

Abstract
The pyrrolyldipyrrin motif is found in several naturally occurring prodigiosin pigments. The potential roles of the interactions of prodigiosins with transition metals and the properties of metal-bound pyrrolyldipyrrins, however, have been difficult to assess because of the very limited number of well-characterized stable complexes. Here, we show that the introduction of a meso-aryl substituent and an ethyl ester group during the sequential assembly of the three heterocycles affords a pyrrolyldipyrrin of enhanced coordinating abilities when compared to that of natural prodigiosins. UV–visible absorption studies indicate that this ligand promptly binds Zn(II) ions with 2:1 ligand-to-metal stoichiometry and Cu(II) ions with 1:1 stoichiometry. Notably, no addition of base is required for the formation of the resulting stable complexes. The crystal structures reveal that whereas the tetrahedral zinc center engages two nitrogen donors on each ligand, the pseudosquare planar copper complex features coordination of all three pyrrolic nitrogen atoms and employs the ester group as a neutral ligand. This first example of coordination of a redox-active transition metal within a fully conjugated pyrrolyldipyrrin framework was investigated spectroscopically by electron paramagnetic resonance to show that the 1:1 metal-to-ligand ratio found in the crystal structure is also maintained in solution.
Short abstract
The pyrrolyldipyrrin motif, which characterizes the prodigiosin natural products of bacterial origin, is engineered to afford a stable tripyrrolic scaffold for coordination of transition metals. Prompt binding of zinc and copper ions occurs in the absence of added bases at room temperature. Full structural and spectroscopic characterization by X-ray diffraction and EPR/ENDOR techniques documents the hitherto elusive coordination of the Cu(II) ion in this ligand framework.
Introduction
Oligopyrrolic compounds are essential to all forms of life and play a critical role in the chemistry of transition metals in living systems. The coordination chemistry of porphyrins and other oligopyrrolic macrocycles has been investigated intensely; however, linear oligopyrroles and particularly dipyrrolic fragments are gaining increasing attention.1,2 Presenting an electron-rich scaffold and an array of pyrrolic nitrogen donors, linear oligopyrrolic fragments are typically well-poised for metal coordination. In addition, these fully or partially conjugated compounds can offer diverse coordinating abilities toward transition metals in multiple oxidation states, as well as highly tunable, potentially redox-active, ligand behavior. Such properties have been observed in studies of reactivity and catalytic applications of complexes of dipyrranes,3,4 dipyrrins,1,5−9 and other linear oligopyrrolic ligands.10−14 Herein, we examine in the context of metal coordination a tripyrrolic scaffold found in several naturally occurring pigments of bacterial origin.
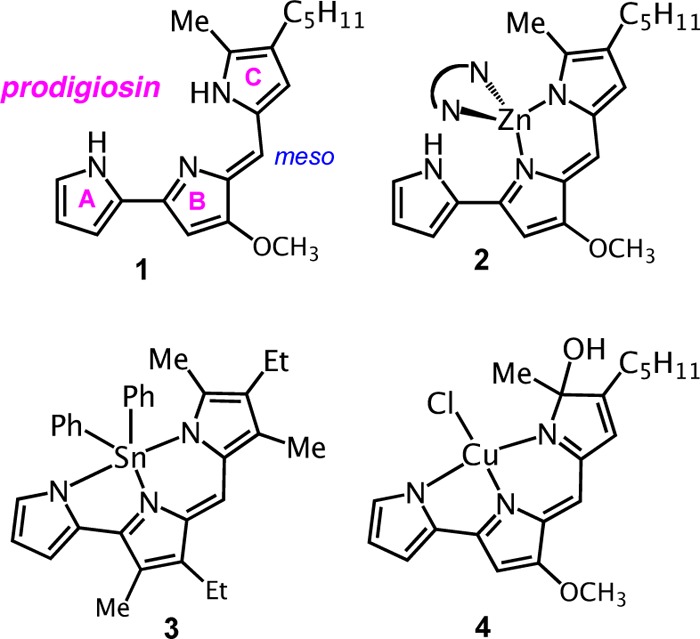
The pyrrolyldipyrrin motif characterizes the natural product prodigiosin (1 in Chart 1, showing the common letter designation of the three pyrrole rings), a red pigment produced by certain Serratia, Streptomyces, and Bacillus bacteria strains.15,16 This tripyrrolic species is the parent compound of a large family of natural and synthetic analogues, termed prodigiosenes (or less commonly prodiginines), which are currently undergoing intense scrutiny in medicinal chemistry studies because of their diverse biological activity profiles. Multiple studies document the efficacy of numerous pyrrolyldipyrrins as immunosuppressive,17,18 antimicrobial,19 antimalarial,20 and anticancer21−24 agents. The reported ability of prodigiosenes to induce apoptosis has been connected to DNA intercalation,25 perturbation of intracellular pH through H+/Cl– symport26 and/or Cl–/HCO3– antiport27,28 mechanisms, and oxidative DNA damage.29−31
Chart 1. Natural Prodigiosin and Selected Metal Complexes of Its Analogues.
Prodigiosin cleaves double-stranded DNA in the presence of O2 and Cu(II) cations without the need of an added reductant.29 Both metal binding and redox interplay between the electron-rich tripyrrole and the redox-active cation, possibly resulting in formation of a ligand-based π-radical cation, have been proposed to explain the observed nuclease activity.29,32 Consistently, modifications of the prodigiosin scaffold that rendered the ligand less coordinating or harder to oxidize decreased DNA cleavage activity.29,32,33 In spite of the interest in the role of metal binding and redox chemistry in the biological activity of prodigiosenes, however, the coordination chemistry of these tripyrrolic fragments remains rather unexplored, and copper complexes of pyrrolyldipyrrins have been elusive.
Bidentate coordination modes to the dipyrrin unit of prodigiosin analogues have been observed in fluorescent boron difluoride complexes,34−36 and two such compounds are available commercially as longer-wavelength members of the BODIPY family of fluorescent dyes (Molecular Probes). Similarly, several homoleptic zinc complexes of pyrrolyldipyrrins, such as 2 (Chart 1),22,23,37 share the characteristics of other complexes within the large family of simple dipyrrins, which coordinate as bidentate monoanionic ligands. The additional pyrrolic ring on the prodigiosin-type scaffolds behaves as a ligand in fluorescent dialkyl and diaryl tin(IV) complexes such as 3 (Chart 1),38 for which structural characterization revealed tridentate coordination of all nitrogen donors on the fully conjugated pyrrolyldipyrrin scaffold. A similar tridentate coordination mode was recently proposed for a Zn(II) complex, which was not characterized fully but was inferred from UV–visible absorption data and computational work.39
Studies aimed at Cu(II) coordination of prodigiosin led to the isolation of complex 4 (Chart 1) following reaction of the free ligand with cupric chloride in the presence of potassium tert-butoxide in tert-butyl alcohol.37 Notably, the tripyrrolic scaffold behaves as a tridentate ligand, but oxidation of pyrrole ring C yielded an sp3-hybridized hydroxyl-bound carbon atom in 4 and confirmed the occurrence of ligand-centered redox reactivity in solution. Additional coordination studies conducted on a C-ring modified analogue of prodigiosin indicated copper–ligand binding interactions of 1:1 and 1:2 stoichiometry, as inferred by spectrophotometric titrations and mass spectrometry, but the resulting copper complexes were not isolated.22 Interestingly, oxidative degradation was also observed during Cu(II) insertion in another tripyrrolic ligand, a tripyrrane that was found to undergo oxidation at the methylene bridges.14
In spite of their electron-rich scaffold featuring an array of three pyrrolic nitrogen donors, pyrrolyldipyrrins are not associated with a well-established coordination chemistry. Herein, we describe the design and synthesis of a pyrrolyldipyrrin ligand of enhanced metal-coordinating ability when compared to that of natural systems and current synthetic analogues. Binding of divalent zinc is observed as well as the prompt and hitherto elusive coordination of divalent copper ions in the absence of bases and without oxidative degradation of the ligand. The spectroscopic and structural characterization of the resulting complexes documents two available coordination modes for the tripyrrolic fragment. The reported synthetic findings will offer access to new classes of prodigiosin analogues; concurrently, our prototype ligand system provides a new platform for the study of metal-bound pyrrolyldipyrrins and their potential applications in medicinal chemistry, small-molecule activation, and catalysis.
