

Abstract
Little is understood about the occurrence of somatic genomic alterations in normal tissues, and their significance in the context of diseases. Here we identified potential somatic copy number alterations (pSCNA) in apparently normal ovarian tissue and peripheral blood of 423 ovarian cancer patients. There were on average 2–4 pSCNAs per sample detectable at a tissue-level resolution, although some individuals had orders of magnitude more. Accordingly, we estimated the lower bound of the rate of pSCNAs per cell division. Older individuals and BRCA mutation carriers had more pSCNAs than others. pSCNAs significantly overlapped with Alu and G-quadruplexes, and the affected genes were enriched for signaling and regulation. Some of the amplification/deletion hotspots in pan-cancer genomes were hotspots of pSCNAs in normal tissues as well-suggesting that those regions might be inherently unstable. Prevalence of pSCNA in peripheral blood predicted survival, implying that mutations in normal tissues might have consequences for cancer patients.
Introduction
Starting at fertilization of the egg, during the course of development and aging, somatic cells accumulate mutations in their genome. Although somatic mutations have been predominantly studied in the context of cancer and aging, increasing evidence suggests that apparently normal cells also carry a considerable burden of somatically acquired mutations, and those mutations might have subtle phenotypic consequences (De, 2011; Poduri et al., 2013; Youssoufian and Pyeritz, 2002). For instance, somatic mutations can contribute to disease onset and ‘missing heritability’ in some complex diseases (Bonnefond et al., 2013; De, 2011; Manolio et al., 2009). The aging-associated burden of somatic mutations is expected to decrease the overall fitness of cells in somatic tissues, facilitating selection for neoplastic cells and increasing cancer incidence in older individuals (DeGregori, 2013). Indeed, two recent population genetics studies by Jacobs et al. and Laurie et al. have shown that detectable clonal mosaicism is linked to cancer risk and aging (Jacobs et al., 2012; Laurie et al., 2012). Although individual somatic cells in a tissue harbor diverse genetic changes, those that are detected at tissue-level i.e. present in a considerable fraction of cells are expected to have noticeable consequences. How common are these somatic mutations? By widely accepted estimates, somatic cells accumulate 10−7 – 10−8 point mutations per base per generation (Araten et al., 2005; Campbell and Eichler, 2013; Lupski, 2007). It was recently suggested that half or more of the point mutations in cancers of self-renewing tissues might originate prior to tumor initiation (Tomasetti et al., 2013). And yet, there are only limited estimates (Jacobs et al., 2012; Laurie et al., 2012; Pham et al., 2014) of the prevalence of other classes of somatic genomic alterations such as amplifications and deletions, available for apparently normal tissue types. Moreover, the effects of somatic genomic alterations in apparently normal tissue in the context of diseases such as cancer are poorly understood. Recently large-scale cancer genomics initiatives (Collins and Barker, 2007; Kanchi et al., 2014; TCGA, 2011, 2012; Zack et al., 2013) have opened up opportunities to test such hypothesis.
Here, we have carried out a large scale, genome-wide survey of potential somatic amplifications and deletions in apparently normal tissues (pSCNAsnorm) of patients with cancer, and assessed their significance towards disease outcome. We chose to focus on the pSCNAsnorm that are detectable by microarrays at tissue-level resolution. We map these genomic changes in apparently normal peripheral blood and ovarian tissue in a large cohort of ovarian cancer patients (TCGA, 2011) by comparing pairs of tumor and matched normal genomes, and (i) provide an estimate of the prevalence of pSCNAsnorm, identifying specific patterns associated with age or germ line BRCA mutations, (ii) study the genomic context of these pSCNAsnorm, (iii) compare and contrast the genome-wide patterns of somatic copy number alterations in normal (pSCNAsnorm) and cancer genomes, and (iv) evaluate whether the burden of somatic mutations in apparently normal tissue predict tumor progression and survival in the same individual.
Results
We obtained genomic and clinical data for 423 ovarian cancer patients from the Cancer Genome Atlas (TCGA, 2011), and inferred the pSCNAsnorm by comparing the paired normal and tumor genomes, after adopting appropriate quality control steps to exclude false positives and remove technical artifacts (Methods and Supplementary Module 1). These pSCNAsnorm were detectable at a tissue-level resolution, indicting either early developmental origin, selection for these genomic alterations, or the effects of random drift. Our final dataset had 279 potential somatic amplifications (pAmpnbl) and 328 potential somatic deletions (pDelnbl) in 314 normal peripheral blood samples (collectively referred to as pSCNAnbl), and 137 potential somatic amplifications (pAmpnov) and 357 potential somatic deletions (pDelnov) in 109 normal ovarian tissue samples (collectively referred to as pSCNAnov).
Prevalence of potential somatic amplifications and deletions
We found that there were typically 4 and 2 detectable pSCNAnorm per ovarian tissue and peripheral blood sample, respectively, although the number of pSCNAnorm varied over two orders of magnitude between the samples (ovary: max:122, min:0; blood: max:53, min:0). The number of pSCNAnorm per sample followed Poisson distributions (pSCNAnov ~Pois(λ =4.53) in ovary; pSCNAnbl ~Pois(λ =1.93) in peripheral blood). The numbers of amplifications and deletions per sample were generally comparable. Anyhow, these are probably very conservative estimates of the number of somatic genomic alterations, since we were unable to detect all pSCNAnorm using our approach (see Methods for details). We also note the potential caveats in Supplementary Module 2. We predicted a parsimonious estimate of the lower bound of the rate of somatic genomic alterations in peripheral blood is 10−5 – 10−6 per locus per somatic cell division (depending on the model chosen, Method, Supplementary Module 2), which is comparable to the germ line estimates (10−4 – 10−6 per locus per generation (Campbell and Eichler, 2013; Lupski, 2007)), and those derived from single cell genome sequencing of cancer cells ((Voet et al., 2013) and Supplementary Module 2).
The burden of detectable pSCNAs increases with age
Integrating the tissue-level pSCNAnbl data together with the age of the patients (TCGA, 2011) (who had no BRCA mutations) we found that the older patients on average had more potential genomic alterations than the younger patients (Figure 1C). For instance, the patients of age 70 years and above had significantly more pSCNAnbl compared to those of age less than 40 years, (Mann Whitney U test; p-value 3.93×10−2); the trend was not apparent between 40 and 70 years. We find similar results for potential somatic amplifications and deletions, independently (Supplementary Module 3). The number of BRCA mutation carriers was too small to warrant a similar analysis only on this select group of patients. In any case, our results concur with recent reports (Jacobs et al., 2012; Laurie et al., 2012), and show that the burden of amplifications and deletions increases with age, a trend that is similar to that reported for point mutations, loss of heterozygosity, and ploidy changes (Maslov et al., 2013; Matsuo et al., 1982; Pedersen et al., 2013a; Tomasetti et al., 2013; Vogelstein et al., 2013). Age-dependent increases in genomic changes could reflect the occurrence of new mutations, alterations in selection (positive selection for some changes and/or reduced purifying selection against others), and/or bottlenecks that lead to reduced clonal diversity.
