

Abstract
Human sulfide:quinone oxidoreductase (SQOR) catalyzes the conversion of H2S to thiosulfate, the first step in mammalian H2S metabolism. SQOR’s inability to produce the glutathione persulfide (GSS–) substrate for sulfur dioxygenase (SDO) suggested that a thiosulfate:glutathione sulfurtransferase (TST) was required to provide the missing link between the SQOR and SDO reactions. Although TST could be purified from yeast, attempts to isolate the mammalian enzyme were not successful. We used bioinformatic approaches to identify genes likely to encode human TST (TSTD1) and its yeast ortholog (RDL1). Recombinant TSTD1 and RDL1 catalyze a predicted thiosulfate-dependent conversion of glutathione to GSS–. Both enzymes contain a rhodanese homology domain and a single catalytically essential cysteine, which is converted to cysteine persulfide upon reaction with thiosulfate. GSS– is a potent inhibitor of TSTD1 and RDL1, as judged by initial rate accelerations and ≥25-fold lower Km values for glutathione observed in the presence of SDO. The combined action of GSS– and SDO is likely to regulate the biosynthesis of the reactive metabolite. SDO drives to completion p-toluenethiosulfonate:glutathione sulfurtransferase reactions catalyzed by TSTD1 and RDL1. The thermodynamic coupling of the irreversible SDO and reversible TST reactions provides a model for the physiologically relevant reaction with thiosulfate as the sulfane donor. The discovery of bacterial Rosetta Stone proteins that comprise fusions of SDO and TSTD1 provides phylogenetic evidence of the association of these enzymes. The presence of adjacent bacterial genes encoding SDO–TSTD1 fusion proteins and human-like SQORs suggests these prokaryotes and mammals exhibit strikingly similar pathways for H2S metabolism.
Hydrogen sulfide (H2S) is the newest member of a small family of gaseous, biological signaling molecules, termed gasotransmitters. Nitric oxide and carbon monoxide are the only other currently known gasotransmitters. H2S biosynthesis in mammals is catalyzed by cystathionine β-synthase, cystathionine γ-lyase, and 3-mercaptopyruvate sulfurtransferase.3,4 H2S signaling is implicated in numerous cellular processes5−7 and plays an especially important role in the cardiovascular system where the gasotransmitter protects against ischemia/reperfusion injury, regulates blood pressure, promotes angiogenesis, and inhibits plaque formation.8−11 H2S also acts as a neuromodulator/neuroprotectant and an oxygen sensor and can induce hibernation-like states in mice.12−16 The emerging paradigm is that H2S signaling is mediated, at least in part, by protein sulfhydration, a covalent modification in which cysteine is converted to a persulfide derivative (CysSS–).17−21
H2S is the only gasotransmitter that is enzymatically metabolized and the only inorganic compound that can be used by mammalian mitochondria to generate ATP.22 H2S metabolism is particularly important because the gasotransmitter is a Janus-faced molecule that can exhibit toxic effects at supraphysiological concentrations. The first step in the mitochondrial metabolism of H2S is catalyzed by sulfide:quinone oxidoreductase (SQOR), an inner mitochondrial membrane-bound flavoenzyme that catalyzes a two-electron oxidation of H2S to sulfane sulfur (S0) using coenzyme Q as an electron acceptor.23−26 The enzyme also requires an acceptor for the sulfane sulfur. Recently, the Jorns lab successfully expressed human SQOR as a membrane-bound protein in Escherichia coli and identified sulfite (SO32–) as the physiological acceptor of the sulfane sulfur.23 This reaction produces thiosulfate (SSO32–) (Scheme 1, step 1), a known intermediate in the oxidation of H2S to sulfate by animals or perfused liver.27−29 Thiosulfate is also a major product of H2S metabolism by colon, a tissue that must detoxify large amounts of H2S produced by sulfate-reducing bacteria.30,31
Scheme 1. Proposed Scheme for Mammalian Hydrogen Sulfide Metabolism.
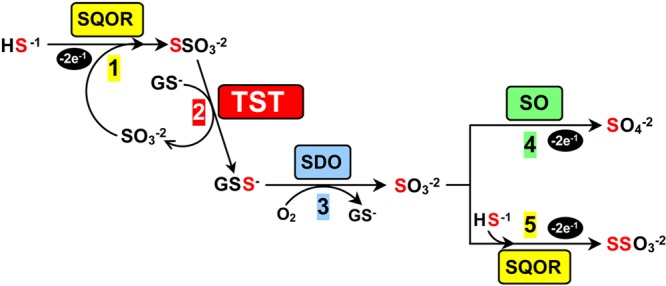
Abbreviations: SQOR, sulfide:quinone oxidoreductase; TST, thiosulfate:glutathione sulfurtransferase; SDO, sulfur dioxygenase; SO, sulfite oxidase.
The identification of thiosulfate as the product of the SQOR reaction requires a major revision of previously suggested pathways for the downstream metabolism of the gasotransmitter.3,24 We propose that thiosulfate acts as a substrate for a glutathione-dependent thiosulfate sulfurtransferase (TST) (Scheme 1, step 2). The TST reaction produces glutathione persulfide (GSS–) and also regenerates the sulfite consumed in step 1. GSS– is a known substrate for sulfur dioxygenase (SDO),1,24,32 an enzyme that catalyzes a four-electron oxidation of the sulfane sulfur in GSS– to produce sulfite (Scheme 1, step 3). The last step of H2S metabolism may proceed via one of two competing alternative reactions, a scenario that can account for observed tissue differences in the final product. In one path, the sulfite produced in step 3 undergoes a two-electron oxidation catalyzed by sulfite oxidase (SO) using cytochrome c as an electron acceptor to produce sulfate (Scheme 1, step 4). This path results in an overall eight-electron oxidation of 1 mol of H2S to sulfate, the major product of H2S metabolism in liver. Alternatively, the sulfite produced in step 3 may be further metabolized by SQOR (Scheme 1, step 5). This path achieves an overall eight-electron oxidation of 2 mol of H2S to 1 mol of thiosulfate, the major metabolic product observed in colon. SQOR and SO exhibit nearly identical catalytic efficiencies for sulfite utilization (kcat/Km sulfite = 2.11 × 106 and 2.4 × 106 M–1 s–1, respectively),23,33 suggesting that the availability of H2S is likely to play a key role in the partitioning of sulfite between the two competing reactions. Consistent with this hypothesis, an elevated level of urinary excretion of thiosulfate is observed under conditions that result in pathologically high levels of H2S (e.g., Down’s syndrome, sublethal environmental exposure to H2S gas, and ethylmalonic encephalopathy32,34,35) and in patients affected by sulfite oxidase deficiency.36 The ability to rapidly convert H2S to thiosulfate is forcefully illustrated by the large increase in blood thiosulfate levels (up to 200-fold) that is observed in fatal cases of H2S poisoning where death occurs virtually instantaneously, before thiosulfate can be detected in the urine.37
The proposed scheme is consistent with key features observed for mammalian metabolism of H2S, including the known catalytic properties of SQOR, SDO, and SO. Little information is, however, currently available regarding the postulated TST. Operationally, TST can be defined as an enzyme that uses thiosulfate to produce sulfite in a glutathione-dependent reaction. The same operational definition has been used in studies with enzymes termed thiosulfate reductases (TRs),38−42 a nomenclature that implies that the reactions also generate H2S and oxidized glutathione (GSSG) (eq 1).
| 1 |
For the sake of simplicity, an enzyme that satisfies the operational definition described above will be termed a TST with the understanding that additional studies are required to justify the rigorous use of this terminology.
Although TST activity is readily detected in liver extracts, attempts to isolate the mammalian enzyme have not been successful.41,42 On the other hand, a TST was purified from Saccharomyces cerevisiae more than 30 years ago; the corresponding gene was not, however, identified.38−40 In this paper, we have used bioinformatics approaches to successfully identify the genes that encode the yeast TST (RDL1) and the corresponding human ortholog (TSTD1). We provide definitive evidence to show that the yeast and human enzymes act as genuine TSTs that synthesize and release GSS– into solution. We present biochemical evidence of the functional interaction of human SDO with the novel human TST and its yeast ortholog. Our discovery of bacterial Rosetta Stone proteins that comprise fusions of human SDO and human TST provides phylogenetic evidence of the association of the two enzymes.
Experimental Procedures
Materials
All enzymes required for ligation-independent cloning were purchased from New England Biolabs. Isopropyl β-d-1-thiogalactopyranoside (IPTG) was purchased from Gold Biotechnology. Thiosulfate, glutathione, d,l-homocysteine, coenzyme A, and d,l-dihydrolipoic acid were purchased from Sigma-Aldrich. Potassium cyanide and dithiothreitol (DTT) were obtained from Fisher. l-Cysteine was obtained from Amresco. Potassium p-toluenethiosulfonate was purchased from TCI scientific.
Expression of Yeast RDL1 in E. coli
Polymerase chain reaction (PCR) was used to amplify the RDL1 gene from S. cerevisiae genomic DNA (EMD chemicals). The reactions were conducted using Taq polymerase (Qiagen) and primers (see Table S1 of the Supporting Information) designed to introduce unique NdeI and XhoI restriction sites, as previously described.43 The Topo TA cloning kit (Invitrogen) was used to insert the PCR product into the pCR2.1-TOPO vector for blue/white screening. The screening was conducted using One Shot TOP10 E. coli cells, supplied with the kit, as the host cell. Plasmid DNA from a white colony was digested with NdeI and XhoI to yield a desired 420 bp fragment that was subcloned between the NdeI and XhoI sites of plasmid pET21b (Novagen). The resulting plasmid, pET21b_rdl1, was used to transform E. coli BL21(DE3) cells to ampicillin resistance and sequenced across the inset (Genewiz, Inc.). A starter culture of E. coli BL21(DE3)/pET21b_rd1l cells was prepared by overnight growth at 37 °C in LB medium containing ampicillin (100 μg/mL) and used to inoculate 2.5 L flasks containing 1 L of TB medium supplemented with ampicillin (100 μg/mL). Cells were grown while being shaken at 30 °C. Expression of yeast RDL1 was induced with IPTG (0.1 mM) when the cell density reached A595 ∼ 0.6. Cells were harvested 4 h after induction (∼20 g from 3 L) and stored at −80 °C.
Purification of Recombinant Yeast RDL1
The enzyme was purified at 4 °C by a modification of a previously described protocol.43 Cells (10 g) were suspended in 15 mL of 20 mM Tris-HCl (pH 8.0) containing 150 mM NaCl and 20 mM imidazole-HCl. The cell suspension was mixed with a nuclease/protease inhibitor cocktail (20 μg/mL DNAase, 20 μg/mL RNAase, 5 mM magnesium sulfate, 12.6 μg/mL soybean trypsin inhibitor, 2 μg/mL aprotinin, 25 μg/mL phenylmethanesulfonyl fluoride, and 3 μg/mL tosyllysine chloromethyl ketone). The cells were disrupted by sonication. Cell debris was removed by centrifugation (10000g for 10 min). The supernatant was mixed with 10 mL of Ni affinity matrix (Talon affinity resin, Qiagen), previously equilibrated with buffer A [20 mM Tris-HCl (pH 8.0) containing 500 mM NaCl and 20 mM imidazole-HCl], and rocked gently for 1 h. The mixture was poured into a column, which was washed with 4 column volumes of buffer A. Yeast RDL1 was eluted using a 120 mL linear gradient from 20 to 250 mM imidazole, which was formed with buffer A and buffer B [20 mM Tris-HCl (pH 8.0) containing 500 mM NaCl and 250 mM imidazole-HCl]. The eluate was dialyzed against 50 mM Tris-HCl (pH 8.0) containing 50 mM NaCl and 5% (w/v) glycerol. The supernatant, obtained after centrifugation (30000g for 10 min), was concentrated using a 5K Macrosep Advance Centrifugal Device (Pall Life Sciences) and then stored in aliquots at −80 °C.
