

Abstract
We report the association of an inherited variant located upstream of the poly(adenosine diphosphate-ribose) polymerase 1 (PARP1) gene (rs2249844), with survival in 11 BioGenoMEL melanoma cohorts. The gene encodes a protein involved in a number of cellular processes including single-strand DNA repair. Survival analysis was conducted for each cohort using proportional hazards regression adjusting for factors known to be associated with survival. Survival was measured as overall survival (OS) and, where available, melanoma-specific survival (MSS). Results were combined using random effects meta-analysis. Evidence for a role of the PARP1 protein in melanoma ulceration and survival was investigated by testing gene expression levels taken from formalin-fixed paraffin-embedded tumors. A significant association was seen for inheritance of the rarer variant of PARP1, rs2249844 with OS (hazard ratio (HR) = 1.16 per allele, 95% confidence interval (CI) 1.04–1.28, p = 0.005, eleven cohorts) and MSS (HR = 1.20 per allele, 95% CI 1.01–1.39, p = 0.03, eight cohorts). We report bioinformatic data supportive of a functional effect for rs2249844. Higher levels of PARP1 gene expression in tumors were shown to be associated with tumor ulceration and poorer OS.
What's New?
Although staging systems predict outcome fairly well for melanoma, survival still varies among individual patients. In this meta-analysis, the authors found that inheritance of a rare genetic variant of PARP1 was associated with improved survival of melanoma patients. Increased expression of PARP1 has been associated with poorer outcome, and depletion of PARP1 may reduce both melanoma growth and angiogenesis. The identification of this and other germline variants that affect survival may help to identify key biological pathways active in host/tumor interactions, which may lead to the discovery of new therapeutic targets for treating advanced melanoma.
Keywords: melanoma, survival, PARP1, genetic determinants of survival, random effects meta-analysis, bioinformatics
The aim of the research consortium BioGenoMEL (www.biogenomel.eu) is to investigate the role of germline (inherited) genetic variation in melanoma survival. Identifying genes that might have an effect on survival by moderation of host/tumor interaction would help us to understand the biology of that interaction, potentially giving rise to novel adjuvant therapies. Previous studies have reported evidence that inherited variation in genes in drug metabolism pathways may affect survival from lung cancer1,2 and that ancestry-related polymorphisms were associated with acute lymphoblastic leukemia relapse.3 We have previously reported evidence that melanocortin receptor 1 (MC1R) variants that influence melanin synthesis and are associated with increased melanoma risk were associated with reduced risk of death from melanoma.4
In our study, we look at the association of a germline variant in the PARP1 gene with outcome. PARP1 is a member of the family of the poly(adenosine diphosphate-ribose) polymerase (PARP) proteins, which are DNA damage sensors, signaling to downstream effectors5 and therefore directly involved in genomic stability, DNA repair and apoptosis. It has been suggested previously that cancer cells can become “addicted” to DNA repair pathways that protect them from lethal levels of DNA damage,6,7 and it has been postulated that PARP1 may play a role in this “addiction.” Increased expression of PARP1 protein has been reported in a number of cancer types, reviewed by Yelamos et al.,5 when increased expression of PARP1 was frequently reported to be associated with a poorer outcome. The role of PARP1 in cancer is said, however, to be pleomorphic, having also been linked to inflammation via its role in upregulation of NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells),8 which has been described as promoting the avoidance of programmed cell death.9 Depletion of PARP1 has been reported to reduce both melanoma growth and angiogenesis, while increasing chemosensitivity in melanoma cells.10 This effect is postulated to be via inhibition of a senescence-induced secretome.11
We report evidence of an association of a single nucleotide polymorphism (SNP) previously shown to be associated with a reduced risk of melanoma12 with outcome. There is, as yet, no direct evidence of a functional effect of this SNP on PARP1 protein levels, so we report an investigation of relevant published bioinformatics data. Although increased expression of PARP1 in melanoma compared to benign melanocytic nevi has been reported using immunohistochemistry,13 we thought it is important to look at melanoma tumors for an association between PARP1 expression and survival as additional evidence that PARP1 might be important in melanoma.
Methods
Data collection
The SNP data are derived from a large Leeds dataset (the Leeds Melanoma Cohort) and from ten smaller datasets within the BioGenoMEL consortium. These cohorts have been described previously4 but details are also provided in Supporting Information (Supporting Information Table S1).
SNP selection process
The PARP1 SNP rs2249844 (A>G) was selected for investigation because of its association with melanoma susceptibility, the minor allele showing a reduced risk of melanoma development. It also has a comparatively high minor allele frequency (MAF), and is in close proximity to the coding sequence of the PARP1 gene. Rs2249844 is in linkage disequilibrium (LD; R2 = 0.96) with the intronic PARP1 SNP rs3219090, which has been shown to be associated with susceptibility to melanoma12 and is itself associated with susceptibility (unpublished data, odds ratio, OR = 0.87; 95% confidence interval, CI 0.81–0.93, p = 0.00005, 2,804 cases, 7,618 controls). Both PARP1 SNPs were typed in the Leeds cohort as part of an initial panel of 23 independent SNPs in selected candidate genes.