Results and Discussion
Ligand Design and Synthesis
Aiming to build a pyrrolyldipyrrin of higher metal-binding affinity when compared to that of natural systems, we introduced two electron-withdrawing groups in the first-generation scaffold H2PD1 (Scheme 1): (i) a phenyl group in the meso-type position and (ii) an ethyl ester group on the C-ring. These substituents were expected to increase the acidity of the pyrrolic N–H protons in order to facilitate deprotonation and coordination of metal cations. In addition, the α-ester functionality was envisioned as an additional ligand to contribute to metal coordination with a neutral oxygen donor, as previously observed for α-substituted dipyrrins.9,40 Further supporting our ligand design featuring two electron-withdrawing substituents, H2PD1 presents a stabilized π system when compared to naturally occurring analogues. As such, we anticipated that such construct would be less prone to the type of oxidative degradation observed in complex 4 (Chart 1) in the presence of redox-active transition metal species such as Cu(II) ions.37
Scheme 1. Synthesis of a meso-Aryl Pyrrolyldipyrrin with an Ester Group on the C-Ring.
meso-Aryl pyrrolyldipyrrin scaffolds have recently appeared in studies on the preparation of pyrrolylBODIPY dyes. Specifically, substitution reactions34,36 on meso-aryl dipyrrin substrates and the one-pot reaction35 of acyl chlorides with excess pyrrole under an oxygen atmosphere afford boron pyrrolyldipyrrins. Demetalation of these dyes can then be employed to prepare free pyrrolyldipyrrin ligands.36,41 Alternatively, a recent synthetic route to this class of compounds requires the low-yielding acylation of 2,2′-bipyrrole, leading to a mixture of products.39 These synthetic methods for the preparation of meso-aryl pyrrolyldipyrrin, however, generally afford modest-to-moderate yields and have not been employed on α-substituted pyrroles. Our stepwise assembly of the three pyrrolic rings is designed to provide flexibility of substitution patterns for the overall structure.
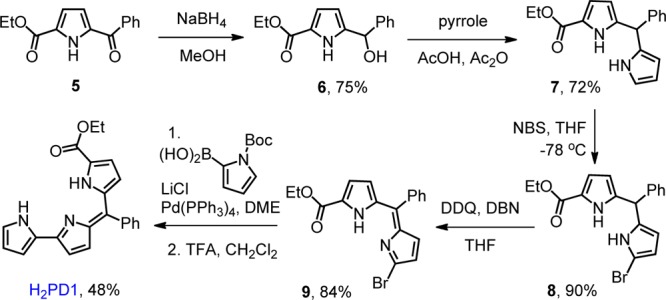
Similar to several reported preparations of C-ring-modified prodigiosenes,22−24 our synthetic pathway begins with the heterocycle that will become the C-ring in the final product. The pyrrolic precursor ethyl 5-benzoyl-1H-pyrrole-2-carboxylate 5, which carries the desired ethyl ester substitution and a benzoyl group for further functionalization, was reduced with NaBH4 to give alcohol 6. This reactive species was utilized promptly upon isolation, and the B-ring was introduced by condensation with excess pyrrole under acidic conditions.42 Alternatively, asymmetric dipyrrane 7 could be prepared via known methods requiring synthesis of a 5-substituted dipyrran and subsequent Grignard-mediated acylation using a pyridyl carbonothioate (Mukaiyama reagent).43 These transformations, however, are typically characterized by low-to-moderate yields and challenging purifications.
Because bromodipyrrins are reliable precursors in the synthesis of prodigiosenes,22−24 dipyrromethane 7 was brominated with N-bromosuccinimide and then oxidized with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) to afford asymmetric bromodipyrrin 9. Compound 9 is susceptible to rapid decomposition under acidic conditions, and the addition of a base during the oxidation reaction proved to be critical. The A-ring completing the pyrrolyldipyrrin scaffold was then introduced using a protected pyrrole-2-boronic acid through a Suzuki–Miyaura coupling reaction, giving the target tripyrrolic pigment H2PD1.
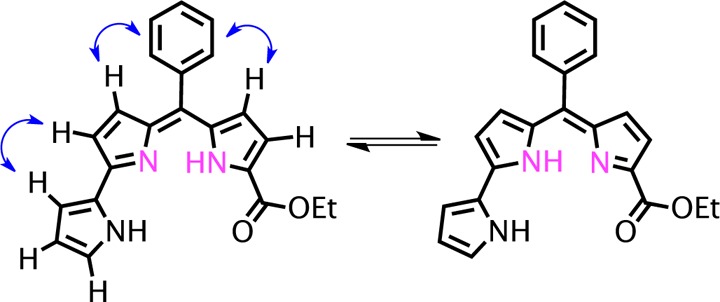
A full assignment of the pyrrolic proton resonances for the pyrrolyldipyrrin ligand was performed by COSY and NOESY 2D NMR techniques (see Supporting Information). These experiments also indicated that free base H2PD1 exists in solution as the rotamer shown in Scheme 2. Although three alternative rotameric structures are available for this scaffold, this pyrrolyldipyrrin is best represented by the structure featuring all three pyrrolic nitrogen atoms on the inner side of the cleft. This rotamer is also the one observed experimentally by 2D NMR experiments and was found to be most stable by DFT analysis in a recent study on a close analogue of natural prodigiosin.44 In addition, our 2D NMR data allowed identification of the NH proton on ring A (Figure S3, Supporting Information) but left undetermined the position of the other NH proton, which was not observed, likely because of rapid exchange equilibrium between the two tautomeric forms of the dipyrrin moiety (Scheme 2). The NMR data summarized above indicate that free base H2PD1 maintains the orientation of pyrrolic nitrogen donors in a tridentate array poised for metal coordination and/or multiple hydrogen-bonding interactions, two aspects of its solution chemistry that have been invoked in the biological mechanisms of action of prodigiosin analogues.
Scheme 2. Tautomeric Equilibrium of H2PD1 in CDCl3 Showing Key NOESY Correlations for the Assignment of the Rotameric Structure.
Metal Binding Studies and Structural Characterization
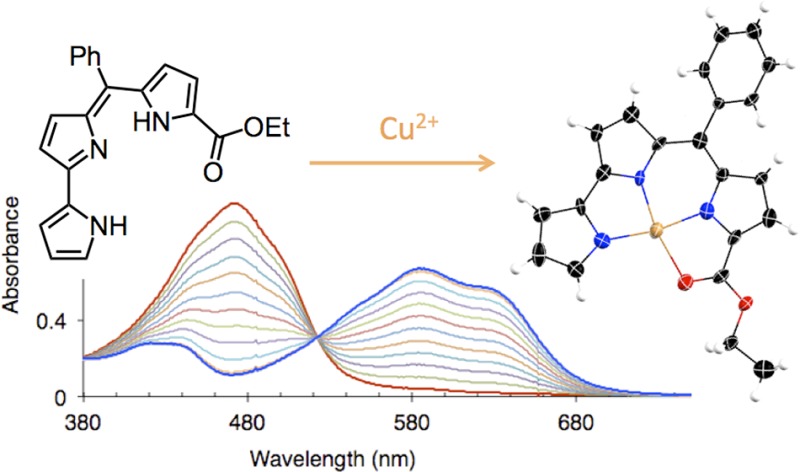
Pyrrolyldipyrrin H2PD1 is a dark red pigment characterized by an intense visible absorption band at λmax 476 nm (ε, 29 600 M–1 cm–1 in CH3OH); thus, the coordination of metal cations could be monitored by UV–vis absorption spectroscopy. Addition of 0.5 equiv of Zn(OAc)2·2H2O to a solution of H2PD1 in methanol (Figure 1) or THF led to prompt formation of a new metal complex featuring two red-shifted absorption bands. Clear isosbesticity was maintained over the course of the metal-binding study, and further additions of zinc salt did not elicit any changes in the absorption spectra; therefore, the formation of a single complex of 2:1 ligand-to-metal stoichiometry was inferred. Because absorbance values changed almost linearly with metal ion additions, and hence the fraction of ligand-bound metal approached 100%, these binding studies of zinc and copper (see below) ions revealed binding stoichiometry but did not allow reliable determination of the high-affinity equilibrium constants.
Figure 1.
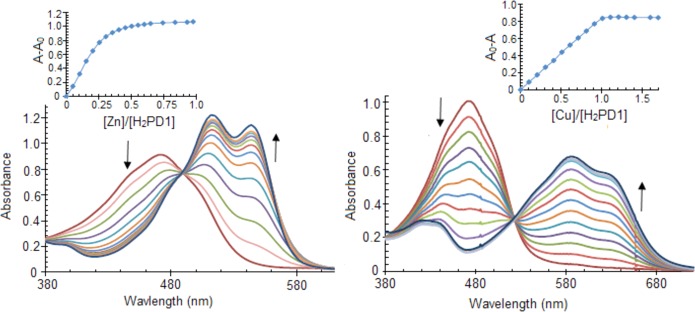
Spectral changes and binding isotherms observed upon addition of Zn(OAc)2·2H2O (left panel) or Cu(OAc)2·H2O (right panel) to pyrrolyldipyrrin H2PD1 (36 μM) in methanol at 298 K.