Expression of Human TSTD1 Isoforms 1–3 in E. coli
A synthetic version of the human gene for each isoform, which had been optimized for expression in E. coli, was obtained from Blue Heron Biotechnology, Inc. (Bothell, WA) (see Figures S1–S3 of the Supporting Information for the sequences of synthetic genes). Ligation-independent cloning was used to add a cleavable (His)6-SUMO tag to the N-terminus of each synthetic gene. The cloning was conducted by using the PCR primers listed in Table S1 of the Supporting Information, plasmid pETHSUL (gift from P. Loll), and a protocol similar to that described by Weeks et al.44 The resulting constructs for isoforms 1–3 (pETHSUL_tstd1IF1, pETHSUL_tstd1IF2, and pETHSUL_tstd1IF3, respectively) were used to transform E. coli BL21(DE3) cells to ampicillin resistance and sequenced across each insert (Genewiz, Inc.).
For expression of isoform 1, a starter culture of E. coli BL21(DE3)/pETHSUL_tstd1IF1 cells was prepared by an overnight growth at 37 °C in LB medium containing ampicillin (100 μg/mL) and used to inoculate 2 L flasks containing 1 L of the same medium. Cells were grown while being shaken at 37 °C. Expression of (His)6-SUMO-tagged isoform 1 was induced with 0.5 mM IPTG when the cell density reached A595 ∼ 0.6. Cells were harvested 2 h after induction (∼35 g of cells from 15 L) and stored at −80 °C.
A similar procedure was used for expression of isoform 2 or 3 except that E. coli cells [BL21(DE3)/pETHSUL_tstd1IF2 or BL21(DE3)/pETHSUL_tstd1IF3, respectively] were induced with 0.5 mM IPTG at A595 ∼ 0.6 and harvested 3 h after induction. The yield of cells was similar to that obtained with isoform 1 (isoform 2, 33 g from 18 L; isoform 3, 31 g from 15 L).
Purification of Recombinant Human TSTD1 Isoforms 1–3
The same procedure was used to purify each isoform, except as noted. All steps were conducted at 4 °C, following a modification of a previously described generic protocol.44 Cells (20 g) were suspended in 35 mL of 25 mM Tris-HCl (pH 8.0) containing 500 mM NaCl, 10% (w/v) glycerol, and 8 mM imidazole-HCl. The cell suspension was mixed with a nuclease/protease inhibitor cocktail, as described above. The cells were disrupted by sonication. Cell debris was removed by centrifugation (39000g for 10 min). The supernatant was mixed with 10 mL of Ni affinity matrix (Talon affinity resin, Qiagen), previously equilibrated with buffer C [25 mM Tris-HCl (pH 8.0) containing 500 mM NaCl, 10% (w/v) glycerol, 2 mM thiosulfate, and 8 mM imidazole-HCl], and rocked gently for 1 h. The mixture was poured into a column, which was washed with 4 column volumes of buffer C. The (His)6-SUMO-tagged TSTD1 isoforms were eluted with a 20 mL linear gradient from 8 to 250 mM imidazole, which was formed with buffer C and buffer D [25 mM Tris-HCl (pH 8.0) containing 500 mM NaCl, 10% (w/v) glycerol, 2 mM thiosulfate, and 250 mM imidazole-HCl]. [In the case of isoform 3, the column eluate was centrifuged (30000g for 10 min) to remove a small precipitate.] The peptide bond between the (His)6-SUMO tag and the N-terminus of each TSTD1 isoform was cleaved using the UD1 domain of the S. cerevisiae Ulp1 peptidase [0.5% (v/v)], an engineered SUMO-specific protease containing a C-terminal (His)6 tag44 (gift from S. Cocklin). After a 6 h incubation with gentle rocking at 4 °C, the reaction mixture was dialyzed against two changes of an ∼60-fold excess of buffer C. The supernatant obtained after centrifugation (30000g for 10 min) was applied to the Ni affinity matrix column described above, which had been re-equilibrated with buffer C. Tag-free TSTD1 isoforms do not bind to the matrix and are recovered in the column flow-through. The purified isoforms were dialyzed against 50 mM Tris-HCl (pH 8.0) containing 50 mM NaCl, 2 mM thiosulfate, and 5% (w/v) glycerol and then centrifuged (30000g for 10 min). The supernatants were stored in aliquots at −80 °C.
Protein Assays
The protein concentration during enzyme purification was assessed by using a Nano Drop 2000 spectrometer (Thermo Scientific). All other absorbance measurements were taken using an Agilent Technologies 8453 diode array spectrophotometer. The concentration of purified RDL1 (ε280 = 25440 M–1 cm–1) and purified TSTD1 isoforms 1–3 (ε280 = 11460, 11460, and 9970 M–1 cm–1, respectively) was determined using extinction coefficients calculated using the ProParam tool (http://web.expasy.org/protparam/).
Catalytic Assays with Thiosulfate as the Sulfane Sulfur Donor and Glutathione as the Acceptor
TST activity during enzyme purification was monitored using a sulfite end point assay, similar to that previously described.40 Briefly, reactions were initiated by the addition of the enzyme to assays containing 50 mM Tris-acetate (pH 9.0), 20 mM thiosulfate, and 20 mM glutathione at 37 °C in a final volume of 500 μL. Assays were quenched after 2 min by addition of mercuric chloride to a final concentration of 115 mM and centrifuged. The supernatant was assayed for sulfite by using a p-rosaniline colorimetric assay (ε570 = 35300 M–1 L–1).
Effect of Sulfur Dioxygenase on Catalytic Assays with Glutathione and Thiosulfate
Assays were conducted at 37 °C in 50 mM Tris-acetate (pH 9.0) or at 25 °C in 50 mM potassium/sodium phosphate (pH 8.0) in the presence of 20 mM thiosulfate and 2.4 or 20 mM glutathione. Reactions in the presence or absence of SDO were initiated by the addition of glutathione, quenched after 2 min, and assayed for sulfite formation, as described above. Recombinant human SDO was expressed in E. coli BL21(DE3)/pMW172ETHE and purified as previously described.32 Plasmid pMW172ETHE was obtained as a gift from V. Tiranti.
Steady-State Kinetic Analysis of the Thiosulfate:Glutathione Sulfurtransferase Reaction
Steady-state kinetic studies were performed by using the sulfite end point assay. Studies were conducted at 37 °C in 50 mM Tris-acetate (pH 9.0) in the absence or presence of SDO, as indicated. Steady-state kinetic parameters were estimated by fitting an equation for a sequential mechanism (eq 2, where A is thiosulfate and B is glutathione) to the data.
| 2 |
Sulfurtransferase Reactions with p-Toluenethiosulfonate as the Sulfane Sulfur Donor and Glutathione as the Acceptor
Reactions using p-toluenethiosulfonate (p-Tol-SO2S–) as the sulfane sulfur donor were conducted using 2 mm cuvettes at 25 °C in 50 mM potassium/sodium phosphate buffer (pH 8.0). Reaction progress was monitored by following the disappearance of p-Tol-SO2S– at 242 nm (Δε242 = 5080 M–1 cm–1), as described in the text. The data are not corrected for absorbance changes due to the conversion of reduced glutathione to glutathione persulfide, which are likely to be small, as judged by the similar extinction coefficients determined for reduced and oxidized glutathione (ε242 = 540 and 410 M–1 cm–1, respectively). Reactions were conducted in the absence or presence of human SDO, as indicated.
Substitution of Glutathione with Alternate Small Molecule Acceptors in Sulfurtransferase Reactions with Thiosulfate as the Sulfane Sulfur Donor
Apparent steady-state kinetic parameters for reactions in which glutathione was replaced with other thiols (cysteine, coenzyme A, and DTT) or cyanide were determined by varying the acceptor concentration at a fixed, saturating concentration of thiosulfate and by varying the thiosulfate concentration at a fixed, saturating concentration of the acceptor. Reactions were conducted at 37 °C in 50 mM Tris-acetate (pH 9.0) and were initiated by addition of yeast RDL1 or human TSTD1. Reaction rates with thiol acceptors were determined by monitoring sulfite formation in an end point assay, as described above. Rates of reaction with cyanide were determined by using a thiocyanate end point assay, similar to that previously described.45 Briefly, reactions were quenched by addition of 5% formaldehyde; thiocyanate was measured on the basis of the formation of a red complex upon addition of an acidic solution of ferric nitrate (ε460 = 4300 M–1 cm–1).
Mutation of the Single Cysteine in Yeast RDL1 or Human TSTD1 Isoform 1
PCR site-directed mutagenesis was used to replace Cys98 in yeast RDL1 or Cys79 in human TSTD1 isoform 1 with Ala or Ser. PCRs were conducted using pET21b_rd1l or pETHSUL_tstd1IF1 as a template, PCR Mix (Amresco), Pfu turbo DNA polymerase (Agilent Technologies), and the primers listed in Table S1 of the Supporting Information. After treatment with DpnI (New England Bio Laboratories) to remove template DNA, the PCR products were used to transform E. coli BL21(DE3) cells to ampicillin resistance and sequenced across the inset (Genewiz, Inc.). Transformants harboring the mutant plasmids (pET21b_rdl1_C98A, pET21b_rdl1_C98S, pETHSUL_tstd1 C79A # 6, and pETHSUL_tstd1 C79S # 1) were used to express and purify the enzyme variants, following the same procedures described for the corresponding wild-type enzymes.
Detection of Cysteine Persulfide in Yeast RDL1 or Human TSTD1
Enzyme samples were incubated for 10 min on ice with 2 mM thiosulfate and then subjected to gel filtration at 4 °C on a Sephadex G-15 column equilibrated with 50 mM potassium phosphate buffer (pH 7.5). The protein concentration in the gel eluate was estimated on the basis of the absorbance at 280 nm. The persulfide concentration in the untreated eluate and in samples incubated for 10 min at room temperature with 7 mM glutathione and 1.2 μM SDO was determined using the cold cyanolysis method, as previously described.46
Results
Use of Bioinformatics To Identify Potential Candidate Gene(s) for Yeast Thiosulfate Sulfurtransferase
Yeast TST is an ∼17000 Da protein that exhibits an isoelectic point of 5.1 and contains a single cysteine residue, as judged by properties observed for the natural enzyme isolated from S. cerevisiae more than 30 years ago.40 We sought to identify potential candidate gene(s) for yeast TST by searching the S. cerevisiae genomic database (http://www.yeastgenome.org) for entries annotated as sulfurtransferases, a term used to describe enzymes that transfer a sulfane sulfur atom from a donor substrate to a thiophilic acceptor molecule. This search retrieved two genes of unknown function (RDL1 and RDL2) and seven other genes of known function (TUM1, UBA4, YCH1, NFS1, LIP5, BIO2, and SLM3) (Table 1). An additional gene of known function (OAC1) was retrieved using thiosulfate as the search term. Searches performed using other terms (e.g., rhodanese, rhodanese-like protein, sulfur transfer, thiosulfate cyanide transsulfurase, and thiosulfate reductase) did not identify any additional candidate genes. Except for RDL1, all of the retrieved genes could be eliminated as potential candidates for yeast TST on the basis of the isoelectric point, molecular weight, and/or cysteine content of the corresponding gene product (Table 1). On the other hand, the protein encoded by the RDL1 gene (RDL1) contains a single cysteine and exhibits other properties (MW = 15.4 kDa; pI = 5.9) remarkably similar to those reported for the natural TST purified from yeast.
Table 1. Potential Candidate Genes for Yeast Thiosulfate Sulfurtransferasea.
| gene |
|||||
|---|---|---|---|---|---|
| function | name | location | molecular weight (Da) | isoelectric point (pI) | no. of cysteine residues |
| thiosulfate sulfurtransferaseb | unknown | unknown | ∼17000 | 5.1 | 1 |
| unknown | RDL1 | YOR285W | 15413 | 5.91 | 1 |
| unknown | RDL2 | YOR286W | 16697 | 9.65 | 1 |
| thiolation tRNA | TUM1 | YOR251C | 34219 | 5.71 | 4 |
| thiolation tRNA | UBA4 | YHR111W | 49361 | 6.12 | 13 |
| phosphatase | YCH1 | YGR203W | 17248 | 7.01 | 2 |
| thiolation tRNA, iron–sulfur cluster biogenesis | NFS1 | YCL017C | 54467 | 8.35 | 6 |
| lipoic acid biosynthesis | LIP5 | YOR196C | 46247 | 9.55 | 10 |
| biotin biosynthesis | BIO2 | YGR286C | 41884 | 8.76 | 10 |
| thiolation tRNA | SLM3 | YDL033C | 47049 | 8.72 | 6 |
| mitochondrial inner membrane transporterc | OAC1 | YKL120W | 35153 | 10.37 | 3 |
Unless otherwise noted, genes were retrieved by searching the S. cerevisiae genome database (http://www.yeastgenome.org) for entries that contained the term “sulfurtransferase” in the gene annotation or description.
