

Abstract
Synthetic Mimics of Antimicrobial Peptides (SMAMPs) imitate natural host-defense peptides, a vital component of the body’s immune system. This work presents a molecular construction kit that allows the easy and versatile synthesis of a broad variety of facially amphiphilic oxanorbornene-derived monomers. Their ring-opening metathesis polymerization (ROMP) and deprotection provide several series of SMAMPs. Using amphiphilicity, monomer feed ratio, and molecular weight as parameters, polymers with 533 times higher selectivitiy (selecitviy = hemolytic concentration/minimum inhibitory concentration) for bacteria over mammalian cells were discovered. Some of these polymers were 50 times more selective for Gram-positive over Gram-negative bacteria while other polymers surprisingly showed the opposite preference. This kind of “double selectivity” (bacteria over mammalian and one bacterial type over another) is unprecedented in other polymer systems and is attributed to the monomer’s facial amphiphilicity.
Introduction
When organisms are attacked by bacterial pathogens, natural antimicrobial peptides (AMPs) are among the first line of defense. These host-defense peptides have broad-spectrum antimicrobial activity.1 Their production in the organism is much faster than that of specific antibodies, and thus they are a vital component of innate immunity. AMPs are found in many species, including humans, animals, plants, and invertebrates.1 Whereas today’s common antibiotics target specific cell structures, AMPs use nonreceptor interactions, including in many cases direct action against the bacteria’s membranes, although other targets have been identified.1,2 The cells of the host organism are less affected; thus AMPs can selectively attack bacteria within a host organism. Bacteria could only become immune to AMPs if they change their entire membrane chemistry or other targets; thus resistance to AMPs is retarded as compared to other antibiotics.3 Due to this promising feature, there has been increasing research in the past few years aimed at the production of synthetic mimics of antimicrobial peptides (SMAMPs). These include the SMAMPs made of α- and β-amino acids,4-11 peptoids,12-14 aromatic oligomers,15-17 and synthetic polymers.18-23
The common feature of most AMPs is their positive charge and facial amphiphilicity. Regardless of their secondary structure, these peptides generally display one hydrophobic and one hydrophilic face along their backbone.1,24,25 Due to positive charges in the hydrophilic part, AMPs bind preferentially to the anionic outer membranes of bacterial pathogens or other anionic targets including proteins and DNA.3,26 In many cases, their facial amphiphilicity allows them to insert into the bacterial membrane and to locally change the membrane’s lipid organization in such a way that transmembrane pores are formed, although other mechanisms of action are also known.1 Several mechanisms of pore formation have been proposed to describe this, including the carpet, barrel-stave, and toroidal pore models.1,2 This interaction may lead to a breakdown of the membrane potential, the leaking of the cytoplasm, and eventually the death of the pathogen cell.27
Learning how to capture the essential biological properties of AMPs in synthetic polymers should teach us which essential chemical features of these natural peptides are required for antibacterial activity. In addition, access to these synthetic polymers may open up new applications, for example, in the materials area, where bacterial infections from medical plastics are a current critical problem in our hospitals. Synthetic polymers can be obtained easily and in large quantities while still presenting facial amphiphilicity and positive charge, the key features of AMPs. Although there have been several recent reports of polymeric SMAMPs, their overall activities and selectivities remain far from optimal. Examples include the following: DeGrado and co-workers reported SMAMPs based on poly(ammonium methylmethacrylate) salts copolymerized with poly(butylmethacrylate) to tune the amphiphilicity;28 Klajnert et al.29 produced dendritic SMAMPs; Liu et al.30 synthesized SMAMPs from poly(maleic acid) linked to peptide tetramers; Makovitzki et al.31 recently made SMAMPs based on lipopeptides; and Gellman and co-workers presented a poly(amide) based polymer with good activities (12.5 μg/mL against E. coli and 3.1 μg/mL against S. aureus) and selectivities up to 32 for bacterial over mammalian cells.23 Tew and coworkers synthesized facially amphiphilic antibacterial polymers basedonarylamides,15 urea,17andpoly(phenyleneethynylene).20,21 SMAMPs based on poly(norbornene) derivatives were previously described by Tew and Coughlin: they reported polymers with facially amphiphilic repeat units that had tunable antimicrobial activity depending on a defined ratio of hydrophobic and hydrophilic moieties in the repeat unit. Their most selective polymer had a hundred times higher activity toward bacteria than against human red blood cells.19 They also very recently reported poly(norbornenes) with quaternary pyridinium groups (selectivities up to 20 against E. coli).32
The previously reported poly(norbornene) based SMAMPs suffered from the fact that each polymer required extensive synthetic effort to tune the amphiphilicity of the repeat units19 or did not allow copolymer synthesis.32 The aim of this work was therefore to develop a ring-opening metathesis polymerization (ROMP) platform that (i) uses a minimum number of building blocks and (ii) allows the easy and independent variation of the hydrophobic and hydrophilic residues on the monomer. Although previous work has shown that antimicrobial activity can also be achieved with random copolymers of hydrophilic and hydrophobic monomers,18,23 we believe that having facially amphiphilic monomers, i.e., monomers with a hydrophilic cationic and a hydrophobic part on the same polymerizable unit, allows for more precise tuning of the antibacterial activity. The key components of our molecular construction kit are highlighted in Figure 1. The hydrophilic (blue) and the hydrophobic component (green) are attached to the polymerizable oxanorbornene group (yellow) and can be varied independently. In this report we have restricted ourselves to varying the hydrophobic component, while holding the hydrophilic “lysine-like” primary amine constant.
Figure 1.
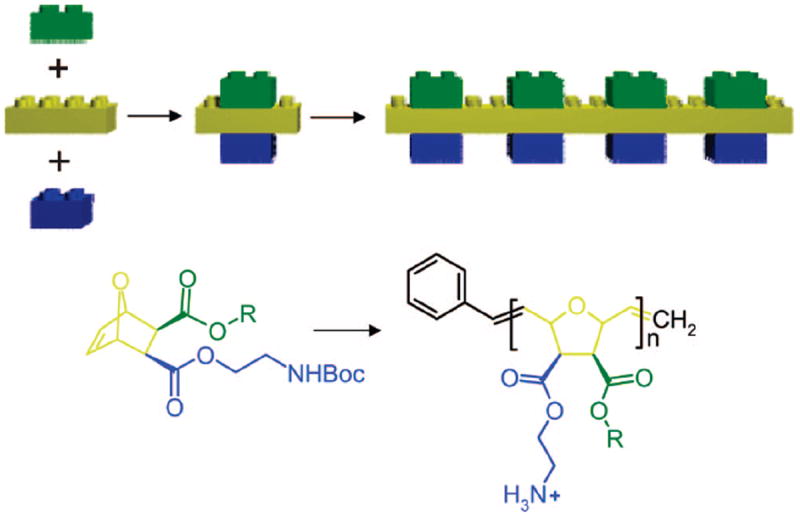
“Construction kit” approach to obtain facially amphiphilic monomers and polymers. Just as with a Lego construction kit, the synthetic approach presented here allows the independent combination of a hydrophilic (blue), a hydrophobic (green), and a polymerizable (yellow) part of the monomer to yield a whole set of antimicrobial polymers with tunable activity and selectivity.