Complex Zn(HPD1)2 was isolated and first characterized by NMR spectroscopy. The proton spectrum displayed a single set of resonances for the tripyrrolic ligand, consistent with a 1:1 stoichiometry of binding or with formation of a symmetric 2:1 complex. Two-dimensional experiments allowed identification of the proton on the pyrrolic nitrogen atom on ring A (Figures S5–S6, Supporting Information), which is shifted downfield with respect to the corresponding resonance in the free ligand (at 9.75 vs 9.35 ppm in H2PD1) and does not participate in metal binding. In addition, a NOESY crosspeak between the α C–H proton on ring A and the phenyl multiplet was identified as a correlation between two ligands held in close proximity by coordination to the zinc center and therefore consistent with 2:1 ligand-to-metal binding stoichiometry. The expected coordination of the zinc(II) ion to two ligands through the bidentate dipyrrin moieties was confirmed in the solid state by X-ray diffraction analysis on a single crystal of complex Zn(HPD1)2 (Figure 2).
Figure 2.
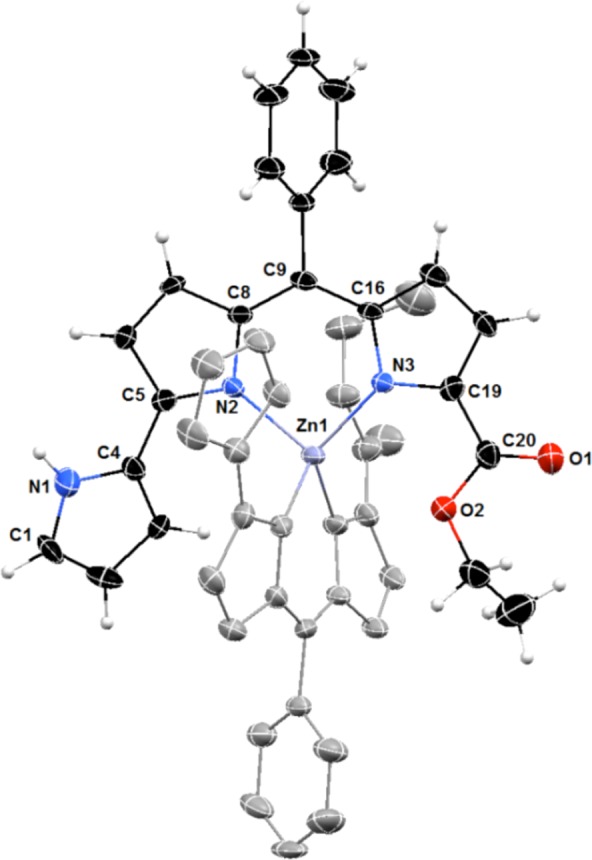
Crystal structure of zinc complex Zn(HPD1)2 showing a partial labeling scheme. For clarity, the two pyrrolyldipyrrin ligands are shown in different colors, and the hydrogen atoms in calculated positions are shown only for one of the ligands. Anisotropic thermal displacement ellipsoids are set at the 50% probability level (CCDC 994299).
The crystallographic metric parameters of complex Zn(HPD1)2 are similar to those of the zinc complex of natural prodigiosin 1,37 in which the zinc center coordinates with tetrahedral geometry to two bidentate monoanionic dipyrrin units. Interestingly, the noncoordinating A-ring of one of the ligands in the structure of Zn(HPD1)2 features a pyrrolic NH group pointing away from the zinc center and hence is in a different rotameric structure when compared to that of the free ligand. This packing effect is attributed to an intermolecular hydrogen-bonding interaction with the carbonyl group of a neighboring complex (Figure S7, Supporting Information) and is not observed in chloroform solution according to our 2D NMR data.
Partly because copper is a biologically relevant metal and partly because of the documented interplay of Cu(II) ions and natural prodigiosin in the cleavage of double-stranded DNA,29,45,46 the copper binding properties of pyrrolyldipyrrin scaffolds have been previously investigated. Nevertheless, copper-bound prodigiosenes have remained elusive, and coordination studies reported oxidative degradation of the ligand in complex 4 (Chart 1)37 or formation of multiple complexes that could not be isolated and fully characterized.22 Because ligand H2PD1 was designed for enhanced metal-binding properties, we sought to investigate its coordination of the Cu(II) cation.
The reaction of H2PD1 with Cu(OAc)2·H2O could be monitored by UV–vis spectrophotometry in CH3OH (Figure 1, right panel) or THF. The spectral changes observed upon addition of the copper salt presented a clear isosbestic point, and full saturation of the ligand was reached when the concentration of Cu(II) ions amounted to 1 equiv, therefore indicating formation of a single copper complex of high affinity and 1:1 binding stoichiometry. H2PD1 is an excellent receptor for copper coordination, and, unlike previously reported binding studies,22,37 complex formation in this case did not require addition of a base to facilitate deprotonation of pyrrolic NH groups.
The solid-state structure of the isolated copper complex Cu(PD1) was investigated by single-crystal X-ray diffraction analysis. Although the refinement was complicated by the presence of disordered solvent molecules (see Experimental Section), this analysis allowed for the determination of the copper coordination mode in the complex (Figure 3). Pyrrolyldipyrrin PD12– behaves as a tetradentate dianionic ligand, and the copper center exhibits a slightly distorted square planar coordination geometry in the resulting neutral complex. All three pyrrolic nitrogen atoms are engaged as donor groups, and the ester group on the C-ring assumes the expected role of neutral ligand through the carbonyl oxygen atom to complete the copper coordination sphere. The Cu–Npyrrole (1.900(8)–1.931(9) Å) and Cu–Ocarbonyl (2.074(7) Å) bond lengths compare well with those found in Cu(II) complexes of prodigiosin37 and α-substituted dipyrrin ligands.9 The copper center is closer to the dipyrrin unit and the Cu–N bond distance to pyrrole ring A (1.931(9) Å) is longer than those to rings B and C (1.909(8) and 1.900(8) Å, respectively). In addition, C–N and C–C bond metric comparisons with free pyrrolyldipyrrin ligands26,36,47,48 and with Zn(II) complex Zn(HPD1)2 confirm a fully conjugated tripyrrolic scaffold in Cu(PD1). Such considerations, together with the absence of counterions, indicate that Cu(II) ions bind to deprotonated ligand PD12– without complications arising from interfering redox events.
Figure 3.
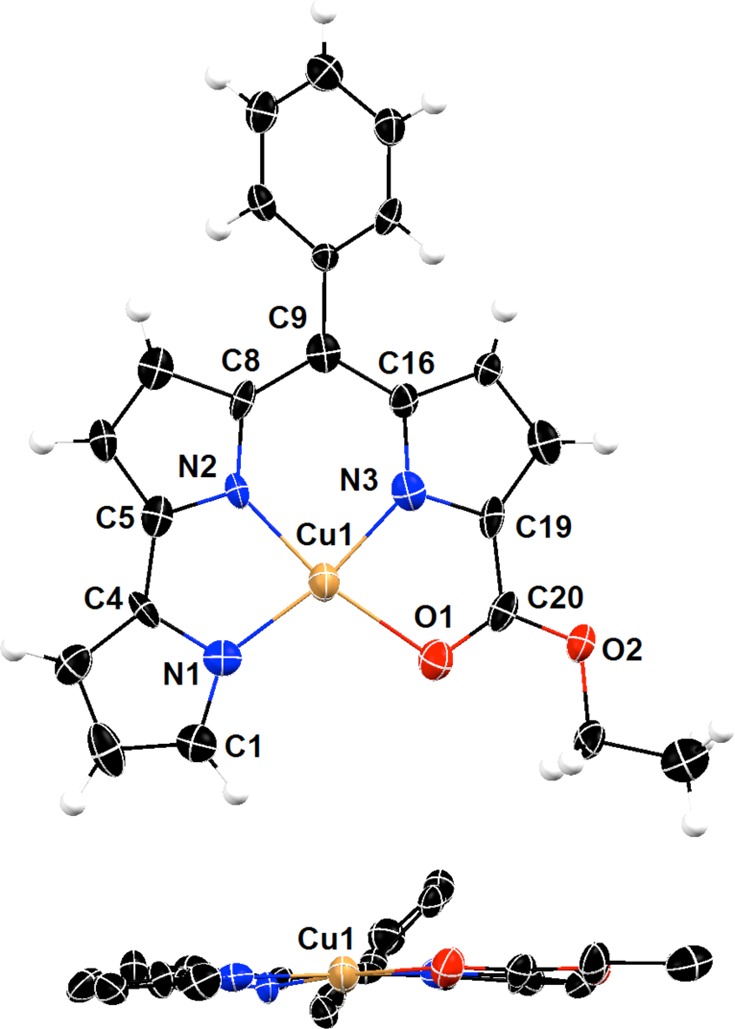
Top and side views of the crystal structure of copper(II) complex Cu(PD1) showing a partial labeling scheme. Anisotropic thermal displacement ellipsoids are scaled to the 50% probability level (CCDC 994298).