Data reported by Uhteg and Westley40 for the enzyme isolated from S. cerevisiae.
Gene retrieved using “thiosulfate” as the search term.
Expression and Purification of Recombinant Yeast RDL1
We used PCR to amplify the RDL1 gene from yeast genomic DNA. The PCR product was subcloned into plasmid pET21b to introduce a C-terminal His tag. Recombinant RDL1 is strongly expressed in E. coli and readily isolated by metal affinity chromatography (Figure 1). A typical preparation yields 50 mg of purified RDL1 from 10 g of cells.
Figure 1.
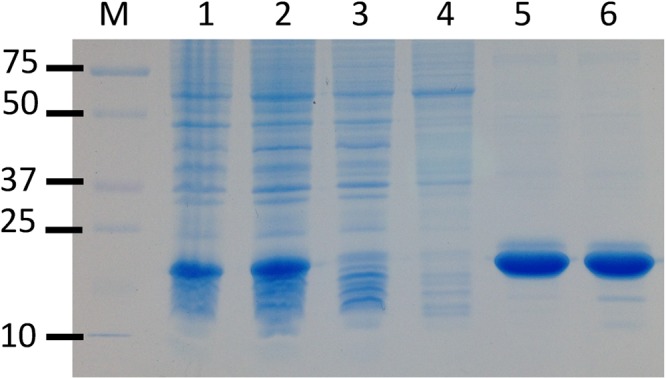
Purification of recombinant yeast RDL1. The SDS–12% polyacrylamide gel was stained for protein with ProSieve Blue Protein Staining Solution (Lonza): lane M, molecular markers; lane 1, whole cell lysate; lane 2, low-speed supernatant; lane 3, Ni affinity column flow-through; lane 4, Ni affinity column wash; lane 5, Ni affinity column eluate; lane 6, dialyzed and concentrated Ni affinity column eluate.
Does Recombinant Yeast RDL1 Exhibit Thiosulfate:Glutathione Sulfurtransferase Activity?
The standard TST assay is performed at pH 9, the optimal pH observed with the natural yeast enzyme.40 This assay is initiated by the addition of the enzyme to reaction mixtures containing 20 mM thiosulfate and 20 mM glutathione. The reaction is quenched after a specified time when sulfite formation is measured using a colorimetric assay. Using this assay, we found that RDL1 catalyzed the formation of sulfite in a reaction that exhibits a linear dependence on time and enzyme concentration (data not shown).
A complete steady-state kinetic analysis of the reaction catalyzed by RDL1 was conducted by measuring turnover rates at various concentrations of thiosulfate and glutathione. Double-reciprocal plots of reaction rate versus thiosulfate at different concentrations of glutathione or versus glutathione at different concentrations of thiosulfate are linear and intersect to the left of the y-axis, just above the x-axis (Figure 2A,B). The observed intersecting line kinetics are in agreement with results obtained in previous studies with TST isolated from yeast.40 Recombinant RDL1 exhibits a 10-fold faster limiting turnover rate (kcat), but other steady-state kinetic parameters are nearly identical to values reported for the natural enzyme isolated from yeast (Table 2). The lower turnover rate observed for the natural enzyme is probably attributable to stability problems encountered during the multiple steps required to purify TST from yeast.40 The results provide compelling evidence that the RDL1 gene encodes yeast TST.
Figure 2.
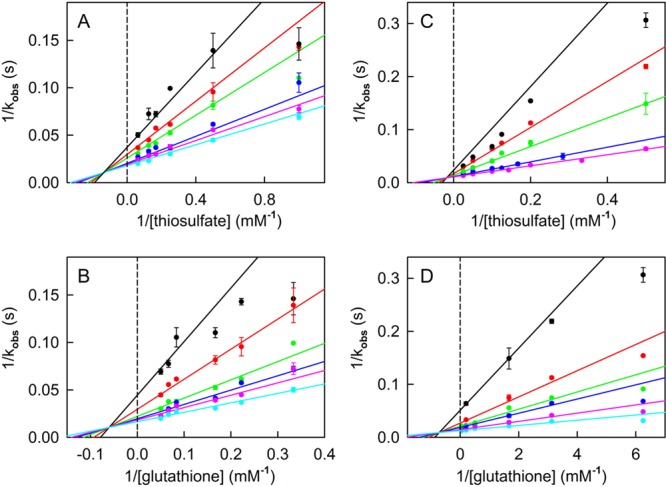
Steady-state kinetic analysis of the thiosulfate:glutathione sulfurtransferase reaction catalyzed by recombinant yeast RDL1. Reactions were conducted at 37 °C in 50 mM Tris-acetate buffer (pH 9.0) containing 0 (A and B) or 1.8 μM SDO (C and D). (A) Data obtained with 3.0, 4.5, 6.0, 12.0, 15.0, and 20.0 mM glutathione are shown by the black, red, green, blue, magenta, and cyan circles, respectively. (B) Data obtained with 1.0, 2.0, 4.0, 6.0, 8.0, and 16.0 mM thiosulfate are shown by the black, red, green, blue, magenta, and cyan circles, respectively. (C) Data obtained with 0.16, 0.32, 0.6, 2, and 15 mM glutathione are shown by the black, red, green, blue, and magenta circles, respectively. (D) Data obtained with 2, 5, 8, 10, 20, and 40 mM thiosulfate are shown by the black, red, green, blue, magenta, and cyan circles, respectively. The solid lines in panels A–D were obtained by fitting eq 2 to the data.
Table 2. Steady-State Kinetic Parameters for Thiosulfate:Glutathione Sulfurtransferase Reactions Catalyzed by Yeast RDL1 or Human TSTD1a.
| enzyme | SDO (μM) | Km for glutathione (mM) | Km for thiosulfate (mM) | Ki for thiosulfate (mM) | kcat (s–1) |
|---|---|---|---|---|---|
| natural yeast TSTb | 0 | 2.9 | 3.7 | 10 | 6.4 |
| yeast RDL1 | 0 | 4.6 ± 0.9 | 2.0 ± 0.5 | 7 ± 2 | 68 ± 6 |
| human TSTD1 | 0 | 1.0 ± 0.2 | 14 ± 2 | 37 ± 13 | 2.7 ± 0.1 |
| yeast RDL1 | 1.8 | 0.2 ± 0.05 | 7.6 ± 0.6 | 54 ± 18 | 94 ± 3 |
| human TSTD1c | 0.8 | <0.04 | 10.7 ± 0.5 | not determined | 1.83 ± 0.04 |
Reactions were conducted at 37 °C in 50 mM Tris-acetate buffer (pH 9.0) in the absence or presence of SDO, as indicated. Unless otherwise noted, steady-state kinetic parameters were obtained upon fitting eq 2 to the data. Values for kcat are determined on the basis of the rate of sulfite formation; data obtained in the presence of SDO are corrected for the fact that the coupled reaction converts 1 mol of thiosulfate into 2 mol of sulfite (eq 3 + 4).
Data for the natural TST isolated from S. cerevisiae, as reported by Uhteg and Westley.40
Apparent steady-state kinetic parameters for the TSTD1 reaction in the presence of SDO were estimated by varying the concentration of thiosulfate at a saturating concentration of glutathione (2 mM).
Use of Bioinformatics To Identify a Potential Candidate Gene for Human Thiosulfate Sulfurtransferase
A BLASTp search of the human genome database using RDL1 as the query sequence retrieved a single promising candidate gene (TSTD1, thiosulfate sulfurtransferase rhodanese-like domain containing 1) for the human ortholog of yeast TST (Table 3). The TSTD1 gene contains four exons and is annotated as having three splice variants (RefSeq mRNAs). The predicted protein isoforms (TSTD1 isoforms 1–3) differ in size (115, 74, and 109 amino acids, respectively) but share a common core of 55 amino acids that contains the single cysteine residue found in each isoform (Figure S4 of the Supporting Information). The TSTD1 isoforms are ∼34% identical (∼52% similar) to yeast RDL1.a The rank ordering of the TSTD1 isoforms by the BLAST algorithm (1 ≫ 3 > 2) is based on the calculated Expect value (E) (see Table 3) that takes into account both the number of conserved residues and the length of the RDL1 query sequence that overlaps with each human isoform.
Table 3. BLASTp Search of the NCBI Database of Human Proteins Using Yeast RDL1 as the Query Sequence.
| gene | description | isoforma | maximal score | total score | query cover (%) | E value | identity (%) |
|---|---|---|---|---|---|---|---|
| TSTD1 | thiosulfate sulfurtransferase/rhodanese domain-containing protein 1 | 1 (115) | 58.5 | 58.5 | 63 | 9 × 10–11 | 34 |
| 3 (109) | 50.1 | 50.1 | 53 | 7 × 10–8 | 36 | ||
| 2 (74) | 43.9 | 43.9 | 43 | 7 × 10–6 | 34 | ||
| TSTD3 | thiosulfate sulfurtransferase/rhodanese domain-containing protein 3 | 35.8 | 35.8 | 47 | 0.011 | 30 | |
| TSTD2 | 3-mercaptopyruvate sulfurtransferase | 33.5 | 33.5 | 53 | 0.22 | 28 | |
| TST | rhodanese | 33.1 | 33.1 | 53 | 0.30 | 26 |
The listed TSTD1 isoforms are encoded by mRNA transcripts with a CCDS (consensus coding sequence) identifier indicative of a well-understood and validated coding sequence. The number of amino acids in each isoform is given in parentheses. Isoforms predicted by automated computational analysis or conceptual translation are not shown.
Expression and Purification of Recombinant Human TSTD1 Isoforms 1–3
The mRNA for the top-ranking isoform 1 contains all four of the exons in the TSTD1 gene. We obtained a synthetic version of the corresponding cDNA that had been optimized for expression of isoform 1 in E. coli. The synthetic gene was subcloned into plasmid pET21b to introduce a C-terminal (His)6 tag. The resulting construct was then used to transform E. coli BL21(DE3) cells, following a strategy similar to that used to successfully express recombinant RDL1. However, no expression of TSTD1 isoform 1 was observed in cell lysates produced under a range of growth conditions, as judged by sodium dodecyl sulfate–polyacrylamide gel electrophoresis analysis. In an attempt to overcome this difficulty, we introduced a (His)6-SUMO tag at the N-terminus of the synthetic gene. The (His)6-SUMO-TSTD1 isoform 1 fusion protein is strongly expressed in E. coli (Figure 3, lane 2) and readily purified by metal affinity chromatography (Figure 3, lane 5). Quantitative cleavage of the fusion protein is achieved by digestion with a SUMO-specific protease (Figure 3, lanes 6 and 7). Rechromatograpy of the digest on a metal affinity column yields pure, tag-free TSTD1 isoform 1 (Figure 3, lanes 8 and 10). We obtained 90 mg of TSTD1 isoform 1 from 20 g of cells.
Figure 3.
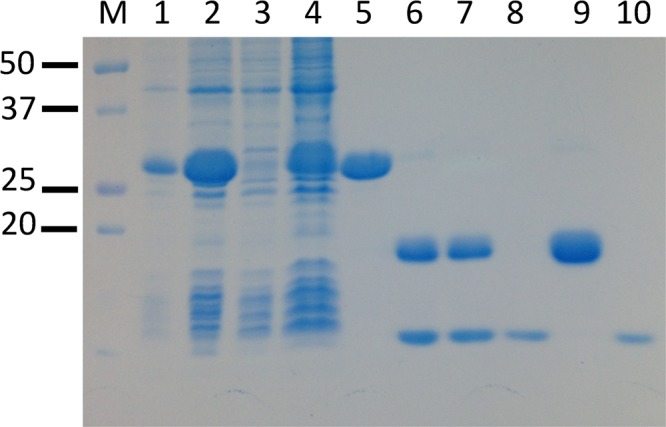
Purification of recombinant human TSTD1 isoform 1 with a cleavable N-terminal His-SUMO tag. The SDS–12% polyacrylamide gel was stained for protein with ProSieve Blue Protein Staining Solution (Lonza): lane M, molecular markers; lane 1, crude cell lysate; lane 2, low-speed supernatant; lane 3, Ni affinity column flow-through; lane 4, Ni affinity column wash; lane 5, Ni affinity column eluate; lane 6, Ni affinity column eluate after cleavage with SUMO hydrolase; lane 7, SUMO hydrolase-treated sample after dialysis; lane 8, second Ni affinity column flow-through; lane 9, second Ni affinity column eluate; lane 10, dialyzed sample from lane 8.