Figure 1.

(A) The pipeline for detecting of somatic amplifications and deletions in apparently normal peripheral blood and ovarian cancer tissue of the TCGA ovarian cancer patients. (B) Summary statistics of somatic amplifications and deletions in peripheral blood and ovarian cancer tissue in the cohort. The BRCA mutation carriers are shown categorically. (C-E) The number of somatic genomic alterations (amplifications and deletions) per (C) peripheral blood sample, grouped according to the age of the individuals, (D) peripheral blood sample, grouped according to BRCA mutation status, and (E) ovarian tissue sample, grouped according to BRCA mutation status. The horizontal line shows the median value across all the samples in respective panel.
BRCA mutation carriers harbor more potential somatic amplifications and deletions
BRCA mutation carriers are at a higher risk of several different types cancer (Friedenson, 2007; Moran et al., 2012). To test whether BRCA mutation carriers have more genomic alterations in apparently normal tissue than non-carriers, we grouped the TCGA samples based on their BRCA mutation status (TCGA, 2011) as those with (i) BRCA (BRCA1 or BRCA2) germ line mutations, (ii) BRCA somatic mutations, and (iii) no BRCA mutations. Comparing the number of pSCNAnbl between the three groups, we found that the number of detectable pSCNAnbl is significantly higher in the samples with BRCA germ line mutation compared to those with no mutations (Mann Whitney U test; p-value: 1.61×10−2). In contrast, individuals with somatic BRCA mutations in their ovarian cancer did not exhibit any increase in pSCNAnbl compared to individuals with no BRCA mutations. We found similar results in ovarian tissue – the number of pSCNAnov was higher in the germline BRCA mutation carriers compared to those with no BRCA mutations, but the statistical significance was modest due to small sample size (Mann Whitney U test; p-value>0.05). We found consistent results when repeating the analysis after grouping the samples by age, and also when analyzing pAmpnbl, pDelnbl, pAmpnov, and pDelnov separately (Supplementary Module 3). Taken together, on average, BRCA mutation carriers harbor more potential somatic amplifications and deletions in apparently normal tissues compared to those with no BRCA mutations. Our findings are consistent with the report that BRCA1 haploinsufficiency promotes genomic instability in non-malignant cells (Konishi et al., 2011), and provide a plausible explanation for higher prevalence of several different cancers in BRCA mutation carriers (Friedenson, 2007; Moran et al., 2012).
Genomic context of potential somatic amplifications and deletions
Next, we generated a genome-wide map of pSCNAnov and pSCNAnbl as shown in Figure 2A-B. Although the pSCNAsnorm were found throughout the genome, some regions had recurrent pSCNAsnorm (clustered in megabase-scale regions). For instance, chr1q32 and chr7q34 had recurrent deletions in both peripheral blood and ovarian tissue, and the trend was independent of BRCA mutation status. Chr14q11.2, which is close to the centromere, had a striking excess of amplifications in ovarian tissue. In contrast, chr7-telomere proximal regions had frequent deletions only in the peripheral blood of BRCA mutation carriers. While most of the individuals had small number of detectable pSCNAnorm, some others had considerable numbers of such events (see Supplementary Module 4 for specific examples). Several of these candidate regions also had similar patterns of copy number alterations in single cell sequencing data (HCC38 cell line; Supplementary Figure 4 of Voet et al.).
Figure 2.

Genome-wide mutational landscape showing pSCNAnorm ( amplifications: blue, deletions: red) in apparently normal (A) ovarian cancer tissue and (B) peripheral blood of the ovarian cancer patients. Each row represents an individual. Chromosomes are indicated below. Faint vertical lines in each chromosome indicate centromere. Only the individuals with at least one detectable pSCNAnorm in the cohort are shown. The individuals with germ line BRCA1 or BRCA2 mutations are shown separately. (C) Comparing the mutation landscape of apparently normal peripheral blood with that of 26 different cancer types combined, as analyzed by Beroukhim et al. Nature. 2010. The balance of blue and red shades indicates the proportion of amplifications and deletions. Heights of the bars indicate prevalence of such events in the genome.
We then surveyed the genomic context of the pSCNAsnorm. We overlaid several different genomic features (Table 1), calculated the overlap with the pSCNAsnorm and then compared the observed overlap with that expected by chance using permutation (Methods). We found that pSCNAsnorm were slightly GC rich compared to the genome-wide average; moreover, the pSCNAsnorm showed enrichment for potential G-quadruplex motifs and Alu elements, but were depleted in evolutionarily conserved and L2 elements (Table 1, Supplementary Module 4). Some of these trends were significant for potential somatic amplifications or deletions only. Given the challenges while combining heterogeneous data types and designing the ideal null model for estimating statistical significance (De et al., 2013), we cautiously interpret the data. Nevertheless, our findings are consistent with the reports that G4 motifs are frequently associated with genomic alterations (Maizels and Gray, 2013; Tarsounas and Tijsterman, 2013), and that Alu and L1 elements are active during early development and contribute to mosaicism (Kano et al., 2009; Macia et al., 2011; van den Hurk et al., 2007). However, pSCNAsnorm, unlike genomic alterations found in cancer genomes (De and Michor, 2011; Durkin and Glover, 2007; Pedersen and De, 2013), did not show any significant preference for fragile sites. That led us to compare the genomic landscape of somatic amplifications and deletions between normal and tumor genomes.
Table 1.
