

Abstract
Despite the fact that DNA polymerases have been investigated for many years and are commonly used as tools in a number of molecular biology assays, many details of the kinetic mechanism they use to catalyze DNA synthesis remain unclear. Structural and kinetic studies have characterized a rapid, pre-catalytic open-to-close conformational change of the Finger domain during nucleotide binding for many DNA polymerases including Thermus aquaticus DNA polymerase I (Taq Pol), a thermostable enzyme commonly used for DNA amplification in PCR. However, little has been done to characterize the motions of other structural domains of Taq Pol or any other DNA polymerase during catalysis. Here, we used stopped-flow Förster resonance energy transfer (FRET) to investigate the conformational dynamics of all five structural domains of the full-length Taq Pol relative to the DNA substrate during nucleotide binding and incorporation. Our study provides evidence for a rapid conformational change step induced by dNTP binding and a subsequent global conformational transition involving all domains of Taq Pol during catalysis. Additionally, our study shows that the rate of the global transition was greatly increased with the truncated form of Taq Pol lacking the N-terminal domain. Finally, we utilized a mutant of Taq Pol containing a de novo disulfide bond to demonstrate that limiting protein conformational flexibility greatly reduced the polymerization activity of Taq Pol.
Keywords: Fluorescence resonance energy transfer (FRET), DNA polymerase, Enzyme kinetics, Protein dynamics, conformational dynamics
Introduction
DNA polymerases are key enzymes in the replication and repair of cellular DNA. Among the six families of DNA polymerases the A-Family enzymes function in DNA replication, repair and the processing of Okazaki fragments during lagging strand synthesis 1. Thermus aquaticus DNA polymerase I (Taq Pol) is a thermostable A-Family DNA polymerase which has been the subject of many structural and kinetic investigations due to its wide spread laboratory use for DNA amplification in PCR. Taq Pol consists of an N-terminal 5′→3′ exonuclease domain and a Klentaq1 domain, analogous to the Klenow fragment of Escherichia coli DNA polymerase I (KF), which contains a polymerase core and an “Intervening” non-functional 3′→5′ proofreading exonuclease domain 2; 3. The polymerase core is further subdivided into Finger, Palm and Thumb domains common to all DNA polymerases. In order to perform the necessary in vivo functions, the functions of the Klentaq1 and N-terminal 5′→3′ exonuclease domains of Taq Pol must be tightly coordinated 4; 5; 6.
Early biochemical studies suggested the existence of a rate-limiting non-covalent step prior to the chemistry step for both KF 7; 8; 9 and another A-family member, T7 DNA polymerase 10; 11. Subsequently a large “closing” conformational change in the Finger domain upon dNTP binding was inferred from comparison between binary (Klentaq1•DNA) and ternary (Klentaq1•DNA•dNTP) structures of Klentaq1 2; 12; 13 and other polymerases including T7 DNA polymerase 14 and human immunodeficiency virus type 1 reverse transcriptase (HIV-1 RT) 15. Solution-state stopped-flow fluorescence studies with KF and Klentaq1 provided further evidence for this conformational change and indicated that the conformational change step was much faster than the observed rate limiting step for correct nucleotide insertion 16; 17; 18; 19. Contrastingly, in stopped-flow measurements of T7 DNA polymerase, the conformational change step was shown to be less than 2-fold faster than the chemistry step for correct incorporation 20. Detailed kinetic analysis of the fluorescence data in combination with the data of 32P-based kinetic assays for both correct and incorrect nucleotide insertion by T7 DNA polymerase suggested a mechanism by which the rate of the reverse conformational change plays a critical role in determining substrate specificity 20; 21. In these studies it was argued that a the finger domain closing upon binding to a correct nucleotide leads to a tight ternary complex committing the substrate to catalysis, while an incorrect nucleotide leads to a misformed ternary complex which favors substrate dissociation over nucleotide incorporation. Similarly, in an investigation with HIV-1 RT, it was shown nucleotide binding was a two-step process involving a conformational change and that rate of the reverse conformational change step relative to the rate of the subsequent chemistry step was a key factor influencing the selective incorporation of a dCTP over a nucleoside analog drug 22. Further investigations have provided evidence for a multi-step nucleotide binding mechanism involving additional rapid conformational changes occurring before Finger domain closing in Klentaq1 and Klenow that may also aid in the selection of correct nucleotides 16; 23, though the structural nature of these steps is not entirely clear. Interestingly, single-molecule evidence has suggested that both open and closed conformations may be sampled by a DNA polymerase even in the absence of DNA and/or nucleotide substrates and that the presence of nucleotide shifts the conformational equilibrium towards the closed state 24; 25; 26; 27. Furthermore, crystallographic 28 and single-molecule 25 investigations have demonstrated the existence of a third conformational state of the Finger domain distinct from the open and closed states which is stabilized in the presence of a mismatched nucleotide. In a recent study, high precision FRET measurements demonstrated how the presence of matched and mismatched nucleotides can alter the conformational equilibrium between these three Finger domain conformations for Klentaq1 27, underscoring the importance of this conformational change as a nucleotide complementarity selection mechanism.
Although there has been extensive study of the closing motion of the Finger domain, structural studies have revealed little to no conformational change beyond the Finger domain 2; 12; 13, and thus little has been done to investigate the solution state dynamics of the other domains of the A-Family DNA polymerases. However, while structural studies of the Y-Family DNA polymerases have revealed no obvious conformational change analogous to the Finger domain closing in other DNA polymerases, our recent studies with Solfolobus Solfataricus DNA polymerase IV (Dpo4), a model Y-Family DNA polymerase, have revealed global conformational changes in all four domains of the enzyme 29; 30. Additionally, Molecular Dynamics simulations have shown that short distance conformational changes throughout the structure of HIV-1 RT accompany the larger finger domain closing during nucleotide binding 31. Previous studies of the conformational dynamics of Klentaq1 did not address the effects of the presence of the N-terminal domain in the full-length Taq Pol. However, neutron spin echo experiments have indicated that the conformational dynamics of the Klentaq1 domain and the N-terminal domain are strongly coupled 32. In addition, biochemical studies have demonstrated that the kinetics of both exonuclease and polymerization activity differ for full-length and truncated forms of the enzyme 33; 34; 35; 36. Therefore, in this study we developed a FRET system to independently monitor the conformational dynamics of all five domains of full-length Taq Pol relative to the DNA substrate during the binding and incorporation of correct dNTPs. Furthermore, we introduced a de novo disulfide bond into Taq Pol to limit the conformational flexibility of the Finger and Thumb domains in order to gain insight into the contribution of this motion to the nucleotide incorporation efficiency of Taq Pol.
RESULTS
Design of a FRET system
In order to investigate the global conformational changes involved in dNTP incorporation by Taq Pol, we designed a FRET system to monitor the motions of residues in each of the five domains of Taq Pol relative to a fluorescently labeled base in a DNA substrate. Since Taq Pol does not contain any native cysteine residues, single cysteine residues were engineered into each domain of Taq Pol individually in order to facilitate site-specific attachment of an Alexa594 acceptor fluorophore (Table S1 and Figure 1). Additionally, each single cysteine mutant also contained a single point mutation (G46D) in order to eliminate 5′→3′ exonuclease activity 34. Sites for mutation to cysteine were chosen at non-conserved solvent exposed residues such that the distance (R) between the donor and acceptor (Table S1) should be within R0 ± 0.5R0 (the Förster radius, R0 = 60 Ǻ for the Alexa488/Alexa594 FRET pair), a requirement for accurate FRET measurements. Additionally residues close to the active site or the DNA binding pocket were not chosen in order to minimize any effect on DNA binding or catalysis. Locations were identified in both α-helical and loop regions of each domain to avoid any bias due to secondary structure and the mutants were subsequently purified and labeled with a thio reactive Alexa 594 dye. Notably, while several sites were identified for mutation in an α-helical region of the N-terminal domain, the mutants could not be expressed in E. coli for unknown reasons and thus the conformational changes of this domain were only examined using the V256C loop mutant. The DNA substrate was then labeled with an Alexa488 donor fluorophore via a C6 amino linker on a dT base (Table 1). As Taq Pol has been shown to interact with up to 7 nucleotides on the primer strand 13, the fluorophore was attached to the 9th base from the 3′ primer terminus in order to minimize both its effect on DNA binding to the enzyme and its interaction with the protein interior which might contribute to non-FRET related changes in fluorescence intensity. In order to distinguish events occurring before and after phosphodiester bond formation, we utilized DNA substrates containing (S-1) or lacking (S-2) a primer 3′-terminal hydroxyl group necessary for covalent dNTP incorporation (Table 1). Single-turnover kinetics showed that point mutations and enzyme dye labeling (Table S2) as well as fluorescent labeling of the DNA substrate (Figure S1) did not alter nucleotide incorporation rate or nucleotide binding affinity more than two-fold.