The mRNA for the smallest isoform 2 lacks exon 2 but contains the other three exons of the TSTD1 gene. A different exon is missing in the mRNA that encodes isoform 3 (exon 4). Additionally, intron 3 is retained in the mRNA for isoform 3 and encodes a predicted decapeptide at the C-terminus of the protein (see Figure S4 of the Supporting Information). Isoforms 2 and 3 were successfully expressed as SUMO fusion proteins and readily isolated, as described in Experimental Procedures and shown in Figures S5 and S6 of the Supporting Information. We obtained 68 mg of TSTD1 isoform 2 and 54 mg of TSTD1 isoform 3 from 20 g of cells.
Do Human TSTD1 Isoforms 1–3 Exhibit Thiosulfate:Glutathione Sulfurtransferase Activity?
A survey was conducted using the standard TST assay to determine whether activity could be detected with any of the human TSTD1 isoforms. Isoform 1 was found to catalyze the conversion of thiosulfate to sulfite in a glutathione-dependent reaction that exhibits a linear dependence on time and enzyme concentration (data not shown). In contrast, neither of the other two TSTD1 isoforms exhibits detectable TST activity. The results with isoform 2 suggest that the peptide encoded by exon 2 is essential for the activity observed with isoform 1. The absence of activity with isoform 3 may reflect the loss of the peptide encoded by exon 4 and/or the translation of retained intron 3. It is worth noting that catalytically inactive isoforms are known to exhibit a regulatory function in metabolism.47 This intriguing possibility is, however, beyond the scope of this investigation. Instead, our studies have focused on isoform 1, as described below.
A complete steady-state kinetic analysis of the thiosulfate:glutathione sulfurtransferase reaction catalyzed by TSTD1 isoform 1 was conducted at pH 9.0 (37 °C). Double-reciprocal plots of reaction rate versus thiosulfate or glutathione intersect to the left of the y-axis, just above the x-axis (Figure 4), as observed with yeast RDL1. The human enzyme exhibits catalytic parameters fairly similar to those of RDL1, except that turnover is ∼25-fold slower (Table 2). The results show that the TSTD1 gene encodes the human ortholog of yeast TST. Catalytically active isoform 1 will henceforth be termed human TSTD1.
Figure 4.
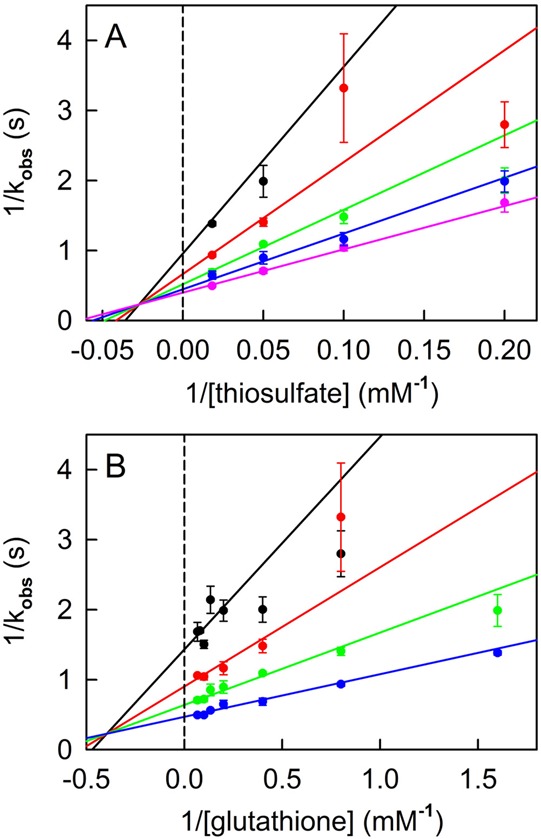
Steady-state kinetic analysis of the thiosulfate:glutathione sulfurtransferase reaction catalyzed by recombinant human TSTD1. Reactions were conducted in 50 mM Tris-acetate buffer (pH 9.0) at 37 °C. (A) Data obtained with 0.63, 1.25, 2.5, 5, and 15 mM glutathione are shown by the black, red, green, blue, and magenta circles, respectively. (B) Data obtained with 5, 10, 20, and 55 mM thiosulfate are shown by the black, red, green, and blue circles, respectively. The solid lines in panels A and B were obtained by fitting eq 2 to the data.
Use of Sulfur Dioxygenase To Distinguish between Thiosulfate:Glutathione Sulfurtransferase and Thiosulfate Reductase Activity
TSTs and TRs both catalyze the glutathione-dependent conversion of thiosulfate to sulfite. The TST reaction is, however, accompanied by the formation of a stoichiometric amount of glutathione persulfide (GSS–). Formation of GSS– as a product is a feature that uniquely distinguishes between TSTs (eq 3) and TRs (eq 1). SDO catalyzes the oxidation of the sulfane sulfur in GSS– to produce sulfite (eq 4).
| 3 |
| 4 |
Consequently, addition of SDO to assays containing thiosulfate and glutathione should cause a 2-fold increase in the rate of sulfite formation observed with an authentic TST (eq 3 + 4)
| 3 + 4 |
whereas the rate observed with a TR should be unaffected.
Indeed, a 2-fold increase in the rate of sulfite formation is observed with TSTD1 or RDL1 upon addition of excess SDO to the standard TST assay (pH 9 and 37 °C) that contains 20 mM thiosulfate and 20 mM glutathione (Figure 5A). A very similar effect is observed in assays containing the same substrate concentrations but conducted at a lower pH (pH 8.0) and a lower temperature (25 °C) (Figure 5B). However, much larger rate increases (4–9-fold) are observed upon addition of SDO to assays containing 2.4 mM glutathione. The greater than anticipated rate enhancements at the lower glutathione concentration are observed in assays with TSTD1 or RDL1 at pH 9 or 8 (Figure 5). The observed increase in the rate of sulfite formation in SDO-coupled assays at 2.4 mM glutathione corresponds to a 2–4.5-fold increase in the rate of the sulfurtransferase reaction (eq 3).
Figure 5.
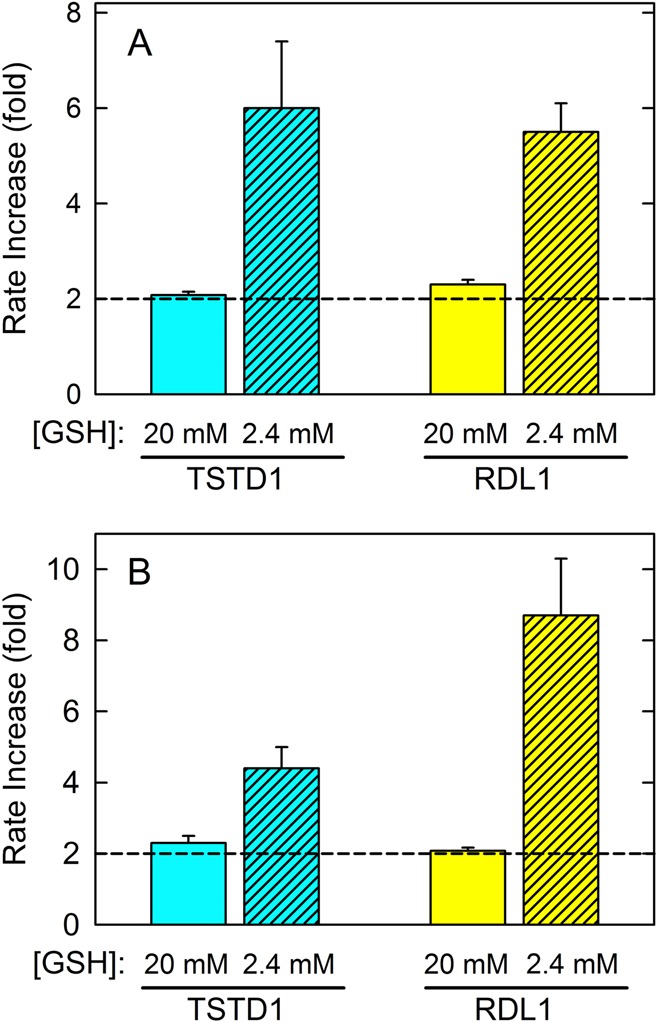
Effect of human SDO on thiosulfate:glutathione sulfurtransferase reactions catalyzed by human TSTD1 or yeast RDL1. Reactions were conducted in 50 mM Tris-acetate buffer (pH 9.0) at 37 °C (A) or 50 mM sodium/potassium phosphate (pH 8.0) at 25 °C (B). Assays containing 20 mM thiosulfate and 20 mM (solid filled bars) or 2.4 mM (diagonally striped bars) glutathione were conducted in the absence or presence of 4 μM (A) or 800 nM (B) SDO. The bar graph shows the rate increase (fold) calculated by dividing values observed in the presence of SDO by those obtained for the same reaction in the absence of SDO.
The results provide definitive evidence that TSTD1 and RDL1 are authentic TSTs that catalyze the formation of GSS– as a product that is released into solution (eq 3). The data also indicate the GSS– is a potent inhibitor of both the human and yeast enzymes. The fact that inhibition by GSS– is observed at lower but not higher glutathione concentrations suggests that the product acts as a competitive inhibitor with respect to glutathione.
Steady-State Kinetics of the Reaction of Yeast RDL1 or Human TSTD1 with Thiosulfate and Glutathione in the Presence of Sulfur Dioxygenase
Intersecting line double-reciprocal plots are obtained for the yeast RDL1 reaction in the presence of SDO (Figure 2C,D), as observed in the absence of SDO. The presence of SDO results in a 25-fold decrease in the Km value obtained for glutathione, whereas only modest changes are observed for other steady-state kinetic parameters (Table 2). A complete steady-state kinetic analysis of the TSTD1 reaction in the presence of SDO is not possible because of a large decrease in the Km value for glutathione (<0.04 mM). It is worth noting that SDO causes only minor changes in the TSTD1 turnover rate or the Km for thiosulfate, as judged by apparent steady-state kinetic parameters obtained by varying the concentration of thiosulfate at a saturating concentration of glutathione (Table 2). The decreased Km value for glutathione observed with RDL1 or TSTD1 in the presence of SDO is consistent with the hypothesis that GSS– is a potent competitive inhibitor with respect to glutathione.
Reaction of Yeast RDL1 or Human TSTD1 with a Chromogenic Substrate as an Alternate Sulfane Sulfur Donor
p-Toluenethiosulfonate (p-Tol-SO2S–) is a more reactive sulfane sulfur donor than thiosulfate.48 The compound exhibits a moderately intense absorption band in the UV region (ε242 = 6760 M–1 cm–1)49 that is lost upon transfer of the sulfane sulfur to an acceptor and formation of p-toluenesulfinate (p-Tol-SO2–) (Figure S7 of the Supporting Information). Consequently, the reaction with p-Tol-SO2S– as the sulfane donor can be monitored by measuring the disappearance of the substrate at 242 nm (Δε242 = 5080 M–1 cm–1). This continuous spectrophotometric assay is less cumbersome than the fixed time point assay used to monitor sulfite formation with thiosulfate as the donor.
To minimize the blank rate with the more reactive donor, assays with p-Tol-SO2S– and glutathione are conducted at a pH (pH 8.0) and temperature (25 °C) lower than those of the standard TST assay with thiosulfate as the donor (pH 9.0 and 37 °C, respectively). Human TSTD1 and yeast RDL1 exhibit p-Tol-SO2S–:glutathione sulfurtransferase activity; the observed initial rates of these reactions are directly proportional to the enzyme concentration (Figure 6A,B). Apparent turnover rates for the TSTD1 and RDL1 reactions were estimated from the slopes of these plots (kcat app = 96 ± 5 and 104 ± 3 s–1, respectively). It is worth noting that replacing thiosulfate with p-Tol-SO2S– appears to eliminate the difference in the turnover rate of the human and yeast enzymes that is observed with the less reactive donor (see Table 2).