Overlap between different genomic features and pSCNAsnorm. For more details, please see Supplementary Figure SF4.2.
| Genomic feature | Data source | Enrichment | q-value of enrichment |
|---|---|---|---|
| 28 way conserved elements | UCSC Genome Browser (Miller et al. 2007) | enriched | <0.05 |
| L2 elements | UCSC Genome Browser (Meyer et al. 2013) | enriched | <0.05 |
| Alu elements | UCSC Genome Browser (Meyer et al. 2013) | depleted | <0.05 |
| G quadruplex motifs | (Hupert and Balasubramanian, 2005) | depleted | <0.05 |
| L1 elements | UCSC Genome Browser (Meyer et al. 2013) | - | >0.05 |
| GC content | UCSC Genome Browser (Meyer et al. 2013) | - | >0.05 |
| Protein coding genes | ENSEML (Flicek et al. 2014) | - | >0.05 |
| Constant early replicating regions | (Hansen et al. 2010) | - | >0.05 |
| Constant late replicating regions | (Hansen et al. 2010) | - | >0.05 |
| Early replicating fragile sites | (Barlow et al. 2013) | - | >0.05 |
| Common fragile sites | (Durkin and Glover, 2007) | - | >0.05 |
Comparing mutational landscapes of tumor and matched normal genomes
We overlaid the sites of frequent amplifications (pAmp) and deletions (pDel) in somatic tissues in our analysis (e.g. chr1q32, chr3q29, chr7q34, chr14q11.2, chr15q11.2, chr17q21) with the pan-cancer GISTIC peaks (sites of significantly recurrent amplifications and deletions in multiple cancer types; Figure 2C), which were identified based on nearly 3000 samples from 26 cancer types (Beroukhim et al., 2010). We found that chr7q34, which harbor T cell receptor locus, was a site of recurrent deletion both in tumor and apparently normal tissue (peripheral blood and also ovarian tissue). Error-prone DNA repair could be a source of genomic instability in these regions. Several of the other sites of frequent amplifications (pAmp) and deletions (pDel) in somatic tissues were proximal to minor GISTIC peaks. For instance, the pan-cancer GISTIC peaks at chr1q32, chr14q11.2, and chr15q11.2 map to cancer associated genes such as MDM4, BCL2L2, and A26B1, while the chr17q21 GISTIC peak marked NGFR, PHB, and CNP (TCGA, 2011). While none of these genes was recurrently affected by pSCNAnorm, frequent genomic alterations in their genomic neighborhood in apparently normal samples might reflect signatures of selection or inherent genomic instability present in these regions.
Genes recurrently amplified or deleted in multiple subjects
Several genes were affected by pSCNAsnorm in multiple individuals as shown in Table 2. The affected genes were enriched for signaling and regulation related function (Hypergeometric test; p-value <0.05); the statistical significance was modest due to the size of the dataset. For instance, PPP1R12B, which encodes for a myosin phosphatase and play a role in interleukin signaling pathway (Bannert et al., 2003) was hemizygously deleted in more than 8% of the normal peripheral blood and ovarian tissue samples. Deletion of complement factor CFHR1 and CFHR3 is known to be associated with defective complement regulation in blood, atypical hemolytic uremic syndrome, and macular degeneration (Hughes et al., 2006; Zipfel et al., 2007). Notably, these genes are consistently deleted, not amplified (FDR corrected p-value using Binomial test in blood; PPP1R12B: 6.81×10−9, CFHR3: 5.65×10−4, CFHR1: 5.6×10−4; Table 2), raising the possibility that these genes might be under directional selection in apparently normal peripheral blood. DUSP22 is a phosphatase known to interact with MAP kinase pathway (Aoyama et al., 2001), and implicated in different cancer types including hematopoietic malignancies. ZBTB34 is a transcriptional repressor broadly expressed in many tissue types and might have a role in recruitment of HDAC (Qi et al., 2006). Protocadherin PCDHA13 plays a role in cell adhesion and signaling. Several other genes that were affected by pSCNAsnorm in >1% of the samples (e.g. LRP5L, PPYR1, SIRPB1 etc) are also involved in signaling. Further work is necessary to ascertain the consequences of these genetic changes in the normal peripheral blood and ovarian tissue, and also to assess whether impaired function of these genes affect tumor-related inflammation response, tumor-maintenance and growth signals.
Table 2. The list of genes affected by recurrent events of somatic amplifications and deletions in apparently normal peripheral blood and ovarian tissue.
Several of these genes are in the defensin gene cluster.
| Gene name | Chromosomal position | Chromosome band | Number of samples with pSCNA | |
|---|---|---|---|---|
| Total | (pAmpbl,pDelbl), (pAmpov,pDelov) | |||
| PPP1R12B | chr1:200584459-200824320:1 | chr1q32.1 | 34 | (0, 26), (0, 9) |
| CFHR3 | chr1:195010553-195031160:1 | chr1q31.3 | 16 | (1, 13), (0, 2) |
| DUSP22 | chr6:237101-296353:1 | chr6p25.3 | 15 | (2, 9), (2, 3) |
| CFHR1 | chr1:195055484-195067940:1 | chr1q31.3 | 12 | (0, 10), (0, 2) |
| ZBTB34 | chr9:128662765-128687978:1 | chr9q33.3 | 7 | (4, 0), (0, 3) |
| PCDHA13 | chr5:140215818-140372113:1 | chr5q31.3 | 7 | (0, 4), (3, 0) |
| LRP5L | chr22:24077424-24131324:-1 | chr22q11.23 | 7 | (4, 0), (3, 0) |
| SIRPB1 | chr20:1493029-1548689:-1 | chr20p13 | 6 | (2, 1), (3, 0) |
| PPYR1 | chr10:46503540-46508326:1 | chr10q11.22 | 6 | (5, 0), (1, 0) |
| NUP210L | chr1:152231790-152394216:-1 | chr1q21.3 | 6 | (0, 2), (0, 4) |
| IGHV1-68 | chr14:106230914-106231208:-1 | chr14q32.33 | 6 | (5, 0), (1, 0) |
| GSTT1 | chr22:22706142-22714271:-1 | chr22q11.23 | 6 | (4, 0), (2, 0) |
| SPAG11 | chr8:7292686-7308602:-1 | chr8p23.1 | 5 | (4, 1), (0, 0) |
| IGHVII-40-1 | chr14:105967906-105967963:-1 | chr14q32.33 | 5 | (0, 5), (0, 0) |
| IGHV3-41 | chr14:105970089-105970538:-1 | chr14q32.33 | 5 | (0, 5), (0, 0) |
| FAM90A7 | chr8:7401070-7406305:-1 | chr8p23.1 | 5 | (4, 1), (0, 0) |
| FAM90A23 | chr8:7424014-7429245:-1 | chr8p23.1 | 5 | (4, 1), (0, 0) |
| FAM90A22 | chr8:7416365-7421601:-1 | chr8p23.1 | 5 | (4, 1), (0, 0) |