FIGURE 1.

Location of the chosen mutants on Taq Pol. The subdomains of Taq pol are shown in N-terminal (yellow and orange), red (intervening), green (Thumb), blue (Finger) and purple (Palm); all of the nine mutation sites (Table S1) are in cyan.
Table 1.
Sequences of DNA substrates*.
| S-1 | 5′–CG AGC CGT CGC ATC CTA CCG C–3′ |
| 3′–GC TCG GCA GCG TAG GAT GGC GAC GTC GTA G–5′ | |
|
| |
| S-2 | 5′–CG AGC CGT CGC ATC CTA CCG C–3′ |
| 3′–GC TCG GCA GCG TAG GAT GGC GAC GTC GTA G–5′ | |
|
| |
| S-3 | 5′–CG AGC CGT CGC ATC CT–3′ |
| 3′–GC TCG GCA GCG TAG GAT GGC GAC GTC GTA G–5′ | |
`T' and `C' denote Alexa488-attached to a 5-C6-amino-2′-deoxythymidine and 2′,3′-dideoxycytidine, respectively.
Steady-state fluorescence analysis
To quantify changes in FRET produced by DNA and dNTP binding, steady-state fluorescence spectra of all mutants were recorded. Steady-state spectra with excitation at the donor maximum absorbance for an N-terminal domain mutant V256C are shown in Figure 2. Upon the addition of labeled enzyme to labeled DNA substrate (black trace), a decrease in donor fluorescence emission at 517 nm and the appearance of an acceptor peak centered at 613 nm (red) were observed, indicating the formation of an efficient FRET pair. Notably, no change in donor fluorescence was observed upon mixing with unlabeled enzyme (data not shown). Subsequent incorporation of correct dTTP into DNA substrate S-1 resulted in a decrease in FRET (green) due to an increase in distance between the fluorophores, likely due to the translocation of Taq Pol by one base pair along the DNA duplex (see below). Interestingly, a smaller decrease in FRET was observed when the experiment was repeated with a non-extendable DNA substrate S-2 (red vs green, Figure 2B). This suggested the existence of pre-catalytic conformational changes which resulted in a change in distance between the N-terminal domain and the DNA substrate upon nucleotide binding. Similar results were obtained with all other mutants as summarized in Table 2, indicating that Taq Pol underwent a global conformational change involving all domains during nucleotide binding and incorporation. It should be noted that, due to uncertainties in the dipole orientation factor and other fluorophore properties, the exact distance between fluorophores cannot be accurately predicated from the apparent FRET efficiency. However, comparison of the magnitude of the FRET efficiency change between each FRET pair is valid. For example, of all mutants, finger domain residue S644C showed the largest change in FRET efficiency (Table 2) which is consistent with crystal structures which show that this residue is near the tip of the helix which undergoes the large Finger domain open-to-close transition 2; 12; 13.
FIGURE 2.
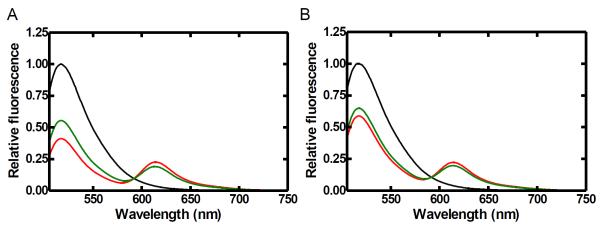
Steady-state fluorescence spectra of N-terminal subdomain mutant (V256C) of Taq Pol at 20 °C. Alexa488-labeled DNA (100 nM, black trace) was excited at a wavelength of 493 nm. The sequential addition of Alexa594-labeled V256C mutant protein (200 nM) and correct dTTP produced the red and green traces, respectively. Spectra were normalized to 1 by using the donor peak for the DNA alone as a reference. Emission spectra are shown for both extendable S-1 (A) and non-extendable S-2 (B) DNA substrates.
Table 2.
Apparent FRET efficiency between the donor Alexa488 on DNA and acceptor Alexa594 on the domains of Taq Pol calculated by steady-state FRET efficiency.
| Domains | Residues* | Apparent FRET efficiency for each FRET paira | Net change in FRET efficiency for each phasee | |||
|---|---|---|---|---|---|---|
| Pre-insertion binaryb (d1) | Insertion ternaryc (d2) | Post-insertion binaryd (d3) | P0+P1 (d2−d1 | P2 (d3−d2) | ||
| N-Terminal | V256C | 0.50 | 0.45 | 0.37 | −0.05 | −0.08 |
| Intervening | K346C | 0.32 | 0.39 | 0.29 | 0.07 | −0.1 |
| R411C | 0.32 | 0.31 | 0.29 | −0.01 | −0.02 | |
| Thumb | E524C | 0.32 | 0.35 | 0.26 | 0.03 | −0.09 |
| E465C | 0.37 | 0.41 | 0.28 | 0.04 | −0.13 | |
| Finger | V649C | 0.37 | 0.40 | 0.33 | 0.03 | −0.07 |
| S644C | 0.32 | 0.53 | 0.28 | 0.21 | −0.25 | |
| Palm | A814C | 0.26 | 0.27 | 0.21 | 0.01 | −0.06 |
| R801C | 0.20 | 0.23 | 0.18 | 0.03 | −0.05 | |
Apparent FRET efficiency for each Alexa594-labeled Taq Pol residue and the Alexa488-labeled primer base was calculated from steady-state spectra using Equation 1 (See Materials and Methods Section).
Calculated from the change in donor peak fluorescence intensity between the binary complex Taq Pol•S-2 and the dideoxy-terminated DNA substrate (S-2) alone at 20 °C.
Calculated from the change in donor peak fluorescence intensity between the ternary complex Taq Pol•S-2•dTTP (Table 1) and the dideoxy-terminated DNA substrate (S-2) alone at 20 °C.
Calculated from the change in donor peak fluorescence intensity between the binary complex Taq Pol•S-1 after dTTP incorporation and DNA (S-1) alone at 20 °C.
Positive or negative change in FRET efficiency values indicate that the Taq Pol residue respectively moves towards or away from DNA.
Real-time conformational dynamics
In order to investigate the global conformational dynamics of Taq Pol in real-time, we performed stopped-flow FRET assays to monitor the motions of each domain during nucleotide incorporation. Control stopped-flow experiments in the absence of dNTPs yielded no fluorescence change (Figure S2). Upon the addition of the correct dTTP, all mutants showed three FRET phases at 20 °C (Figure 3). Similar results were obtained at 37 °C and above, however the first two FRET phases were too rapid to be easily observed (data not shown). Notably, fluorescence changes are shown on an arbitrary scale and thus the absolute change in FRET efficiency cannot be calculated from these data as it can for the steady-state fluorescence data (Table 2). In contrast to the global conformational dynamics previously observed with Dpo4 by us 29; 30, the observed FRET phases for mutants of Taq Pol all followed the same trend, indicating that all monitored positions moved in the same direction relative to the DNA substrate. The phases were characterized by a very rapid acceptor fluorescence intensity decrease (P0 = 11.5–36 s−1), followed by a slow increase (P1 = 0.08–0.26 s−1) and then a slower decrease phase (P2 = 0.006–0.017 s−1) (Table 3). When using a non-extendable substrate S-2, the general trend of P0 and P1 remained unchanged, while P2 was no longer observed, indicating that the first two phases resulted from processes occurring prior to phosphodiester bond formation, while the third phase occurred either during or after the chemistry step (Figure 4). Notably, since P1 and P2 were not well separated temporally, for most mutants, fitting of P1 in the absence of P2 using the non-extendable DNA substrate leads to slightly different rates (Table 3). The similarity of P1 and P2 for all mutants suggested that the open-to-close transition described previously for the Finger domain of Klentaq1 17; 23 occurred concurrently with a global conformational change involving all of the other domains of the full-length Taq Pol. The rapid FRET decrease (P0) observed for all mutants of Taq Pol was not previously detected for the Finger domain in Klentaq1 17. However, a similar rapid FRET increase was previously proposed to represent a rapid one base pair translocation of the DNA substrate induced by nucleotide binding to Dpo4 29; 30. Likewise, P0 may represent a similar rapid DNA motion in Taq Pol though an additional protein conformational change cannot be ruled out.