EPR Characterization of Cu(PD1)
The coordination environment of the copper center in Cu(PD1) was investigated in solution by electron paramagnetic resonance (EPR) spectroscopy. The X-band (∼9.5 GHz) continuous-wave (CW) EPR and the Ka-band (∼30 GHz) electron spin echo (ESE) field-sweep spectra (Figure 4) are characterized by nearly axial g and ACu tensors (where ACu denotes the hyperfine interaction (hfi) of the central Cu nucleus) with (g∥, g⊥) = (2.188, 2.043) and (ACu∥, ACu⊥) ≈ (17.6, <4) mT, indicative of the unpaired electron predominantly localized in the dx2–y2 orbital.
Figure 4.
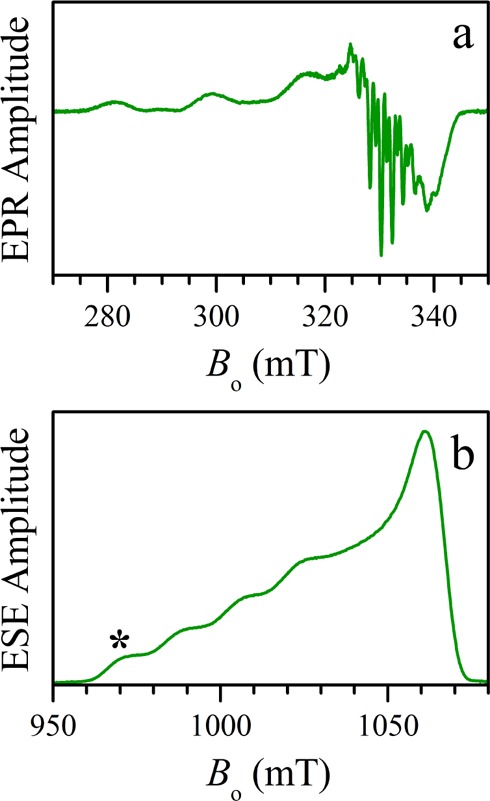
(a) X-band CW EPR and (b) Ka-band two-pulse ESE field-sweep spectra of a Cu(PD1) solution in toluene. The asterisk in panel b indicates the EPR position where the pulsed ENDOR measurements (Figure 5) were performed. Experimental conditions: (a) Microwave frequency, 9.450 GHz; microwave power, 2 mW; magnetic field modulation amplitude, 0.2 mT; temperature, 77 K. (b) Microwave frequency, 30.360 GHz; microwave pulses, 24 and 42 ns; time interval between microwave pulses, τ = 400 ns; temperature, 15 K.
The 14N hyperfine splittings in the CW EPR spectrum (Figure 4a) are not sufficiently resolved to permit the determination of the number and detailed parameters of the 14N ligands. In order to reveal the (relative) number of copper-bound nitrogen atoms in Cu(PD1) in solution, we employed a pulsed electron–nuclear double resonance (ENDOR) technique due to Davies,49 which is particularly suitable for detecting the strong (tens of megahertz) hfi of 14N in Cu(II) complexes.
Because we were mostly interested in quantification of the 14N nuclei, we performed only the measurements at the low-field g∥ turning point of the EPR spectrum (marked by an asterisk in Figure 4), which corresponds to a single-crystal-like situation and to the highest resolution in the ENDOR spectra. The relevant theoretical background and the experimental details are given in the Experimental Section. Here, we will mention only that the microwave (mw) pulses used were sufficiently long to make the Davies ENDOR response independent of the hfi constants of the detected 14N nuclei. In addition, to minimize the dependence of the 14N ENDOR line amplitudes on the transition probabilities, the experiment was performed in a 2D fashion (Figure S8, Supporting Information): radiofrequency (RF) versus the RF pulse length, tRF, and then the 2D set was integrated over tRF to obtain the 1D spectrum.
The obtained 14N Davies ENDOR spectrum (Figure 5) shows three pairs of features attributable to 14N nuclei (labeled Na, Nb, and Nc in Figure 5), with the splitting within each pair equal to twice the Zeeman frequency of 14N: 2νN ∼ 6 MHz in the applied magnetic field, B0 ∼ 1 T. The smaller quadrupole splittings are poorly resolved because of the line broadening. These three pairs of lines are centered at the frequencies of 12.6, 21.9, and 30.2 MHz, resulting in the 14N hfi constants AN∥ = 25.2, 43.8, and 60.4 MHz, respectively.
Figure 5.
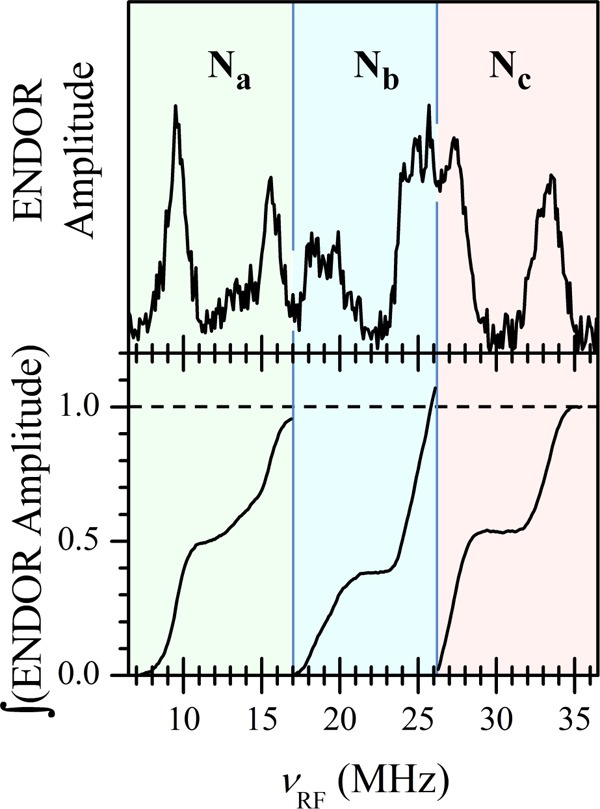
14N Davies ENDOR spectrum of a Cu(PD1) solution in toluene (top panel) and integrals under the ENDOR features belonging to different 14N ligand nuclei (bottom panel). The experiment was performed in a 2D fashion, νRF vs the RF pulse length, tRF, and then the 2D set was integrated over tRF to obtain the 1D spectrum shown in the top panel. Experimental conditions: microwave frequency, 30.360 GHz; magnetic field, B0 = 970 mT (marked by an asterisk in Figure 4b); microwave pulses, 160, 80, and 160 ns; time interval between the first and second microwave pulses, 36 μs; time interval between the second and third microwave pulses, 400 ns; tRF variation range, 2–32 μs; temperature, 15 K.
In order to estimate the relative numbers of nitrogen nuclei contributing to each pair of 14N ENDOR features, we have integrated the spectrum within the regions occupied by each line group (Figure 5, bottom panel). The similar areas under each feature correspond to three types of copper-bound nitrogen donors in equal numbers. We conclude, therefore, that the solution structure of Cu(PD1) is similar to that determined by single-crystal X-ray diffraction analysis (Figure 3), with a single PD12– ligand coordinated in tetradentate fashion to the Cu(II) center. Alternative structures formally having three types of nitrogen ligands in equal numbers can be discarded: three-coordinate Cu(II) complexes are quite unusual, and they exhibit ACu∥ ∼ 9–13 mT,50,51 significantly lower than that found in our case, whereas Cu(II) complexes with six nitrogen ligands have g∥ values in excess of 2.23,52 significantly greater than that observed in this work.
The hfi constants AN∥ found in this work are within the range known for 14N ligands in Cu(II) complexes.53 The largest hfi constant, AN∥ = 60.4 MHz (Nc in Figure 5), is close to those found in Cu(II) complexes of tetraphenylporphyrin (TPP) (four equivalent nitrogen donors with AN∥ = 54.2 MHz)54 and N-confused TPP (NcTPP) (two of the larger hfi constants AN∥ = 60 MHz).55 For pyrrolic nitrogens coordinated trans to oxygen ligands, hfi constants are reduced to ∼40 MHz,9,56 and AN∥ of Nb is of similar magnitude (43.8 MHz). The remaining AN∥ = 25.2 MHz (Na) found in this work appears to be much smaller than the hfi constants found for pyrrole or imidazole ligands of Cu(II) in nitrogen–oxygen coordination environments53 to be explained by electronic factors only. Because the Cu–N1 bond distance is longer than the other nitrogen contacts in the crystal structure of Cu(PD1) (see structural characterization and Figure 3 above), we tentatively assign Na to N1, whereas Nb and Nc are assigned to N2 and N3, respectively.