Experimental Section
All experimental procedures, including monomer and polymer synthesis, as well as the biological assays, are included in the Supporting Information.
Results and Discussion
Monomer Synthesis
To obtain new synthetic antimicrobial polymers via ROMP, the first task was to design an easy and modular synthetic pathway toward facially amphiphilic monomers (Figure 1). The three-step approach taken to obtain these monomers is presented in Scheme 1. In the first step, furan and maleic anhydride underwent a Diels–Alder reaction, yielding exclusively the exoadduct in accordance with the literature.34 This facile step provided compound 1 containing a polymerizable oxanorbornene group and a cyclic anhydride that allowed 2-fold and unsymmetrical functionalization. The anhydride 1 was ring-opened with an alcohol to introduce the desired hydrophobic moiety R, which was varied from methyl to hexyl, yielding a series of half-monomers 2a–f with different hydrophobicities. All compounds were crystallizable and thus easy to purify. In the last step, the designated cationic group was attached. As ROMP usually does not tolerate the presence of unprotected amines due to their ligating properties,35 the desired hydrophilic group (NH3+) was introduced in its protected tertbutyl carbamate (NHBoc) form. The half-monomers 2a–f were reacted with the Boc-protected 2-amino ethanol by DCC coupling, yielding a series of masked amphiphilic monomers 3a–f (Scheme 1). This last step required purification by column chromatography to yield pure products. The overall yield of all three reaction steps was ~40% (see Supporting Information for all details).
Scheme 1.
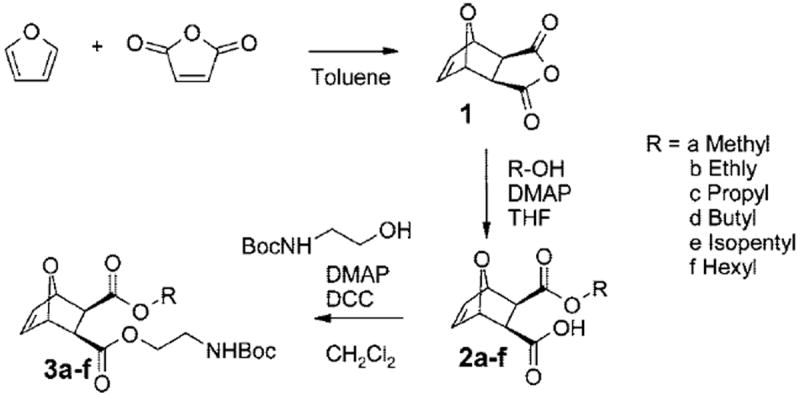
Monomer Synthesisa
a The hydrophobic component of the facially amphiphilic monomer is introduced in the second reaction step (R = methyl, ethyl, propyl, butyl, isopentyl, or hexyl), and the protected hydrophilic moiety is attached in the last step.
Homopolymer Synthesis and Molecular Weight Characterization
From the monomers 3a–f, two series of homopolymers with molecular weights of 3000 g/mol (= 3k series) and 10 000 g/mol (= 10 k series) were synthesized. The polymerization of the monomers, shown in Scheme 2, was carried out using the third generation Grubbs catalyst (G3)36 and yielded the precursor polymers 4a–f. The facially amphiphilic SMAMPs 5a–f were obtained by polymer analogous deprotection: the Boc protecting group was completely removed with trifluoroacetic acid according to NMR. Depending on the alkyl residue, the resulting polymers were water-soluble or dispersible after workup. Details on the workup procedure can be found in the Supporting Information. For the reader’s convenience, instead of referring to these polymers by compound number, for example 5a–f, a polymer with R = propyl and a molecular weight of 3000 g/mol will be referred to as Propyl_3k. The organosoluble precursor polymers were analyzed by gel permeation chromatography (GPC) in DMF (Table 1a). The molecular weights obtained from GPC using polystyrene standards for calibration are significantly larger than the ones expected from the reaction stoichiometry. It is well-known from the literature that the G3 catalyst initiates quantitatively,35 so incomplete initiation is unlikely to be responsible for this disparity. Analysis of the Boc-protected Propyl_3k sample (Mn, GPC = 9200 g/mol) by MALDI-TOF MS (Matrix Assisted Laser Ionization and Desorption Time Of Flight Mass Spectrometry) yields a distribution with a maximum intensity at m/z = 3095, which is in excellent agreement with the targeted molecular weight. This means that GPC overestimates the molecular weight for these structures, which is further supported by the oligomer data discussed in more detail below.
Scheme 2.
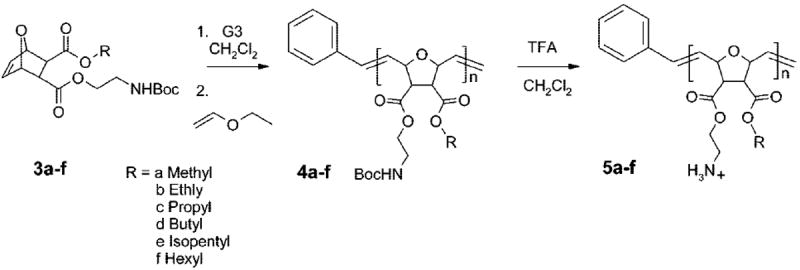
Polymer Synthesisa
a ROMP polymerization is followed by polymer analogous hydrolysis with trifluoroacetic acid to yield the facially amphiphilic polymer.
Table 1.