FIGURE 3.

Stopped-flow kinetics of dTTP incorporation into S-1 DNA at 20 °C. A pre-incubated solution of Taq Pol mutant (200 nM) and DNA substrate S-1 (100 nM) was rapidly mixed with dTTP (0.5 mM) and fluorescence was monitored using a stopped-flow apparatus. Donor (black) and acceptor (gray) traces are shown for (A) the intervening (K346C and R411C), (B) Thumb (E524C and E465C), (C) Finger (V649C and S644C), (D) Palm (A814C and R801C), and (E) the N-terminal (V256C) domain mutants.
Table 3.
The phase rate (s−1) from stopped-flow fluorescence of dTTP binding and incorporation by Taq Pol mutants at 20 °C.
| Domain | Residue | DNA | Phase rate (s−1)* | |||||
|---|---|---|---|---|---|---|---|---|
| Donor | Acceptor | |||||||
| P0 | P1 | P2 | P0 | P1 | P2 | |||
| N-terminal | V256C | S-1 | 11.5 ± 0.8 | 0.11 ± 0.01 | 0.008 ± 0.003 | 12.0 ± 0.9 | 0.088 ± 0.005 | 0.009 ± 0.005 |
| S-2 | 13.1 ± 0.5 | 0.217 ± 0.009 | 13.1 ± 0.6 | 0.24 ± 0.12 | ||||
| Intervening | K346C | S-1 | 18 ± 3 | 0.10 ± 0.05 | 0.009 ± 0.006 | 36 ± 6 | 0.077 ± 0.004 | 0.009 ± 0.005 |
| S-2 | 20 ± 4 | 0.24 ± 0.01 | 34 ± 6 | 0.233 ± 0.003 | ||||
| R411C | S-1 | 15 ± 3 | 0.112 ± 0.008 | 0.011 ± 0.007 | 18 ± 3 | 0.15 ± 0.03 | 0.009 ± 0.002 | |
| S-2 | 12.7 ± 0.7 | 0.25 ± 0.05 | 14 ± 2 | 0.26 ± 0.05 | ||||
| Thumb | E524C | S-1 | 17 ± 2 | 0.12 ± 0.04 | 0.011 ± 0.007 | 33 ± 3 | 0.105 ± 0.005 | 0.013 ± 0.005 |
| S-2 | 21 ± 2 | 0.17 ± 0.01 | 28 ± 4 | 0.085 ± 0.008 | ||||
| E465C | S-1 | 29 ± 1 | 0.09 ± 0.05 | 0.011 ± 0.006 | 27 ± 2 | 0.08 ± 0.04 | 0.011 ± 0.009 | |
| S-2 | 19.9 ± 08 | 0.21 ± 0.01 | 25 ± 3 | 0.18 ± 0.02 | ||||
| Finger | V649C | S-1 | 17 ± 2 | 0.08 ± 0.01 | 0.008 ± 0.002 | 24 ± 4 | 0.09 ± 0.01 | 0.007 ± 0.001 |
| S-2 | 18 ± 3 | 0.24 ± 0.03 | 22 ± 2 | 0.22 ± 0.02 | ||||
| S644C | S-1 | 22 ± 2 | 0.13 ± 0.02 | 0.006 ± 0.001 | 17.3 ± 0.9 | 0.21 ± 0.03 | 0.017 ± 0.003 | |
| S-2 | 14 ± 1 | 0.14 ± 0.01 | 20 ± 3 | 0.16 ± 0.08 | ||||
| Palm | A814C | S-1 | 24 ± 3 | 0.13 ± 0.03 | 0.010 ± 0.009 | 15 ± 3 | 0.09 ± 0.02 | 0.007 ± 0.006 |
| S-2 | 18 ± 2 | 0.23 ± 0.09 | 17 ± 5 | 0.14 ± 0.05 | ||||
| R801C | S-1 | 19 ± 2 | 0.15 ± 0.06 | 0.015 ± 0.005 | 17 ± 4 | 0.11 ± 0.02 | 0.016 ± 0.007 | |
| S-2 | 22 ± 1 | 0.22 ± 0.09 | 22 ± 3 | 0.23 ± 0.08 | ||||
The rates are the average from multiple independent experiments and reported as mean ± standard deviation.
FIGURE 4.

Stopped-flow kinetics of dTTP binding to the Taq Pol•DNA binary complex at 20 °C. A pre-incubated solution of a Taq Pol mutant (200 nM) and non-extendable DNA substrate S-2 (100 nM) was rapidly mixed with dTTP (0.5 mM) and fluorescence was monitored using a stopped-flow apparatus. Donor (black) and acceptor (gray) traces are shown for (A) the intervening (K346C and R411C), (B) Thumb (E524C and E465C), (C) Finger (V649C and S644C), (D) Palm (A814C and R801C), and (E) the N-terminal (V256C) domain mutants.
Surprisingly, the rate of the pre-catalytic motion was measured to be ~50 fold slower than that observed for the closing of the Finger domain of the truncated Klentaq1 also measured at 20 °C in a previous study 17. In order to further investigate this potential difference between the full-length and truncated forms of this DNA polymerase, we performed stopped-flow assays with several Klentaq1 mutants labeled at the same positions as in the full-length Taq Pol. Notably, due to a significantly decreased DNA binding affinity of the truncated protein 34, a higher concentration of Klenaq1 (600 nM compared to 200 nM for full-length Taq Pol) was used in order to obtain reliable FRET signals. Stopped-flow traces with Klentaq1's Finger, Palm and Intervening domain mutants showed three FRET phases analogous to those observed with the full-length enzyme (Figure 5), but with greatly increased rates (P0 = 50–170 s−1 vs. 11.5–36 s−1; P1 = 1.7–4.9 s−1 vs. 0.08–0.26 s−1; P2 = 0.05–0.17 s−1 vs. 0.007–0.017 s−1) (Tables 3 and 4). As observed for the full-length Taq Pol mutants, only the first two phases were observed when using the non-extendable S-2 DNA substrate (data not shown). It is therefore evident that the presence of the N-terminal domain had a significant impact on the conformational dynamics of Taq Pol, greatly limiting the rate of conformational change.
FIGURE 5.

Stopped-flow kinetics of dTTP incorporation into S-1 DNA catalyzed by Klentaq1 at 20 °C. A pre-incubated solution of Klentaq1 mutant (600 nM) and DNA substrate S-1 (100 nM) was rapidly mixed with dTTP (0.5 mM) and fluorescence was monitored using a stopped-flow apparatus. Donor (black) and acceptor (gray) traces are shown for (A) the intervening (K346C), (B) Finger (V649C), and (C) Palm (A814C).
Table 4.
The phase rate (s−1) from stopped-flow fluorescence of dTTP binding and incorporation by each Klentaql mutant at 20 °C.
| Domains | Residues | Phase rate (s−1)* | |||||
|---|---|---|---|---|---|---|---|
| Donor | Acceptor | ||||||
| P0 | P1 | P2 | P0 | P1 | P2 | ||
| Intervening | K346C | 70 ± 10 | 3.8 ± 0.2 | 0.13 ± 0.01 | 77 ± 9 | 3.6 ± 0.3 | 0.05 ± 0.02 |
| Finger | V649C | 50 ± 3 | 2.7 ± 0.2 | 0.17 ± 0.05 | 80 ± 30 | 1.7 ± 0.3 | 0.11 ± 0.01 |
| Palm | A814C | 68 ± 5 | 4.9 ± 0.2 | 0.17 ± 0.03 | 170 ± 70 | 4.5 ± 0.6 | 0.08 ± 0.01 |
The rates are the average from multiple independent experiments and reported as mean ± standard deviation.