Together with the visible absorption data, the EPR and ENDOR spectroscopic analysis of the paramagnetic complex Cu(PD1) indicated that prodigiosin analogue H2PD1 coordinates Cu(II) ions with 1:1 stoichiometry, employing all three nitrogen donors on the ligand in the absence of any added base in organic solvents.
Conclusions
The electron-rich tripyrrolic scaffold and preorganized array of nitrogen donor groups of pyrrolyldipyrrin motifs have made them long-standing candidates for binding of transition metals. In spite of these features, these oligopyrrolic fragments are not characterized by a rich coordination chemistry. Here, we report a molecular design of the substitution pattern on this tripyrrolic motif that leads to the construction of an effective platform for metal coordination. Specifically, the addition of a meso-aryl group and an ester group on the C-ring resulted in ligand system H2PD1, which not only maintains the known monoanionic bidentate binding mode shown in complex Zn(HPD1)2 but also offers an unprecedented dianionic tetradentate coordination mode for Cu(II) in a pyrrolyldipyrrin complex. The latter was established by X-ray crystallography in the solid state and confirmed in solution by pulsed ENDOR. The described spectroscopic analysis provides a basis for the study of metal-bound pyrrolyldipyrrins in other paramagnetic complexes.
The modular construction of meso-substituted pyrrolyldipyrrins described herein is anticipated to produce a class of ligands featuring high tunability of donor capacity and redox potentials. In addition, lacking the rigid structure of porphyrin and other pyrrole-based macrocycles, tripyrrolic ligands enable the formation of complexes in which the metal center is possibly more accessible for substrate coordination in catalytic applications. These expectations reflect recent reports on the coordination compounds of several linear oligopyrroles that testify to the rich redox chemistry3,9 and catalytic applications8 of transition metal complexes of this class of ligands. Our findings offer new opportunities in the construction and untapped reactivity of metal complexes of pyrrolyldipyrrin ligands. These studies could provide insight into the involvement of transition metals in the biological activities of prodigiosin compounds and their synthetic analogues.
Experimental Section
Materials and Methods
All reactions were carried out under an inert (N2 or Ar) atmosphere using dry solvents unless otherwise noted. Tetrahydrofuran (THF), methanol (MeOH), pentane, diethyl ether (Et2O), and dichloromethane (CH2Cl2) were dried by passage through a Vacuum Atmospheres solvent purifier. 1,2-Dimethoxyethane (DME) was freshly distilled from CaH2. Flash column chromatography was carried out using SiliaFlash P60 silica (40–63 μm particle size, 230–400 mesh, SiliCycle) or Brockmann grade I neutral aluminum oxide (58 Å, 60 mesh, Alfa Aesar). Reactions were monitored by thin-layer chromatography (TLC) on silica gel plates (aluminum-backed, 60 W F254s, EMD Millipore). All other reagents were obtained commercially and used as received.
1H and 13C NMR spectra were recorded at the University of Arizona NMR Facility on Bruker DRX-600, DRX-500, or AVIII-400 instruments and calibrated using residual undeuterated solvent or tetramethylsilane as an internal reference. Low- and high-resolution mass spectra were acquired at the University of Arizona Mass Spectrometry Facility. Elemental analyses were performed by Numega Resonance Laboratories, San Diego, CA. UV–vis spectra were recorded on an Agilent 8453 UV–vis spectrophotometer, and solutions were freshly prepared in MeOH. The EPR measurements were performed at the University of Arizona EPR facility (see the section below for details).
Ethyl 5-(Hydroxy(phenyl)methyl)-1H-pyrrole-2-carboxylate (6)
Ethyl 5-benzoyl-1H-pyrrole-2-carboxylate57,58 (1.72 g, 7.07 mmol) was dissolved in MeOH (15 mL) in a round-bottomed flask at 0 °C. NaBH4 (0.802 g, 21.2 mmol) was added to the flask in three portions over 30 min. The reaction mixture was warmed to room temperature and stirred for 8 h. The reaction mixture was then cooled to 0 °C and carefully quenched by adding saturated aqueous NaHCO3. The aqueous layer was extracted three times with ethyl acetate (20 mL), and the combined organic layers were washed with brine (10 mL) and dried over anhydrous Na2SO4. Following solvent evaporation under reduced pressure, crude product 6 was used directly in the next step without further purification (1.47 g, 6.01 mmol, 75%). 1H NMR (500 MHz, CDCl3, δ): 9.69 (s, 1H), 7.44–7.33 (m, 5H), 6.85 (dd, J = 3.8, 2.6 Hz, 1H), 5.98–5.96 (m, 1H), 5.92 (d, J = 4.1 Hz, 1H), 4.29 (q, J = 7.1 Hz, 2H), 3.23 (d, J = 4.1 Hz, 1H), 1.35 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3, δ): 161.57, 141.74, 139.21, 128.71, 128.30, 126.60, 122.31, 115.80, 108.36, 60.46, 14.46. LRMS-ESI+m/z (relative intensity): 228.0 (100%).
Ethyl 5-(Phenyl(pyrrol-2-yl)methyl)-1H-pyrrole-2-carboxylate (7)
Compound 6 (1.47 g, 6.01 mmol) was dissolved in glacial acetic acid (68 mL) and acetic anhydride (6.6 mL) in a round-bottomed flask. Pyrrole (2.1 mL, 30.1 mmol) was added, and the reaction mixture was refluxed for 8 h. Following solvent evaporation under reduced pressure (to remove the acetic acid), the crude compound was purified by flash chromatography (silica gel, 25% ethyl acetate in hexanes) to yield 7 as an orange-brown oil (1.27 g, 4.33 mmol, 72%). 1H NMR (500 MHz, CDCl3, δ): 8.84 (s, 1H), 7.92 (s, 1H), 7.39–7.30 (m, 3H), 7.25–7.21 (m, 2H), 6.88 (dd, J = 3.7, 2.5 Hz, 1H), 6.75 (ddd, J = 2.7, 1.6, 0.7 Hz, 1H), 6.20 (dd, J = 6.1, 2.7 Hz, 1H), 6.00 (ddd, J = 3.6, 2.7, 0.7 Hz, 1H), 5.97 (dddd, J = 3.7, 2.5, 1.6, 0.9 Hz, 1H), 5.51 (s, 1H), 4.30 (q, J = 7.1 Hz, 2H), 1.36 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3, δ): 161.11, 140.83, 138.15, 131.08, 128.88, 128.31, 127.30, 122.15, 117.74, 115.54, 109.82, 108.56, 107.74, 60.27, 44.16, 14.94. LRMS-ESI+m/z (relative intensity): 294.0 (4%), 228.0 (100%).
Ethyl 5-[(5-Bromo-1H-pyrrol-2-yl)(phenyl)methyl]-1H-pyrrole-2-carboxylate (8)
Compound 7 (0.100 g, 0.340 mmol) was dissolved in THF (6 mL) at −78 °C. N-Bromosuccinimide (61 mg, 0.340 mmol) was added to the reaction flask in one portion, and the mixture was stirred at −78 °C for 30 min. The reaction mixture was diluted with hexanes (5 mL) and with water (5 mL). The aqueous layer was extracted three times with ethyl acetate (20 mL), and the combined organic layers were washed with brine (10 mL) and dried over anhydrous Na2SO4. Solvent evaporation under reduced pressure afforded compound 8 as a brown oil (0.114 g, 0.241 mmol, 90%), which was used directly in the next step without further purification. (Note that in order to obtain the desired compound in high yields and free of side products, this reaction could not be conducted on more than 0.150 g of 7.) 1H NMR (500 MHz, CDCl3, δ): 8.79 (s, 1H), 7.82 (s, 1H), 7.40–7.32 (m, 3H), 7.24–7.20 (m, 2H), 6.87 (dd, J = 3.7, 2.5 Hz, 1H), 6.11 (dd, J = 3.6, 2.7 Hz, 1H), 6.01 (ddd, J = 3.6, 2.7, 0.7 Hz, 1H), 5.88 (ddd, J = 3.7, 2.5, 0.9 Hz, 1H), 5.44 (s, 1H), 4.31 (q, J = 7.1 Hz, 2H), 1.36 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3, δ): 161.71, 140.36, 137.84, 132.19, 128.65, 128.14, 127.34, 122.29, 116.01, 110.47, 109.97, 109.45, 97.54, 60.47, 44.21, 14.35. LRMS-ESI+m/z (relative intensity): 372.1 (33%), 327.0 (100%).