Homopolymers
| (a) Precursor polymer characterization by GPCa
| ||||
|---|---|---|---|---|
| sample | monomer | Mn. Target g mol−1 | GPC Mn g mol−1 | Mw/Mn |
| Methyl_3k | methyl | 3000 | 11 200 | 1.08 |
| Ethyl_3k | ethyl | 3000 | 9200 | 1.10 |
| Propyl_3k | propyl | 3000 | 9200 | 1.10 |
| Butyl_3k | butyl | 3000 | 11 500 | 1.08 |
| Isopentyl_3k | isopentyl | 3000 | 11 400 | 1.11 |
| Hexyl_3k | hexyl | 3000 | 11 000 | 1.08 |
| Methyl_10k | methyl | 10 000 | 49 000 | 1.05 |
| Ethyl_10k | ethyl | 10 000 | 11 300 | 1.10 |
| Propyl_10k | propyl | 10 000 | 28 200 | 1.07 |
| Butyl_10k | butyl | 10 000 | 26 700 | 1.07 |
| Isopentyl_8k | isopentyl | 8000 | 22 600 | 1.06 |
| Isopentyl_10k | isopentyl | 10 000 | 51 500 | 1.16 |
| Hexyl_10k | hexyl | 10 000 | 27 600 | 1.04 |
| (b) Characterization of the antimicrobial homopolymers by biological assaysb
| |||||
| sample | MIC90
μg mL−1
|
HC50 μg mL−1 | selectivty
|
||
| E. coli | S. aureus | E. coli | S. aureus | ||
|
| |||||
| Methyl_3k | >200 | 100 | 2000 | <10 | 20 |
| Ethyl_3k | 50 | 50 | 1400 | 28 | 28 |
| Propyl_3k | 6.25 | 15 | 50 | 8.3 | 3.3 |
| Butyl_3k | 15 | 25 | <50 | <3.3 | <2.0 |
| Isopentyl_3k | 12.5 | 50 | <50 | <4.0 | <1.0 |
| Hexyl_3k | >200 | >200 | <50 | <0.3 | <0.3 |
| Methyl_10k | >200 | >200 | 50 | <10 | <10 |
| Ethyl_10k | >200 | >200 | 1250 | <6.3 | <6.3 |
| Propyl_10k | 3.75 | 200 | <50 | 13 | <0.25 |
| Butyl_10k | 20 | >200 | <50 | <2.5 | <0.3 |
| Isopentyl_8k | 50 | >200 | <50 | <0.3 | <0.3 |
| Isopentyl_10k | 50 | 200 | <50 | <0.3 | <0.3 |
| Hexyl_10k | 100 | >200 | n.d. | n.d. | n.d. |
DMF, 0.01 mol L−1 LiCl, PS standards.
Inhibitory activity towards bacterial growth of E. coli and S. aureus bacteria. (MIC90 = minimal inhibitory concentration preventing 90% bacterial growth) and hemolytic activity towards red blood cells (HC50 = concentration lysing 50% of blood cells).
Biological Activity of Homopolymers
The biological properties of the homopolymers, i.e., their antibacterial activity, MIC90, toward growth of Escherichia coli and Staphylococcus aureus bacteria, and their hemolytic activity, HC50, toward red blood cells, were tested as described previously.37 HC50/MIC90 quantifies the antimicrobial selectivity. The biological data obtained for these homopolymers are included in Table 1b. The MIC90 and HC50 values are also plotted in Figure 2a and 2b. As can be seen from this set of data, the polymer with the highest selectivity for bacterial over mammalian cells is Ethyl_3k, with a good selectivity of 28 for both E. coli and S. aureus. For comparison, MSI-78, a derivative of the natural host-defense peptide magainin, has a selectivity of 10.38 For the 3k series shown in Figure 2a, the following trend is observed: starting with the nontoxic (HC50 = 2000 μg/mL) and inactive (MIC90 > 200 μg/mL) Methyl_3k, the antibacterial activity peaks for the Propyl_3k (MIC90 = 6.25 μg/mL for E. coli), and there the polymers also start becoming more hemolytic (HC50 ≤ 50 μg/mL). From Butyl_3k to Hexyl_3k the activity decreases again until the polymers become inactive (MIC90 > 200 μg/mL) but this time becoming strongly hemolytic. The MIC90’s for S. aureus show the same trend; however the activities are generally lower. The data for the 10k series are compiled in Figure 2b. The MIC90’s for the 10k polymers are similar to the ones for the 3k series, with a maximum activity at Propyl_10k, but they are generally less active against E. coli than the 3k polymers and inactive against S. aureus.
Figure 2.
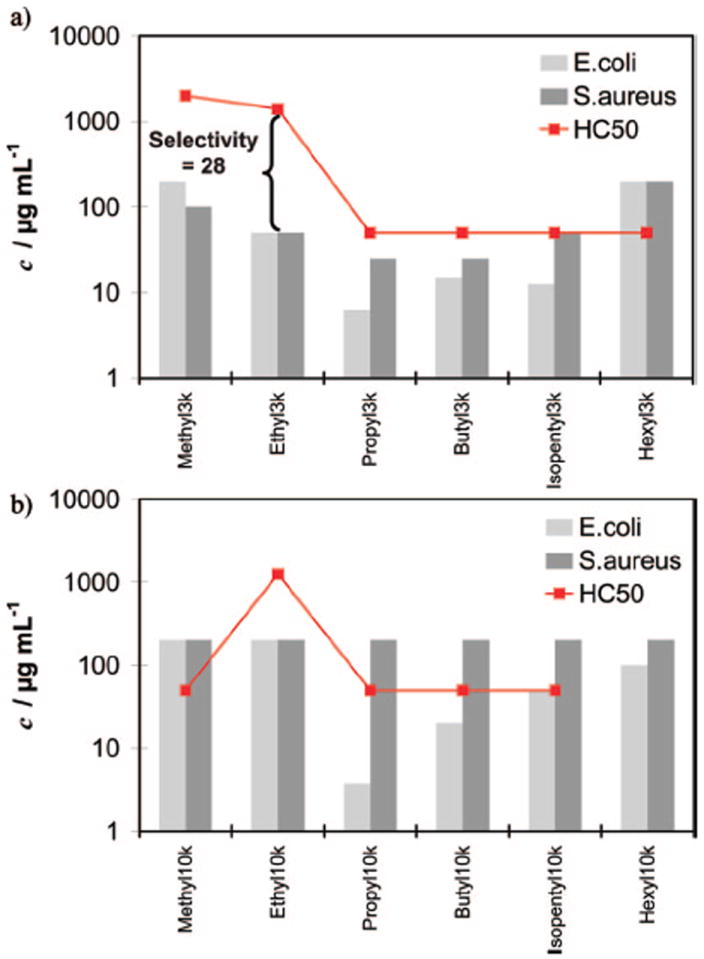
Biological data (MIC90 for E. coli and S. aureus, and HC50 for red blood cells) for the homopolymers. (a) 3000 g/mol series, (b) 10 000 g/mol series.