| DEFB4 | chr8:7789609-7791647:1 | chr8p23.1 | 5 | (4, 1), (0, 0) |
| DEFB107B | chr8:7340778-7354243:1 | chr8p23.1 | 5 | (4, 1), (0, 0) |
| DEFB107A | chr8:7706652-7710648:-1 | chr8p23.1 | 5 | (4, 1), (0, 0) |
| DEFB106B | chr8:7327436-7331319:-1 | chr8p23.1 | 5 | (4, 1), (0, 0) |
| DEFB106A | chr8:7720104-7723985:1 | chr8p23.1 | 5 | (4, 1), (0, 0) |
| DEFB105B | chr8:7332649-7334483:1 | chr8p23.1 | 5 | (4, 1), (0, 0) |
| DEFB105A | chr8:7716940-7718774:-1 | chr8p23.1 | 5 | (4, 1), (0, 0) |
| DEFB104B | chr8:7315236-7320014:-1 | chr8p23.1 | 5 | (4, 1), (0, 0) |
| DEFB104A | chr8:7731403-7736178:1 | chr8p23.1 | 5 | (4, 1), (0, 0) |
| DEFB103B | chr8:7776136-7777515:1 | chr8p23.1 | 5 | (4, 1), (0, 0) |
| DEFB103A | chr8:7273901-7275280:-1 | chr8p23.1 | 5 | (4, 1), (0, 0) |
Patients with more pSCNAs have poor survival
It is poorly understood whether somatic genomic alterations in apparently normal tissue can impact cancer development (e.g. as by influencing selection for driver mutations or otherwise influencing evolutionary trajectories of cancer genomes), which would influence overall patient survival. Somatic genomic alterations in normal tissues could be symptomatic of systemic issues, from generalized genomic instability, immune dysfunction to impaired tissue fitness, which could substantially alter cancer evolution. The frequencies of genomic alterations in normal tissue did not immediately predict the burden of genomic alterations (i.e. point mutations, copy number alterations, and LOH events; Supplementary Module 5) in the matched tumor genomes. However, analyzing somatic point mutations of known cancer genes, we found that the individuals with no detectable pSCNAnbl in peripheral blood had more cancer gene mutations compared to those with a higher frequency (≥4 pSCNAnbl; p-value: 5.85×10−2) of detectable pSCNAnbl. Moreover, mutations in RB1, MLL3 and CREBBP were present in >5% of the ovarian tumor samples with no detectable pSCNAnbl, but rarely occurred in the tumor samples with an excess of pSCNAsnbl (Figure 3A; sample size is insufficient for statistical testing). The results were consistent irrespective of the cut-off chosen and other potential covariates (Supplementary Module 5).
Figure 3.
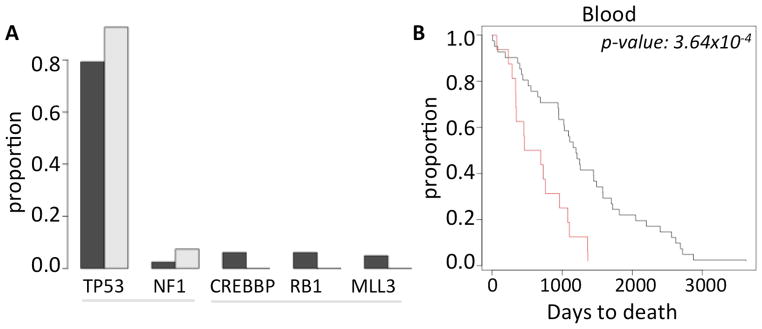
(A) Frequency of the common cancer gene mutations in ovarian cancer patients who had no pSCNAbl (black) and those who have ≥4 pSCNAbl (grey). (B) Kaplan Meier curve showing difference in the survival patterns between the ovarian cancer patients who have no pSCNAbl (black) and those who have ≥4 pSCNAbl (red). The difference was statistically significant ( log rank test, p-value 3.64×10−4).
We then compared the survival characteristics of the ovarian cancer patients with high number of amplifications and deletions in apparently normal peripheral blood (pSCNAnbl ≥4) against those who have apparently more normal, diploid genome (no detectable pSCNAnbl), after excluding the patients with BRCA mutations. There was a significant negative correlation between the number of pSCNAnbl events and survival (Spearman rank correlation coefficient: -0.20; p-value: 1.92×10−2). Using Kaplan Meier survival analysis, we found that the patients with ≥4 pSCNAsnorm in peripheral blood had significantly shorter survival (log rank test; p-value: 3.64×10−4; Figure 3B) compared to those with no pSCNAs detected in blood. The results were not biased by age, stage, tumor purity, and remained consistent for alternative pSCNA thresholds (Supplementary Module 5). Taken together, our findings suggest that the burden of somatic amplifications and deletions in normal peripheral blood predicts clinical outcome.
Analysis of TCGA lung cancer dataset
We extended the key analyses to the TCGA lung squamous cell carcinoma dataset (TCGA, 2012). There were 110 samples that had copy number status interrogated using two independent centers; we analyzed these samples in our study. Of these, 18 and 92 samples had peripheral blood and lung tissue as the matched normal tissues, respectively. We found that, the number of somatic genomic alterations detectable at a tissue-level resolution was comparable to that reported in Figure 1, although slightly lower; this is probably due to the fact that the patients in this cohort are relatively younger compared to the ovarian cancer cohort (Supplementary Module 6). Furthermore, the number of pSVNAnorm increased with age, and some genomic regions (e.g. chr1q32, chr15q11, and chr17q21) had clusters of pSCNAnorm in this cohort as well (Supplementary Module 6).
Discussion
Our analysis provides one of the first generation surveys of the patterns of somatic amplifications and deletions in apparently normal human tissue types of patients with cancer. We found, on average 2–4 potential somatic amplifications and deletions per normal sample detectable at a tissue-level resolution, although there were considerable inter-individual variations. The burden of such genomic changes increased with age, and BRCA mutation carriers harbored such events at a greater frequency than non-carriers. It is possible that germ-line mutations in other genes (e.g. ATM, RAD51, TP53) also increase the prevalence of potential somatic genomic alterations in apparently normal tissues, but the TCGA dataset was not suitable to examine that possibility systematically. Some genomic regions have clusters of recurrent genomic alterations, the footprint of which could be found even in the genomes of several different types of cancer – indicating that genomic alterations in these regions might predate tumor initiation. Many of the genes affected by recurrent somatic amplifications and deletions (pSCNAsnorm) were associated with signaling and regulation. Interestingly, the frequency of pSCNAsnorm significantly predicted survival patterns of these cancer patients. We propose that somatic genomic are common in apparently normal tissues, and have implications for complex diseases.