(Z)-Ethyl 5-[(5-Bromo-2H-pyrrol-2-ylidene)(phenyl)methyl]-1H-pyrrole-2-carboxylate (9)
Compound 8 (0.114 g, 0.241 mmol) was dissolved in THF (6.7 mL) at 0 °C. 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone (DDQ, 55 mg, 0.241 mmol) was dissolved in THF (1.4 mL) and added dropwise. 1,5-Diazabicyclo(4.3.0)non-5-ene (DBN, 30 μL, 0.241 mmol) was added, and the mixture was stirred at room temperature for 7 h. The reaction mixture was quenched by adding saturated aqueous NaHCO3 (10 mL), and the aqueous layer was extracted three times with CH2Cl2 (10 mL). The combined organic layers were washed with brine (10 mL) and dried over anhydrous Na2SO4. Solvent evaporation under reduced pressure resulted in a brown oil. The crude product was redissolved in 1:1 mixture of ethyl acetate/hexanes and filtered through a silica pad in order to give compound 9 (75 mg, 0.202 mmol, 84%) as a yellow oil. 1H NMR (500 MHz, CDCl3, δ): 7.55–7.45 (m, 5H), 6.87 (dd, J = 4.1, 2.4 Hz, 1H), 6.76 (d, J = 4.5 Hz, 1H), 6.52 (d, J = 4.5 Hz, 1H), 6.33 (dd, J = 4.1, 2.4 Hz, 1H), 4.43 (q, J = 7.1 Hz, 2H), 1.46 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3, δ): 160.54, 150.17, 149.00, 139.47, 137.00, 135.65, 134.87, 130.65, 129.57, 128.39, 127.99, 121.63, 115.54, 60.76, 14.26. LRMS-ESI+m/z (relative intensity): 370.9 (19%), 325.1 (100%).
(Z)-Ethyl 5-[1H,5′H(2,2′-Bipyrrol]-5′-ylidene(phenyl)methyl]-1H-pyrrole-2-carboxylate (H2PD1)
Compound 9 (75 mg, 0.202 mmol), LiCl (30 mg, 0.707 mmol), pyrrole-2-boronic acid (64 mg, 0.303 mmol), and Pd(PPh3)4 (23 mg, 10 mol %, 20.2 μmol) were dissolved in 1,2-dimethoxyethane (6.1 mL), and the solution was degassed for 2 h. A degassed solution of aqueous Na2CO3 (2 M, 0.5 mL) was added dropwise, and the mixture was refluxed for 2 h. The reaction mixture was cooled to room temperature and diluted with water (10 mL). The aqueous layer was extracted three times with CH2Cl2 (10 mL). The combined organic layers were washed with brine (10 mL) and dried over anhydrous Na2SO4. Solvent evaporation under reduced pressure afforded the crude product, which was dissolved in CH2Cl2 (10 mL). The solution was cooled to 0 °C, and trifluoroacetic acid (0.8 mL) was added dropwise. The mixture was stirred at room temperature for 8 h, cooled to 0 °C, and carefully quenched with saturated aqueous NaHCO3 (20 mL). The mixture was extracted three times with CH2Cl2 (10 mL), and the combined organic layers were washed with brine (10 mL) and dried over anhydrous Na2SO4. Solvent evaporation followed by flash chromatography (neutral alumina, 10–30% ethyl acetate in hexanes) gave pyrrolyldipyrrin H2PD1 as a dark red solid (38 mg, 0.107 mmol, 48%). (Note that this reaction gives lower yields when conducted on more than 0.100 g of 9.) 1H NMR (400 MHz, CDCl3, TMS δ): 9.35 (bs, 1H), 7.49–7.41 (m, 5H), 7.10 (bs, 1H), 6.92 (d, J = 4.7 Hz, 1H), 6.90 (d, J = 4.7 Hz, 1H), 6.85–6.83 (m, 2H), 6.40–6.37 (m, 1H), 6.24 (d, J = 4.0 Hz, 1H), 4.41 (q, J = 7.1 Hz, 2H), 1.46 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3, δ): 160.30, 153.05, 138.25, 137.70, 136.68, 132.58, 132.11, 131.51, 131.23, 129.92, 128.69, 125.46, 122.69, 122.24, 122.15, 117.59, 113.82, 61.48, 14.40. HRMS-MALDI (m/z): [M + H]+ calcd for [C22H20N3O2], 358.1555; found, 358.1536. UV–vis (MeOH) λmax (ε), 476 nm (29 600 M–1 cm–1).
Zn(II) Complex Zn(HPD1)2
Pyrrolyldipyrrin H2PD1 (20 mg, 5.6 μmol) was dissolved in THF (6 mL), and Zn(OAc)2·2H2O (6.0 mg, 2.8 μmol) was added. The reaction progress was monitored by UV–vis spectroscopy. Upon completion, the solvent was removed under high vacuum, and the resulting crude solid was dissolved in dichloromethane (0.5 mL). Filtration through a Celite pad to remove any unreacted zinc salt followed by evaporation of the solvent and recrystallization from THF/pentane gave the zinc complex as a dark orange/red powder (10 mg, 1.3 μmol, 46%). Single crystals for X-ray diffraction analysis and elemental analysis were grown from THF/pentane mixtures. 1H NMR (400 MHz, CDCl3, TMS δ): 9.75 (bs, 2H), 7.58–7.48 (m, 10H), 6.96 (d, J = 4.0 Hz, 2H), 6.85 (d, J = 4.7 Hz, 2H), 6.79 (d, J = 4.7 Hz, 2H), 6.67 (ddd, J = 3.8, 2.5, 1.4 Hz, 2H), 6.58 (td, J = 2.7, 1.4 Hz, 2H), 6.45 (d, J = 4.0 Hz, 2H), 6.14 (dt, J = 3.7, 2.4 Hz, 2H), 4.05–3.98 (m, 4H), 0.98 (t, J = 7.1 Hz, 6H). HRMS (m/z) [M + H]+ calcd for [C22H17N3O2Zn], 777.2168; found, 777.2163. UV–vis (MeOH) λmax (ε): 512 nm (56 607 M–1 cm–1), 544 nm (50 200 M–1 cm–1). Anal. Calcd for C44H37N6O4Zn·2THF·4H2O: C, 62.80; H, 6.08; N, 8.45%. Found: C, 62.20; H, 5.56; N, 8.70%.
Cu(II) Complex Cu(DP1)
Pyrrolyldipyrrin H2PD1 (20 mg, 5.6 μmol) was dissolved in THF (6 mL), and Cu(OAc)2·H2O (11 mg, 5.6 μmol) was added. The reaction progress was monitored by UV–vis spectroscopy. Upon completion, the solvent was removed under high vacuum and the resulting crude solid was dissolved in THF (0.5 mL). Filtration through a Celite pad followed by slow evaporation of the solvent gave the copper complex as a dark blue microcrystalline powder (12 mg, 2.9 μmol, 52%). Crystals for X-ray diffraction analysis and elemental analysis were grown in Et2O/n-hexane mixtures. HRMS-MALDI (m/z): [M + H]+ calcd for [C22H17N3O2Cu], 418.0617; found, 418.0611. UV–vis (MeOH) λmax (ε): 586 nm (18 200 M–1c m–1). Anal. Calcd for C44H37N6O4Cu·n-hexane·Et2O·2H2O: C, 62.70; H, 6.04; N, 8.13%. Found: C, 62.80; H, 5.92; N, 7.87%.
X-ray Diffraction Analysis
Data were collected at the University of Arizona X-ray Diffraction Facility. Crystals were mounted onto a MiTeGen micromount under a protective film of Paratone oil, and diffraction data for both crystals were measured using a Bruker Kappa APEX II DUO diffractometer with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) generated by a sealed tube and an APEX II CCD area detector. The diffractometer was fitted with an Oxford Cryostream low-temperature device, and the data sets were collected using the APEX2 software package.65 The data were corrected for absorption effects using a multiscan method in SADABS.66 Experimental details of the structure determinations are given in Table S1 (see Supporting Information). All structures were solved by direct methods (SHELXS-97), and developed by full least-squares refinement based on F2 (SHELXL).59 Crystallographic figures were prepared using Mercury60 and POV-Ray.67
Structure Refinement of Zn(HPD1)2.
Data were collected, solved, and refined in the triclinic space group P–1. The asymmetric unit contains one full complex molecule with no other significant peaks in the Fourier map. All non-hydrogen atoms were located in the Fourier map and refined anisotropically, whereas all hydrogen atoms were placed in calculated positions and refined using a riding model. The highest residual Fourier peak is 0.7 e Å–3, located 1.0 Å from Zn(1), and the deepest Fourier hole is −0.45 e Å–3, located 0.93 Å from Zn(1).