SNP genotyping
Genotyping for the datasets from Leeds, both Vienna cohorts, Stockholm, Lund, Athens and Riga was performed in Leeds using Taqman technology (Applied Biosystems, Foster City, CA). A total of 2 µl polymerase chain reactions (PCRs) were performed in 384-well plates using 10 ng of DNA (dried), 0.05 µl assay mix and 1 µl Universal Master Mix (Applied Biosystems) according to the manufacturer's instructions. End point reading of the genotypes was performed using an ABI 7900HT Real-time PCR system (Applied Biosystems). All genotypes were double scored by an independent analyst. The PARP1 SNP rs2249844 was genotyped using the Taqman assay C__34511379_10 (Applied Biosystems).
Genotyping in the additional datasets
Genotyping for the Barcelona, Valencia and Essen datasets was performed in Heidelberg using a PCR-based allelic discrimination method (KBiosciences, UK) in 384-well plate format. In each plate 8% of wells were assigned for quality control. Genotypes in amplified products were determined by differences in VIC and FAM fluorescent level in plate read operation on ABI PRISM 7900HT (Applied Biosystems) using SDS 1.2 Software. Postoperation data were transferred as Microsoft Excel files and converted into genotype information. Genotypes in random samples were validated by DNA sequencing.
DNA samples for genotyping for the Tampa and Vienna FFPE cohorts were extracted from healthy skin removed during wide local excision procedures.
Gene expression analysis
Complementary analyses were run examining the association of gene expression levels with outcome in primary and metastatic formalin-fixed paraffin-embedded tumor samples. Tumor blocks were obtained from patients recruited to a Leeds retrospective study (Predicting Response to Chemotherapy) allowing identification of gene expression profiles associated with primary tumor features and overall survival (OS). Patients recruited to our study had stage IV melanoma and were treated with palliative dacarbazine or temozolomide as part of clinical trials or routine clinical care. Clinical data relating to survival and characteristics of the primary tumor were obtained from treating clinicians and diagnostic histopathology reports.
PARP1 gene expression in the tumors was explored in relation to survival but, as the intent was to study the putative effects of PARP1 on the tumor microenvironment, we also examined the relationship of PARP1 gene expression with tumor ulceration and angiolymphatic invasion. Further details can be found in Supporting Information and Supporting Information Table S2.
Statistical analysis for the SNP data
Link-anonymized data on date of diagnosis and date of death, and prognostic indicators such as Breslow thickness, tumor site, age at diagnosis and sex were additionally centralized in Leeds from the nine European BioGenoMEL cohorts. Analyses were carried out independently using the same procedure by the Tampa group on their dataset as this approach was compliant with their IRB approval.
Survival time was defined as the period between the date of surgical excision of the primary and date of death or last date of follow-up (at which point records were censored). Multivariable survival analyses were performed using Cox's proportional hazards model in R 2.10.1. Hazard ratio estimates were calculated for the effect of each of the SNPs on OS adjusted for sex, site (head/neck, trunk, limbs or other) and age of diagnosis and then additionally for Breslow thickness. Melanoma-specific survival (MSS) data were not available from all groups. Therefore, to reduce the number of nonmelanoma-related deaths reported, we truncated OS time at 8 years of follow-up. An additive genetic model was assumed.
Relevant per-allele effect estimates and standard errors for each study were taken from the fitted Cox's proportional hazards models. These data were then used to carry out a random-effects meta-analysis in R. Models are reported adjusted for age of diagnosis, sex and tumor site, with and without Breslow thickness. We also compared the association of the SNP for OS and MSS in the seven cohorts where cause of death was available. The proportional hazards assumption was tested by fitting Schoenfeld residuals and testing for a significant association with time. Sensitivity analyses were conducted to ensure that no single cohort skewed the results. Additional details regarding the statistical analysis, sensitivity analysis and testing of the proportional hazards assumption can be found in Supporting Information, Supporting Information Table S3, and Figures S1 and S2.
Bioinformatic analysis
The rs2242844 SNP is in close proximity to the 5′ end of the PARP1 gene but is not known to have a functional effect on the gene itself or to be in strong LD with a known functional variant. We undertook several bioinformatic analyses to determine whether rs2249844 or a linked genetic variant is associated with a putative functional effect that may affect outcome in melanoma cases.