We note the advantages and potential caveats of our study design to provide a balanced perspective. We chose to focus on the pSCNAsnorm that are detectable at tissue-level resolution, since these genetic changes might have noticeable effects on tissue-level function. The copy number calls using two independent arrays led to detection of high confidence pSCNAnorm events, but those with inconsistent aCGH calls were missed. Additionally, we could not detect the events that were small (~102 bp or smaller), had low signal to noise ratio, occurred in a minor subpopulation of cells, or were masked by copy number changes in matched tumor samples. Therefore, the frequencies of somatic genomic alterations reported here probably represent the lower bound of such events in apparently normal tissues. Future assessments based on genome sequencing would be able to overcome many of these limitations. Even then, our estimates of the absolute frequency and the rate of such events per locus per somatic cell division were consistent with that reported elsewhere, including single cell-based estimates (Campbell and Eichler, 2013; Jacobs et al., 2012; Laurie et al., 2012; Lupski, 2007; Voet et al., 2013). We could not determine whether the pSCNAsnorm represent different subclones leading to genetic heterogeneity in the normal tissue samples. While the genomic alterations in peripheral blood were accumulated over the lifetime of the individual, those in the ovarian tissue were accrued after the time of separation of the normal and tumor stem cell, which is unknown and could precede or follow the first driver event in tumorigenesis. Therefore, we recommend caution when analyzing and interpreting the pSCNAsnov data.
Our findings concur with the emerging concept that apparently normal somatic tissues also accumulate considerable burden of somatic mutations (Abyzov et al., 2012; Biesecker and Spinner, 2013; De, 2011), and that genomic alterations in some chromosomal regions might predate tumor initiation (Konishi et al., 2011; Tomasetti et al., 2013). We found that the pSCNAsnorm were enriched for G4 motifs and Alu elements (and also showed weak preference for L1 elements), but depleted for evolutionarily conserved elements. Both L1 and Alu elements are known to be active in embryonic stem cell and during early development in the human genome, (Macia et al., 2011; van den Hurk et al., 2007), giving rise to mosaicism (De, 2011; Kano et al., 2009). Alu retrotransposition is mediated by active L1 elements, suggesting that these two mutagenic processes in somatic cells could be linked (Dewannieux et al., 2003). Emerging reports show that G4 structures are stable and detectable in the human genome (Lam et al., 2013), and that these elements play roles in genomic alterations (Kruisselbrink et al., 2008; Maizels and Gray, 2013; Tarsounas and Tijsterman, 2013). In the light of these reports, it is tempting to propose the likely origins of the pSCNAsnorm; but further work needs to be done to infer causality beyond correlation. During development and aging, random drift can also lead to a scenario where a small number of clones contribute to the bulk of the cells, as reported elsewhere (Clemente et al., 2011; Elson et al., 2001). Given their poor overlap with evolutionarily conserved elements, most of the pSCNAsnorm, especially those outside the hotspots, are expected to be neutral. Recurrent genomic changes, especially those that are detectable at a tissue-level resolution (e.g. pSCNAnbl) and occur near genes involved in signaling and regulation, might indicate potential natural selection promoting the clones that harbor these changes. Of course, these different factors can also operate in combination to lead to the observed genome-wide patterns. In any case, the fact that some of the genomic regions amplified and deleted in tumor samples were also recurrently and independently altered in normal somatic tissue is likely to bring new challenges for diagnosis, drug development and prognosis. For instance, in liquid biopsies it might introduce additional difficulties to ascertain the cell of origin of the copy of cell-free DNA carrying certain mutations. Furthermore, our findings raise a fresh debate regarding what should be considered as a reference normal tissue.
While decades of research has predominantly focused on tumor cells or microenvironment in their immediate vicinity, we present a provocative hypothesis that genomic landscape of apparently normal tissue such as peripheral blood might also have implications for the course of tumor progression and associated clinical outcome. Individuals with more somatic genomic alterations are at a greater cancer risk as reported elsewhere (Jacobs et al., 2012; Laurie et al., 2012), and have poorer survival than others, as shown here. One might argue that increased frequencies of pSCNAs in normal tissues reflect a general genomic instability (e.g. in ovary in the current study), which impacts tumor development; e.g. increased cancer risk and an excess of somatic genomic alterations in apparently normal tissue in BRCA mutation carriers (Friedenson, 2007; Konishi et al., 2011; Moran et al., 2012) support this concept. Another possible explanation for the correlation between pSCNAnorm and the cancer phenotype could be that abnormal signaling/function in blood or peripheral tissue might impair normal anti-cancer defenses (such as immunity) (Hanahan and Weinberg, 2011); genomic alterations involving signaling genes in peripheral blood are consistent with this idea. Finally, an alternative (and not necessarily mutually exclusive) hypothesis is that increased frequencies of pSCNAnorm reflect reductions in the overall fitness of somatic tissues, which can increase selection for particular adaptive mutations, thus facilitating clonal selection for neoplastic cells with these adaptive driver mutations (contributing to poorer survival and increased incidence of cancer in older individuals) (DeGregori, 2011, 2013)). We suspect that any individual patient probably experiences a combination of these effects. Taken together, our findings suggest that somatic amplifications and deletions are common in apparently normal human tissues, and can have consequences for complex diseases such as cancer.
Methods
Datasets
Data on genomic alterations and clinical parameters for the lung and ovarian cancer patients were obtained from the Cancer Genome Atlas (TCGA) (TCGA, 2011, 2012). In the TCGA initiative, copy number status for ovarian tumor-normal pairs was determined using different arrays in two genome analysis centers: Agilent HG-CGH-415K_G4124A and HG-CGH-244A arrays at Harvard Medical School, and Agilent CGH-1x1M_G4447A array at MSKCC. We analyzed the aCGH-based copy number status of 423 serous ovarian cancer samples and matched normal tissues (healthy ovarian tissue or peripheral blood), for which copy number calls were available using two independent arrays. Of them, 109 and 314 samples had normal ovarian tissue and peripheral blood as matched normals, respectively. In the cohort 27, 10, 20, and 9 patients had BRCA1 germ line mutations, BRCA1 somatic mutations, BRCA2 germ line mutations, and BRCA2 somatic mutations, respectively. Of them the samples, TCGA-13-1512 had germ line mutations in both BRCA1 and BRCA2, while TCGA-23-1026 had somatic mutation in BRCA1 and germ line mutation in BRCA2. These patients were of age 26–89 years, with a median of 58 years.
Detection of somatic copy number status
The arrays used in the TCGA initiative had kb-level resolution, and thus smaller amplifications and deletions were not detected. We determined somatic genomic alterations in apparently normal human tissues as follows: (i) a genomic region was flagged to have somatic amplification in a normal sample if this region had log2 signal-to-noise ratio >0.2 in the normal tissue, and log2 signal-to-noise ratio <0.1 (copy neutral or deletion) in tumor tissue, supported by both the arrays. (ii) Conversely, a genomic region was flagged to have somatic deletion in a normal sample if it had log2 signal-to-noise ratio < -0.2 in the normal tissue, but log2 signal-to-noise ratio > -0.1 (copy neutral or amplification) in tumor tissue, again supported by both the arrays. Sex chromosomes were not analyzed. Moreover, these arrays typically do not cover the centromere regions and the tips of the telomeres, so we could not assess those regions for copy number status. Therefore, our estimation probably presents very conservative estimates of the number of somatic genomic alterations in these samples.