Nucleotide incorporation kinetic studies
In order to investigate the functional implications of the observed conformational changes, we conducted rapid chemical quench kinetic assays with Taq Pol to determine and the rate-limiting step of nucleotide incorporation (kp) and the steady state rate which is limited by the slow dissociation of the enzyme-DNA binary complex at 20 °C. Interestingly, the kp for each mutant was measured to be 0.076–0.136 s−1 (Table S2), at most only 2-fold lower than the rate of P1 (0.08–0.26 s−1). In direct contrast to what has been previously observed for Klentaq1 16; 17; 19; 23, this finding indicated that the pre-catalytic conformational change was at least partially rate-limiting for the full-length Taq Pol. Furthermore, as the rate of P2 (0.006–0.017 s−1) was ~10-fold lower than the rate-limiting step of nucleotide incorporation (0.076–0.136 s−1), this conformational change must be limited by a slow process occurring after the chemistry step. Previous studies with Klentaq1 19 or Dpo4 29; 30 have found the analogous post-catalytic motion to be limited only by the rate-limiting step of nucleotide incorporation. Consistently, our kinetic assays here show that the kp values for the Klentaq mutants (0.22–0.33 s−1, Table S2) are much slower than the P1 rates (1.7–4.9 s−1) and only slightly faster than the P2 rates (0.05–0.17 s−1) (Table 4). It is therefore apparent that the post-catalytic conformational change is also significantly slowed by the presence of the N-terminal domain. Interestingly, the rate of P2 matched well with steady-state rate of polymerization measured by 32P-based kinetic assay (0.011 ± 0.002 s−1 for wild-type Taq and 0.009 ± 0.001 s−1 for mutant E524C, Figure S3), indicating that these two steps may be rate limited by the same mechanistic step. However, it is unclear if the change in FRET associated with P2 is directly due to the dissociation of the enzyme from the DNA. In the stopped-flow assays, the enzyme is in excess of the DNA substrate, and thus any DNA which dissociates from the enzyme after nucleotide incorporation will be rapidly bound again, resulting in no observable net change in FRET unless the enzyme rebinds the DNA in a different conformation. Notably, the rate of P2 for the Klentaq1 mutants (0.05–0.17 s−1, Table 3) was faster than the steady-state rate of polymerization (0.023 ± 0.002 s−1 for wild-type Klentaq1 and 0.020 ± 0.001 s−1 for mutant A814C, Figure S3), suggesting that the nature of this conformational change may differ for the full length and truncated forms of Taq Pol.
Global fitting of kinetic and stopped-flow FRET data
In order to perform a more rigorous kinetic analysis of our results, we performed simultaneous global fitting of the data from both stopped-flow and 32P-based kinetic assays using KinTek Explorer global fitting software 37. As an example, data obtained with Taq Pol mutant E524C are shown in Figure 6. Simultaneous fitting of data from 32P-based kinetic assays showing the rate of nucleotide incorporation under single turnover conditions (Figure 6C), and steady-state conditions (Figure 6D) along with stopped-flow fluorescence data showing conformational changes using regular or dideoxy-terminated DNA (Figures 6A,B) show that all results for an individual mutant can be fit to the minimal mechanism in Figure 6E. Including a step to account for the P0 phase in the model did not appreciably change any of other kinetic parameters or the fits for the 32P-based kinetic data and thus the early data points in the stopped flow traces showing the P0 phase were omitted from the global fitting analysis in order to allow for more accurate fitting of the other kinetic rate constants. Notably, the global fitting yielded kinetic parameters (Figure 6E) for the apparent nucleotide binding affinity (KD ~ 50 μM, step 2), the conformational change step (0.15 s−1, step 3) and dissociation of the enzyme from the product following catalysis (0.007 s−1, step 5) which were similar to the rates obtained from fitting each data set individually (Table 3, Table S2, and Figure S3).
FIGURE 6.

Simultaneous global fitting of 32P-based kinetic and stopped-flow fluorescence data for Taq Pol mutant E524C. In panels A–D, the black lines show the best fit obtained by nonlinear regression in fitting all data simultaneously to the model shown in panel E using KinTek Explorer software 37. Stopped-flow fluorescence experiment show acceptor fluorescence upon mixing dTTP (0.5 mM) with a pre-incubated solution of a Taq Pol mutant (200 nM) and either (A) extendable DNA substrate S-1 or (B) non-extendable DNA substrate S-2 (100 nM). (C) Pre-steady-state kinetic experiment in which a pre-incubated solution of a Taq Pol mutant (60 nM) and 5′-32P labeled DNA substrate S-2 (30 nM) was rapidly mixed with various concentrations of dTTP (5, 25, 50, 100, 250 and 500 μM), and was quenched at various times with 0.37 M EDTA. (D) Steady-state kinetic experiment in which a pre-incubated solution of Taq Pol mutant (1.5 nM) and 5′-32P-labeled DNA substrate S-1 (250 nM) was rapidly mixed with dTTP (500 μM) and quenched at various times with 0.37 M EDTA. (E) Minimal model showing the kinetic parameters obtained from the global fitting.
Unfortunately, the rate of step 4 in Figure 6E, which includes the chemistry step as well as any potential active site rearrangement steps, was not well constrained by the data and only a lower limit of 0.4 s−1 could be obtained from the global fitting. However, stopped-flow fluorescence data with the non-extendable DNA substrate (Figure 4) showing that the P1 phase represents a pre-catalytic conformational change justifies the inclusion of this step in the model and the global fitting required this step to be at least slightly faster than the preceding conformational change step. The reverse rate of the conformational change was also not well defined by the data, but an upper limit of 0.3 s−1 was established. Nevertheless, the global fitting analysis provides additional evidence that the conformational change step is relevant for catalysis.
Effects of limited conformational flexibility
In order to further investigate the role that the conformational dynamics of Taq Pol plays in nucleotide incorporation, we sought to generate a double cysteine mutant to create a de novo disulfide bond which may be capable of limiting the conformational motions of Taq Pol. Crystal structures show that the largest change in distance between the binary and ternary complex of Klentaq1 occurs at the N-terminal end of the O-helix and that this region of the Finger domain is much closer to the Thumb domain in the closed conformation than in the open conformation2,10,11. While no residue pairs between the Thumb and the O-helix of the Finger domain are close enough to form a disulfide bond if mutated to cysteine based on the reported closed ternary complex of Klentaq113, it is possible that Taq Pol is flexible enough to allow such a bond to form in the solution state. In this context, such a disulfide bond should limit Taq Pol to a conformation resembling the closed conformation. In order to find a potential site for introducing a disulfide bond, we utilized the sGal program 38 to determine residue pairs on the Thumb and Finger domains which were within 10–13 Ǻ of each other in the ternary complex but which were separated by more than 20 Å in the binary complex. sGal chooses residues based on exposed surface area and discards pairs if other protein residues would sterically clash with a disulfide bond between the chosen sites 38. From this analysis, the T588/M658 pair was identified as the top candidate as these residues formed the closest suitable pair in the closed conformation but were separated by greater than 25 Å in the open conformation. We subsequently purified the T586C/M566C mutant in order to determine if the proposed disulfide bond could be formed and how it would affect the catalytic activity of Taq Pol. To investigate the extent of the disulfide bond formation in this mutant, we performed an Ellman's reagent assays to quantify the amount of free sulfhydryl group in this mutant (See Methods). Our results indicated that the concentration of free sulfhydryl group was only ~2% of the total protein concentration, suggesting that the majority of the protein in solution contained the disulfide bond as predicted by the sGal program. We then performed a processivity assay in the presence of all four dNTPs to determine the effect of the introduced disulfide bond on DNA polymerization activity (Figure 7). Interestingly, the T586C/M656C mutant containing an inter-domain disulfide bond showed significantly reduced processive polymerization activity compared to the wild-type enzyme (Figures 7A(−) and 7B(−)). However, after incubation with β-mercaptoethanol (β-ME) for 30 min to reduce the disulfide bond, the activity of this mutant was restored to the wild-type level (Figures 7A(+) and 7B(+)). This suggests that the lack of polymerization activity was indeed due to the presence of a disulfide bond rather than the mutations alone. To further test if the loss of activity from the disulfide bond formation was due to inhibition of polymerase translocation between consecutive nucleotide incorporations or inhibition of individual nucleotide incorporations, we performed similar experiments to monitor the incorporation of a single dTTP. Our data show that single nucleotide incorporation activity of mutant T586C/M656C (Figure 7D(−)), but not of wild-type Taq Pol (Figure 7C(−)), was significantly slowed by the disulfide bond formation and the effect could again be reversed by incubation with β-ME (Figures 7C(+) and 7D(+)). These results indicated that the polymerization efficiency of Taq Pol could be greatly reduced by limiting the conformational flexibility of the enzyme.