Identification of noncoding regulatory elements
To identify potentially important noncoding regulatory regions we queried experimentally derived data on sites of DNAse hypersensitivity deposited in public databases; these regions are of particular interest because they indicate open stretches of DNA available to bind transcription factors that are therefore potentially transcriptionally active. We then attempted to further characterize these regions by looking at additional experimentally derived data such as observed CpG and histone methylation patterns, observed transcription factor binding and sequence conservation. We used pattern recognition algorithms and software such as ChromHMM,14 which predicts functional elements (such as enhancers) based on the chromatin state of a region (derived from patterns learnt from experimental data), and JASPAR,15 which predicts potential transcription factor-binding sites by matching the sequence to a database of transcription factor motif patterns. We investigated the region in LD with rs2249844 (r2 ≥ 0.6) for putative regulatory elements using HaploReg v2 (http://www.broadinstitute.org/mammals/haploreg/haploreg.php),16 the Roadmap Epigenomics project (http://www.roadmapepigenomics.org/) and the ENCclopedia Of DNA Elements (ENCODE; http://genome.ucsc.edu/ENCODE/).17,18
Identification of functional elements using SNPinfo
We used the SNPinfo (http://snpinfo.niehs.nih.gov/)19 function prediction tool to predict the function of SNPs in PARP1 listed in HapMap (CEU population) up to 10 kb downstream and 150 kb upstream. SNPinfo uses several computational algorithms to predict potential transcription factor-binding sites, splice sites, miRNA-binding sites, nonsynonymous SNPs, potential stop codon mutations, sequence conservation, regulatory potential and severity of the effect of a mutation (using Polyphen, http://genetics.bwh.harvard.edu/pph2/20 or SNPs3D, http://www.snps3d.org/21).
Investigation of expression quantitative trait loci associated with PARP1
We investigated the association of PARP1 expression with SNPs in close proximity (within 1 Mb to rs2249844) using the expression quantitative trait loci (eQTL) data (http://www.sanger.ac.uk/resources/software/genevar/)22 and the cis-eQTL—Gene analysis option in the GeneVar software package. We subsequently investigated the association of rs2249844 with expression of genes in close proximity to PARP1 using the cis-eQTL—SNP analysis option in GeneVar. We would have preferred to use tissue-specific expression data (MuTHER resource NCBI36/ReMOAT 1.0.0, TwinsUK-S skin data, n = 85623); however, we found no data for PARP1 expression in this dataset at the time of writing so we subsequently ran the same analysis using the MuTHER pilot study data (Twin1-S, Twin2-S, n = 98).24 We also investigated data deposited in the Chicago eQTL browser for evidence of eQTLs in the PARP1 region. Further details can be found in Supporting Information.
Analysis of the gene expression data from melanoma tumors
Differences in gene expression associated with tumor ulceration and presence of angiolymphatic invasion were tested using Mann–Whitney U-tests. Fold change differences in median gene expression were calculated between ulcerated and nonulcerated tumors and tumors with or without angiolymphatic invasion. Analysis of the putative association between ulceration and PARP1 expression was then adjusted for age, sex and Breslow thickness.
Analysis was performed to identify gene expression patterns associated with OS. The Cox proportional hazards model was used to calculate HRs and 95% CIs for each gene using log-transformed data (log2). Survival analysis was conducted in the same manner as described above for the SNP survival analysis.
Results
Descriptive statistics
A total of 8,599 cases were recruited across the 11 cohorts; 3,965 of these were included in the analysis after dropping ineligible cases (Table1). As has been reported previously,4 there were no major differences between cohorts in age distribution (Fig. 1). Breslow thicknesses were similar across most of the cohorts with the exception of the Riga and Essen cohorts, where cases have been recruited with thicker tumors on average, or were recruited from a clinic serving as a referral center, respectively. As all cases in the Vienna FFPE cohort (recruited between 1997 and 2002) had a sentinel node biopsy this cohort also had a bias toward tumors thicker than 1 mm. There was considerable variation in the time period in which cases were recruited across the cohorts though the majority of cases had been recruited within the last 10 years.
Table 1.
Cases eligible for analysis in the 11 cohorts that comprise the meta-analysis of the association of the rs2249844 SNP (A>G) with outcome
| Whole cohort size | Incident case (recruited <2 years after diagnosis) | Cases genotyped for the SNP | Number of cases with a single melanoma | Cases with complete data on adjusting covariates |
Number with Breslow > 0.75 mm | Cases with complete follow-up | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Center | Age | Sex | Site | Breslow | ||||||
| Leeds | 2,180 | 2,131 | 1,696 | 1,648 | 1,648 | 1,648 | 1,648 | 1,608 | 1,422 | 1,419 |
| Vienna | 1,085 | 389 | 173 | 173 | 164 | 164 | 152 | 151 | 140 | 140 |
| Vienna FFPE | 302 | 302 | 295 | 287 | 284 | 284 | 284 | 280 | 268 | 268 |
| Stockholm | 870 | 605 | 264 | 264 | 264 | 264 | 264 | 264 | 240 | 240 |
| Lund | 355 | 355 | 342 | 342 | 342 | 342 | 321 | 321 | 166 | 166 |
| Athens | 200 | 200 | 197 | 197 | 197 | 197 | 197 | 197 | 191 | 190 |
| Riga | 243 | 242 | 226 | 224 | 224 | 224 | 224 | 194 | 175 | 175 |
| Barcelona | 398 | 358 | 345 | 304 | 290 | 289 | 287 | 278 | 269 | 269 |
| Valencia | 1,440 | 1,248 | 647 | 647 | 649 | 649 | 633 | 579 | 394 | 393 |
| Essen | 941 | 643 | 615 | 599 | 563 | 563 | 482 | 384 | 298 | 298 |
| Tampa | 585 | 585 | 422 | 422 | 422 | 422 | 422 | 420 | 407 | 407 |
| Total | 8,599 | 7,058 | 5,222 | 5,107 | 5,047 | 5,046 | 4,914 | 4,676 | 3,970 | 3,965 |
Cases in each column also meet the criteria of all conditions to the left of it.