As detailed in the Supplementary Module 1, we performed extensive quality control steps, (i) excluding the tumor-normal pairs for which there were poor agreements in copy number calls between the pairs of aCGH arrays, (ii) excluding those genomic alterations that overlap with the common CNVs present in the human population, and (iii) excluding outlier samples (e.g. TCGA-13-0797) that had an excess of genomic alteration calls in somatic tissue. The excess of somatic amplification or deletion calls in these samples could be genuine – indicating extensive DNA damage and/or defects in DNA repair; alternately their copy number calls could be affected by unique patterns of amplifications and deletions in the cancer genome, or technical problems associated with the copy number calls in those samples. We were unable to differentiate between these possibilities, and thus chose to exclude these outlier samples. Anyhow, exclusion of these samples minimized the concern that individual outliers could bias our overall results. We applied additional filters to ensure that these events were not due to compensatory genomic alterations in the tumor genome (Supplementary Module 1). Furthermore, tumor purity had only minor effects on our analysis (Supplementary Module 1). Our filtered dataset had 607 potential somatic amplifications and deletions in 314 normal peripheral blood samples, and 494 somatic amplifications and deletions in 109 normal ovarian tissue samples.
Mutation rate estimation
We estimated the rate of somatic amplification and deletion in the blood using two models. In the first model, which follows a discrete-time pure birth stochastic process (Galton-Watson process with zero death rate), the mutation rate per locus per symmetric division r is roughly estimated as: , where N and L are the frequency and median length of the detectable somatic copy number alterations per blood sample, and α is the fraction of cells where the genomic alterations were detected.
In the second model, we consider the possibility of cellular death and relax the assumption of simultaneous generations. In this model, which uses a continuous-time binary branching process, the mutation rate per locus per symmetric division r is roughly estimated as: , where b and d are the birth and death rate of the hematopoietic stem cell. Both models were basic, in a sense that we did not consider tissue composition, challenges while detecting somatic copy number status using arrays, and complex developmental trajectories of various cell-types, which are often poorly understood. Please see Supplementary Module 2 for details of the calculation and the underlying assumptions.
Genomic context analysis
We also obtained the genomic co-ordinates of genes and other genomic features based on the human reference genome version hg18 from the UCSC Genome Browser (Meyer et al., 2013). In particular, we analyzed data for protein coding genes (Flicek et al., 2014), evolutionarily conserved elements (UCSC Genome Browser, 28_way_conservaton track (Miller et al., 2007)), repeat elements (Meyer et al., 2013), potential G-quadruplex (G4) forming motifs (Huppert and Balasubramanian, 2005), early and late DNA replication patterns conserved across tissue types (Hansen et al., 2010), common and early replicating fragile sites (Barlow et al., 2013; Durkin and Glover, 2007); these features were previously shown as being associated with local mutation patterns in the human genome (Pang et al., 2013; Podlaha et al., 2012; Yang et al., 2013). Some genomic regions replicate early (or late) in all human cell-types, while other genomic regions show variable replication timing across different cell types (Hansen et al., 2010). We have only analyzed genomic regions whose replication timing patterns were deemed cell-type invariant (Hansen et al., 2010), but we can’t rule out the possibilities that some of those regions might have different replication timing in certain blood or ovarian cell type. We obtained the UCSC data using CruzDB (Pedersen et al., 2013b), and calculated the extent of overlap between these genomic features and pSCNAnorm, and their likely statistical significance using different scripts in Bedtools (Quinlan and Hall, 2010). For each normal sample, we first calculated the extent of overlap using IntersectBed after masking selected regions: 1Mb centering centromeres, 500kb from the tip of the telomeres, and also the genomic regions that underwent copy number changes in its matched tumor genome (and thus was not assessed for copy number status in the paired normal sample). We then permuted the pSCNAnorm within respective chromosomes using ShuffleBed, while keeping the location and higher order organization of genomic features unchanged, and after masking the same selected regions in each sample. We repeated the permutation for 103 times, counting the number (n) of simulated overlap greater than the observed one, after aggregating the results over the dataset, and converted that to q- value (q-value = n/103).
While comparing the landscape of genomic alterations in tumor and normal genomes, we divided the human reference genome into 1Mb non-overlapping blocks, and counted the number of amplification and deletion events in each block. We also collected the pan-cancer GISTIC peaks identified based on nearly 3000 samples from 26 cancer types (Beroukhim et al., 2010). We plotted a scaled version of the −log(p-value) of significance of these GISTIC peaks as a proxy for the abundance of amplifications and deletions in cancer genomes.
Cancer gene mutation analysis
We obtained data on somatic point mutations in protein coding genes for the ovarian cancer samples from the TCGA (TCGA, 2011). The variants were identified using Illumina GaII and ABI SOLiD sequencing, and then comparing tumor and matched normal samples as a part of the TCGA initiative. We analyzed the potentially functional mutations i.e. missense, non-sense, frameshift, and splice-site mutations that occurred in the set of 121 classic cancer genes (definition: the COSMIC database). A vast majority of these were missense mutations.
Survival analysis
We obtained survival data (days_to_death) for these samples from the TCGA (TCGA, 2011). Survival analysis was performed using Kaplan Meier plot and log-rank test. The samples for which survival data was not available were censored.
Supplementary Material
Acknowledgments
The authors thank Rich White, Brent Pedersen, Madan Babu, Vinod Yadav and the anonymous reviewers for providing helpful comments on the manuscript. SD gratefully acknowledges support from the American Cancer Society (ACS IRG 57-001-53), Lung Cancer Colorado Fund, and United Against Lung Cancer grant (sponsored by Elliot’s Legacy and Joan’s Legacy). JD gratefully acknowledges support from the NIH (R01CA180175).