Figure 6.
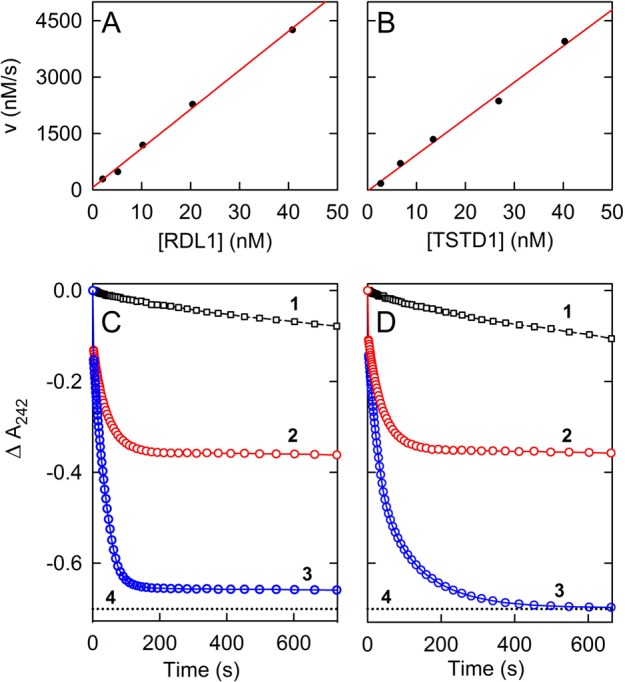
Reaction of recombinant yeast RDL1 or human TSTD1 with p-toluenethiosulfonate (p-Tol-SO2S–) and glutathione as the sulfane sulfur donor and acceptor, respectively. Reactions were conducted at 25 °C using 2 mm cuvettes in 50 mM potassium/sodium phosphate buffer (pH 8.0) containing 0.75 mM p-Tol-SO2S– and 2.4 mM glutathione, with enzyme(s), as indicated. (A and B) The plots show the effect of the concentration of yeast RDL1 or human TSTD1 on the velocity observed during the initial 20 s of reactions conducted in the absence of SDO. The data are corrected for the blank rate observed in the absence of enzyme. (C and D) The plots show the effect of SDO (368 nM) on the extent of the reaction observed with 40.8 nM RDL1 and 80.3 nM TSTD1, respectively. In each panel, curve 1 is the blank reaction, curves 2 and 3 were obtained for the reactions with RDL1 and TSTD1 in the absence and presence of SDO, respectively, and the dotted line (curve 4) shows the absorbance change calculated for 100% conversion of p-Tol-SO2S– to p-Tol-SO2–. The first data point in curves 2 and 3 in panels C and D corresponds to the absorbance of p-Tol-SO2S–, observed immediately prior to the addition of enzyme. The apparent gap between the first and subsequent data points is due to the enzyme reaction that occurs during mixing (<5 s), before readings can be taken.
A 2.3- or 2.0-fold increase in the initial rate of disappearance of p-Tol-SO2S– is observed when assays with RDL1 or TSTD1, respectively, are conducted in the presence of SDO (Figure 6C,D). The observed rate enhancements are similar to those seen in assays at the same glutathione concentration (2.4 mM) with thiosulfate as the sulfane sulfur donor. In addition to the effect on initial rates, SDO causes a dramatic change in the extent of the reactions observed with p-Tol-SO2S–. In the absence of SDO, the reactions with RDL1 and TSTD1 appear to be complete in ∼200 s, as judged by the observed plateau in the progress curves (Figure 6C,D, curve 2). However, the maximal ΔA242 observed with either enzyme is only 50% of that expected for the complete consumption of p-Tol-SO2S–, as indicated by the dotted lines (curve 4) in panels C and D of Figure 6. The observed progress curves suggested that the reactions may have reached equilibrium after 200 s (eq 5).
| 5 |
Consistent with this hypothesis, a quantitative conversion of p-Tol-SO2S– to p-Tol-SO2– is observed when GSS– is oxidatively decomposed, as judged by the reaction traces obtained for the RDL1 and TSTD1 reactions in the presence of SDO (Figure 6C,D, curve 3).
Reaction of Yeast RDL1 or Human TSTD1 with Alternate Sulfane Sulfur Acceptors
We investigated the specificity of yeast RDL1 and human TSTD1 with respect to the sulfane sulfur acceptor by determining whether alternate acceptors could substitute for glutathione. We surveyed four physiologically relevant thiols and two nonphysiological compounds in reactions conducted at pH 9.0 with thiosulfate as the sulfane sulfur donor. Activity was observed with RDL1 or TSTD1 using l-cysteine, coenzyme A, DTT, or cyanide as the acceptor. No activity was, however, detected with d,l-homocysteine or d,l-dihydrolipoic acid.
Apparent steady-state kinetic parameters for reactions observed with different acceptors were determined by varying the acceptor concentration in the presence of a saturating concentration of thiosulfate and vice versa. The observed velocities exhibit a hyperbolic dependence on the concentration of the varied substrate except for the reaction with RDL1 when thiosulfate is varied at a saturating concentration of coenzyme A. In this case, a sigmoidal dependence on the thiosulfate concentration is observed. Varying the nature of the acceptor causes only modest changes (≤3.5-fold) in the apparent turnover rate or the Km for thiosulfate, as judged by results obtained for the RDL1 or TSTD1 reactions with glutathione and four alternate acceptors (Table 4). Similar Km values are observed for l-cysteine and glutathione with RDL1 or TSTD1. The Km value obtained for coenzyme A with RDL1 or TSTD1 is, however, 40- or 20-fold lower, respectively, than the Km value observed for glutathione. The potential significance of the observed Km values will be discussed.
Table 4. Apparent Steady-State Kinetic Parameters Observed for Yeast RDL1 or Human TSTD1 Reactions with Various Sulfane Sulfur Acceptors and Thiosulfate as the Donora.
| yeast
RDL1 |
human
TSTD1 |
|||||
|---|---|---|---|---|---|---|
|
Km (mM) |
Km (mM) |
|||||
| acceptor | thiosulfate | acceptor | kcat (s–1) | thiosulfate | acceptor | kcat (s–1) |
| glutathione | 3.1 ± 0.3 | 5.6 ± 0.8 | 60 ± 2 (60 ± 3) | 11 ± 1 | 1.7 ± 0.3 | 1.91 ± 0.06 (1.9 ± 0.1) |
| cysteine | 5.5 ± 0.7 | 2.9 ± 0.3 | 49 ± 4 (45 ± 1) | 10.4 ± 0.7 | 2.5 ± 0.2 | 1.67 ± 0.04 (2.25 ± 0.05) |
| coenzyme A | 4.3 ± 0.8 | 0.15 ± 0.05 | 29 ± 3 (21 ± 2) | 36 ± 3 | 0.09 ± 0.01 | 1.53 ± 0.06 (1.32 ± 0.05) |
| DTT | 3.9 ± 0.6 | 0.16 ± 0.03 | 27 ± 1 (40 ± 2) | 14 ± 2 | 0.12 ± 0.02 | 2.8 ± 0.1 (2.82 ± 0.07) |
| cyanide | 4.0 ± 0.4 | 2.6 ± 0.3 | 38 ± 1 (36 ± 1) | 12 ± 1 | 0.22 ± 0.02 | 2.41 ± 0.08 (2.05 ± 0.06) |
Apparent steady-state kinetic parameters were determined at 37 °C in 50 mM Tris-acetate buffer (pH 9.0) by varying the thiosulfate concentration at a saturating concentration of the acceptor and by varying the acceptor concentration at a saturating concentration of thiosulfate. The values obtained for kcat by varying the acceptor concentration are given in parentheses.
Role of the Single Cysteine Residue in Yeast RDL1 or Human TSTD1
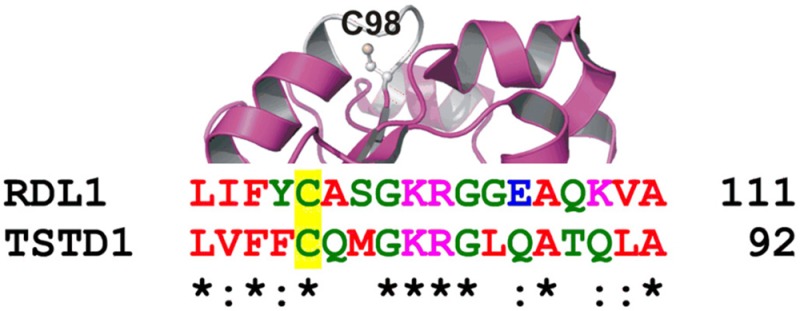
Yeast RDL1 and human TSTD1 exhibit a signature motif characteristic of a rhodanese homology domain (RHOD). This αβ fold domain is found in other sulfurtransferases and is also observed in the crystal structure of RDL1, which was determined as part of a structural genomics initiative and published as Protein Data Bank (PDB) entry 3D1P. An invariant cysteine occupies the first position of a six-amino acid active site loop observed in other RHOD-containing sulfurtransferases.50 The single cysteine in yeast RDL1 (Cys98) occupies the same location within an active site loop found in the crystal structure of the protein (Figure 7, top panel). A sequence alignment indicates that the putative catalytic cysteine and three other residues in the active site loop of yeast RDL1 are conserved in human TSTD1 (Figure 7, bottom panel).
Figure 7.
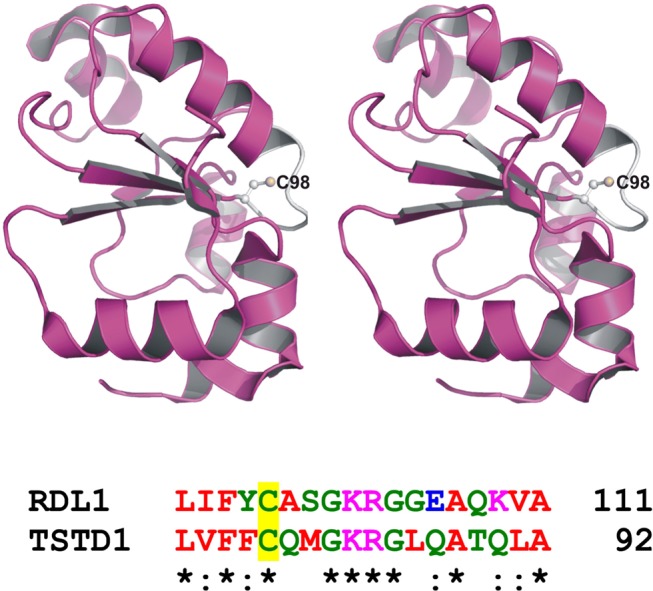
Active site loop and putative catalytic cysteine in RDL1 and TSTD1. The top panel is a stereo ribbon drawing of yeast RDL1 (PDB entry 3D1P), which is shown as a magenta ribbon, except for the white active site loop. Cys98 is shown in ball and stick form. The bottom panel shows a region of a sequence alignment of RDL1 and TSTD1 around the putative catalytic cysteine (Cys98 and Cys79, respectively), which is located at the first position of the six-amino acid active site loop.
To evaluate the possible catalytic role of Cys98 in yeast RDL1 and Cys79 in human TSTD1, we mutated each residue to Ala or Ser. The mutations did not affect protein expression. However, each of the purified mutant enzymes was found to be catalytically inactive. The results provide compelling evidence that the single cysteine in the yeast and human enzymes is catalytically essential. In contrast, Chancey and Westley concluded that the cysteine was not required for catalysis by the yeast enzyme based on the failure of iodoacetate to inactivate the enzyme.39 Unfortunately, the postulated site of iodoacetate incorporation was not identified in this study.
The mutagenesis results suggested that catalysis by yeast RDL1 and human TSTD1 might occur via a double-displacement mechanism involving transfer of the sulfane sulfur from thiosulfate to the active site cysteine (eq 6) and subsequent reaction of the persulfide-containing intermediate with glutathione (eq 7).