These observations can be rationalized by taking the different compositions of mammalian and bacterial membranes into account. Human red blood cells (RBC) are predominantly composed of cholesterol and phosphatidylcholine (outer leaflet). In contrast, the membranes of Gram-negative bacteria like E. coli consist mostly of phosphatidylethanolamine and anionically charged phosphatidylglycerol (PG) while Gram-positive bacteria like S. aureus have membranes that consist mainly of anionically charged PG and cardiolipin. Thus bacterial membranes are more negatively charged than RBC membranes.39 Just as observed for natural AMPs,1 the anionic surface charge may attract the positively charged SMAMP, and then due to their hydrophobic component they can subsequently penetrate the lipid bilayer if the SMAMP’s facial amphiphilicity is rightly balanced. An overly hydrophilic SMAMP, like Methyl_3k, is not able to penetrate the hydrophobic core of the lipid bilayer and is therefore inactive. Alternatively, such a hydrophilic molecule may prefer to remain in solution as opposed to being adsorbed to the membrane. Their high hydrophilicity also prevents Methyl_3k and Ethyl_3k from lysing RBCs; only the more hydrophobic homologues (Propyl_3k onward) cause significant hemolysis (Figure 2a). An overly hydrophobic SMAMP like Hexyl_3k has strong membrane activity and as a result is very hemolytic. These observations lead to the maximum activity against E. coli and S. aureus for the 3k series was from the Propyl_3k polymer because it seems to have the optimal facial amphiphilicity to penetrate the bacterial membrane.
While Propyl_3k was also found to be the most toxic 3k polymer against S. aureus, the 10k polymers were all inactive against this pathogen (Figure 2b). This may be rationalized as follows: Gram-positive bacteria have a 15–80 nm thick negatively charged murein layer around the cell membrane. As is well-known from the polyelectrolyte literature, complexation of one polyion with an oppositely charged polyion (symplex formation) is essentially irreversible, while complexes of a polyion with a less charged species are reversible.40 Thus, assuming that the negatively charged murein layer forms a polyion–polyion complex with the positively charged SMAMPs, the dissociation of such a complex becomes increasingly more difficult with increasing molecular weight (more charges on the SMAMP). Thus the higher molecular weight SMAMPs get stuck in the murein layer of Gram-positive bacteria before reaching the plasma membrane and as a result do not kill S. aureus cells irrespective of their amphiphilicity. This would suggest that the appropriately hydrophobic polymers would be active against Gram-negative bacteria because they have a much thinner (2 nm) murein layer which is located between the outer and cytoplasmic membranes. Figure 2b shows that the data are consistent with this hypothesis. For example, Propyl_10k has an MIC90 of 3.75 μg/mL against E. coli while its MIC90 against S. aureus is 200 μg/mL, resulting in a polymer that is doubly selective; it is >50 times more active against E.coli than against S. aureus and 13 times more active against E.coli than against RBCs.
Oligomer Synthesis and Molecular Weight Impact on Biological Activity
The finding that biological activity is different for the 3k and 10k series led to the following questions: (i) At what molecular weight does the biological activity start, and (ii) can tuning of the molecular weight be further exploited to selectively target S. aureus or E. coli?
To answer the first question, all monomers were deprotected (with trifluoroacetic acid as described for the polymers), subjected to MIC90 testing, and found to be inactive (MIC90’s > 200 μg/mL). This shows that a minimum chain length is necessary to obtain any antibacterial activity, consistent with the proposed idea that a facially amphiphilic structure and not a simple surfactant is required for antibacterial activities in the μg/mL range. Consequently, choosing the propyl monomer as an example, small molecular weight oligomers were synthesized and analyzed (see Table 2a). The GPC traces for the oligomers (selected samples for clarity), along with the monomer, are shown in Figure 3a. The peak maxima of all samples are in the expected order: the higher molecular weight oligomers elute before the lower moleculartk;2 weight ones. MALDI-TOF MS was used to determine the actual oligomer molecular weight. The peaks of the MALDI-TOF mass spectra are shown in Figure 3b as a distribution function (normalized intensity vs m/z). Peaks below m/z = 1500 g/mol could not be used as those peaks were lost in the background noise. From the relative peak intensities and m/z ratio of each peak, the number average molecular weight, Mn, and the polydispersity tk;1of the samples were calculated. Those distributions were monomodal, unlike some the GPC traces which appear to be multimodal due to the column resolution. These data are included in Table 2a. The results show once more that GPC curves calibrated with poly(styrene) standards heavily overestimate the molecular weight, while the average number of repeat units nMALDI is in much better agreement with the calculated number of repeat units. However, the smallest oligomer obtained had a degree of polymerization of 3.7 instead of the target of 2 due to the reaction kinetics of this particular solvent/catalyst system.
Table 2.
Oligomers
| (a) Precursor oligomer characterization by GPCa
| |||||||
|---|---|---|---|---|---|---|---|
| sample | Mn Target g mol−1 | GPC Mn g mol−1 | nGPC | GPC Mw/Mn | MALDI Mn g mol−1 | MALDI Mw/Mn | nMALDI |
| monomer (3c, hydrolyzed) | 370 | 1080 | 2.9 | 1.04 | – | – | – |
| Oligo 1 | 740 | 3300 | 8.9 | 1.08 | 1529 | 1.11 | 3.7 |
| Oligo 2 | 1110 | 4300 | 11.6 | 1.09 | 2151 | 1.09 | 5.3 |
| Oligo 3 | 1480 | 5800 | 15.7 | 1.07 | 2642 | 1.07 | 6.7 |
| Oligo 4 | 1850 | 5900 | 16.0 | 1.09 | 2873 | 1.11 | 7.0 |
| Oligo 5 | 2220 | 6900 | 18.6 | 1.10 | 3260 | 1.08 | 8.2 |
| Oligo 6 | 2590 | 7400 | 20 | 1.08 | 3935 | 1.09 | 9.7 |
| (b) Characterization of the antimicrobial oligomers by biological assaysb
| |||||
| sample | MIC90
μg mL−1
|
HC50 μg mL−1 | selectivty
|
||
| E. coli | S. aureus | E. coli | S. aureus | ||
|
| |||||
| monomer (3c, hydrolyzed) | >200 | >200 | n.d. | n.d. | n.d. |
| Oligo 1 | 200 | <3.75 | 1050 | 5.25 | 280 |
| Oligo 2 | 100 | 6.25 | 800 | 8.0 | 128 |
| Oligo 3 | 200 | 25 | 1250 | 6.3 | 50 |
| Oligo 4 | >200 | 50 | >2000 | >10 | >40 |
| Oligo 5 | 100 | 50 | 1000 | 10 | 20 |
| Oligo 6 | 50 | 25 | 150 | 3.0 | 6.0 |
| Propyl_3k | 6.25 | 15 | 50 | 8.3 | 3.3 |
| Propyl_10k | 3.75 | 200 | <50 | <13 | <0.25 |
DMF, 0.01 mol L−1 LiCl, PS standards.