References
- Abyzov A, Mariani J, Palejev D, Zhang Y, Haney MS, Tomasini L, Ferrandino AF, Rosenberg Belmaker LA, Szekely A, Wilson M, et al. Somatic copy number mosaicism in human skin revealed by induced pluripotent stem cells. Nature. 2012;492:438–442. doi: 10.1038/nature11629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyama K, Nagata M, Oshima K, Matsuda T, Aoki N. Molecular cloning and characterization of a novel dual specificity phosphatase, LMW-DSP2, that lacks the cdc25 homology domain. The Journal of biological chemistry. 2001;276:27575–27583. doi: 10.1074/jbc.M100408200. [DOI] [PubMed] [Google Scholar]
- Araten DJ, Golde DW, Zhang RH, Thaler HT, Gargiulo L, Notaro R, Luzzatto L. A quantitative measurement of the human somatic mutation rate. Cancer research. 2005;65:8111–8117. doi: 10.1158/0008-5472.CAN-04-1198. [DOI] [PubMed] [Google Scholar]
- Bannert N, Vollhardt K, Asomuddinov B, Haag M, Konig H, Norley S, Kurth R. PDZ Domain-mediated interaction of interleukin-16 precursor proteins with myosin phosphatase targeting subunits. The Journal of biological chemistry. 2003;278:42190–42199. doi: 10.1074/jbc.M306669200. [DOI] [PubMed] [Google Scholar]
- Barlow JH, Faryabi RB, Callen E, Wong N, Malhowski A, Chen HT, Gutierrez-Cruz G, Sun HW, McKinnon P, Wright G, et al. Identification of early replicating fragile sites that contribute to genome instability. Cell. 2013;152:620–632. doi: 10.1016/j.cell.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biesecker LG, Spinner NB. A genomic view of mosaicism and human disease. Nature reviews Genetics. 2013;14:307–320. doi: 10.1038/nrg3424. [DOI] [PubMed] [Google Scholar]
- Bonnefond A, Skrobek B, Lobbens S, Eury E, Thuillier D, Cauchi S, Lantieri O, Balkau B, Riboli E, Marre M, et al. Association between large detectable clonal mosaicism and type 2 diabetes with vascular complications. Nature genetics. 2013 doi: 10.1038/ng.2700. [DOI] [PubMed] [Google Scholar]
- Campbell CD, Eichler EE. Properties and rates of germline mutations in humans. Trends in genetics : TIG. 2013 doi: 10.1016/j.tig.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente MJ, Wlodarski MW, Makishima H, Viny AD, Bretschneider I, Shaik M, Bejanyan N, Lichtin AE, Hsi ED, Paquette RL, et al. Clonal drift demonstrates unexpected dynamics of the T-cell repertoire in T-large granular lymphocyte leukemia. Blood. 2011;118:4384–4393. doi: 10.1182/blood-2011-02-338517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins FS, Barker AD. Mapping the cancer genome. Pinpointing the genes involved in cancer will help chart a new course across the complex landscape of human malignancies. Scientific American. 2007;296:50–57. [PubMed] [Google Scholar]
- De S. Somatic mosaicism in healthy human tissues. Trends in genetics : TIG. 2011;27:217–223. doi: 10.1016/j.tig.2011.03.002. [DOI] [PubMed] [Google Scholar]
- De S, Michor F. DNA secondary structures and epigenetic determinants of cancer genome evolution. Nature structural & molecular biology. 2011;18:950–955. doi: 10.1038/nsmb.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De S, Pedersen BS, Kechris K. The dilemma of choosing the ideal permutation strategy while estimating statistical significance of genome-wide enrichment. Briefings in bioinformatics. 2013 doi: 10.1093/bib/bbt053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGregori J. Evolved tumor suppression: why are we so good at not getting cancer? Cancer research. 2011;71:3739–3744. doi: 10.1158/0008-5472.CAN-11-0342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGregori J. Challenging the axiom: does the occurrence of oncogenic mutations truly limit cancer development with age? Oncogene. 2013;32:1869–1875. doi: 10.1038/onc.2012.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewannieux M, Esnault C, Heidmann T. LINE-mediated retrotransposition of marked Alu sequences. Nature genetics. 2003;35:41–48. doi: 10.1038/ng1223. [DOI] [PubMed] [Google Scholar]
- Durkin SG, Glover TW. Chromosome fragile sites. Annual review of genetics. 2007;41:169–192. doi: 10.1146/annurev.genet.41.042007.165900. [DOI] [PubMed] [Google Scholar]
- Elson JL, Samuels DC, Turnbull DM, Chinnery PF. Random intracellular drift explains the clonal expansion of mitochondrial DNA mutations with age. American journal of human genetics. 2001;68:802–806. doi: 10.1086/318801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, et al. Ensembl 2014. Nucleic acids research. 2014;42:D749–755. doi: 10.1093/nar/gkt1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedenson B. The BRCA1/2 pathway prevents hematologic cancers in addition to breast and ovarian cancers. BMC cancer. 2007;7:152. doi: 10.1186/1471-2407-7-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hansen RS, Thomas S, Sandstrom R, Canfield TK, Thurman RE, Weaver M, Dorschner MO, Gartler SM, Stamatoyannopoulos JA. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:139–144. doi: 10.1073/pnas.0912402107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AE, Orr N, Esfandiary H, Diaz-Torres M, Goodship T, Chakravarthy U. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nature genetics. 2006;38:1173–1177. doi: 10.1038/ng1890. [DOI] [PubMed] [Google Scholar]
- Huppert JL, Balasubramanian S. Prevalence of quadruplexes in the human genome. Nucleic acids research. 2005;33:2908–2916. doi: 10.1093/nar/gki609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs KB, Yeager M, Zhou W, Wacholder S, Wang Z, Rodriguez-Santiago B, Hutchinson A, Deng X, Liu C, Horner MJ, et al. Detectable clonal mosaicism and its relationship to aging and cancer. Nature genetics. 2012;44:651–658. doi: 10.1038/ng.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanchi KL, Johnson KJ, Lu C, McLellan MD, Leiserson MD, Wendl MC, Zhang Q, Koboldt DC, Xie M, Kandoth C, et al. Integrated analysis of germline and somatic variants in ovarian cancer. Nature communications. 2014;5:3156. doi: 10.1038/ncomms4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano H, Godoy I, Courtney C, Vetter MR, Gerton GL, Ostertag EM, Kazazian HH., Jr L1 retrotransposition occurs mainly in embryogenesis and creates somatic mosaicism. Genes & development. 2009;23:1303–1312. doi: 10.1101/gad.1803909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi H, Mohseni M, Tamaki A, Garay JP, Croessmann S, Karnan S, Ota A, Wong HY, Konishi Y, Karakas B, et al. Mutation of a single allele of the cancer susceptibility gene BRCA1 leads to genomic instability in human breast epithelial cells. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:17773–17778. doi: 10.1073/pnas.1110969108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruisselbrink E, Guryev V, Brouwer K, Pontier DB, Cuppen E, Tijsterman M. Mutagenic capacity of endogenous G4 DNA underlies genome instability in FANCJ-defective C. elegans. Current biology : CB. 2008;18:900–905. doi: 10.1016/j.cub.2008.05.013. [DOI] [PubMed] [Google Scholar]