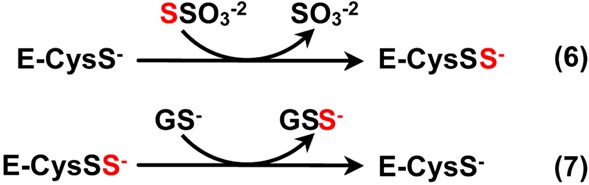 |
6 |
Evidence for evaluating this hypothesis was sought by determining whether cyanolyzable sulfur could be detected in the enzyme isolated by gel filtration after a short incubation with thiosulfate. We found that ∼10 or ∼20% of the cysteine in the column eluate of RDL1 or TSTD1, respectively, is present as cysteine persulfide. The persulfide is largely eliminated upon incubation of the isolated intermediates with glutathione in the presence of SDO, which was included to oxidize the cyanolyzable sulfur in the GSS– product (Table 5).
Table 5. Detection of Cysteine Persulfide in RDL1 or TSTD1 Isolated by Gel Filtration after Reaction with Thiosulfatea.
| persulfide
content (%) |
||
|---|---|---|
| enzyme | as isolated | GSH-treated |
| RDL1 | 8.9 ± 0.8 | 1.3 ± 0.8 |
| TSTD1 | 19 ± 2 | 3.4 ± 0.8 |
Samples were isolated and analyzed as described in Experimental Procedures.
Identification of Bacterial SDO–TSTD1 Fusion Proteins
Rosetta Stone proteins are fusion proteins consisting of two nonhomologous proteins that are found as separate proteins in another genome. The fusion is thought to be maintained by selection because it facilitates a functional interaction between proteins, such as the kinetic coupling of consecutive enzymes in a pathway.51 Six bacterial proteins were previously classified as fusions of SDO with rhodanese, a thiosulfate:cyanide sulfurtransferase encoded by the TST gene.32,52 The functional interaction between human TSTD1 and human SDO observed in this study led us to question the validity of this assignment. Accordingly, we performed BLASTp searches of the human genome database using each of the putative SDO–rhodanese fusion proteins as the query sequence. Contrary to the previous classification, TSTD1 was the top-scoring BLAST hit for the C-terminal domain in all six proteins (Table 6, entries 2, 4, 10, 11, 14, and 15). Furthermore, rhodanese was not found among the lower-scoring BLAST hits for the C-terminal domain. On the other hand, SDO was the highest-scoring BLAST hit for the N-terminal domain in each query sequence. The results indicate that all six bacterial proteins are more appropriately classified as fusions of SDO and TSTD1.
Table 6. BLASTp Search of the NCBI Database of Human Proteins Using Bacterial Fusion Proteins as the Query Sequencea.
| C-terminal TSTD1-like domain |
N-terminal ETHE1 (SDO)-like domain |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| bacterial query sequence (GenBank accession number) | human gene | query cover (%) | E value | identity (%) | human gene | query cover (%) | E value | identity (%) | |
| 1 | Nitrosococcus oceani (WP_011330831.1) | TSTD1 | 26 | 3 × 10–11 | 33 | ETHE1 | 63 | 9 × 10–75 | 52 |
| 2 | Nitrosococcus watsonii (WP_013220760.1)b | TSTD1 | 26 | 2 × 10–8 | 34 | ETHE1 | 63 | 3 × 10–73 | 51 |
| 3 | Methylocella silvestris (WP_012591638.1) | TSTD1 | 26 | 3 × 10–8 | 28 | ETHE1 | 65 | 2 × 10–70 | 49 |
| 4 | Methylobacter tundripaludum (WP_006892756.1)b | TSTD1 | 26 | 2 × 10–7 | 31 | ETHE1 | 62 | 2 × 10–68 | 50 |
| 5 | Methylomicrobium album (WP_005371248.1) | TSTD1 | 26 | 9 × 10–7 | 33 | ETHE1 | 63 | 9 × 10–69 | 49 |
| 6 | Methyloglobulus morosus (WP_023495230.1) | TSTD1 | 25 | 2 × 10–6 | 31 | ETHE1 | 60 | 2 × 10–65 | 48 |
| 7 | Methylocystis rosea (WP_018407856.1) | TSTD1 | 25 | 5 × 10–6 | 31 | ETHE1 | 66 | 3 × 10–63 | 46 |
| 8 | Methylosarcina fibrata (WP_020565272.1) | TSTD1 | 24 | 8 × 10–6 | 30 | ETHE1 | 63 | 1 × 10–67 | 49 |
| 9 | Bradyrhizobiaceae bacterium (WP_009737228.1) | TSTD1 | 25 | 1 × 10–5 | 29 | ETHE1 | 65 | 2 × 10–64 | 47 |
| 10 | Afipia broomeae (WP_006021739.1)b | TSTD1 | 24 | 4e–05 | 33 | ETHE1 | 65 | 1 × 10–63 | 47 |
| 11 | γ-proteobacterium HTCC2148 (WP_007229491.1)b | TSTD1 | 23 | 1 × 10–4 | 36 | ETHE1 | 64 | 5 × 10–80 | 54 |
| 12 | Rhizobium giardinii (WP_018330152.1) | TSTD1 | 26 | 2 × 10–4 | 32 | ETHE1 | 66 | 7 × 10–64 | 46 |
| 13 | Mesorhizobium amorphae (WP_006203523.1) | TSTD1 | 26 | 6 × 10–4 | 28 | ETHE1 | 66 | 9 × 10–67 | 48 |
| 14 | Polaromonas sp. JS666 (WP_011485703.1)b | TSTD1 | 29 | 0.005 | 30 | ETHE1 | 60 | 5 × 10–33 | 37 |
| 15 | Burkholderia cepacia (WP_014900869.1)b | TSTD1 | 21 | 0.31 | 29 | ETHE1 | 64 | 3 × 10–87 | 59 |
We reasoned that the six SDO–TSTD1 fusion proteins might be part of a larger group of homologous Rosetta Stone proteins. To evaluate this hypothesis, the fusion protein from M. tundripaludum (Table 6, entry 4) was used as the query sequence in a BLASTp search against all nonredundant GenBank CDS translations, with the maximal number of target sequences set to 500. The search retrieved 116 bacterial proteins that exhibit 96–99% coverage with the query sequence and low E values (3 × 10–65 and 9 × 10–165), properties expected for homologues of the M. tundripaludum SDO–TSTD1 fusion protein.b We performed BLASTp searches against the human genome using query sequences from a selected subset of the 116 hits that included no more than one representative species from within each genus. Satisfyingly, SDO and TSTD1 were the highest-scoring BLAST hits obtained for the N- and C-terminal domains, respectively, in each tested query. The newly identified fusion proteins were rank-ordered with respect to the E values obtained for the TSTD1 hits; those exhibiting E values of ≤6 × 10–4 are listed in Table 6.
Are Human-like SQORs Found in Bacteria That Express SDO–TSTD1 Fusion Proteins?
H2S and thiosulfate are the most abundant reduced inorganic sulfur species in the environment.2 Bacteria that express SDO–TSTD1 fusion proteins might utilize exogenously derived thiosulfate as the substrate for the TSTD1 reaction. Alternatively, these bacteria might contain human-like SQORs that initiate the oxidation of H2S and produce an endogenous source of thiosulfate for the TSTD1 reaction, analogous to that proposed for mammalian H2S metabolism (see Scheme 1).
The proteomes of 12 of the 13 bacteria that harbor top-scoring SDO–TSTD1 fusion proteins are available in the NCBI database (Table 6, entries 1–10 and 12–13). We conducted BLASTp searches of these proteomes using human SQOR as the query sequence. The searches identified SQOR homologues in 11 of the 12 searchable proteomes (Table S2 of the Supporting Information). Complete genomic data are available for two of the 11 bacteria that contain both SQOR and SDO–TSTD1 fusion proteins (N. oceani and N. watsonii); whole-genome shotgun contigs are available for the other nine bacteria. The genes for SQOR and the SDO–TSTD1 fusion protein in N. oceani and N. watsonii are transcribed in the same direction and separated by just 73 nucleotides. Although not predicted to lie in the same operon, the genes for the two proteins in N. oceani are considered to be functionally related as part of a regulon cluster, according to MicrobesOnline (http://www.microbesonline.org/operons/). Genomic distances could also be determined in four other bacteria where the genes for SQOR and the SDO–TSTD1 fusion protein were found within the same contig. In these cases, the genes were separated by 21–246 nucleotides (Table S2 of the Supporting Information).
Seven of the 11 identified bacterial SQORs, including the N. oceani and N. watsonii enzymes, are highly similar to human SQOR (E ≤ 5 × 10–90) (see Table S2 of the Supporting Information). SQORs have been classified into six types.25 A phylogenetic tree shows that the seven top-scoring bacterial enzymes cluster with type II SQORs, a category that includes all known eukaryotic SQORs and a subset of prokaryotic enzymes (Figure 8). The lower-scoring hits include one that clusters with type I bacterial SQORs and three that did not cluster with representatives of other known types of bacterial SQORs (not shown).
Figure 8.

Phylogenetic tree of SQOR homologues. A multiple-sequence alignment of 31 homologues was performed using COBALT (http://www.ncbi.nlm.nih.gov/tools/cobalt/). The alignment included 11 SQORs produced in bacteria that express SDO–TSTD1 fusion proteins (see Table S2 of the Supporting Information) and 20 previously identified type I and type II SQORs.25 The phylogenetic tree was rendered using the Newick file generated by COBALT and the online PHY·FI application63 (http://cgi-www.cs.au.dk/cgi-chili/phyfi/go). The red boxes mark the 11 SQOR homologues found in bacteria that express SDO–TSTD1 fusion proteins. Starting from the upper left-hand corner of the tree, the bacterial sequences include enzymes from Bradyrhizobiaceae bacterium, Afipia broomeae, Mesorhizobium amorphae, Methylocystis rosea, Rhodobacter capsulatus, Acidithiobacillus ferrooxidans, Aquifex aeolicus, Pseudomonas putida, Rhizobium giardinii, Ralstonia solanacearum, Nitrosococcus oceani, Nitrosococcus watsonii, Methylobacter tundripaludum, Methyloglobulus morosus, Methylomicrobium album, Methylosarcina fibrata, Bacillus anthracis, Bacillus cereus, and Staphylococcus aureus. The eukaryotic sequences include SQORs from Schizosaccharomyces pombe, Arenicola marina (lugworm), and various higher eukaryotic animals, as indicated.
Discussion
The first step in mammalian H2S metabolism was previously postulated to produce the GSS– substrate for SDO, an enzyme thought to catalyze the second step in the pathway.3,24 This hypothesis was contradicted by the discovery that glutathione does not act as an acceptor of the sulfane sulfur generated during H2S oxidation by human SQOR. Instead, SQOR catalyzes the oxidative conversion of H2S to thiosulfate, a reaction in which sulfite acts as the sulfane sulfur acceptor from an enzyme persulfide intermediate (E–CysSS– + SO32– ⇒ E–CysS– + SSO32–).23 The inability of SQOR to produce GSS– led others to suggest that SDO might directly oxidize the sulfane sulfur in the persulfide intermediate (E–CysSS– + O2 + H2O ⇒ E–CysS– + SO32– + 2H+).1 However, human SDO cannot replace sulfite in the SQOR reaction, as judged by its inability to accelerate the slow rate of H2S oxidation observed in the absence of sulfite.c,d The results strongly suggested that a thiosulfate:glutathione sulfurtransferase was required to provide the missing link between the SQOR and SDO reactions.