FIGURE 7.

Activities of a double cysteine mutant of Taq Pol under various reaction conditions at 20 °C. A pre-incubated solution of wild-type Taq Pol (A,C) or its double cysteine mutant T586C/M656C (B,D) (100 nM) and 5′-32P labeled DNA substrate S-3 (A,B) or S-1 (C,D) (100 nM) was rapidly mixed with either 200 μM each of all four dNTPs (A,B) or 200 μM dTTP (C,D) and the reaction mixture was quenched at various times with 0.37 M EDTA. Reactions were performed either in the absence (−) or presence (+) of 5 mM β-ME.
DISCUSSION
While many studies have been carried out to characterize the conformational dynamics of the motions of the Finger domain of the truncated Klentaq1, our results represent a major step forward in understanding the global conformational changes of Taq Pol and highlight significant and unexpected differences between the full-length and truncated forms of this enzyme. Notably, as with previous studies on Klentaq1 23, our studies were performed at 20 °C as distinct fluorescence phases were difficult to be resolved at higher temperatures. We were able to detect a FRET change in the Thumb, Palm, Intervening and N-terminal domains relative to the DNA analogous to those in the Finger domain of Klentaq1 17; 19; 23 and thus provide the first evidence to suggest that nucleotide incorporation catalyzed by Taq Pol involves a coordinated global conformational change that is not limited to only the Finger domain. This result is surprising due to the lack of crystallographic evidence for conformational changes outside of the Finger domain 2; 12; 13. Except the conformational change detected in the Finger domain, the observed motions in this study were on the order of only a few angstroms as indicated by the small net changes in FRET efficiency in Table 2. Thus, it is possible that the changes in distances between the DNA and the Thumb, Palm, Intervening and N-terminal domains may be too small to be resolved in crystal structures. Notably, the reported binary crystal structure of Klentaq1 was obtained by soaking a pre-formed E•DNA•dNTP ternary complex crystal in a buffer lacking dNTP thus allowing the dNTP to diffuse out of the active site13. The crystal state may therefore suppress any potential small conformational changes occurring in the E•DNA complex as the nucleotide diffuses out of the active site in the ternary complex crystal.
The similarity in the kinetic rates observed for mutants in all domains (Table 3) suggest that the Finger domain conformational change represents the same mechanistic step as the conformational change observed for other domains. One potential hypothesis is that all five domains of Taq Pol close in towards the DNA substrate in a manner similar to the Finger domain, only with a much smaller overall change in distance. However, with the lack of high resolution, solution-phase structures of Taq Pol, the exact nature of the conformational change cannot be explicitly determined, and it is possible that the observed change in FRET may also involve a change in DNA-labeling site and/or the position of the DNA substrate in the protein which contribute to the shortening of the distance between each residue and the donor fluorophore during the P1 phase. However, since the labeling site on the DNA is located in the rigid double-helix, it is unlikely that the conformational change would be due to a local change in the DNA substrate near the fluorophore labeled base. In addition, it seems unlikely that the DNA could move enough to yield the changes in FRET observed in this study without disrupting the position of the primer 3′ end within the polymerase active site. Future investigations of the solution-phase structure via protein NMR or molecular dynamics simulation may help provide additional information on the structural nature of this global conformational change.
Based on the comparison between our kinetic studies and stopped-flow FRET assays and the global fitting analysis, we proposed a catalytic mechanism of Taq Pol to include the observed conformational changes (Figure 8). Our results with Klentaq1 are consistent with previous studies 17; 19; 23 and indicate that the conformational change during the P1 phase (Figure 8, step 4) was faster than the rate-limiting step of nucleotide incorporation (kp), which is instead likely limited by the rate of a slower and more subtle active site rearrangement (Figure 8, step 5). However, the transition during P1 was at least partially rate-limiting in the full-length Taq Pol. Consequently, as shown by the global fitting (Figure 6), our data does not necessarily require the inclusion of a slow active site rearrangement (Figure 8, step 5) which is distinct from the global conformational transition (Figure 8, step 4) or the actual chemistry step (Figure 8, step 9). In addition, the P2 FRET phase (Figure 8, Step 7 and/or 8) was also significantly slowed, and occurred at rate (0.006–0.017 s−1) much slower than the rate limiting step of nucleotide incorporation (0.076–0.136 s−1) measured by 32P-based single-turnover assays. For Klentaq1, the P2 rates (average 0.12 s−1, Table 4) are only slightly lower than the kp measured (average 0.26 s−1, Table S2). The remarkable 10-fold difference between the full-length and truncated forms of the enzyme in the rates of P0, P1 and P2 suggested that the N-terminal 5′→3′ exonuclease domain interacted significantly with either the rest of the protein or with the DNA substrate or both, and strongly affected the conformational dynamics involved in nucleotide incorporation. The slowed P2 phase may be the result of slowing of either the domain opening itself (Step 8) or the preceding reverse active site rearrangement (Step 7) or both. Furthermore, since the rate of P2 (0.006–0.017 s−1) is similar to the measured steady-state rate of nucleotide incorporation (~0.01 s−1, Figure S3), the slowing of these steps (Figure 8, Step 7 and Step 8) may be acting as a rate determining process during multiple enzyme turnovers. We cannot, however, rule out the possibility that, after catalysis, Taq Pol remains bound to the DNA in a closed conformation until it dissociates from the DNA and does not adopt the open conformation until its rebinding to the DNA. In either case, the global data fitting analysis demonstrated that the fluorescence change in P2 and the steady-state nucleotide incorporation rate can be fit to the same step in a minimal kinetic model in Figure 6E.
FIGURE 8.
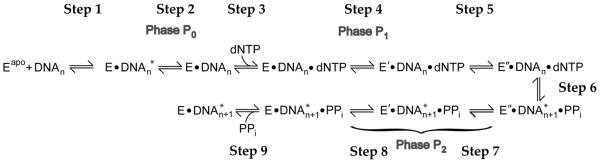
Proposed catalytic mechanism of Taq Pol. P0, P1, and P2 are the three FRET phases observed in the stopped-flow traces. DNA* and DNA denotes the position of the templating base either in the pre-insertion site, stacked with Y671 of Taq Pol, or in the insertion site, correctly positioned for base pairing with an incoming dNTP. Eapo, E, E′ and E″ represent four distinct conformations of Taq Pol. PPi indicates pyrophosphate.
Crystallographic studies reveal two distinct conformations of the N-terminal domain relative to the polymerase domain, either with the N-terminal domain extending into solution 2; 39 or a compact form with the N-terminal domain associated closely with the polymerase domain 40. A more recent study employing solution state X-ray scattering and analytical ultracentrifugation has indicated that in solution, the relative position of the N-terminal domain in apo state more closely resembles a more extended form, but is likely somewhat different from the extended conformation observed in the crystal structures 41. Interestingly, neutron scattering experiments suggest that upon DNA binding, the N-terminal domain shifts towards the more compact state and interacts with the Thumb and Palm domains 42. Our results suggest that these interactions may lead to the slowing of the conformational dynamics of the full-length Taq Pol relative to the truncated Klentaq1. This modulation of the conformational dynamics involved in DNA polymerization by the N-terminal domain may help coordinate the function of the exonuclease and polymerase activities of Taq Pol in vivo.
Interestingly, the full-length Taq Pol has been shown to introduce mutations two-fold more frequently during DNA replication in vitro than the Klentaq1 truncation 33. Notably, for Klentaq1, matched and mismatched dNTPs are known to alter the kinetics of the conformational change steps 17; 27, which confirms the notion that the pre-catalytic conformational change step is a major factor influencing polymerase fidelity 21; 43. It is thus possible that difference in the conformational dynamics observed here contribute to the difference in nucleotide incorporation fidelity between the full-length and truncated forms of Taq Pol. However, further investigation on the kinetics and conformational dynamics of misincorporation by full-length and truncated Taq Pol is necessary to fully explore this possibility.