Structure Refinement of Cu(PD1)
Although multiple crystallizations were attempted under different conditions, the best single crystals obtained for this complex diffracted significantly only to 2θmax = 40° and presented substantial solvent disorder, which further complicated the analysis. Data were collected, solved, and refined in the monoclinic space group P21/c. The asymmetric unit contains one full complex molecule and a solvent channel running parallel to (100) filled with electron density that refined reasonably as three partially occupied oxygen atoms of a water molecule (labeled O(3A), O(3B), and O(3C)). Two of these were modeled with molar occupancies of 20% and one with 15%. Alternative solvent molecules of crystallization were tentatively fitted in this solvent channel but failed to offer a stable refinement possibly because of the low observed data/parameter ratio (6.8).
All fully occupied, crystallographically ordered non-hydrogen atoms were refined anisotropically. Carbon atom C(10) was refined with a rigid bond restraint because this atom continually refined as nonpositive definite without restraints. All hydrogen atoms were placed in calculated positions and refined using a riding model.
A SQUEEZE analysis61,62 of a model containing only the complex molecule identified 330 Å3 of total void space, equivalent to 16.3% of the unit cell volume. These voids contained 73 electrons per unit cell, or approximately 18 per asymmetric unit. This could equate approximately to one molecule of hexane (50 electrons each) and one-half molecule of diethyl ether (42 electrons each) or seven molecules of water (10 electrons each) per unit cell, or a combination of all three with partial molar occupancies. Because only three partially occupied molecules of water could be positively identified in the Fourier map, a formula of complex·1.8(H2O) was used for the density and F(000) calculations. The highest residual Fourier peak of 0.9 e A–3 is located within the solvent accessible void, and the deepest residual Fourier hole of −0.5 e A–3 is located 1.1 Å from N(3).
EPR and Pulsed ENDOR Spectroscopy
The X-band (∼9 GHz) CW EPR experiments were performed at room temperature and 77 K using a Bruker E500 (Elexsys) spectrometer. The pulsed EPR measurements were performed on a Ka-band (26–40 GHz) pulsed EPR spectrometer63 at 15 K. In order to establish the relative number of copper-bound nitrogen ligands in Cu(PD1), Ka-band pulsed (Davies) ENDOR spectroscopy was used.
Here, we briefly describe the ENDOR spectra expected for 14N ligands in Cu(II) complexes under our experimental conditions. The 14N transition lines in such spectra are located at the frequencies
| 1 |
where AN is the diagonal part of the 14N hfi (predominantly isotropic), νN ∼ 3 MHz is the 14N Zeeman frequency in the applied magnetic field, B0 ∼ 1 T, and QN is the diagonal part of the 14N nqi: QN ∼ −0.9 MHz for the pyrrole 14N at g∥.54 For the nitrogen ligands in Cu(II) complexes, AN is on the order of tens of megahertz. Therefore, under our experimental conditions, the relationship between the various terms in eq 1 is AN/2 > νN > QN. Without the nqi, the ENDOR pattern for the 14N nucleus would consist of two lines centered at ν = AN/2, with the splitting between them equal to 2νN ∼ 6 MHz. The nqi will split each of these lines into a doublet, with the splitting equal to 2QN (∼1.8 MHz at g∥). However, a broadening of the individual lines caused by even an insignificant degree of structural disorder can result in a partial or complete loss of the quadrupolar splitting and observation of only two broader lines for each 14N nucleus at the frequencies ν = AN/2 ± νN. Such a situation is observed in the spectrum of Cu(PD1) (Figure 5).
In order to make the Davies ENDOR response independent of the hfi constants of the detected nuclei, one has to ensure that the amplitudes of the mw pulses were much smaller than that of the hfi constants while keeping the spin flip angles close to optimal (π for the preparation (inversion) pulse and π/2 and π for the two-pulse detection sequence).64 The hfi constants of 14N ligands in Cu(II) complexes are on the order of tens of megahertz and therefore this requirement is easily satisfied for mw pulses with durations ∼100 ns (the mw amplitude ∼5 MHz).
Because of the strong hfi and non-negligible nuclear quadrupole interaction (nqi), the probabilities of transitions of nonequivalent 14N nuclei, and even different transitions of the same 14N nucleus, induced by the RF field are expected to be noticeably different. Therefore, to approximately equalize the contributions of different nitrogens to the ENDOR spectrum, a 2D experiment was performed, with one dimension being the radiofrequency, and the other being the RF pulse duration. The 2D data set was then integrated over the RF pulse duration to obtain the 1D ENDOR spectrum with the relative intensities of the 14N lines reflecting relative numbers of nuclei rather then relative transition probabilities. The 2D ENDOR data set (from which the 1D spectrum in Figure 5 was obtained) is shown in Figure S8 (Supporting Information).
Acknowledgments
We are grateful to Drs. Elizabeth Ilardi and Jonathan Loughrey for assistance with the purification of H2PD1 and Zn(HPD1)2, respectively, and to Drs. Jonathan Loughrey and Sue Roberts for assistance with the acquisition and analysis of X-ray diffraction data. We thank the University of Arizona and the Donors of the American Chemical Society Petroleum Research Fund (grant 51754-DNI3 to E.T.) for financial support. A.V.A. gratefully acknowledges NSF (DBI-0139459, DBI-9604939, and BIR-9224431) and NIH (S10RR020959 and S10RR026416-01) grants for the development of the EPR facility at the University of Arizona.
Supporting Information Available
1H NMR and ENDOR spectroscopic data, X-ray crystallographic details, and CIF files. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
† These authors contributed equally to this work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Wood T. E.; Thompson A. Chem. Rev. 2007, 107, 1831–1861. [DOI] [PubMed] [Google Scholar]
- Gryko D. T.; Gryko D.; Lee C.-H. Chem. Soc. Rev. 2012, 41, 3780–3789. [DOI] [PubMed] [Google Scholar]
- Katayev E. A.; Severin K.; Scopelliti R.; Ustynyuk Y. A. Inorg. Chem. 2007, 46, 5465–5467. [DOI] [PubMed] [Google Scholar]
- Reid S. D.; Wilson C.; Blake A. J.; Love J. B. Dalton Trans. 2010, 39, 418–425. [DOI] [PubMed] [Google Scholar]
- Halper S. R.; Cohen S. M. Inorg. Chem. 2005, 44, 486–488. [DOI] [PubMed] [Google Scholar]
- King E. R.; Betley T. A. Inorg. Chem. 2009, 48, 2361–2363. [DOI] [PubMed] [Google Scholar]
- King E. R.; Sazama G. T.; Betley T. A. J. Am. Chem. Soc. 2012, 134, 17858–17861. [DOI] [PubMed] [Google Scholar]
- Hennessy E. T.; Betley T. A. Science 2013, 340, 591–595. [DOI] [PubMed] [Google Scholar]
- Thoi V. S.; Stork J. R.; Niles E. T.; Depperman E. C.; Tierney D. L.; Cohen S. M. Inorg. Chem. 2008, 47, 10533–10541. [DOI] [PubMed] [Google Scholar]