Although TST could be purified from S. cerevisiae,38−40 attempts to isolate the mammalian enzyme were not successful.41,42 We used bioinformatic approaches to identify the genes that encode human TST (TSTD1) and its yeast ortholog (RDL1). Both genes produce small RHOD-containing proteins. Recombinant yeast RDL1 catalyzes the glutathione-dependent conversion of thiosulfate to sulfite in a reaction that proceeds via a ternary complex mechanism and exhibits steady-state kinetic parameters similar to those reported for the natural yeast enzyme.40 The human TSTD1 gene contains four exons and has three splice variants. TSTD1 isoform 1 is produced from a transcript that contains all four exons and exhibits TST activity; the smaller TSTD1 isoforms are catalytically inactive. TSTD1 (isoform 1) exhibits steady-state kinetic parameters similar to those of RDL1, except that turnover of the human enzyme is ∼25-fold slower. Proteomics studies indicate that human TSTD1 is highly expressed in various tissues, including liver and heart.53,54 Data for the individual isoforms are unavailable except in tumor cell lines where only isoform 1 is detected by Western blotting.55
Yeast RDL1 and human TSTD1 contain a single cysteine (Cys98 and Cys79, respectively) that is catalytically essential, as judged by the loss of activity observed when the cysteines are replaced with serine or alanine. The detection of albeit modest amounts of cysteine persulfide in the human or yeast enzyme isolated after reaction with thiosulfate suggests that catalysis occurs via a double-displacement mechanism involving the formation of a cysteine persulfide-containing intermediate (see eqs 6 and 7), as observed with other RHOD-containing sulfurtransferases.4,50,56,57 Formation of the persulfide intermediate may be rate-limiting during catalysis by human TSTD1, as judged by the substantial increase in turnover rate observed when thiosulfate is replaced with a more reactive sulfane donor, p-Tol-SO2S–. Coenzyme A and l-cysteine can act as alternate sulfane sulfur acceptors for the yeast and mammalian enzymes. Human TSTD1 exhibits Km values for glutathione and coenzyme A that are below or within the normal range reported for the concentrations of these thiols in animal cells.58,59 The Km value obtained for l-cysteine is, however, 1 order of magnitude higher than the l-cysteine level in liver cells.60 The results suggest that coenzyme A, but not l-cysteine, may be a possibly significant alternate acceptor for TSTD1 in mammalian cells. It is worth noting that coenzyme A persulfide is the only currently known alternate (albeit poor) substrate for human SDO.1
Functional Interaction between Human SDO and Human TSTD1 or Yeast RDL1
The operational definition of a TST as an enzyme that catalyzes a glutathione-dependent conversion of thiosulfate to sulfite does not distinguish between an authentic TST that produces GSS– as a product and a TR that catalyzes a glutathione-dependent reduction of thiosulfate to sulfite and H2S. Definitive evidence that TSTD1 and RDL1 are authentic TSTs is provided by studies that show that the reaction product is oxidized by human SDO, as evidenced by an expected doubling of the rate of sulfite formation upon addition of SDO to assays at a saturating glutathione concentration.e The greater than expected rate accelerations observed when SDO is added to assays at lower glutathione concentrations indicate that GSS– is an inhibitor of the yeast and human enzymes. The presence of SDO results in a ≥25-fold decrease in the Km value obtained for glutathione with RDL1 or TSTD1, suggesting that GSS– is a potent competitive inhibitor with respect to glutathione. SDO also enhances the initial rate of glutathione-dependent sulfurtransferase reactions observed with p-Tol-SO2S– as the sulfane sulfur donor. The observed inhibition of the TST reactions by GSS–, which is circumvented in the presence of human SDO, provides an apparent mechanism for regulating mammalian GSS– biosynthesis and preventing the accumulation of a highly reactive metabolite.
The reactions observed with p-Tol-SO2S– and TSTD1 or RDL1 in the absence of SDO reach equilibrium when ∼50% of the substrate has been consumed. A quantitative conversion of p-Tol-SO2S– to p-Tol-SO2– is, however, observed when GSS– is oxidatively decomposed in the presence of SDO. The ability of human SDO to drive the reaction with a reactive sulfane sulfur donor to completion provides a paradigm for the thermodynamic coupling of the irreversible SDO reaction with the less favorable, but physiologically relevant, sulfurtransferase reaction with thiosulfate as the sulfane donor.
The effect of SDO on the extent of the reaction with p-Tol-SO2S– is similar to that reported in a previous study with the natural yeast TST for the effect of cyanide on the extent of a reaction observed with glutathione and benzenethiosulfonate (Ph-SO2S–).38 However, the authors attributed the observed formation of a stoichiometric amount of thiocyanate to the reaction of cyanide with GSS– that had been released into solution. This interpretation is rendered problematic by the fact that cyanide is an alternate substrate for yeast TST, as shown by results obtained in this study and in a previous survey of the thiosulfate:cyanide sulfurtransferase activity exhibited by RHOD-containing yeast proteins.43
Phylogenetic Association of Human TSTD1 and Human SDO
Using bioinformatics approaches, we identified a group of bacterial SDO–TSTD1 fusion proteins that includes six members previously misclassified as SDO–rhodanese fusions. The C-terminal TSTD1-like domain in the fusion proteins is connected to the N-terminal SDO-like domain by a variable-length linker region (Figure S8 of the Supporting Information). Catalytically important residues and signature motifs are conserved between the respective bacterial domains and human TSTD1 or SDO, as judged by multiple-sequence alignments of the bacterial domains with the human proteins (Figure S9 of the Supporting Information). The phylogenetic data reinforce the biochemical evidence obtained for the functional interaction of human TSTD1 and SDO.
It is worth noting that at least some of the bacteria that express SDO–TSTD1 fusion proteins also contain an adjacent gene that encodes a human-like SQOR. The results suggest that these organisms metabolize H2S via a pathway that is strikingly similar to the first three steps proposed for mammalian H2S metabolism (see Scheme 1).f The metabolic similarity is not surprising from an evolutionary perspective given that the genes for H2S metabolism were acquired by eukaryotic cells from an endosymbiotic bacterial ancestor. Nevertheless, bacteria and mammals exhibit considerable divergence with respect to the role of H2S metabolism. Bacterial metabolism of environmental H2S provides an important source of energy and reducing equivalents. In contrast, the primary physiological significance of H2S metabolism in mammals is closely tied to the acquired role of endogenously produced H2S as an important signaling molecule.
Concluding Remarks
In summary, we describe a novel human sulfurtransferase that catalyzes the formation of a central intermediate in mammalian H2S metabolism. We propose that human TSTD1 constitutes the hitherto missing link between the reactions catalyzed by SQOR and SDO. The biosynthesis of glutathione persulfide by human TSTD1 provides a rare example of a mammalian enzyme that catalyzes the biosynthesis of a reactive sulfane sulfur donor that is released into solution.g In this regard, it is worth noting that glutathione persulfide and polysulfides have been found to mediate protein sulfhydration.21,61 It has recently been suggested that persulfides and other reactive sulfane sulfur donors may be the actual signaling molecules that implement many of the biological effects previously attributed to H2S.21,61,62 Additional studies are clearly required to evaluate the intriguing possibility that H2S metabolism may have an unanticipated direct role in H2S signaling.
Acknowledgments
For their generous gifts of plasmids and other reagents (in parentheses), we thank Drs. Patrick Loll (plasmid pETHSUL), Simon Cocklin (UD1 domain of the S. cerevisiae Ulp1 peptidase), and Valeria Tiranti (plasmid pMW172ETHE). We also thank Nadish Goyal for initial studies on yeast RDL1.
Glossary
Abbreviations
- SQOR
sulfide:quinone oxidoreductase
- TST
thiosulfate sulfurtransferase
- GSS–
glutathione persulfide
- SDO
sulfur dioxygenase
- SO
sulfite oxidase
- TR
thiosulfate reductase
- IPTG
isopropyl β-d-1-thiogalactopyranoside
- DTT
dithiothreitol
- p-Tol-SO2S–
p-toluenethiosulfonate
- RHOD
rhodanese homology domain.
Supporting Information Available
Primers used for PCRs (Table S1), BLASTp search of bacterial proteomes that contain SDO–TSTD1 fusion proteins using human SQOR as the query sequence (Table S2), sequences of the synthetic genes used to express recombinant TSTD1 isoforms 1–3 (Figures S1–S3, respectively), alignment of the sequences of human TSTD1 isoforms 1–3 (Figure S4), purification of recombinant TSTD1 isoform 2 (Figure S5) and isoform 3 (Figure S6), spectral properties of p-toluenethiosulfonate and p-toluenesulfonite (Figure S7), multiple-sequence alignment of selected SDO–TSTD1 bacterial fusion proteins (Figure S8), and multiple-sequence alignment of human SDO or human TSTD1 with the N-terminal SDO-like or C-terminal TSTD1-like domain, respectively, in selected SDO–TSTD1 bacterial fusion proteins (Figure S9). This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
S.L.M. and M.R.J. contributed equally to this work.
This research work was supported by National Institutes of Health Grant R01 GM107389 (M.S.J.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Footnotes
Calculated values were obtained in pairwise local sequence alignments using EMBOSS Water (http://www.ebi.ac.uk/Tools/psa/emboss_water/).
The group of 116 retrieved proteins included each of the six previously identified SDO–TSTD1 fusion proteins. The remainder of the 500 retrieved sequences are SDO homologues that align only with the N-terminal domain of the query fusion protein (60–66% query coverage) with E values between 1 × 10–65 and 6 × 10–109.
K. Acharya and M. S. Jorns, unpublished observations.
Similar negative results were obtained with coenzyme A, which was tested because the corresponding persulfide is an alternate, albeit poor, substrate for SDO.1
Oxidation of GSS– by SDO can also be detected by measuring oxygen consumption using an oxygen electrode-based TSTD1–SDO coupled assay (S. L. Melideo and M. S. Jorns, unpublished observations).