In addition to the differences between the full-length and truncated forms of Taq Pol, our stopped-flow studies provided direct evidence for a step occurring prior to the transition in P1. Crystal structures of the Klentaq1-DNA binary complex indicate that the templating base is located in a “pre-insertion site”, where it is stacked with a conserved tyrosine residue (Y671) 13. This stacking arrangement must then be relieved to allow the templating base to move into the “insertion site” where it is then positioned to base pair with the incoming nucleotide. In light of this information, it is possible that the P0 phase observed in our results could involve a rapid repositioning of the DNA substrate in the enzyme active site which allowed for dNTP binding and occurred prior to the open-to-close transition and subsequent events (Figure 8, Step 2). A similar shift of the template base from a pre-insertion to an insertion site was also proposed in a structural study of BF, a homolog of KF and Taq Pol 44. More recently, a structural study of the BF ternary complex with DNA and mismatched dNTP has captured the Finger domain in an “ajar” conformational state, intermediate between the open and closed conformations 28. This state resembles the open conformation more closely than the closed conformation, but with the template base in the insertion site, thus allowing for an interaction with the mismatched nucleotide as observed previously for an RNA polymerase 45. Stopped-flow FRET studies of Klentaq1 have also indicated that there is an equilibrium between two DNA binding modes which were suggested to correspond to whether the templating base is positioned to form a base pair with an incoming nucleotide, or is flipped out of the active site in the enzyme•DNA binary complex 23. Two analogous DNA binding states have been observed in HIV-1 reverse transcriptase as well 46; 47; 48. A similar stopped-flow study has demonstrated the existence of a rapid DNA motion in which nucleotide complementarity is checked prior to the open-to-close domain transition for KF 16. Binding of dNTP likely induces this rapid DNA motion or shifts the equilibrium towards the state with the templating base correctly positioned for base pairing, thus leading to the decrease in FRET of P0 (Figure 8, Step 2) prior to the P1 closing transition (Step 4). Surprisingly, despite the fact that the labeled DNA base in our investigation is far from the active site, P0 was observed for all domains of Taq Pol. This suggests that this mechanistic step may involve a repositioning of the entire DNA substrate. However, we cannot rule out that an additional global change in the protein conformation, rather than only a local rearrangement of the templating base and active site residues, leads to the fluorescence change in P0. It will be interesting to determine whether a mismatched dNTP can also induce this FRET change, thus indicating that this step allows the templating base to preview the incoming nucleotide prior to proceeding to the closed conformation as proposed previously 16; 23; 43; 45.
Recently there has been some debate as to how important conformational changes are to the catalytic activity of DNA polymerases 49; 50; 51. In order to further address the role of conformational dynamics in the processive insertion of nucleotides, we generated a Taq Pol construct with a disulfide bond introduced between the Finger and Thumb domains. This disulfide bond should essentially lock the Finger domain in a closed conformation and thus significantly inhibit the conformational flexibility. Our results showed that this disulfide bond mutant was able to catalyze nucleotide incorporation, but with greatly reduced activity compared to the wild-type enzyme. This is direct evidence that conformational flexibility contributes substantially to polymerase activity, but that a significant limitation of protein conformational dynamics is not sufficient to completely inhibit catalysis. Alternatively, the protein conformational dynamics may be altered and slowed by this disulfide bond. Future biochemical and structural studies with this mutant may provide critical insights into the role that conformational changes play in fidelity and efficiency of nucleotide incorporation.
In summary, our stopped-flow FRET and chemical quench experiments revealed evidence for global conformational changes beyond the Finger domain of Taq Pol and showed that the presence of the N-terminal domain greatly affected the rates of these changes. Furthermore, our results have shed light on how protein conformational flexibility can affect polymerase activity.
Materials and Methods
Preparation of full-length Taq Pol mutants and DNA substrates
A plasmid pTTQ18 encoding the Taq Pol gene was generously provided by Dr. Venkat Gopalan (The Ohio State University). The full-length Taq Pol gene (1–832 amino acids) was PCR amplified and subcloned into the pET26B vector (Novagen) using NdeI and SalI restriction sites, which has a sequence encoding an N-terminal His6-Tag. The starting plasmid for this study contained the G46D mutation to inactivate the 5′→3′ exonuclease without any significant effect on polymerase activity 34. For brevity, the N-His6-Tagged G46D genotype present in all mutants of the full-length Taq Pol in this study is not listed, and the mutant proteins are described simply by their cysteine substitutions and/or fluorophore modifications. Based on this recombinant expression plasmid, a truncated form of Taq Pol, Klentaq1 (residues 281–832) with a four amino acid N-terminal extension (MNSQ), was constructed using the Stratagene QuickChange Kit. Using the N-His6-Tagged plasmid as a template, single or double cysteine mutations (Table S1) were introduced individually into the domains of Taq Pol or Klentaq1 by site directed mutagenesis and were confirmed by DNA sequencing. Proteins were purified by nickel affinity and ion exchange chromatography. Protein concentrations were determined spectrophotometrically by the absorbance at 280 nm using the calculated extinction coefficient of 70,360 M−1 cm−1 for Klentaq1 and 11,2760 M−1 cm−1 for Taq Pol. Single cysteine mutants were labeled with Alexa594 (Molecular Probes, Invitrogen) according to the manufacturer's protocol. The labeling efficiencies were typically 90% or greater as determined by absorbance at 280 nm and at 590 nm, the absorption maximum of Alexa 594 (Bio-Rad).
Oligonucleotides (Table 1) were purchased from Integrated DNA Technologies. Alexa488 (Molecular Probes, Invitrogen) was attached to a 5-C6-Amino-2′-deoxythymidine on the ninth base from the primer 3′-end of the DNA substrates. Alexa488-labeled DNA was purified according to the manufacturer's protocol and annealed as described previously 29; 30.
Activity assays with a Taq Pol double cysteine mutant
The percentage of free sulfhydryl group in each mutant was measured by the Ellman's reagent assay according to the manufacturer's protocol (Thermo Scientific). The processivity assays of a double cysteine mutant were performed in buffer R (50 mM Tris-HCl, pH 7.5 at 20 °C, 50 mM NaCl, 2 mM MgCl2, and 10% glycerol) by manual chemical quench. A pre-incubated Taq Pol (100 nM) and DNA (100 nM) mixture was rapidly reacted with four dNTPs (0.2 mM each) or dTTP only (0.2 mM). Aliquots of the reaction mixtures were quenched at various times using 0.37 M EDTA and resolved using sequencing gel electrophoresis (17% acrylamide, 8 M Urea) and visualized using autoradiography (Typhoon TRIO Variable Mode Imager, GE Healthcare). To measure the processivity of the cross-linked double cysteine mutant in the presence of β-ME, the mutant was pre-incubated with 1 mM β-ME at room temperature for one hour in order to reduce the disulfide bond.
Kinetic assays
Steady-state fluorescence assays (Fluoromax-4, Jobin Yvon Horiba), stopped-flow kinetic assays (Applied Photophysics SX20) and rapid chemical-quench kinetic assays (KinTek) were carried out under the same conditions in buffer R. Fluorescence spectra were recorded at the excitation maximum of Alexa488 (493 nm). The apparent FRET efficiency between each Alexa594-labeled Taq Pol residue (acceptor) and the Alexa488-labelled primer base (donor) was calculated using Equation 1:
| (1) |
where ET is the FRET efficiency; FAD and FD are the donor peak fluorescence intensity in the presence and absence of an acceptor, respectively. For stopped-flow experiments, a pre-incubated solution of 200 nM Taq Pol mutant (or 600 nM Klentaq1) and 100 nM DNA was rapidly mixed with 0.5 mM correct dTTP. Upon excitation of the donor Alexa488 at 493 nm, both donor and acceptor fluorescence signals were recorded separately by using band pass filters XF3084 for Alexa488 and XF3028 for Alexa594 (band pass range: 510–570 nm and 615–650 nm respectively, Omega Optical). The stopped-flow fluorescence traces were normalized and reported in arbitrary units with the donor and acceptor traces offset for clarity. The fluorescence changes are shown in arbitrary units. The rates for each fluorescence phase were determined by fitting stopped-flow fluorescence traces using the KinTek Explorer global fitting software 37. The steady-state and single-turnover measurements to determine the steady-state polymerization rate (kss), maximum dNTP incorporation rate (kp) and equilibrium dissociation constant (Kd) were performed as described previously 52. In single-turnover experiments, a plot of product concentration versus reaction time for each nucleotide concentration was fit to Equation 2 using KaleidaGraph (Synergy Software) to obtain an observed rate (kobs),
| (2) |
where A represents the reaction amplitude. The kp and apparent Kd values for a single nucleotide incorporation were then obtained by fitting a plot of the extracted kobs as a function of dNTP concentration to Equation 3,
| (3) |
Data from the steady-state experiment were fit to Equation 4
| (4) |
where kss is the steady-state rate of dNTP incorporation and E0 is the initial active enzyme concentration.