Figure 1.

Box plots showing variation of Breslow thickness (>0.75mm), age of diagnosis and date of study entry in each of the ten cohorts. Individual patient data were not available for the Tampa cohort. For the Lund and Athens cohorts date of study entry was not available so date of diagnosis is presented; date of study entry was within 2 years of diagnosis.
Supporting Information Figure S1 shows Kaplan–Meier estimates for survival in each cohort. The curves for most cohorts are similar, except for Essen and Riga for which we see worse prognosis, consistent with the recruitment, on average, of cases with thicker tumors in both of these cohorts.
Analysis of the association of the PARP1 rs2249844 SNP with survival
Figures 2a and 2b and Table2 show the association of rs2249844 with OS in the 11 BioGenoMEL cohorts. Overall, in a random effects meta-analysis there was a significant association seen across the 11 cohorts (HR = 1.17, 95% CI 1.06–1.30, p = 0.003, adjusted for age, sex and tumor site); the significant association persisted when the large Leeds cohort was omitted (HR = 1.20, 95% CI 1.06–1.35, p = 0.003). Additional adjustment for Breslow thickness did not appear to attenuate this association (HR = 1.17, 95% CI 1.04–1.33, p = 0.01). There was no evidence of significant heterogeneity among the cohorts (I2 = 0%, Cochran's Q, p = 0.7).
Figure 2.
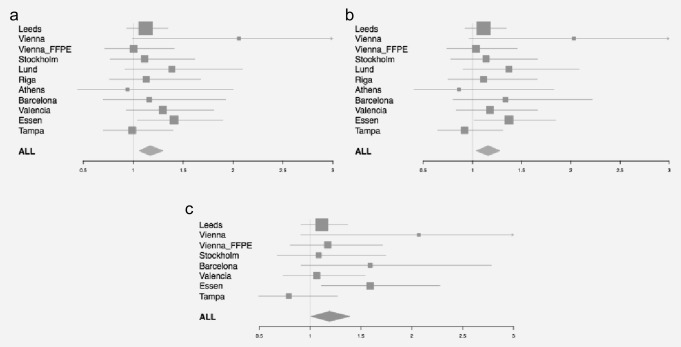
Forest plot of the association of the rs2249844 SNP (A>G) with overall survival in 11 cohorts. Adjusted for (a) age, sex, site, (b) additionally for Breslow thickness and (c) using melanoma-specific survival as the end point.
Table 2.
Association of the rs2249844 SNP (A>G) with overall survival (truncated at 8 years of follow-up) in each of the eight melanoma cohorts
| Center | Cases | Minor allele frequency1 | No. of deaths | HR (95% CI)2 | p-Value | HR (95% CI)3 | p-Value |
|---|---|---|---|---|---|---|---|
| Leeds | 1,419 | 0.32 | 258 | 1.12 (0.93–1.35) | 0.2 | 1.11 (0.92–1.34) | 0.3 |
| Vienna | 140 | 0.26 | 15 | 2.06 (0.99–4.28) | 0.05 | 2.03 (0.97–4.27) | 0.06 |
| Vienna FFPE | 268 | 0.33 | 77 | 1.00 (0.72–1.41) | 1 | 1.04 (0.74–1.46) | 0.8 |
| Stockholm | 240 | 0.40 | 65 | 1.12 (0.77–1.62) | 0.6 | 1.14 (0.78–1.66) | 0.5 |
| Lund | 166 | 0.33 | 45 | 1.39 (0.92–2.10) | 0.1 | 1.37 (0.90–2.08) | 0.1 |
| Athens | 190 | 0.31 | 17 | 0.94 (0.44–2.00) | 0.9 | 0.86 (0.41–1.83) | 0.7 |
| Riga | 175 | 0.33 | 56 | 1.13 (0.76–1.68) | 0.5 | 1.11 (0.75–1.66) | 0.6 |
| Barcelona | 269 | 0.29 | 41 | 1.16 (0.70–1.93) | 0.6 | 1.34 (0.80–2.22) | 0.3 |
| Valencia | 393 | 0.30 | 77 | 1.30 (0.93–1.81) | 0.1 | 1.18 (0.83–1.66) | 0.4 |
| Essen | 298 | 0.32 | 96 | 1.41 (1.04–1.90) | 0.03 | 1.37 (1.02–1.85) | 0.04 |
| Tampa | 407 | 0.33 | 82 | 0.99 (0.70–1.40) | 0.97 | 0.92 (0.65–1.31) | 0.7 |
| Combined− Leeds4 | 2,546 | 571 | 1.20 (1.06–1.35) | 0.003 | 1.17 (1.04–1.33) | 0.01 | |
| Combined+ Leeds4 | 3,965 | 829 | 1.17 (1.06–1.30) | 0.003 | 1.16 (1.04–1.28) | 0.005 |
Cox proportional hazard models were fitted assuming an additive effect.
