodendrocytes in the subventricular zone of the adult brain. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2006;26:7907–7918. doi: 10.1523/JNEUROSCI.1299-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu X, Bergles DE, Nishiyama A. NG2 cells generate both oligodendrocytes and gray matter astrocytes. Development. 2008;135:145–157. doi: 10.1242/dev.004895. [DOI] [PubMed] [Google Scholar]
- 31.Emery B, et al. Myelin gene regulatory factor is a critical transcriptional regulator required for CNS myelination. Cell. 2009;138:172–185. doi: 10.1016/j.cell.2009.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou Q, Wang S, Anderson DJ. Identification of a novel family of oligodendrocyte lineage-specific basic helix-loop-helix transcription factors. Neuron. 2000;25:331–343. doi: 10.1016/s0896-6273(00)80898-3. [DOI] [PubMed] [Google Scholar]
- 33.Schuller U, et al. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell. 2008;14:123–134. doi: 10.1016/j.ccr.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu WP, Collarini EJ, Pringle NP, Richardson WD. Embryonic expression of myelin genes: evidence for a focal source of oligodendrocyte precursors in the ventricular zone of the neural tube. Neuron. 1994;12:1353–1362. doi: 10.1016/0896-6273(94)90450-2. [DOI] [PubMed] [Google Scholar]
- 35.Lappe-Siefke C, et al. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet. 2003;33:366–374. doi: 10.1038/ng1095. [DOI] [PubMed] [Google Scholar]
- 36.Dugas JC, et al. Dicer1 and miR-219 Are required for normal oligodendrocyte differentiation and myelination. Neuron. 2010;65:597–611. doi: 10.1016/j.neuron.2010.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaga Y, et al. Mice with conditional inactivation of fibroblast growth factor receptor-2 signaling in oligodendrocytes have normal myelin but display dramatic hyperactivity when combined with Cnp1 inactivation. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2006;26:12339–12350. doi: 10.1523/JNEUROSCI.3573-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paintlia MK, Paintlia AS, Singh AK, Singh I. Synergistic activity of interleukin-17 and tumor necrosis factor-alpha enhances oxidative stress-mediated oligodendrocyte apoptosis. J Neurochem. 2011;116:508–521. doi: 10.1111/j.1471-4159.2010.07136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Furuta S, et al. IL-25 causes apoptosis of IL-25R-expressing breast cancer cells without toxicity to nonmalignant cells. Sci Transl Med. 2011;3:78ra31. doi: 10.1126/scitranslmed.3001374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simon C, Gotz M, Dimou L. Progenitors in the adult cerebral cortex: cell cycle properties and regulation by physiological stimuli and injury. Glia. 2011;59:869–881. doi: 10.1002/glia.21156. [DOI] [PubMed] [Google Scholar]
- 41.Nishiyama A, Chang A, Trapp BD. NG2+ glial cells: a novel glial cell population in the adult brain. J Neuropathol Exp Neurol. 1999;58:1113–1124. doi: 10.1097/00005072-199911000-00001. [DOI] [PubMed] [Google Scholar]
- 42.Lee Y, et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. 2012;487:443–448. doi: 10.1038/nature11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Funfschilling U, et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature. 2012;485:517–521. doi: 10.1038/nature11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Butts BD, Houde C, Mehmet H. Maturation-dependent sensitivity of oligodendrocyte lineage cells to apoptosis: implications for normal development and disease. Cell Death Differ. 2008;15:1178–1186. doi: 10.1038/cdd.2008.70. [DOI] [PubMed] [Google Scholar]
- 45.Franklin RJ, Ffrench-Constant C. Remyelination in the CNS: from biology to therapy. Nat Rev Neurosci. 2008;9:839–855. doi: 10.1038/nrn2480. [DOI] [PubMed] [Google Scholar]
- 46.Lev N, Barhum Y, Melamed E, Offen D. Bax-ablation attenuates experimental autoimmune encephalomyelitis in mice. Neurosci Lett. 2004;359:139–142. doi: 10.1016/j.neulet.2004.01.076. [DOI] [PubMed] [Google Scholar]
- 47.Hisahara S, Okano H, Miura M. Caspase-mediated oligodendrocyte cell death in the pathogenesis of autoimmune demyelination. Neurosci Res. 2003;46:387–397. doi: 10.1016/s0168-0102(03)00127-5. [DOI] [PubMed] [Google Scholar]
- 48.Mc Guire C, Beyaert R, van Loo G. Death receptor signalling in central nervous system inflammation and demyelination. Trends Neurosci. 2011;34:619–628. doi: 10.1016/j.tins.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 49.Hara H, Nanri Y, Tabata E, Mitsutake S, Tabira T. Identification of astrocyte-derived immune suppressor factor that induces apoptosis of autoreactive T cells. J Neuroimmunol. 2011;233:135–146. doi: 10.1016/j.jneuroim.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 50.Patel J, Balabanov R. Molecular mechanisms of oligodendrocyte injury in multiple sclerosis and experimental autoimmune encephalomyelitis. Int J Mol Sci. 2012;13:10647–10659. doi: 10.3390/ijms130810647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qian Y, et al. Act1, a negative regulator in CD40- and BAFF-mediated B cell survival. Immunity. 2004;21:575–587. doi: 10.1016/j.immuni.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 52.Bajenaru ML, et al. Astrocyte-specific inactivation of the neurofibromatosis 1 gene (NF1) is insufficient for astrocytoma formation. Mol Cell Biol. 2002;22:5100–5113. doi: 10.1128/MCB.22.14.5100-5113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frugier T, et al. Nuclear targeting defect of SMN lacking the C-terminus in a mouse model of spinal muscular atrophy. Human molecular genetics. 2000;9:849–858. doi: 10.1093/hmg/9.5.849. [DOI] [PubMed] [Google Scholar]
- 54.Pedraza CE, Monk R, Lei J, Hao Q, Macklin WB. Production, characterization, and efficient transfection of highly pure oligodendrocyte precursor cultures from mouse embryonic neural progenitors. Glia. 2008;56:1339–1352. doi: 10.1002/glia.20702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen Y, et al. Isolation and culture of rat and mouse oligodendrocyte precursor cells. Nat Protoc. 2007;2:1044–1051. doi: 10.1038/nprot.2007.149. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.