Global data fitting
Data from stopped-flow and 32P-based kinetic assays were fit simultaneously using KinTek explorer software 37 as described previously for HIV-RT1 22. Initial estimates for the kinetic rate constants were determined from the results of the traditional data fitting of individual experiments, and in the global fitting, parameters were identified which reproduced the results of the conventional fitting. Kinetic parameters which were not well defined by the global fitting were not listed in Figure 6E. Notably, the polymerase-DNA binding step (Figure 6E, step 1) was not well constrained by the data, but a rapid equilibrium at this step was necessary to accommodate the results from the steady-state kinetics experiment (Figure 6D) which monitors multiple enzyme turnover events.
Supplementary Material
Highlights.
Taq DNA polymerase (Taq Pol) uses conformational changes during catalysis
Global transition of all five domains of Taq Pol during dNTP incorporation
Conformational dynamics play a critical role in the enzymatic function of Taq Pol
Conformational change kinetics of truncated Taq Pol differ from full length enzyme
All DNA polymerases may use global conformational dynamics during polymerization
Acknowledgement
This work was supported by an National Science Foundation Grant (MCB0960961) and an National Institutes of Health Grant (GM079403) to ZS. B.A.M. was supported by a Presidential Fellowship at The Ohio State University.
Abbreviations
- β-ME
β-mercaptoethanol
- Dpo4
Solfolobus Solfataricus DNA polymerase IV
- KF
Klenow Fragment of Escherichia coli DNA polymerase I
- FRET
Förster resonance energy transfer
- Klentaq1
truncated Thermus aquaticus DNA polymerase I lacking an N-terminal 5′→3′ exonuclease domain
- Taq Pol
Thermus aquaticus DNA polymerase I
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Kornberg A, Baker TA. DNA replication. 2nd edit W.H. Freeman; New York: 1992. [Google Scholar]
- 2.Eom SH, Wang J, Steitz TA. Structure of Taq polymerase with DNA at the polymerase active site. Nature. 1996;382:278–81. doi: 10.1038/382278a0. [DOI] [PubMed] [Google Scholar]
- 3.Tindall KR, Kunkel TA. Fidelity of DNA synthesis by the Thermus aquaticus DNA polymerase. Biochemistry. 1988;27:6008–13. doi: 10.1021/bi00416a027. [DOI] [PubMed] [Google Scholar]
- 4.Xu Y, Grindley ND, Joyce CM. Coordination between the polymerase and 5'-nuclease components of DNA polymerase I of Escherichia coli. J Biol Chem. 2000;275:20949–55. doi: 10.1074/jbc.M909135199. [DOI] [PubMed] [Google Scholar]
- 5.Lyamichev V, Brow MA, Dahlberg JE. Structure-specific endonucleolytic cleavage of nucleic acids by eubacterial DNA polymerases. Science. 1993;260:778–83. doi: 10.1126/science.7683443. [DOI] [PubMed] [Google Scholar]
- 6.Kaiser MW, Lyamicheva N, Ma W, Miller C, Neri B, Fors L, Lyamichev VI. A comparison of eubacterial and archaeal structure-specific 5'-exonucleases. J Biol Chem. 1999;274:21387–94. doi: 10.1074/jbc.274.30.21387. [DOI] [PubMed] [Google Scholar]
- 7.Eger BT, Benkovic SJ. Minimal kinetic mechanism for misincorporation by DNA polymerase I (Klenow fragment) Biochemistry. 1992;31:9227–36. doi: 10.1021/bi00153a016. [DOI] [PubMed] [Google Scholar]
- 8.Kuchta RD, Mizrahi V, Benkovic PA, Johnson KA, Benkovic SJ. Kinetic mechanism of DNA polymerase I (Klenow) Biochemistry. 1987;26:8410–7. doi: 10.1021/bi00399a057. [DOI] [PubMed] [Google Scholar]
- 9.Mizrahi V, Henrie RN, Marlier JF, Johnson KA, Benkovic SJ. Rate-limiting steps in the DNA polymerase I reaction pathway. Biochemistry. 1985;24:4010–8. doi: 10.1021/bi00336a031. [DOI] [PubMed] [Google Scholar]
- 10.Patel SS, Wong I, Johnson KA. Pre-steady-state kinetic analysis of processive DNA replication including complete characterization of an exonuclease-deficient mutant. Biochemistry. 1991;30:511–25. doi: 10.1021/bi00216a029. [DOI] [PubMed] [Google Scholar]
- 11.Wong I, Patel SS, Johnson KA. An induced-fit kinetic mechanism for DNA replication fidelity: direct measurement by single-turnover kinetics. Biochemistry. 1991;30:526–37. doi: 10.1021/bi00216a030. [DOI] [PubMed] [Google Scholar]
- 12.Li Y, Waksman G. Crystal structures of a ddATP-, ddTTP-, ddCTP, and ddGTP- trapped ternary complex of Klentaq1: insights into nucleotide incorporation and selectivity. Protein Sci. 2001;10:1225–33. doi: 10.1110/ps.250101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Y, Korolev S, Waksman G. Crystal structures of open and closed forms of binary and ternary complexes of the large fragment of Thermus aquaticus DNA polymerase I: structural basis for nucleotide incorporation. Embo J. 1998;17:7514–25. doi: 10.1093/emboj/17.24.7514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doublie S, Tabor S, Long AM, Richardson CC, Ellenberger T. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 A resolution. Nature. 1998;391:251–8. doi: 10.1038/34593. [DOI] [PubMed] [Google Scholar]
- 15.Huang H, Chopra R, Verdine GL, Harrison SC. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science. 1998;282:1669–75. doi: 10.1126/science.282.5394.1669. [DOI] [PubMed] [Google Scholar]
- 16.Joyce CM, Potapova O, Delucia AM, Huang X, Basu VP, Grindley ND. Fingers-closing and other rapid conformational changes in DNA polymerase I (Klenow fragment) and their role in nucleotide selectivity. Biochemistry. 2008;47:6103–16. doi: 10.1021/bi7021848. [DOI] [PubMed] [Google Scholar]
- 17.Rothwell PJ, Mitaksov V, Waksman G. Motions of the fingers subdomain of klentaq1 are fast and not rate limiting: implications for the molecular basis of fidelity in DNA polymerases. Mol Cell. 2005;19:345–55. doi: 10.1016/j.molcel.2005.06.032. [DOI] [PubMed] [Google Scholar]
- 18.Purohit V, Grindley ND, Joyce CM. Use of 2-aminopurine fluorescence to examine conformational changes during nucleotide incorporation by DNA polymerase I (Klenow fragment) Biochemistry. 2003;42:10200–11. doi: 10.1021/bi0341206. [DOI] [PubMed] [Google Scholar]
- 19.Allen WJ, Rothwell PJ, Waksman G. An intramolecular FRET system monitors fingers subdomain opening in Klentaq1. Protein Sci. 2008;17:401–8. doi: 10.1110/ps.073309208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsai YC, Johnson KA. A new paradigm for DNA polymerase specificity. Biochemistry. 2006;45:9675–87. doi: 10.1021/bi060993z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson KA. Role of induced fit in enzyme specificity: a molecular forward/reverse switch. J Biol Chem. 2008;283:26297–301. doi: 10.1074/jbc.R800034200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kellinger MW, Johnson KA. Nucleotide-dependent conformational change governs specificity and analog discrimination by HIV reverse transcriptase. Proc Natl Acad Sci U S A. 2010;107:7734–9. doi: 10.1073/pnas.0913946107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rothwell PJ, Waksman G. A pre-equilibrium before nucleotide binding limits fingers subdomain closure by Klentaq1. J Biol Chem. 2007;282:28884–92. doi: 10.1074/jbc.M704824200. [DOI] [PubMed] [Google Scholar]