Frequency in CEU population in HapMap = 0.36.
Cases adjusted for age, sex, site of primary and a single primary melanoma recruited no more than 2 years after diagnosis.
Cases additionally adjusted for Breslow thickness data > 0.75 mm.
Meta-analysis results assume a random effects model.
Observed MAF of rs2249844 ranged from 0.26 to 0.40 in the 11 cohorts. To determine whether the observed frequencies were significantly different from the frequency of the SNP recorded for the CEU population in HapMap (MAF = 0.36) we compared the counts of the SNP in each cohort with those in the CEU population. No significant difference was seen for any cohort at the 5% level though a borderline difference was observed for the Austrian cohort (MAF = 0.26, Fisher's exact test p = 0.06) (data not shown).
By using OS as the outcome we could potentially be reporting an association of the PARP1 SNP with a nonmelanoma-related outcome. Therefore, we investigated the association of rs2249844 with MSS in the eight cohorts for which we had data on cause of death (Fig. 2c, Supporting Information Table S4). Overall there was little difference in the association of the variant for MSS (HR for melanoma-specific death 1.19, 95% CI 1.01–1.39, p = 0.03) compared to OS for the same cohorts (HR for death 1.15, 95% CI 1.03–1.29, p = 0.01). This suggests that our OS data are a reasonable proxy for MSS in the cohorts for which these data are unavailable.
Determination of potential functional elements associated with rs2242844 using bioinformatics tools
Identification of noncoding regulatory elements
We found evidence of three DNAse-sensitive regions in foreskin melanocyte primary cells, one of which corresponds to the transcription initiation region (suggesting that in melanocytes the PARP1 gene is actively expressed), one located in the first intron and one located 12 kb upstream of the gene (Supporting Information Fig. S3).
The upstream peak is predicted by the ChromHMM algorithm to contain a weak enhancer region, which suggests it could potentially have a role in increasing PARP1 transcription levels, whereas the intron peak is predicted to contain an insulator region, which could potentially protect the gene by acting as a “barrier” that prevents silencing of the gene through advance of heterochromatin formation.25,26 To find clues as to what kind of transcription factors may bind to these regions we queried the JASPAR database of regulatory motifs with the sequence of each peak and found that JASPAR predicted the best scoring motif to be RREB1 (ras-responsive element binding protein 1) and RUNX1 (Runt-related transcription factor 1) for the upstream peak (Supporting Information Table S5). The best scoring motifs for the intronic peak were for NF-κB and TATA-binding proteins (Supporting Information Table S6). It should be stressed that JASPAR has poor sensitivity and these predictions require confirmation through experimental data. We did, however, see some experimental evidence of NF-κB binding in the region of the intron peak from ENCODE ChiP-seq data (Supporting Information Fig. S4).
We found experimental evidence in other cell lines for the existence of a Maf transcription factor-binding site in close proximity to the SNP (details are given in Supporting Information and Supporting Information Figs. S5–S10). There is no evidence to show that this region is active in normal foreskin melanocyte primary cells but it could potentially play an active role in melanoma cells.
Identification of functional elements using SNPinfo
We used the SNPinfo prediction tool to predict the function of SNPs in PARP1 10 kb downstream and 150 kb upstream listed for the CEU population in HapMap. We saw no evidence that the SNP itself was associated with any transcription factor-binding sites, miRNA-binding sites or that it is particularly well conserved (see Supporting Information). There was no evidence of a common coding SNP within the gene that would have a large effect on PARP1 function. There was some evidence of miRNA binding seen in the 3′ tip of the gene, which may have functional relevance. Two SNPs (rs11541664 and rs1059040) were predicted by the PolyPhen feature of SNPinfo to be potentially damaging to PARP1 functionality. Both of these occur in regions that SNPinfo predicts are exonic splicing silencers, which could disrupt splicing activity. These are, therefore, both theoretically good functional candidates; however, these variants appear to be very rare (no variant alleles were seen for rs11541664 in the HapMap dataset and no information on allele frequency is available for rs1059040). In practice it is unlikely that these two variants alone could explain the association of rs2242844 with outcome.
Investigation of eQTL associated with PARP1
We used Genevar to investigate whether rs2242844 was associated with expression of genes in the region around PARP1. We found no evidence to support a significant association of this SNP with expression of any expression probe in the region, in the full published MuTHER Twins UK Skin data (Supporting Information Figs. S11 and S12), although we could not test PARP1 directly as no probe data for it were present. No significant association of the SNP was seen in the smaller MuTHER Twins pilot data in which, curiously, PARP1 probes were present (Supporting Information Figs. S13–S15). From the Chicago eQTL browser one significant QTL was identified in the region around PARP1: a variant 4 kb downstream of PARP1 was found to be significantly associated with DNAseI sensitivity.27 DNAse sensitivity is a marker of regions of open chromatin, but there is no complimentary evidence of an association of an eQTL with the tagged variant (Supporting Information Fig. S16).