Inhibitory activity towards bacterial growth of E. coli and S. aureus bacteria (MIC90 = minimal inhibitory concentration preventing 90% bacterial growth) and hemolytic activity towards red blood cells (HC50 = concentration lysing 50% of blood cells).
Figure 3.
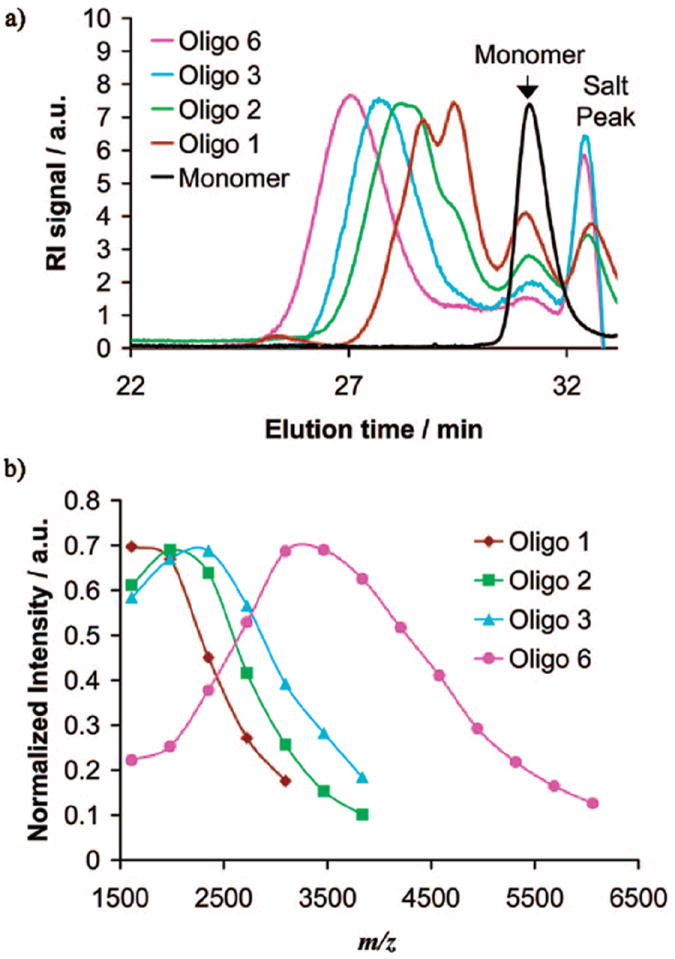
Characterization of propyl oligomers. (a) GPC traces and (b) MALDI-TOF MS peaks and distributions.
The biological data for the propyl series are summarized in Figure 4 and Table 2b: within the error limits of the method, all oligomers are equally nonhemolytic up to Oligo 5. Hemolytic activity increases by 1 order of magnitude with Oligo 6 (HC50 = 150 μg/mL) and increases further for Propyl_3k (HC50 = 50 μg/mL) and Propyl_10k (HC50 > 50 μg/mL). Figure 4 shows that the small molecular weight oligomers are inactive against E. coli. Starting with Oligo 5, a marked increase in activity against E. coli (MIC90 = 100 μg/mL) is observed, which reaches a maximum for Propyl_10k with an MIC90 of 3.75 μg/mL. For S. aureus, the opposite trend is observed (Figure 4). The best activities were found for the small oligomers (MIC90’s = 6.25 μg/mL and < 3.75 μg/mL for Oligo 1 and Oligo 2, respectively). Activity is progressively lost as the molecular weight increases up to Propyl_10k (MIC90 = 200 μg/mL). This is in line with the above-discussed hypothesis that the larger SMAMPs get stuck at the murein layer due to symplex formation. The results of the molecular weight dependent propyl series show that molecular weight can be used as a parameter to selectively target either E. coli or S. aureus, and possibly Gram-negative or Gram-positive bacteria in general.
Figure 4.
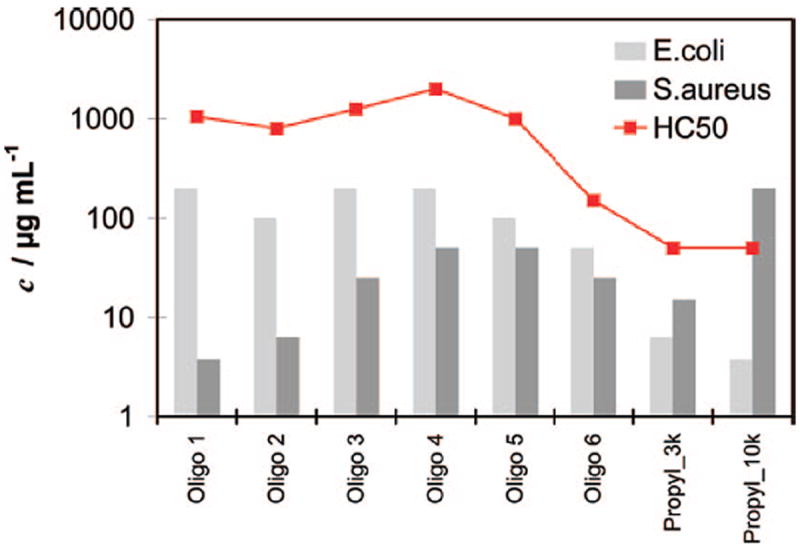
Biological data (MIC90 for E. coli and S. aureus, and HC50 for red blood cells) for the propyl oligomer series.
Previous research indicated that some SMAMPs have only slight molecular weight dependent activity. Ilker et al.19 concluded for their system that the biological activity is independent of molecular weight (for Mn = 1600 to 137 000 g/mol); poly2 and poly4 (MIC90’s of 200 μg/mL against E. coli) did not show a molecular weight dependence. Poly3 was reported with an Mn ranging from 1650 g/mol to 57 200 g/mol and MIC90’s of 25 μg/mL to 80 μg/mL, respectively. DeGrado18 reported poly(methacrylate) copolymers with an Mn around 1600 g/mol having an MIC90 of 16 μg/mL while polymers above 7900 g/moL exhibited an MIC90 of 80 μg/mL against E. coli. In contrast, Oligo 1 has an Mn equal to 740 g/mol and MIC90’s < 3.75 μg/mL against S. aureus and 200 μg/mL against E. coli, while Propyl_10k has MIC90’s of 200 μg/mL against S. aureus and 3.75 μg/mL against E. coli. This leads to the conclusion that antimicrobial activity is molecular weight dependent for these SMAMPs.