- 24.Luo G, Wang M, Konigsberg WH, Xie XS. Single-molecule and ensemble fluorescence assays for a functionally important conformational change in T7 DNA polymerase. Proc Natl Acad Sci U S A. 2007;104:12610–5. doi: 10.1073/pnas.0700920104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berezhna SY, Gill JP, Lamichhane R, Millar DP. Single-molecule Forster resonance energy transfer reveals an innate fidelity checkpoint in DNA polymerase I. J Am Chem Soc. 2012;134:11261–8. doi: 10.1021/ja3038273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maxwell BA, Suo Z. Single-molecule Investigation of Substrate Binding Kinetics and Protein Conformational Dynamics of a B-family Replicative DNA Polymerase. J Biol Chem. 2013;288:11590–600. doi: 10.1074/jbc.M113.459982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rothwell PJ, Allen WJ, Sisamakis E, Kalinin S, Felekyan S, Widengren J, Waksman G, Seidel CA. dNTP-dependent conformational transitions in the fingers subdomain of Klentaq1 DNA polymerase: insights into the role of the “nucleotide-binding” state. J Biol Chem. 2013;288:13575–91. doi: 10.1074/jbc.M112.432690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu EY, Beese LS. The structure of a high fidelity DNA polymerase bound to a mismatched nucleotide reveals an “ajar” intermediate conformation in the nucleotide selection mechanism. J Biol Chem. 2011;286:19758–67. doi: 10.1074/jbc.M110.191130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maxwell BA, Xu C, Suo Z. DNA Lesion Alters Global Conformational Dynamics of Y-family DNA Polymerase during Catalysis. J Biol Chem. 2012;287:13040–7. doi: 10.1074/jbc.M112.345835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu C, Maxwell BA, Brown JA, Zhang L, Suo Z. Global conformational dynamics of a Y-family DNA polymerase during catalysis. PLoS Biol. 2009;7:e1000225. doi: 10.1371/journal.pbio.1000225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kirmizialtin S, Nguyen V, Johnson KA, Elber R. How conformational dynamics of DNA polymerase select correct substrates: experiments and simulations. Structure. 2012;20:618–27. doi: 10.1016/j.str.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bu Z, Biehl R, Monkenbusch M, Richter D, Callaway DJ. Coupled protein domain motion in Taq polymerase revealed by neutron spin-echo spectroscopy. Proc Natl Acad Sci U S A. 2005;102:17646–51. doi: 10.1073/pnas.0503388102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barnes WM. The fidelity of Taq polymerase catalyzing PCR is improved by an N-terminal deletion. Gene. 1992;112:29–35. doi: 10.1016/0378-1119(92)90299-5. [DOI] [PubMed] [Google Scholar]
- 34.Brandis JW, Edwards SG, Johnson KA. Slow rate of phosphodiester bond formation accounts for the strong bias that Taq DNA polymerase shows against 2',3'-dideoxynucleotide terminators. Biochemistry. 1996;35:2189–200. doi: 10.1021/bi951682j. [DOI] [PubMed] [Google Scholar]
- 35.Ma WP, Kaiser MW, Lyamicheva N, Schaefer JJ, Allawi HT, Takova T, Neri BP, Lyamichev VI. RNA template-dependent 5' nuclease activity of Thermus aquaticus and Thermus thermophilus DNA polymerases. J Biol Chem. 2000;275:24693–700. doi: 10.1074/jbc.M002268200. [DOI] [PubMed] [Google Scholar]
- 36.Lyamichev V, Brow MA, Varvel VE, Dahlberg JE. Comparison of the 5' nuclease activities of taq DNA polymerase and its isolated nuclease domain. Proc Natl Acad Sci U S A. 1999;96:6143–8. doi: 10.1073/pnas.96.11.6143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson KA, Simpson ZB, Blom T. Global Kinetic Explorer: A new computer program for dynamic simulation and fitting of kinetic data. Analytical Biochemistry. 2009;387:20–29. doi: 10.1016/j.ab.2008.12.024. [DOI] [PubMed] [Google Scholar]
- 38.Woolley GA, Lee ES, Zhang F. sGAL: a computational method for finding surface exposed sites in proteins suitable for Cys-mediated cross-linking. Bioinformatics. 2006;22:3101–2. doi: 10.1093/bioinformatics/btl530. [DOI] [PubMed] [Google Scholar]
- 39.Kim Y, Eom SH, Wang J, Lee DS, Suh SW, Steitz TA. Crystal structure of Thermus aquaticus DNA polymerase. Nature. 1995;376:612–6. doi: 10.1038/376612a0. [DOI] [PubMed] [Google Scholar]
- 40.Urs UK, Murali R, Krishna Murthy HM. Structure of taq DNA polymerase shows a new orientation for the structure-specific nuclease domain. Acta Crystallogr D Biol Crystallogr. 1999;55:1971–7. doi: 10.1107/s090744499901135x. [DOI] [PubMed] [Google Scholar]
- 41.Joubert AM, Byrd AS, LiCata VJ. Global conformations, hydrodynamics, and X-ray scattering properties of Taq and Escherichia coli DNA polymerases in solution. J Biol Chem. 2003;278:25341–7. doi: 10.1074/jbc.M302118200. [DOI] [PubMed] [Google Scholar]
- 42.Ho DL, Byrnes WM, Ma WP, Shi Y, Callaway DJ, Bu Z. Structure-specific DNA-induced conformational changes in Taq polymerase revealed by small angle neutron scattering. J Biol Chem. 2004;279:39146–54. doi: 10.1074/jbc.M404565200. [DOI] [PubMed] [Google Scholar]
- 43.Joyce CM, Benkovic SJ. DNA polymerase fidelity: kinetics, structure, and checkpoints. Biochemistry. 2004;43:14317–24. doi: 10.1021/bi048422z. [DOI] [PubMed] [Google Scholar]
- 44.Johnson SJ, Taylor JS, Beese LS. Processive DNA synthesis observed in a polymerase crystal suggests a mechanism for the prevention of frameshift mutations. Proc Natl Acad Sci U S A. 2003;100:3895–900. doi: 10.1073/pnas.0630532100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Temiakov D, Patlan V, Anikin M, McAllister WT, Yokoyama S, Vassylyev DG. Structural basis for substrate selection by t7 RNA polymerase. Cell. 2004;116:381–91. doi: 10.1016/s0092-8674(04)00059-5. [DOI] [PubMed] [Google Scholar]
- 46.Rothwell PJ, Berger S, Kensch O, Felekyan S, Antonik M, Wohrl BM, Restle T, Goody RS, Seidel CA. Multiparameter single-molecule fluorescence spectroscopy reveals heterogeneity of HIV-1 reverse transcriptase:primer/template complexes. Proc Natl Acad Sci U S A. 2003;100:1655–60. doi: 10.1073/pnas.0434003100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wohrl BM, Krebs R, Goody RS, Restle T. Refined model for primer/template binding by HIV-1 reverse transcriptase: pre-steady-state kinetic analyses of primer/template binding and nucleotide incorporation events distinguish between different binding modes depending on the nature of the nucleic acid substrate. J Mol Biol. 1999;292:333–44. doi: 10.1006/jmbi.1999.3057. [DOI] [PubMed] [Google Scholar]
- 48.Sarafianos SG, Clark AD, Jr., Das K, Tuske S, Birktoft JJ, Ilankumaran P, Ramesha AR, Sayer JM, Jerina DM, Boyer PL, Hughes SH, Arnold E. Structures of HIV-1 reverse transcriptase with pre- and post-translocation AZTMP-terminated DNA. Embo J. 2002;21:6614–24. doi: 10.1093/emboj/cdf637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prasad BR, Kamerlin SCL, Florian J, Warshel A. Prechemistry barriers and checkpoints do not contribute to fidelity and catalysis as long as they are not rate limiting. Theor Chem Acc. 2012;131:1288. [Google Scholar]
- 50.Mulholland AJ, Roitberg AE, Tunon I. Enzyme dynamics and catalysis in the mechanism of DNA polymerase. Theor Chem Acc. 2012;131:1286. [Google Scholar]
- 51.Schlick T, Arora K, Beard WA, Wilson SH. Perspective: pre-chemistry conformational changes in DNA polymerase mechanisms. Theor Chem Acc. 2012;131:1287. doi: 10.1007/s00214-012-1287-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maxwell BA, Suo Z. Kinetic Basis for the Differing Response to an Oxidative Lesion by a Replicative and a Lesion Bypass DNA Polymerase from Solfolobus solfataricus. Biochemistry. 2012;51:3485–96. doi: 10.1021/bi300246r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.