Expression data
To investigate the association of tumor gene expression levels with outcome, RNA was extracted from tumor blocks from the Leeds Predicting Response to Chemotherapy Study. Higher levels of PARP1 expression were seen where the primary tumors had been ulcerated (Supporting Information Table S7, n = 152, fold change 1.31, p = 0.007), although this effect was restricted to primary (n = 51) rather than metastatic tumors (n = 58). The association between ulceration and PARP1 expression remained significant in logistic regression analysis when adjusted for age, sex and Breslow thickness (Table3, n = 109, OR = 1.99 per log2 expression, 95% CI 1.02–3.88, p = 0.04; unadjusted values, Supporting Information Table S8). In multivariable survival analysis (Table4; unadjusted values, Supporting Information Table S9 and Fig. S17), increased PARP1 expression levels were associated with poorer outcome (HR for death = 1.61 per log2 unit of expression, 95% CI 1.20–2.15, p = 0.002).
Table 3.
The association of PARP1 gene expression taken from formalin-fixed paraffin-embedded tissue samples with ulceration
| PARP1 gene expression (per log2 expression unit) |
|||
|---|---|---|---|
| n | OR (95% CI) | p-Value | |
| Overall | 109 | 1.99 (1.02–3.88) | 0.04 |
| Primary tumors | 51 | 7.25 (1.69–31.08) | 0.008 |
| Metastatic tumors | 58 | 1.00 (0.46–2.35) | 1 |
A significant association of greater ulceration with greater PARP1 expression is seen for these data in both primary tumors and metastatic tumors. Analyses are adjusted for age, sex and Breslow thickness.
Table 4.
The association of PARP1 gene expression taken from formalin-fixed paraffin-embedded tissue samples with overall survival
| PARP1 gene expression (per log2 expression unit) |
|||
|---|---|---|---|
| n | HR (95% CI) | p-Value | |
| Overall | 149 | 1.61 (1.20–2.15) | 0.002 |
| Primary tumors | 67 | 1.95 (1.20–3.15) | 0.007 |
| Metastatic tumors | 82 | 1.73 (1.15–2.58) | 0.008 |
A significant association of poorer outcome with greater PARP1 expression is seen for these data in both primary tumors and metastatic tumors. Analyses are adjusted for age, sex and Breslow thickness.
Discussion
We have investigated the association of rs2249844 with survival across 11 melanoma cohorts. We have demonstrated a significant association with increased risk of death for individuals with the minor allele. Rs2249844 is located 5.3 kb 5′ of PARP1 and is in LD (R2 = 0.96) with the intronic PARP1 SNP rs3219090, which has been shown to be associated with reduced risk of melanoma.12 We have also presented additional evidence for a role of PARP1 in melanoma, as elevated PARP1 gene expression in tumors was associated with poorer outcome in primary tumors of individuals who ultimately developed stage IV disease and were associated with ulceration in primary tumors. The observed association was not demonstrated to be strong enough to reach genome-wide significance. However, prior knowledge of the functional roles of PARP1 and its previous association with melanoma risk increases the confidence we have that the association observed is a true positive.
The strengths of our study are the size of the Leeds cohort, combined with ten melanoma cohorts from Europe and North America. Although some of these cohorts are small, we have demonstrated that combination in a meta-analysis gives sufficient power to identify significant associations with survival.
A weakness of the study was that we were only able to use OS as the end point across all cohorts. However, MSS was available for the majority of cohorts, and in a direct comparison of cohorts that have both MSS and OS data there was no overall difference in the estimated association of the variant with both outcome measures. We applied sensitivity analysis, but showed no evidence that any one cohort unduly influenced the observed association. A second weakness of the study is that there are no published data demonstrating whether the PARP1 SNP results in lower or higher expression of PARP1. PARP1 fulfills a plethora of cellular roles, including the regulation of inflammation,28 differentiation29 and control of the secretome,11 which might have effects on the tumor or the normal tissue responses to the tumor, and we cannot postulate which of those effects is most biologically relevant.
A third weakness was that we did not exchange samples for quality control; however, samples were screened in three centers only (Leeds, Heidelberg and Tampa) and we have exchanged samples in the past within the consortium and demonstrated very high concordance for CDKN2A mutation detection30 and MC1R variant detection.4 Finally, we may have introduced a small amount of survival bias into the study by allowing recruitment for up to 2 years after diagnosis. However, SEER 13 data (generated from white melanoma cases, 1992–2009) show that overall less than 5% of cases die within the first 2 years.
Our findings have potential clinical significance. The tumor gene expression data (see below) suggest that higher levels of PARP1 are associated with poorer outcome. A recent article by Rodríguez et al. supports the notion that PARP1 plays a role in melanoma progression; PARP inhibition interfered with the endothelial to mesenchymal transition, suppressed vasculogenic mimicry and protected against lung metastasis in mice models.31
Where cells have become overly reliant or “addicted” to a DNA repair pathway, therapeutic efficacy may result from targeted disruption of the pathway. Our observations of increased expression of PARP1 in poor prognosis tumors do lend support to the view that PARP inhibitors might play a role in combination therapies for melanoma patients.