Tuning the Selectivities by Copolymer Synthesis
It was shown previously that the incorporation of highly active but hemolytic repeat units into an otherwise nonactive and nonhemolytic polymer can increase the selectivity of that polymer tremendously, making the best of both worlds.19 In our compound library, the Methyl_3k homopolymer qualifies as inactive and nontoxic, whereas the Propyl_3k homopolymer is active and hemolytic. The Ethyl_3k homopolymer is neither most active nor toxic but has the highest selectivity. Thus, methyl, ethyl, and propyl monomers were chosen for copolymerization. Three series of copolymers with a target molecular weight around 3000 g/mol and a varying monomer feed ratio were synthesized and studied (Table 3a). The copolymer composition was checked by NMR spectroscopy, which revealed that the polymer composition matched with the monomer feed ratio.
Table 3.
Copolymers
| (a) Precursor polymer characterization by GPCa
| ||||
|---|---|---|---|---|
| sample | monomers | Mn Target g mol−1 | GPC Mn g mol−1 | Mw/Mn |
| E1:P9 | ethyl-propyl | 3500 | 11 600 | 1.08 |
| E1:P2 | ethyl-propyl | 3400 | 7000 | 1.34 |
| E1:P1 | ethyl-propyl | 3000 | 11 000 | 1.08 |
| E2:P1 | ethyl-propyl | 3500 | 10 200 | 1.15 |
| E9:P1 | ethyl-propyl | 3400 | 7600 | 1.30 |
| M9:E1 | methyl-ethyl | 3400 | 13 600 | 1.10 |
| M1:E1 | methyl-ethyl | 3500 | 14 400 | 1.08 |
| M1:E9 | methyl-ethyl | 3500 | 14 500 | 1.08 |
| M9:P1 | methyl-propyl | 3400 | 15 300 | 1.07 |
| M1:P1 | methyl-propyl | 3600 | 15 900 | 1.07 |
| M1:P9 | methyl-propyl | 3700 | 14 700 | 1.07 |
| (b) Characterization of the antimicrobial copolymers by biological assaysb
| |||||||
| sample | monomer | Mn. Target g mol−1 | MIC90
μgmL−1
|
HC50 μg mL−1 | selectivty
|
||
| E. coli | S. aureus | E. coli | S. aureus | ||||
|
| |||||||
| Propyl_3k | propyl | 3000 | 6.25 | 15 | 50 | 8.0 | 3.3 |
| E1:P9 | ethyl:propyl | 3500 | 15 | 15 | <50 | <3.3 | <3.3 |
| E1:P2 | ethyl:propyl | 3400 | 15 | 15 | <50 | <3.3 | <3.3 |
| E1:P1 | ethyl:propyl | 3000 | 100 | 200 | 1000 | 10 | 5.0 |
| E2:P1 | ethyl:propyl | 3500 | >200 | 200 | >2000 | 10 | >10 |
| E9:P1 | ethyl:propyl | 3400 | >200 | 50 | 500 | <2.5 | 10 |
| Ethyl_3k | ethyl | 3000 | 50 | 50 | 1400 | 28 | 28 |
|
| |||||||
| Methyl_3k | methyl | 3000 | >200 | 100 | 2000 | <10 | 20 |
| M9:E1 | methyl-ethyl | 3400 | >200 | 6.25 | 700 | <3.5 | 112 |
| M1:E1 | methyl-ethyl | 3500 | >200 | 12.5 | 1200 | <6.0 | 96 |
| M1:E9 | methyl-ethyl | 3500 | >200 | 12.5 | 1500 | <7 | 120 |
| Ethyl_3k | ethyl | 3000 | 50 | 50 | 1400 | 28 | 28 |
|
| |||||||
| Methyl_3k | methyl | 3000 | >200 | 100 | 2000 | 10 | 20 |
| M9:P1 | methyl-propyl | 3400 | >200 | <3.75 | >2000 | 10 | >533 |
| M1:P1 | methyl-propyl | 3600 | >200 | <3.75 | >2000 | 10 | >533 |
| M1:P9 | methyl-propyl | 3700 | >200 | <3.75 | >2000 | 10 | >533 |
| Propyl_3k | propyl | 3000 | 6.25 | 15 | 50 | 8.3 | 3.3 |
DMF, 0.01 mol L−1 LiCl, PS standards.
Inhibitory activity towards bacterial growth of E. coli and S. aureus bacteria (MIC90 = minimal inhibitory concentration preventing 90% bacterial growth) and hemolytic activity towards red blood cells (HC50 = concentration lysing 50% of blood cells). Sample labeling: e.g., E1:P9 is a copolymer with 10 mol% ethyl and 90 mol% propyl monomer.
The biological data are compiled in Table 3b and in Figure 5a to 5c. For the ethyl-propyl series, none of the copolymers reached the high selectivity of the parent Ethyl_3k polymer: with increasing propyl content, the ethyl-propyl copolymers become more active but also more toxic. Both series of methyl copolymers showed the expected trend: they become extremely active even with little amount (10 mol%) of the more active monomer but stay nonhemolytic. An amazing and unexpected result however is that they are only potently active against S. aureus (MIC90 < 3.75 μg/mL) and remain totally inactive against E. coli (MIC90 >200 μg/mL)! These copolymers are again doubly selective: >533 for bacterial over mammalian cells, and >53 for S. aureus over E. coli. This double selectivity is inverse to the one described earlier. The high selectivity of >533 for S. aureus is unprecedented in the SMAMP literature.
Figure 5.