- Patra A. K.; Dube K. S.; Sanders B. C.; Papaefthymiou G. C.; Conradie J.; Ghosh A.; Harrop T. C. Chem. Sci. 2012, 3, 364–369. [Google Scholar]
- Sanders B. C.; Patra A. K.; Harrop T. C. J. Inorg. Biochem. 2013, 118, 115–127. [DOI] [PubMed] [Google Scholar]
- Bröring M.; Köhler S.; Pietzonka C. J. Porphyrins Phthalocyanines 2012, 16, 641–650. [Google Scholar]
- Bröring M.; Köhler S.; Ostapowicz T.; Funk M.; Pietzonka C. Eur. J. Inorg. Chem. 2009, 3628–3635. [Google Scholar]
- Sessler J. L.; Gebauer A.; Král V.; Lynch V. Inorg. Chem. 1996, 35, 6636–6637. [DOI] [PubMed] [Google Scholar]
- Bennett J. W.; Bentley R. Adv. Appl. Microbiol. 2000, 47, 1–32. [DOI] [PubMed] [Google Scholar]
- Fürstner A. Angew. Chem., Int. Ed. 2003, 42, 3582–3603. [DOI] [PubMed] [Google Scholar]
- D’Alessio R.; Bargiotti A.; Carlini O.; Colotta F.; Ferrari M.; Gnocchi P.; Isetta A.; Mongelli N.; Motta P.; Rossi A.; Rossi M.; Tibolla M.; Vanotti E. J. Med. Chem. 2000, 43, 2557–2565. [DOI] [PubMed] [Google Scholar]
- Fürstner A.; Grabowski J.; Lehmann C. W.; Kataoka T.; Nagai K. ChemBioChem. 2001, 2, 60–68. [DOI] [PubMed] [Google Scholar]
- Marchal E.; Uddin M. I.; Smithen D. A.; Hawco C. L. A.; Lanteigne M.; Overy D. P.; Kerr R. G.; Thompson A. RSC Adv. 2013, 3, 22967–22971. [Google Scholar]
- Papireddy K.; Smilkstein M.; Kelly J. X.; Shweta; Salem S. M.; Alhamadsheh M.; Haynes S. W.; Challis G. L.; Reynolds K. A. J. Med. Chem. 2011, 54, 5296–5306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Tomas R.; Vinas M. Curr. Med. Chem. 2010, 17, 2222–2231. [DOI] [PubMed] [Google Scholar]
- Regourd J.; Ali A. A.-S.; Thompson A. J. Med. Chem. 2007, 50, 1528–1536. [DOI] [PubMed] [Google Scholar]
- Díaz R. I. S.; Bennett S. M.; Thompson A. ChemMedChem. 2009, 4, 742–745. [DOI] [PubMed] [Google Scholar]
- Smithen D. A.; Forrester A. M.; Corkery D. P.; Dellaire G.; Colpitts J.; McFarland S. A.; Berman J. N.; Thompson A. Org. Biomol. Chem. 2013, 11, 62–68. [DOI] [PubMed] [Google Scholar]
- Melvin M. S.; Ferguson D. C.; Lindquist N.; Manderville R. A. J. Org. Chem. 1999, 64, 6861–6869. [DOI] [PubMed] [Google Scholar]
- Sessler J. L.; Eller L. R.; Cho W.-S.; Nicolaou S.; Aguilar A.; Lee J. T.; Lynch V. M.; Magda D. J. Angew. Chem., Int. Ed. 2005, 44, 5989–5992. [DOI] [PubMed] [Google Scholar]
- Davis J. T.; Gale P. A.; Okunola O. A.; Prados P.; Iglesias-Sánchez J. C.; Torroba T.; Quesada R. Nat. Chem. 2009, 1, 138–144. [DOI] [PubMed] [Google Scholar]
- Busschaert N.; Gale P. A. Angew. Chem., Int. Ed. 2013, 52, 1374–1382. [DOI] [PubMed] [Google Scholar]
- Melvin M. S.; Tomlinson J. T.; Saluta G. R.; Kucera G. L.; Lindquist N.; Manderville R. A. J. Am. Chem. Soc. 2000, 122, 6333–6334. [Google Scholar]
- Melvin M. S.; Wooton K. E.; Rich C. C.; Saluta G. R.; Kucera G. L.; Lindquist N.; Manderville R. A. J. Inorg. Biochem. 2001, 87, 129–135. [DOI] [PubMed] [Google Scholar]
- Díaz R. I. S.; Regourd J.; Santacroce P. V.; Davis J. T.; Jakeman D. L.; Thompson A. Chem. Commun. 2007, 2701–2703. [DOI] [PubMed] [Google Scholar]
- Fürstner A.; Grabowski J. E. ChemBioChem. 2001, 2, 706–709. [DOI] [PubMed] [Google Scholar]
- Melvin M. S.; Tomlinson J. T.; Park G.; Day C. S.; Saluta G. R.; Kucera G. L.; Manderville R. A. Chem. Res. Toxicol. 2002, 15, 734–741. [DOI] [PubMed] [Google Scholar]
- Rao M. R.; Tiwari M. D.; Bellare J. R.; Ravikanth M. J. Org. Chem. 2011, 76, 7263–7268. [DOI] [PubMed] [Google Scholar]
- Zhang M.; Hao E.; Xu Y.; Zhang S.; Zhu H.; Wang Q.; Yu C.; Jiao L. RSC Adv. 2012, 2, 11215–11218. [Google Scholar]
- Zhang M.; Hao E.; Zhou J.; Yu C.; Bai G.; Wang F.; Jiao L. Org. Biomol. Chem. 2012, 10, 2139–2145. [DOI] [PubMed] [Google Scholar]
- Park G.; Tomlinson J. T.; Melvin M. S.; Wright M. W.; Day C. S.; Manderville R. A. Org. Lett. 2003, 5, 113–116. [DOI] [PubMed] [Google Scholar]
- Crawford S. M.; Ali A. A.-S.; Cameron T. S.; Thompson A. Inorg. Chem. 2011, 50, 8207–8213. [DOI] [PubMed] [Google Scholar]
- Hong T.; Song H.; Li X.; Zhang W.; Xie Y. RSC Adv. 2014, 4, 6133–6140. [Google Scholar]
- Murakami Y.; Matsuda Y.; Sakata K.; Martell A. E. J. Chem. Soc., Dalton Trans. 1973, 1729–1734. [Google Scholar]
- Smithen D. A.; Baker A. E. G.; Offman M.; Crawford S. M.; Cameron T. S.; Thompson A. J. Org. Chem. 2012, 77, 3439–3453. [DOI] [PubMed] [Google Scholar]
- Wallace D. M.; Leung S. H.; Senge M. O.; Smith K. M. J. Org. Chem. 1993, 58, 7245–7257. [Google Scholar]
- Ptaszek M.; McDowell B. E.; Lindsey J. S. J. Org. Chem. 2006, 71, 4328–4331. [DOI] [PubMed] [Google Scholar]
- García-Valverde M.; Alfonso I.; Quiñonero D.; Quesada R. J. Org. Chem. 2012, 77, 6538–6544. [DOI] [PubMed] [Google Scholar]
- Melvin M. S.; Calcutt M. W.; Noftle R. E.; Manderville R. A. Chem. Res. Toxicol. 2002, 15, 742–748. [DOI] [PubMed] [Google Scholar]
- Rastogi S.; Marchal E.; Uddin I.; Groves B.; Colpitts J.; McFarland S. A.; Davis J. T.; Thompson A. Org. Biomol. Chem. 2013, 11, 3834–3845. [DOI] [PubMed] [Google Scholar]
- Yu C.; Jiao L.; Tan X.; Wang J.; Xu Y.; Wu Y.; Yang G.; Wang Z.; Hao E. Angew. Chem., Int. Ed. 2012, 51, 7688–7691. [DOI] [PubMed] [Google Scholar]
- Jenkins S.; Incarvito C. D.; Parr J.; Wasserman H. H. CrystEngComm 2009, 11, 242–245. [Google Scholar]
- Davies E. R. Phys. Lett. A 1974, 47, 1–2. [Google Scholar]
- Holland P. L.; Tolman W. B. J. Am. Chem. Soc. 1999, 121, 7270–7271. [Google Scholar]
- Holland P. L.; Tolman W. B. J. Am. Chem. Soc. 2000, 122, 6331–6332. [Google Scholar]
- Pap J. S.; Kripli B.; Bányai V.; Giorgi M.; Korecz L.; Gajda T.; Árus D.; Kaizer J.; Speier G. Inorg. Chim. Acta 2011, 376, 158–169. [Google Scholar]
- Iwaizumi M.; Kudo T.; Kita S. Inorg. Chem. 1986, 25, 1546–1550. [Google Scholar]
- Brown T. G.; Hoffman B. M. Mol. Phys. 1980, 39, 1073–1109. [Google Scholar]
- Mitrikas G.; Calle C.; Schweiger A. Angew. Chem., Int. Ed. 2005, 44, 3301–3303. [DOI] [PubMed] [Google Scholar]
- Heinze K.; Reinhart A. Inorg. Chem. 2006, 45, 2695–2703. [DOI] [PubMed] [Google Scholar]
- Bailey D. M.; Johnson R. E.; Albertson N. F. Org. Synth. 1971, 51, 100. [Google Scholar]
- Fang Q. K.; Hopkins S.; Heffernan M.; Chytil M.. Pyrrole and Pyrazole DAAO Inhibitors. US Patent 7,488,747, 2009.
- APEX2; Bruker AXS Inc.: Madison, WI, 2007.
- Sheldrick G. M.SADABS; University of Göttingen: Göttingen, Germany, 1997.
- Sheldrick G. M. Acta Crystallogr. 2008, A64, 112–122. [DOI] [PubMed] [Google Scholar]
- Macrae C. F.; Bruno I. J.; Chisholm J. A.; Edgington P. R.; McCabe P.; Pidcock E.; Rodriguez-Monge L.; Taylor R.; van de Streek J.; Wood P. A. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar]
- POV-Ray, version 3.6; Persistence of Vision Raytracer Pty. Ltd.: Williamstown, Victoria, Australia, 2004.
- Spek A. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar]
- van der Sluis P.; Spek A. Acta Crystallogr. 1990, A46, 194–201. [Google Scholar]
- Astashkin A. V.; Enemark J. H.; Raitsimring A. Concepts Magn. Reson., Part B 2006, 29B, 125–136. [Google Scholar]
- Schweiger A.; Jeschke G.. Principles of Pulse Electron Paramagnetic Resonance; Oxford University Press: New York, 2001. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.