We report an association between PARP1 expression in tumors and ulceration, which was independent of Breslow thickness. Ulceration is a powerful prognostic indicator and is recognized as such in the AJCC staging system.32 Its presence rather paradoxically also appears to predict benefit from adjuvant interferon therapy, and therefore it is possible that it is a biomarker of a specific biological difference between melanomas.33 We have previously reported that ulceration is associated with evidence of macrophage-driven tumor inflammation and lymphatic invasion34 and the data reported here suggest that PARP1 may play a role in determining this phenotype.
Inheritance of the minor allele in the PARP1 SNP rs2249844 was associated with a reduced risk of melanoma in a genome-wide association study12 but increased risk of death from melanoma. Intuitively, we might expect that in general alleles associated with greater risk would be associated with poorer survival, but examples of the opposite relationship have been previously documented in the literature.4,35 To understand the biological implications of this observation, the functional correlates of the inherited variant SNP would require correlation between germline genotyping and expression data from normal tissues of the same individual, which is beyond the scope of this article. Bioinformatic analyses may offer tentative indicators of mechanisms by which a gene's cellular activity may be altered but are greatly limited by a paucity of existing data. Our bioinformatic analyses show some evidence that rs2249844 lies in melanocytes within a transcriptionally active region, which contains putative binding sites for transcription factors such as RREB1 and NF-κB that are involved in signaling pathways known to be important in melanoma.
The study has provided further support for the view that inherited variation may moderate survival expectations for cancer patients and reveal biological pathways of importance in host/tumor interaction.
Acknowledgments
Additional support for the analyses conducted at the H. Lee Moffitt Cancer Center & Research Institute, Tampa was provided by Hyun Park (technical laboratory support) from the Department of Cancer Epidemiology; Jane L. Messina (dermatopathologist) and Vernon K. Sondak (program leader) from the Department of Cutaneous Oncology. The authors thank Dr. Katerina Kypreou (biochemist) and Dr. Elizabeth Kodela (molecular biologist) for collecting and preparing the biological samples of the Athens cohort (A. Sygros Hospital) and Dr. Dimitrios Bafaloukos (medical oncologist, Oncology Department, Metropolitan Hospital, Athens) for assisting in patient recruitment.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supplementary Information
Supplementary Information
References
- 1.Joerger M, Burgers SA, Baas P, et al. Germline polymorphisms in patients with advanced nonsmall cell lung cancer receiving first-line platinum-gemcitabine chemotherapy: a prospective clinical study. Cancer. 2012;118:2466–2475. doi: 10.1002/cncr.26562. [DOI] [PubMed] [Google Scholar]
- 2.Shiraishi K, Kohno T, Tanai C, et al. Association of DNA repair gene polymorphisms with response to platinum-based doublet chemotherapy in patients with non-small-cell lung cancer. J Clin Oncol. 2010;28:4945–4952. doi: 10.1200/JCO.2010.30.5334. [DOI] [PubMed] [Google Scholar]
- 3.Yang JJ, Cheng C, Devidas M, et al. Ancestry and pharmacogenomics of relapse in acute lymphoblastic leukemia. Nat Genet. 2011;43:237–241. doi: 10.1038/ng.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davies JR, Randerson-Moor J, Kukalizch K, et al. Inherited variants in the MC1R gene and survival from cutaneous melanoma: a BioGenoMEL study. Pigment Cell Melanoma Res. 2012;25:384–394. doi: 10.1111/j.1755-148X.2012.00982.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yelamos J, Farres J, Llacuna L, et al. PARP-1 and PARP-2: new players in tumor development. Am J Cancer Res. 2011;1:328–346. [PMC free article] [PubMed] [Google Scholar]
- 6.Shaheen M, Allen C, Nickoloff JA, et al. Synthetic lethality: exploiting the addiction of cancer to DNA repair. Blood. 2011;117:6074–6082. doi: 10.1182/blood-2011-01-313734. [DOI] [PubMed] [Google Scholar]
- 7.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hassa PO, Hottiger MO. A role of poly (ADP-ribose) polymerase in NF-kappaB transcriptional activation. Biol Chem. 1999;380:953–959. doi: 10.1515/BC.1999.118. [DOI] [PubMed] [Google Scholar]
- 9.Stilmann M, Hinz M, Arslan SC, et al. A nuclear poly(ADP-ribose)-dependent signalosome confers DNA damage-induced IkappaB kinase activation. Mol Cell. 2009;36:365–378. doi: 10.1016/j.molcel.2009.09.032. [DOI] [PubMed] [Google Scholar]