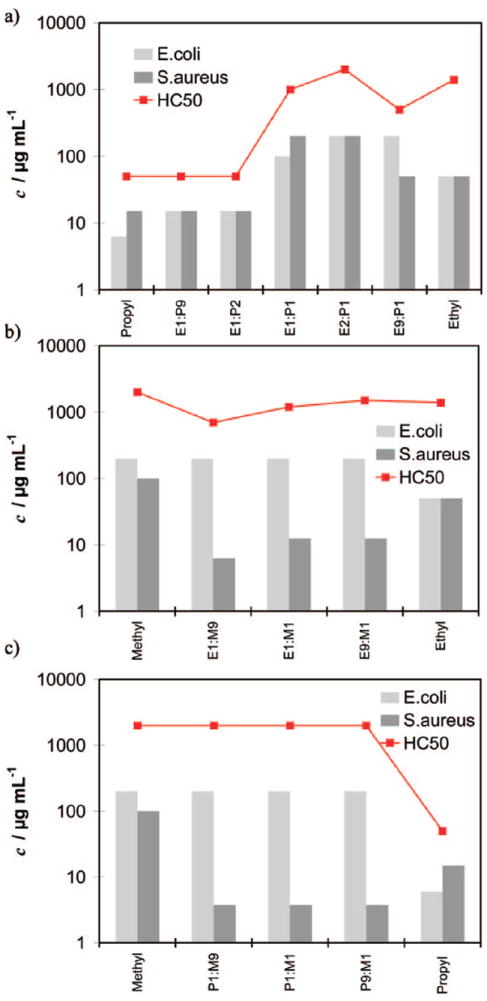
Biological data (MIC90 for E. coli and S. aureus, and HC50 for red blood cells) for the copolymers. (a) Ethyl-propyl series, (b) methylethyl series, and (c) methyl-propyl series. Sample labeling: e.g., E1/P9 is a copolymer with 10 mol% ethyl and 90 mol% propyl monomer.
The reasons for this amazing and surprising double selectivity are not yet understood and are currently under further investigation. An up to 10-fold selectivity for Gram-positive B. subtilis over Gram-negative E. coli has been observed for peptoids12 and β-peptides;41 however this trend was not explained. One attempt toward an explanation is that E. coli has an outer membrane around its actual plasma membrane, while S. aureus has only one plasma membrane below its murein layer. This means that the SMAMPs need to rupture two membranes in order to kill E. coli cells, which intuitively might require a higher SMAMP concentration. On the other hand, if the SMAMPs are able to diffuse unmolested through the murein layer of S. aureus (i.e., with no absorption or complexation), only little material is required to disintegrate its plasma membrane.
In summary, the selectivity of our polymers appears to be affected by the overall hydrophobic/philic balance as previously demonstrated by us32,19 and other groups18 and can be further enhanced by synthesizing an optimal molecular weight. Notably though, we have only seen this kind of high double selectivity (bacteria over mammalian cells and one bacterial type over another) with the specific polymers presented here, in which the monomers themselves are facially amphiphilic. We therefore believe that facial amphiphilicity of the monomers is an important design principle for SMAMPs with precisely tunable activity and selectivity. Only very recently, Shambhy et al. reported two series of copolymers with tunable amphiphilicity. In the first series, one repeat unit carried the cationic group and the other the hydrophobic group (“separate centers” in the authors’ terminology). The second series contained one facially amphiphilic comonomer (“same centers” in the authors’ terminology), and the other was chosen such that the same molecular formula as in the previous series was obtained. The facially amphiphilic polymers have much higher selectivities (up to 34) than the comparable copolymers of the other series.33 This very elegant comparison supports our hypothesis.
Conclusion
The molecular construction kit approach described allows the synthesis of a whole library of synthetic facially amphiphilic antibacterial homopolymers and copolymers with tunable activity and selectivity. The hydrophilic and hydrophobic components can be varied independently, allowing future synthesis of an even more extensive library of antibacterial polymers. The 3k homopolymers thus obtained show a remarkable antibacterial activity against E. coli and S. aureus, which could be tuned over 2 orders of magnitude by variation of the hydrophobic residue R. Some of the 10k homopolymers showed a double selectivity of E. coli over mammalian cells as well as over S. aureus. It was further demonstrated that molecular weight can be used as a parameter to tune the antibacterial activity. The optimum activity against S. aureus was found to be at 750–1100 g/mol, and that against E. coli, at 3000 g/mol and 10000 g/mol. We were able to push the selectivities of S. aureus over red blood cells as high as >533 by copolymerizing an inactive/nonhemolytic monomer with an active/hemolytic monomer. These polymers were also doubly selective with a >533 times higher selectivity of S. aureus over red blood cells and a >53 times more selective for S. aureus over E. coli. Such a high selectivity against S. aureus is unprecedented. This is particularly exciting as the Methicillin-resistant form of S. aureus, MRSA, is an antibiotic-resistant “superbug” and one of today’s most serious health threats for hospital patients.
This is the clearest example thus far of how the biological properties of cationic amphiphilic polymers can be tuned to, first, increase the selectivity of bacteria over mammalian cells and, second, increase the selectivity toward one bacteria family over another. This discrimination is important for antimicrobial agents since many bacterial types are in fact beneficial to the body. It has yet to be shown that our doubly selective molecules operate by the same membrane-disrupting mechanism as natural AMPs. A detailed analysis of the interaction of these remarkably active molecules with model membranes, for example using dyeleakage experiments, might give insight to their mechanism of action. These investigations are currently in progress and will be reported in due course. In future work, we will further exploit the versatility of this molecular construction kit approach by varying the hydrophilic component of the monomer and determining its influence on the antimicrobial activity of the resulting polymers. This will hopefully lead to the discovery of polymers whose activities and selectivities will surpass even the ones reported in this paper.
Supplementary Material
Acknowledgments
Dr. Abhigyan Som and Dr. Gregory J. Gabriel are acknowledged for helpful and challenging discussions. Mass spectral data were obtained by Dr. Steve Eyles at the University of Massachusetts Amherst Mass Spectrometry Facility which is supported, in part, by the National Science Foundation. Funding by the German Research Foundation (DFG), MRSEC, and ONR is gratefully acknowledged.