- Lam EY, Beraldi D, Tannahill D, Balasubramanian S. G-quadruplex structures are stable and detectable in human genomic DNA. Nature communications. 2013;4:1796. doi: 10.1038/ncomms2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurie CC, Laurie CA, Rice K, Doheny KF, Zelnick LR, McHugh CP, Ling H, Hetrick KN, Pugh EW, Amos C, et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nature genetics. 2012;44:642–650. doi: 10.1038/ng.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR. Genomic rearrangements and sporadic disease. Nature genetics. 2007;39:S43–47. doi: 10.1038/ng2084. [DOI] [PubMed] [Google Scholar]
- Macia A, Munoz-Lopez M, Cortes JL, Hastings RK, Morell S, Lucena-Aguilar G, Marchal JA, Badge RM, Garcia-Perez JL. Epigenetic control of retrotransposon expression in human embryonic stem cells. Molecular and cellular biology. 2011;31:300–316. doi: 10.1128/MCB.00561-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maizels N, Gray LT. The G4 genome. PLoS genetics. 2013;9:e1003468. doi: 10.1371/journal.pgen.1003468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslov AY, Ganapathi S, Westerhof M, Quispe-Tintaya W, White RR, Van Houten B, Reiling E, Dolle ME, van Steeg H, Hasty P, et al. DNA damage in normally and prematurely aged mice. Aging cell. 2013;12:467–477. doi: 10.1111/acel.12071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo M, Kaji K, Utakoji T, Hosoda K. Ploidy of human embryonic fibroblasts during in vitro aging. Journal of gerontology. 1982;37:33–37. doi: 10.1093/geronj/37.1.33. [DOI] [PubMed] [Google Scholar]
- Meyer LR, Zweig AS, Hinrichs AS, Karolchik D, Kuhn RM, Wong M, Sloan CA, Rosenbloom KR, Roe G, Rhead B, et al. The UCSC Genome Browser database: extensions and updates 2013. Nucleic acids research. 2013;41:D64–69. doi: 10.1093/nar/gks1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller W, Rosenbloom K, Hardison RC, Hou M, Taylor J, Raney B, Burhans R, King DC, Baertsch R, Blankenberg D, et al. 28-way vertebrate alignment and conservation track in the UCSC Genome Browser. Genome research. 2007;17:1797–1808. doi: 10.1101/gr.6761107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran A, O’Hara C, Khan S, Shack L, Woodward E, Maher ER, Lalloo F, Evans DG. Risk of cancer other than breast or ovarian in individuals with BRCA1 and BRCA2 mutations. Familial cancer. 2012;11:235–242. doi: 10.1007/s10689-011-9506-2. [DOI] [PubMed] [Google Scholar]
- Pang AW, Migita O, Macdonald JR, Feuk L, Scherer SW. Mechanisms of formation of structural variation in a fully sequenced human genome. Human mutation. 2013;34:345–354. doi: 10.1002/humu.22240. [DOI] [PubMed] [Google Scholar]
- Pedersen BS, De S. Loss of heterozygosity preferentially occurs in early replicating regions in cancer genomes. Nucleic acids research. 2013;41:7615–7624. doi: 10.1093/nar/gkt552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen BS, Konstantinopoulos PA, Spillman MA, De S. Copy neutral loss of heterozygosity is more frequent in older ovarian cancer patients. Genes, chromosomes & cancer. 2013a;52:794–801. doi: 10.1002/gcc.22075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen BS, Yang IV, De S. CruzDB: software for annotation of genomic intervals with UCSC genome-browser database. Bioinformatics. 2013b;29:3003–3006. doi: 10.1093/bioinformatics/btt534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham J, Shaw C, Pursley A, Hixson P, Sampath S, Roney E, Gambin T, Kang SH, Bi W, Lalani S, et al. Somatic mosaicism detected by exon-targeted, high-resolution aCGH in 10 362 consecutive cases. European journal of human genetics : EJHG. 2014 doi: 10.1038/ejhg.2013.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podlaha O, Riester M, De S, Michor F. Evolution of the cancer genome. Trends in genetics : TIG. 2012;28:155–163. doi: 10.1016/j.tig.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poduri A, Evrony GD, Cai X, Walsh CA. Somatic mutation, genomic variation, and neurological disease. Science. 2013;341:1237758. doi: 10.1126/science.1237758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi J, Zhang X, Zhang HK, Yang HM, Zhou YB, Han ZG. ZBTB34, a novel human BTB/POZ zinc finger protein, is a potential transcriptional repressor. Molecular and cellular biochemistry. 2006;290:159–167. doi: 10.1007/s11010-006-9183-x. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarsounas M, Tijsterman M. Genomes and G-quadruplexes: for better or for worse. Journal of molecular biology. 2013;425:4782–4789. doi: 10.1016/j.jmb.2013.09.026. [DOI] [PubMed] [Google Scholar]
- TCGA. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TCGA. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasetti C, Vogelstein B, Parmigiani G. Half or more of the somatic mutations in cancers of self-renewing tissues originate prior to tumor initiation. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:1999–2004. doi: 10.1073/pnas.1221068110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Hurk JA, Meij IC, Seleme MC, Kano H, Nikopoulos K, Hoefsloot LH, Sistermans EA, de Wijs IJ, Mukhopadhyay A, Plomp AS, et al. L1 retrotransposition can occur early in human embryonic development. Human molecular genetics. 2007;16:1587–1592. doi: 10.1093/hmg/ddm108. [DOI] [PubMed] [Google Scholar]
- Voet T, Kumar P, Van Loo P, Cooke SL, Marshall J, Lin ML, Zamani Esteki M, Van der Aa N, Mateiu L, McBride DJ, et al. Single-cell paired-end genome sequencing reveals structural variation per cell cycle. Nucleic acids research. 2013;41:6119–6138. doi: 10.1093/nar/gkt345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Luquette LJ, Gehlenborg N, Xi R, Haseley PS, Hsieh CH, Zhang C, Ren X, Protopopov A, Chin L, et al. Diverse mechanisms of somatic structural variations in human cancer genomes. Cell. 2013;153:919–929. doi: 10.1016/j.cell.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youssoufian H, Pyeritz RE. Mechanisms and consequences of somatic mosaicism in humans. Nature reviews Genetics. 2002;3:748–758. doi: 10.1038/nrg906. [DOI] [PubMed] [Google Scholar]
- Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhang CZ, Wala J, Mermel CH, et al. Pan-cancer patterns of somatic copy number alteration. Nature genetics. 2013;45:1134–1140. doi: 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipfel PF, Edey M, Heinen S, Jozsi M, Richter H, Misselwitz J, Hoppe B, Routledge D, Strain L, Hughes AE, et al. Deletion of complement factor H-related genes CFHR1 and CFHR3 is associated with atypical hemolytic uremic syndrome. PLoS genetics. 2007;3:e41. doi: 10.1371/journal.pgen.0030041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.