The SDO–TSTD1 fusion proteins may also function in the bacterial oxidation of environmental thiosulfate, providing an alternative to the widely distributed Sox (sulfur oxidizing) system.2
Very recent studies show that a reactive sulfane sulfur donor is produced by two other mammalian enzymes (cystathione β-synthase and cystathionine γ-lyase), which catalyze the conversion of cystine to cysteine persulfide.64
Supplementary Material
References
- Kabil O.; Banerjee R. (2012) Characteriation of patient mutations in human persulfide dioxygenase (ETHE1) involved in H2S catabolism. J. Biol. Chem. 287, 44561–44567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamford V. A.; Bruno S.; Rasmussen T.; Ayme C.; Cheesman M. R.; Berks B. C.; Hemmings A. M. (2002) Structural basis for the oxidation of thiosulfate by a sulfur cycle enzyme. J. Biol. Chem. 21, 5599–5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabil O.; Banerjee R. (2010) Redox biochemistry of hydrogen sulfide. J. Biol. Chem. 285, 21903–21907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav P. K.; Yamada K.; Chiku T.; Koutmos M.; Banerjee R. (2013) Structure and kinetic analysis of H2S production by human mercaptopyruvate sulfurtransferase. J. Biol. Chem. 288, 20002–20013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C. (2012) Roles of hydrogen sulfide in the pathogenesis of diabetes mellitus and its complications. Antioxid. Redox Signaling 17, 68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. (2012) Physiological implications of hydrogen sulfide: A whiff exploration that blossomed. Physiol. Rev. 92, 791–896. [DOI] [PubMed] [Google Scholar]
- Zhu X. Y.; Gu H.; Ni X. (2011) Hydrogen sulfide in the endocrine and reproductive systems. Expert Rev. Clin. Pharmacol. 4, 75–82. [DOI] [PubMed] [Google Scholar]
- Predmore B. L.; Lefer D. J. (2011) Hydrogen sulfide-mediated myocardial pre- and post-conditioning. Expert Rev. Clin. Pharmacol. 4, 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G.; Wu L.; Jiang B.; Yang W.; Qi J.; Cao K.; Meng Q.; Mustafa A. K.; Mu W.; Zhang S. (2008) H2S as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine γ-lyase. Science 322, 587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papapetropoulos A.; Pyriochou A.; Altaany Z.; Yang G.; Marazioti A.; Zhou Z.; Jeschke M. G.; Branski L. K.; Herndon D. N.; Wang R. (2009) Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 106, 21972–21977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn E. G.; Austin R. C. (2011) Hydrogen sulfide in the pathogenesis of atherosclerosis and its therapeutic potential. Expert Rev. Clin. Pharmacol. 4, 97–108. [DOI] [PubMed] [Google Scholar]
- Kimura H. (2002) Hydrogen sulfide as a neuromodulator. Mol. Neurobiol. 26, 13–19. [DOI] [PubMed] [Google Scholar]
- Kimura Y.; Kimura H. (2004) Hydrogen sulfide protects neurons from oxidative stress. FASEB J. 18, 1165–1167. [DOI] [PubMed] [Google Scholar]
- Lee M.; Sparatore A.; Del Soldato P.; Mcgeer E.; McGeer P. L. (2010) Hydrogen sulfide-releasing NSAIDs attenuate neuroinflammation induced by microglial and astrocytic activation. Glia 58, 103–113. [DOI] [PubMed] [Google Scholar]
- Penga Y.-J.; Nanduria J.; Raghuramana G.; Souvannakittia D.; Gadallab M. M.; Kumara G. K.; Snyder S. H.; Prabhakara N. R. (2010) H2S mediates O2 sensing in the carotid body. Proc. Natl. Acad. Sci. U.S.A. 107, 10719–10724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackstone E.; Morrison M.; Roth M. B. (2005) H2S induces a suspended animation-like state in mice. Science 308, 518. [DOI] [PubMed] [Google Scholar]
- Mustafa A. K.; Gadalla M. M.; Sen N.; Kim S.; Mu W. T.; Gazi S. K.; Barrow R. K.; Yang G. D.; Wang R.; Snyder S. H. (2009) H2S signals through protein S-sulfhydration. Sci. Signaling 2, ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen N.; Paul B. D.; Gadalla M. M.; Mustafa A. K.; Sen T.; Xu R.; Kim S.; Snyder S. H. (2012) Hydrogen sulfide-linked sulfhydration of NF-κB mediates its antiapoptotic actions. Mol. Cell 45, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa A. K.; Sikka G.; Gazi S. K.; Steppan J.; Jung S. M.; Bhunia A. K.; Barodka V. M.; Gazi F. K.; Barrow R. K.; Wang R.; Amzel L. M.; Berkowitz D. E.; Snyder S. H. (2011) Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ. Res. 109, I259–I268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan N.; Fu C. X.; Pappin D. J.; Tonks N. K. (2011) H2S-induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Sci. Signaling 4, ra86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greiner R.; Palinkas Z.; Bäsell K.; Becher D.; Antelmann H.; Nagy P.; Dick T. P. (2013) Polysulfides link H2S to protein thiol oxidation. Antioxid. Redox Signaling 19, 1749–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong R.; Searcy D. G. (2001) Sulfide oxidation coupled to ATP synthesis in chicken liver mitochondria. Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol. 129, 129–137. [DOI] [PubMed] [Google Scholar]
- Jackson M. R.; Melideo S. L.; Jorns M. S. (2012) Human sulfide:quinone oxidoreductase catalyzes the first step in hydrogen sulfide metabolism and produces a sulfane sulfur metabolite. Biochemistry 51, 6804–6815. [DOI] [PubMed] [Google Scholar]
- Hildebrandt T. M.; Grieshaber M. K.; Westley J. (2008) Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J. 275, 3352–3361. [DOI] [PubMed] [Google Scholar]
- Marcia M.; Ermler U.; Peng G.; Michel H. (2010) A new structure-based classification of sulfide:quinone oxidoreductases. Proteins 78, 1073–1083. [DOI] [PubMed] [Google Scholar]
- Lagoutte E.; Mimoun S.; Andriamihaja M.; Chaumontet C.; Blachier F.; Bouillaud F. (2010) Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochim. Biophys. Acta 1797, 1500–1511. [DOI] [PubMed] [Google Scholar]
- Koj A.; Frendo J.; Janik Z. (1967) [35S]-Thiosulphate oxidation by rat liver mitochondria in the presence of glutathione. Biochem. J. 103, 791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczepkowski T. W.; Skarzynski B.; Weber M. (1961) The metabolic state of thiosulfate. Nature 189, 1007–1008. [DOI] [PubMed] [Google Scholar]
- Bartholomew T. C.; Powell G. M.; Dodgson K. S.; Curtis C. G. (1980) Oxidation of sodium sulphide by rat liver, lungs and kidney. Biochem. Pharmacol. 29, 2431–2437. [DOI] [PubMed] [Google Scholar]
- Levitt M. D.; Furne J.; Springfield J.; Suarez F.; DeMaster E. (1999) Detoxification of hydrogen sulfide and methanethiol in the cecal mucosa. J. Clin. Invest. 104, 1107–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furne J.; Springfield J.; Koenig T.; DeMaster E.; Levitt M. D. (2001) Oxidation of hydrogen sulfide and methanethiol to thiosulfate by rat tissues: A specialized function of the colonic mucosa. Biochem. Pharmacol. 62, 255–259. [DOI] [PubMed] [Google Scholar]
- Tiranti V.; Viscomi C.; Hildebrandt T.; Di Meo I.; Mineri R.; Tiveron C.; Levitt M. D.; Prelle A.; Fagiolari G.; Rimoldi M.; Zeviani M. (2009) Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat. Med. 15, 200–205. [DOI] [PubMed] [Google Scholar]
- Johnson-Winters K.; Nordstrom A. R.; Emesh S.; Astashkin A. V.; Rajapakshe A.; Berry R. E.; Tollin G.; Enemark J. H. (2010) Effects of interdomain tether length and flexibility on the kinetics of intramolecular electron transfer in human sulfite oxidase. Biochemistry 49, 1290–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamoun P.; Belardinelli M. C.; Chabli A.; Lallouchi K.; Chadefaux-Vekemans B. (2003) Endogenous hydrogen sulfide overproduction in Down syndrome. Am. J. Med. Genet. 116A, 310–311. [DOI] [PubMed] [Google Scholar]
- Kangas J.; Savolainen H. (1987) Urinary thiosulphate as an indicator of exposure to hydrogen sulphide vapour. Clin. Chim. Acta 164, 7–10. [DOI] [PubMed] [Google Scholar]
- Grings M.; Moura A. P.; Parmeggiani B.; Marcowich G. F.; Amaral A. U.; Wyse A. T. D.; Wajner M.; Leipnitz G. (2013) Disturbance of brain energy and redox homeostasis provoked by sulfite and thiosulfate: Potential pathomechanisms involved in the neuropathology of sulfite oxidase deficiency. Gene 531, 191–198. [DOI] [PubMed] [Google Scholar]
- Kage S.; Ikeda H.; Ikeda N.; Tsujita A.; Kudo K. (2004) Fatal hydrogen sulfide poisoning at a dye works. Leg. Med. 6, 182–186. [DOI] [PubMed] [Google Scholar]
- Chauncey T. R.; Westley J. (1983) The catalytic mechanism of yeast thiosulfate reductase. J. Biol. Chem. 258, 15037–15045. [PubMed] [Google Scholar]
- Chauncey T. R.; Westley J. (1983) Improved purification and sulfhydryl analysis of thiosulfate reductase. Biochim. Biophys. Acta 744, 304–311. [DOI] [PubMed] [Google Scholar]
- Uhteg L. C.; Westley J. (1979) Purification and steady-state kinetic analysis of yeast thiosulfate reductase. Arch. Biochem. Biophys. 195, 211–222. [DOI] [PubMed] [Google Scholar]
- Koj A.; Frendo J. (1967) Oxidation of thiosulphate to sulphate in animal tissues. Folia Biol. (Krakow, Pol.) 15, 49–65. [PubMed] [Google Scholar]
- Koj A. (1968) Enzymic reduction of thiosulphate in preparations from beef liver. Acta Biochim. Pol. 15, 161–169. [PubMed] [Google Scholar]
- Foster M. W.; Forrester M. T.; Stamler J. S. (2009) A protein microarray-based analysis of S-nitrosylation. Proc. Natl. Acad. Sci. U.S.A. 106, 18948–18953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks S. D.; Drinker M.; Loll P. J. (2007) Ligation independent cloning vectors for expression of SUMO fusions. Protein Expression Purif. 53, 40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorbo B. (1955) Rhodanese. Methods Enzymol. 2, 334–337. [Google Scholar]
- Wood J. L. (1987) Sulfane sulfur. Methods Enzymol. 143, 25–29. [DOI] [PubMed] [Google Scholar]
- Seelan R. S.; Lakshmanan J.; Casanova M. F.; Parthasarathy R. N. (2009) Identification of myo-inositol-3-phosphate synthase isoforms. J. Biol. Chem. 284, 9443–9457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintel R.; Westley J. (1966) The rhodanese reaction mechanism of sulfur-sulfur bond cleavage. J. Biol. Chem. 241, 3381–3385. [PubMed] [Google Scholar]
- Sorbo B. (1962) On the acceptor specificity of rhodanese. Acta Chem. Scand. 16, 243–245. [Google Scholar]
- Cipollone R.; Ascenzi P.; Visca P. (2007) Common themes and variations in the rhodanese superfamily. UBMB Life 59, 51–59. [DOI] [PubMed] [Google Scholar]
- Galperin M. Y.; Koonin E. V. (2000) Who’s your neighbor? New computational approaches for functional genomics. Nat. Biotechnol. 18, 609–613. [DOI] [PubMed] [Google Scholar]
- Zhang L. T.; Liu X. L.; Liu J. G.; Zhang Z. F. (2013) Characteristics and function of sulfur dioxygenase in echiuran worm Urechis unicinctus. PLoS One 8, e81885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolker E.; Higdon R.; Haynes W.; Welch D.; Broomall W.; Lancet D.; Stanberry L.; Kolker N. (2012) MOPED: Model organism protein expression database. Nucleic Acids Res. 40, D1093–D1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M.; Weiss M.; Simonovic M.; Haertinger G.; Schrimpf S. P.; Hengartner M. O.; von Mering C. (2012) PaxDb, a database of protein abundance averages across all three domains of life. Mol. Cell. Proteomics 11, 492–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel K.; Felix S. B.; Flachmeier C.; Heere P.; Schulze W.; Grunewald I.; Pankow H.; Hewelt A.; Scherneck S.; Bauer D. (2003) Identification and characterization of KAT, a novel gene preferentially expressed in several human cancer cell lines. Biol. Chem. 384, 763–775. [DOI] [PubMed] [Google Scholar]
- Gliubich F.; Gazerro M.; Zanotti G.; Delbonoi S.; Bombieri G.; Rodolfo Berni R. (1972) Effect of sulfur binding on rhodanese fluorescence. Eur. J. Biochem. 28, 89–93.5065706 [Google Scholar]
- Ploegman J. H.; Drent G. É.; Kalk K. H.; Hol W. G. (1979) The structure of bovine liver rhodanese: II. The active site in the sulfur-substituted and the sulfur-free enzyme. J. Mol. Biol. 127, 149–162. [DOI] [PubMed] [Google Scholar]
- Griffith O. W. (1999) Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radical Biol. Med. 27, 922–935. [DOI] [PubMed] [Google Scholar]
- Leonardi R.; Zhang Y.; Rock C. O.; Jackowski S. (2005) Coenzyme A: Back in action. Prog. Lipid Res. 44, 125–153. [DOI] [PubMed] [Google Scholar]
- Tateishi N.; Higashi T.; Shinya S.; Naruse A.; Sakamoto Y. (1974) Studies on the regulation of glutathione level in rat liver. J. Biochem. 75, 93–103. [DOI] [PubMed] [Google Scholar]
- Francoleon N. E.; Carrington S. J.; Fukuto J. M. (2011) The reaction of H2S with oxidized thiols: Generation of persulfides and implications to H2S biology. Arch. Biochem. Biophys. 516, 146–153. [DOI] [PubMed] [Google Scholar]
- Kimura Y.; Mikami Y.; Osumi K.; Tsugane M.; Oka J.; Kimura H. (2013) Polysulfides are possible H2S-derived signaling molecules in rat brain. FASEB J. 27, 2451–2457. [DOI] [PubMed] [Google Scholar]
- Fredslund J. (2006) PHY·FI: Fast and easy online creation and manipulation of phylogeny color figures. BMC Bioinf. 7, 315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ida T.; Sawa T.; Ihara H.; Tsuchiya Y.; Watanabe Y.; Kumagai Y.; Suematsu M.; Motohashi H.; Fujii S.; Matsunaga T.; Yamamoto M.; Ono K.; Devarie-Baez N. O.; Xian M.; Fukuto J. M.; Akaike T. (2014) Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc. Natl. Acad. Sci. U.S.A. 111, 7606–7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.