Footnotes
Supporting Information Available: Experimental details (monomer and polymer synthesis, all spectroscopic data). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Brodgen KA. Nat Rev Microbiol. 2005;3:238–250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 2.Yang L, Mishra A, Purdy K, Som A, Tew GN, Wong GCL. J Am Chem Soc. 2007;129:12141. doi: 10.1021/ja072310o. [DOI] [PubMed] [Google Scholar]
- 3.Zasloff M. Nature. 2002;415:389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 4.Zasloff M. Proc Natl Acad Sci U S A. 1987;84:5449. doi: 10.1073/pnas.84.15.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Castro MS, Cilli EM, Fontes W. Curr Protein Peptide Sci. 2006;7:473. doi: 10.2174/138920306779025648. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y, Mant CT, Farmer SW, Hancock REW, Vasil ML, Hodges RS. J Biol Chem. 2005;280:12316. doi: 10.1074/jbc.M413406200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Won H-S, Jung S-J, Kim HE, Seo M-D, Lee B-J. J Biol Chem. 2004;279:14784. doi: 10.1074/jbc.M309822200. [DOI] [PubMed] [Google Scholar]
- 8.Hamuro Y, Schneider JP, DeGrado WF. J Am Chem Soc. 1999;121:12200. [Google Scholar]
- 9.Porter EA, Wang X, Lee H-S, Weisblum B, Gellman SH. Nature. 2000;404:565. doi: 10.1038/35007145. [DOI] [PubMed] [Google Scholar]
- 10.Liu D, DeGrado WF. J Am Chem Soc. 2001;123:7553. doi: 10.1021/ja0107475. [DOI] [PubMed] [Google Scholar]
- 11.Epand RF, Raguse TL, Gellman SH, Epand RM. Biochemistry. 2004;43:9527. doi: 10.1021/bi049414l. [DOI] [PubMed] [Google Scholar]
- 12.Patch JA, Barron AE. J Am Chem Soc. 2003;125:12092. doi: 10.1021/ja037320d. [DOI] [PubMed] [Google Scholar]
- 13.Brouwer CPJM, Bogaards SJP, Wulferink M, Velders MP, Welling MM. Peptides. 2006;27:2585. doi: 10.1016/j.peptides.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 14.Haynie SL, Crum GA, Doele BA. Antimicrob Agents Chemother. 1995;39:301. doi: 10.1128/aac.39.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tew GN, Liu D, Chen B, Doerksen RJ, Kaplan J, Carroll PJ, Klein ML, DeGrado WF. Proc Natl Acad Sci U S A. 2002;99:5110. doi: 10.1073/pnas.082046199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu D, Choi S, Chen B, Doerksen RJ, Clements DJ, Winkler JD, Klein ML, De Grado WF. Angew Chem Int Ed. 2004;43:1158. doi: 10.1002/anie.200352791. [DOI] [PubMed] [Google Scholar]
- 17.Tang HZ, Doerksen RJ, Tew GN. Chem Commun. 2005;12:1537. doi: 10.1039/b413679a. [DOI] [PubMed] [Google Scholar]
- 18.Kuroda K, DeGrado WF. J Am Chem Soc. 2005;127:4128. doi: 10.1021/ja044205+. [DOI] [PubMed] [Google Scholar]
- 19.Ilker MF, Nusslein K, Tew GN, Coughlin EB. J Am Chem Soc. 2004;126:15870. doi: 10.1021/ja045664d. [DOI] [PubMed] [Google Scholar]
- 20.Arnt L, Tew GN. J Am Chem Soc. 2002;124:7664. doi: 10.1021/ja026607s. [DOI] [PubMed] [Google Scholar]
- 21.Arnt L, Tew GN. Langmuir. 2003;19:2404. [Google Scholar]
- 22.Arnt L, Nusslein K, Tew GN. J Polym Sci Part A: Polym Chem. 2004;42:3860. [Google Scholar]
- 23.Mowery BP, Lee SE, Kissounko DA, Epand RF, Epand RM, Weisblum B, Stahl SS, Gellman SH. J Am Chem Soc. 2007;129:15474. doi: 10.1021/ja077288d. [DOI] [PubMed] [Google Scholar]
- 24.Boman HG. Immunol Rev. 2000;173:5. doi: 10.1034/j.1600-065x.2000.917301.x. [DOI] [PubMed] [Google Scholar]
- 25.Hancock REW, Lehrer R. Trends Biotechnol. 1998;16:82. doi: 10.1016/s0167-7799(97)01156-6. [DOI] [PubMed] [Google Scholar]
- 26.Yeaman MR, Yount NY. Pharmacol Rev. 2003;55:27. doi: 10.1124/pr.55.1.2. [DOI] [PubMed] [Google Scholar]
- 27.Yount NY, Bayer AS, Xiong YQ, Yeaman MR. Biopolymers (Peptide Sci) 2006;84:435. doi: 10.1002/bip.20543. [DOI] [PubMed] [Google Scholar]
- 28.Kuroda K, DeGrado WF. Polym Prepr (Am Chem Soc Div Polym Chem) 2004;45:610. [Google Scholar]
- 29.Klajnert B, Janiszewska J, Urbanczyk-Lipkowska Z, Bryszewska M, Shcharbin D, Labieniec M. Int J Pharm. 2006;309:208. doi: 10.1016/j.ijpharm.2005.10.039. [DOI] [PubMed] [Google Scholar]
- 30.Liu Z, Deshazer H, Rice AJ, Chen K, Zhou C, Kallenbach NR. J Med Chem. 2006;49:3436. doi: 10.1021/jm0601452. [DOI] [PubMed] [Google Scholar]
- 31.Makovitzki A, Avrahami D, Shai Y. Proc Natl Acad Sci U S A. 2006;103:15997. doi: 10.1073/pnas.0606129103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eren T, Som A, Rennie JR, Nelson CF, Urgina Y, Nüsslein K, Coughlin EB, Tew GN. Macromol Chem Phys. 2008;209:516. [Google Scholar]
- 33.Sambhy V, Peterson RT, Sen A. Angew Chem Int Ed. 2008;47:1250. doi: 10.1002/anie.200702287. [DOI] [PubMed] [Google Scholar]
- 34.Mantovani G, Lecolley F, Tao L, Haddleton DM, Clerx J, Cornelissen JJLM, Velonia K. J Am Chem Soc. 2005;127:2966. doi: 10.1021/ja0430999. [DOI] [PubMed] [Google Scholar]
- 35.Slugovc C. Macromol Rapid Commun. 2004;25:1283. [Google Scholar]
- 36.Dichloro-di(3-bromopyridino)-N,N′-dimesitylenoimidazolino-RudCHPh, c.f.: Love JA, Morgan JP, Trnka TM, Grubbs RH. Angew Chem Int Ed. 2002;41:4035. doi: 10.1002/1521-3773(20021104)41:21<4035::AID-ANIE4035>3.0.CO;2-I.
- 37.MIC90 = minimal inhibitory concentration preventing 90% bacterial growth, HC50 = hemolytic concentration lysing 50% of blood cells, c.f.: Rennie J, Arnt L, Tang H, Nuesslein K, Tew GN. J Industrial Microbiol Biotechnol. 2005;32:296. doi: 10.1007/s10295-005-0219-0.
- 38.Gabriel GJ, Som A, Madkour AE, Eren T, Tew Gregory N. Mater Sci Eng Rev. 2007;57:28. doi: 10.1016/j.mser.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Som A, Tew GN. J Phys Chem B. 2008;112:3495. doi: 10.1021/jp077487j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dautzenberg H, Jaeger W, Kötz J, Philipp B, Seidel C, Stscherbina D. Polyelectrolytes. Hanser/Gardner; Munich: 1994. [Google Scholar]
- 41.Porter EA, Weisblum B, Gellman SH. J Am Chem Soc. 2002;124:7324. doi: 10.1021/ja0260871. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.