- 10.Tentori L, Lacal PM, Muzi A, et al. Poly(ADP-ribose) polymerase (PARP) inhibition or PARP-1 gene deletion reduces angiogenesis. Eur J Cancer. 2007;43:2124–2133. doi: 10.1016/j.ejca.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 11.Ohanna M, Giuliano S, Bonet C, et al. Senescent cells develop a PARP-1 and nuclear factor-{kappa}B-associated secretome (PNAS) Genes Dev. 2011;25:1245–1261. doi: 10.1101/gad.625811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Macgregor S, Montgomery GW, Liu JZ, et al. Genome-wide association study identifies a new melanoma susceptibility locus at 1q21.3. Nat Genet. 2011;43:1114–1118. doi: 10.1038/ng.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Staibano S, Pepe S, Lo Muzio L, et al. Poly(adenosine diphosphate-ribose) polymerase 1 expression in malignant melanomas from photoexposed areas of the head and neck region. Hum Pathol. 2005;36:724–731. doi: 10.1016/j.humpath.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 14.Ernst J, Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nat Methods. 2012;9:215–216. doi: 10.1038/nmeth.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sandelin A, Alkema W, Engstrom P, et al. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004;32:D91–D94. doi: 10.1093/nar/gkh012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40:D930–D934. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kent WJ, Sugnet CW, Furey TS, et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosenbloom KR, Sloan CA, Malladi VS, et al. ENCODE Data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res. 2013;41:D56–D63. doi: 10.1093/nar/gks1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu ZL, Taylor JA. SNPinfo: integrating GWAS and candidate gene information into functional SNP selection for genetic association studies. Nucleic Acids Res. 2009;37:W600–W605. doi: 10.1093/nar/gkp290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yue P, Melamud E, Moult J. SNPs3D: candidate gene and SNP selection for association studies. BMC Bioinformatics. 2006;7:166. doi: 10.1186/1471-2105-7-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang TP, Beazley C, Montgomery SB, et al. Genevar: a database and Java application for the analysis and visualization of SNP-gene associations in eQTL studies. Bioinformatics. 2010;26:2474–2476. doi: 10.1093/bioinformatics/btq452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grundberg E, Small KS, Hedman AK, et al. Mapping cis- and trans-regulatory effects across multiple tissues in twins. Nat Genet. 2012;44:1084–1089. doi: 10.1038/ng.2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nica AC, Parts L, Glass D, et al. The architecture of gene regulatory variation across multiple human tissues: the MuTHER study. PLoS Genet. 2011;7:e1002003. doi: 10.1371/journal.pgen.1002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun FL, Elgin SCR. Putting boundaries on silence. Cell. 1999;99:459–462. doi: 10.1016/s0092-8674(00)81534-2. [DOI] [PubMed] [Google Scholar]
- 26.West AG, Gaszner M, Felsenfeld G. Insulators: many functions, many mechanisms. Genes Dev. 2002;16:271–288. doi: 10.1101/gad.954702. [DOI] [PubMed] [Google Scholar]
- 27.Degner JF, Pai AA, Pique-Regi R, et al. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature. 2012;482:390–394. doi: 10.1038/nature10808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ba XQ, Garg NJ. Signaling mechanism of poly (ADP-ribose) polymerase-1 (PARP-1) in inflammatory diseases. Am J Pathol. 2011;178:946–955. doi: 10.1016/j.ajpath.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hemberger M, Nozaki T, Winterhager E, et al. Parp1-deficiency induces differentiation of ES cells into trophoblast derivatives. Dev Biol. 2003;257:371–381. doi: 10.1016/s0012-1606(03)00097-6. [DOI] [PubMed] [Google Scholar]
- 30.Harland M, Goldstein AM, Kukalizch K, et al. A comparison of CDKN2A mutation detection within the Melanoma Genetics Consortium (GenoMEL) Eur J Cancer. 2008;44:1269–1274. doi: 10.1016/j.ejca.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rodríguez MI, Peralta-Leal A, O'Valle F, et al. PARP-1 regulates metastatic melanoma through modulation of vimentin-induced malignant transformation. PLoS Genet. 2013;9:e1003531. doi: 10.1371/journal.pgen.1003531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Balch CM, Gershenwald JE, Soong SJ, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27:6199–6206. doi: 10.1200/JCO.2009.23.4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eggermont AM, Spatz A, Lazar V, et al. Is ulceration in cutaneous melanoma just a prognostic and predictive factor or is ulcerated melanoma a distinct biologic entity? Curr Opin Oncol. 2012;24:137–140. doi: 10.1097/CCO.0b013e32834fcb0d. [DOI] [PubMed] [Google Scholar]
- 34.Storr SJ, Safuan S, Mitra A, et al. Objective assessment of blood and lymphatic vessel invasion and association with macrophage infiltration in cutaneous melanoma. Mod Pathol. 2012;25:493–504. doi: 10.1038/modpathol.2011.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang D, Khan S, Sun Y, et al. Association of BRCA1 and BRCA2 mutations with survival, chemotherapy sensitivity, and gene mutator phenotype in patients with ovarian cancer. JAMA. 2011;306:1557–1565. doi: 10.1001/jama.2011.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Supplementary Information